

Abstract
Prolonged skeletal muscle inactivity causes muscle fibre atrophy. Redox imbalance has been considered one of the major triggers of skeletal muscle disuse atrophy, but whether redox imbalance is actually the major cause or simply a consequence of muscle disuse remains of debate. Here we hypothesized that a metabolic stress mediated by PGC-1α down-regulation plays a major role in disuse atrophy. First we studied the adaptations of soleus to mice hindlimb unloading (HU) in the early phase of disuse (3 and 7 days of HU) with and without antioxidant treatment (trolox). HU caused a reduction in cross-sectional area, redox status alteration (NRF2, SOD1 and catalase up-regulation), and induction of the ubiquitin proteasome system (MuRF-1 and atrogin-1 mRNA up-regulation) and autophagy (Beclin1 and p62 mRNA up-regulation). Trolox completely prevented the induction of NRF2, SOD1 and catalase mRNAs, but not atrophy or induction of catabolic systems in unloaded muscles, suggesting that oxidative stress is not a major cause of disuse atrophy. HU mice showed a marked alteration of oxidative metabolism. PGC-1α and mitochondrial complexes were down-regulated and DRP1 was up-regulated. To define the link between mitochondrial dysfunction and disuse muscle atrophy we unloaded mice overexpressing PGC-1α. Transgenic PGC-1α animals did not show metabolic alteration during unloading, preserving muscle size through the reduction of autophagy and proteasome degradation. Our results indicate that mitochondrial dysfunction plays a major role in disuse atrophy and that compounds inducing PGC-1α expression could be useful to treat/prevent muscle atrophy.
Key points
Oxidative stress is widely considered a major cause of muscle loss not only in disuse but also in most chronic diseases, triggering carbonylation of proteins and activation of catabolic pathways involved in their degradation.
Here we show that administration of an antioxidant prevents redox imbalance, but does not prevent activation of catabolic pathways and muscle atrophy.
We indicate that alterations of oxidative metabolism, occurring in slow soleus muscle, are not just a consequence of disuse, but a major cause of activation of catabolic pathways and loss of mass.
This conclusion is confirmed by the observation that muscle-specific overexpression of PGC-1α, a master regulator of mitochondrial biogenesis, prevents activation of catabolic systems and disuse muscle atrophy.
These findings contribute to a better mechanistic understanding of disuse muscle loss.
Introduction
The maintenance of muscle mass is critical for health and quality of life. It is widely recognized that loss of muscle mass can impair the ability to perform daily tasks, increase the incidence of injuries, prolong the period of rehabilitation and be a major risk factor of chronic diseases. Prolonged skeletal muscle inactivity results in the loss of muscle protein, fibre atrophy and impaired muscle function. Understanding of the signalling pathways responsible for the imbalance between protein synthesis and degradation leading to disuse muscle atrophy is an important step toward the development of therapeutic strategies to delay or prevent skeletal muscle atrophy.
Prolonged periods of contractile inactivity lead to increased production of reactive oxygen species (ROS) in muscle fibres, suggesting that oxidative stress could be a major trigger of disuse muscle atrophy (Powers et al. 2005, 2010; Moylan & Reid, 2007). However, whether ROS play a causal role in disuse atrophy of limb muscles is still debated (Brocca et al. 2010; Pellegrino et al. 2011; Powers et al. 2012). Recent findings challenge the role of oxidative stress in disuse atrophy (Desaphy et al. 2010; Glover et al. 2010; Kuwahara et al. 2010). The ineffectiveness of antioxidant treatments to prevent muscle atrophy (Ikemoto et al. 2002; Koesterer et al. 2002; Servais et al. 2007), including our recent observation that antioxidant trolox prevents alterations in antioxidant defence systems and protein carbonylation, but fails to prevent soleus atrophy (Brocca et al. 2010), casts further doubts on the causative role of oxidative stress.
Interestingly, alterations in mitochondrial function characterize different conditions of muscle wasting such as ageing (Mishra & Misra, 2003), diabetes (Patti et al. 2003; Mootha et al. 2004), chronic obstructive pulmonary disease (Balasubramanian & Varkey, 2006) and muscular dystrophies (Bernardi & Bonaldo, 2008). Mitochondria are highly dynamic organelles that change in shape and function as a consequence of physical activity. The mitochondrial pro-fusion or fission shaping proteins and autophagy–lysosome system regulate mitochondrial size and network. Emerging evidence suggests that mitochondrial dynamics related to muscle atrophy play a critical role in the regulation of signalling pathways controlling muscle mass. Recently it has been shown that an imbalance of mitochondrial dynamics, i.e. an overexpression of fission factors, contributes to the development of muscle atrophy induced by denervation, fasting and FoxO3 overexpression (Romanello et al. 2010).
A key player controlling mitochondrial shape and content is peroxisome proliferative activated receptor-γ coactivator 1α (PGC-1α), a master regulator of mitochondrial biogenesis (Lin et al. 2002). PGC-1α is a transcriptional coactivator affected by muscle activity and energy stress (Brault et al. 2010). An early decrease of PGC-1α in muscle has been found in denervation-induced atrophy (Sandri et al. 2006) and in several catabolic states such as diabetes (Patti et al. 2003; Roberts-Wilson et al. 2010) and ageing (Anderson & Prolla, 2009). Forced expression of PGC-1α in cultured mammalian cells, in muscles of transgenic mice and in transfected adult muscle fibres inhibits muscle atrophy induced by denervation (Sandri et al. 2006; Brault et al. 2010), and inhibits amyotrophic lateral sclerosis (Da Cruz et al. 2012) by directly interfering with a FoxO3-dependent pathway.
We formulated the hypothesis that (i) metabolic stress mediated by PGC-1α down-regulation plays a major role in disuse atrophy and (ii) oxidative stress is not a major cause of muscle atrophy. To test this hypothesis we studied two different time points (3 and 7 days) in the early phases of disuse muscle atrophy in soleus (antigravitary muscle, preferentially affected by unloading) hindlimb unloaded mice with and without antioxidant treatment. In an attempt to better define the link between mitochondrial dysfunction and muscle atrophy we unloaded muscle-specific PGC-1α transgenic mice. Our data show that mitochondrial dysfunction induced by hindlimb unloading (HU) plays a critical role in the onset of muscle atrophy activating catabolic systems. Elevated PGC-1α levels in muscle significantly mitigate soleus atrophy induced by HU, prompting an attractive therapeutic strategy for maintaining muscle mass during disuse.
Methods
Ethical approval
Experiments were approved by the Italian Health Department and complied with the Italian guidelines for the use of laboratory animals, and conform to the principles of UK regulations, as described by Drummond (2009).
Animal care and hindlimb unloading
Six-month-old male C57BL/6 mice (Charles River Laboratories, Wilmington, MA, USA) and transgenic mice overexpressing PGC-1α (TgPGC-1α) in skeletal muscle, previously described by Lin et al. (2002), were used. Mice were unloaded for 3 and 7 days as previously described (Brocca et al. 2010). Briefly, animals were suspended individually in special cages by thin string tied at one end to the tail and at the other end to the top of the cage; the length of the string was adjusted to allow the animals to move freely on their forelimbs, while the body was inclined at 30–40 deg from the horizontal plane. All mice had access to water and food ad libitum. The ground C57BL/6 and TgPGC-1α mice were maintained free in single cages and killed after 3 or 7 days. A hindlimb-suspended group was treated with an antioxidant, trolox (HU-3-Tr). These mice received daily an intraperitoneal injection of 0.25 ml of a 1 m NaHCO3 solution containing 5 g l−1 trolox, corresponding to ∼45 mg kg−1 day−1 for 10 days (Brocca et al. 2010), commencing 1 week before HU and continuing for the 3 days of HU. Furthermore, a placebo treated mice group (HU-3-Pl) received an intraperitoneal injection of 1 m NaHCO3 solution with a timeline identical to HU-3-Tr mice. For each experimental group six animals were used.
The animals of all experimental groups were killed at 10.00 h after 2 h without food with cervical dislocation to allow removal of soleus muscle. Muscles were immediately frozen in liquid nitrogen and stored at −80°C. Six muscle samples were used for each analysis.
Cross-sectional area (CSA) analysis
CSA of individual muscle fibres was determined in the mid-belly region of soleus muscle as previously described (Brocca et al. 2010). Briefly, muscle serial transverse sections (10 μm thick) were immunostained (see list of antibodies below). Images of the stained sections were captured from a light microscope (Leica DMLS) and transferred to a personal computer using a video camera (Leica DFC 280). Fibre CSAs were measured with Image J analysis software (NIH, Bethesda, MD, USA) and expressed in μm2.
Detection of superoxide by dihydroethidium (DHE) fluorescence staining
DHE is a commonly used indicator of ROS production both in vitro (Benov et al. 1998) and in vivo (Robinson et al. 2006). It is oxidized by superoxide, forming ethidium bromide, which fluoresces red when intercalated with DNA (Benov et al. 1998). Muscle transverse sections (10 μm thick) were collected and incubated with DHE (5 μm) in a light-protected place at 37°C for 30 min. After rinsing with PBS, fluorescence was assessed using fluorescence microscopy.
Hydrogen peroxide quantification
Frozen muscle samples were pulverized in a steel mortar with liquid nitrogen to obtain a powder that was immediately re-suspended in the assay buffer provided with the kit (Abcam, Cambridge, MA, USA). After protein quantification, samples were cleared of all proteins. A standard curve with known H2O2 concentration was generated following the supplied protocol. Samples and H2O2 standards were incubated at room temperature for 10 min in HRP and OxiRed Probe that reacted with the H2O2 to produce red fluorescence (Ex/Em = 535/587 nm), which was measured with a micro-plate reader (Tecan Infinite 200 Pro) (Li et al. 2012).
Analysis of myosin heavy chain (MHC) isoform content
The MHC isoform content was determined using an electrophoretic approach previously described in detail (Brocca et al. 2010). Briefly, about 6 μg of each muscle sample were dissolved in lysis buffer (20 mm Tris-HCl, 1% Triton X-100, 10% glycerol, 150 mm NaCl, 5 mm EDTA, 100 mm NaF, 2 mm NaPPi, 1× inhibitor protease phosphatase (Protease Inhibitor Cocktail, Sigma-Aldrich, St Louis, MO, USA) and 1 mm phenylmethylsulfonyl fluoride (PMSF)). The lysates were loaded onto 8% polyacrylamide SDS-PAGE gels. Electrophoresis was run for 2 h at 200 V and then for 24 h at 250 V; the gels were stained with Coomassie Blue. Four bands could be separated in the region of MHC isoforms. Densitometric analysis of MHC bands was performed to assess the relative proportion of the four MHC isoforms, MHC-I, MHC-IIA, MHC-IIX and MHC-IIB, in each sample (Pellegrino et al. 2003)
Western blot analysis
Frozen muscle samples were pulverized and immediately re-suspended in a lysis buffer (20 mm Tris-HCl, 1% Triton X-100, 10% glycerol, 150 mm NaCl, 5 mm EDTA, 100 mm NaF and 2 mm NaPPi supplemented with 1× protease, phosphatase inhibitors (Sigma-Aldrich) and 1 mm PMSF). The lysate was left for 20 min in ice and the homogenate obtained was centrifuged at 18000 g for 20 min at 4°C. Protein concentration was determined on the supernatant using the RC DCTM protein assay kit (BioRad, Hercules, CA, USA). The supernatant was stored at –80°C until ready to use.
Equal amounts of muscle samples were loaded on gradient precast gels purchase from BioRad (AnyKd). Proteins were electro-transferred to polyvinylidene fluoride (PVDF) membranes at 35 mA overnight. The membranes were probed with specific primary antibodies (see below). Thereafter, the membranes were incubated in HRP-conjugated secondary antibody. The protein bands were visualized by an enhanced chemiluminescence method. The content of each protein investigated was assessed by determining the brightness–area product of the protein band as previously described (Gondin et al. 2011).
Oxyblot analysis
Muscle samples previously stored at −80°C were pulverized and homogenized at 4°C in an antioxidant buffer containing protease inhibitors, 25 mm imidazole and 5 mm EDTA, pH 7.2 adjusted with NaOH as previously described in detail (Brocca et al. 2010). The lysate was left for 20 min in ice and the homogenate obtained was centrifuged at 18000 g for 20 min at 4°C. Protein concentration was determined on the supernatant using the RC DCTM protein assay kit (BioRad). The supernatant was stored at −80°C until ready to use.
The protein carbonylation level was detected using the OxyBlot Kit (AbNova, Taipei City, Taiwan), which provides reagents for sensitive immunodetection of these carbonyl groups. The carbonyl groups in the protein side chains are derivatized to 2,4-dinitrophenylhydrazone (DNP hydrazone) by reaction with 2,4-dinitrophenylhydrazine (DNPH); 6 μg of the DNP-derivatized protein samples were separated by PAGE (15% SDS-polyacrylamide gels) and then blotted for 2 h at 100 V to a nitrocellulose membrane. Membranes obtained were stained with Ponceau Red and then scanned. Membranes were incubated with primary antibody, specific to the DNP moiety of the proteins and subsequently with an HRP–antibody conjugate directed against the primary antibody (secondary antibody: goat anti-rabbit IgG). Blots were developed by using an enhanced chemiluminescence method. Positive bands emitting light were detected by short exposure to photographic films. Protein oxidation was quantified by defining the oxidative index (OI), i.e. the ratio between densitometric values of the oxyblot bands and those stained with Ponceau Red.
Gene expression analysis
Total RNA, from skeletal samples, was extracted using the Promega SV Total RNA isolation kit; the concentration of RNA were evaluated by using a NanoDrop instrument (Thermo Scientific, Waltham, MA, USA) and 300 ng was reverse-transcribed with SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA, USA) to obtain cDNA. The cDNA was analysed by real time-PCR (AB 7500) with the SYBR Green PCR kit (Applied Biosystems, Foster City, CA, USA) and the data were normalized to HPRT rRNA (hypoxanthine-guanine phosphoribosyl transferase).
Primers
Forward (FP) and reverse (RP) primers used for RT-PCR were: MuRF1 FP: ACCTGCTGGTGGAAAACATC, RP: CTTCGTGTTCCTTGCACATC; Atrogin-1 FP: GCAAACACTGCCACATTCTCTC, RP: CTTGAGGGGAAAGTGAGACG, p62 FP: CCCAGTGTCTTGGCATTCTT, RP: AGGGAAAGCAGAGGAAGCTC; Beclin1 FP: GCTCCTGAGGCATGGAGGGGTCT, RP: GGTTTCGCCTGGGCTGTGGTAA; NRF2 FP: TTCTTTCAGCAGCATCCTCTCCAC, RP: ACAGCCTTCAATAGTCCCGTCCA; SOD1 FP: GAGACCTGGGCAATGTGACT, RP: GTTTACTGCGCAATCCCAAT; Catalase FP: CACTGACGAGATGGCACACTTTG, RP: TGGAGAACCGAACGGCAATAGG; PGC-1α FP: ACCCCAGAGTCACCAAATGA, RP: CGAAGCCTTGAAAGGGTTATC; HPRT: Quanti Tect Primer Assay (Qiagen, Valencia, CA, USA)
Antibodies
Antibodies used were: anti-rabbit superoxide dismutase 1 (Abcam); anti-rabbit catalase (Abcam); anti-rabbit α tubulin (Sigma-Aldrich); anti-mouse OXPHOS complexes (Abcam); anti-rabbit PGC-1α (Abcam); anti-rabbit DRP1 (Cell Signaling Technology, Inc., Danvers, MA, USA); anti-rabbit p-AKT(ser473) (Cell Signaling); anti-rabbit AKT (Cell Signaling); anti-rabbit p-S6Rp(ser235/236) (Cell Signaling); anti-rabbit S6Rp (Cell Signaling); anti-rabbit p-4EBP1(thr37/46) (Cell Signaling); anti-rabbit 4EBP1 (Cell Signaling); anti-mouse myosin heavy chain 1 isoform (BA-F8); anti-mouse myosin heavy chain 2A isoform (SC-71); anti-mouse IgG (Dako North America Inc., Carpinteria, CA, USA); and anti-rabbit IgG (Cell Signaling).
Statistical analysis
Data were expressed as mean ± SEM. Statistical significance of the differences between means was assessed by one-way ANOVA followed by Student–Newman–Keuls test. A probability of less than 5% was considered significant (P < 0.05).
Results
To define the mechanism responsible for the loss of muscle mass, all experiments were performed in the early phases of disuse (3 and 7 days following HU) on soleus muscle, which is well known to go through disuse atrophy in the early phases of HU.
CSA is reduced and MHC isoform distribution does not change in the early phases of HU
To assess the impact of disuse on muscle size, CSA of skeletal muscle fibres was analysed in the mid-belly region of muscles in control and HU mice on cryosections stained with monoclonal antibodies specific for MHC-1 and MHC-2A isoforms (Fig. 1). At both 3 and 7 days of HU, myofibre CSA was significantly lower in slow fibres (17.2% at 3 days and 20% at 7 days) as well as in type 2A fast fibres (15.4% at 3 days and 14.9% at 7 days). MHC isoform composition was unchanged (Table 1).
Figure 1. CSA is reduced in soleus in the early phases of HU.
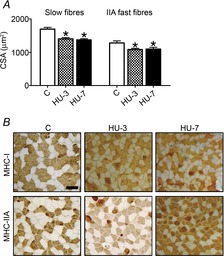
A, mean (±SEM) CSA of slow and fast fibres of soleus in control (C) and following 3 (HU-3) and 7 (HU-7) days of unloading; *significantly different from control (P < 0.05). B, representative cross-cryosections of soleus muscles using anti-MHC-I (BA-F8) and anti-MHC-IIA (SC-71) monoclonal antibodies. Scale bar: 100 μm.
Table 1.
Myosin heavy chain (MHC) isoform composition does not change in the early phases of HU
| MHC isoform | C | HU-3 | HU-7 |
|---|---|---|---|
| MHC-I | 28.27 ± 6.31 | 40.06 ± 13.34 | 32.29 ± 12.5 |
| MHC-IIA | 50.82 ± 7.12 | 46.75 ± 13.42 | 36.72 ± 14.47 |
| MHC-IIX | 7.78 ± 3.49 | 9.17 ± 7.25 | 17.82 ± 9.65 |
| MHC-IIB | 13.13 ± 7.67 | 4.02 ± 2.23 | 13.17 ± 11.12 |
MHC isoform composition of soleus muscle of control mice (C), following 3 days (HU-3) and 7 days (HU-7) of HU. MHC composition was determined by SDS-PAGE and densitometric analysis of MHC bands. Data are expressed as mean ± SD.
Redox imbalance occurs following HU
Based on evidence from the literature and from our previous findings (Brocca et al. 2010, 2012), we hypothesized that redox imbalance occurs in the HU mice model, but it does not trigger the early activation of catabolic systems. We first assessed whether oxidative stress occurred by studying: (i) NRF2 (nuclear factor, erythroid derived 2, like 2, Nfe2l2) transcription factor and antioxidant defence system adaptations, (ii) superoxide and H2O2 content and (iii) protein carbonylation.
Figure 2A shows the mRNA expression levels of NRF2, the major sensor of cell redox balance, SOD1 (superoxide dismutase1) and catalase, the major enzymes of ROS scavenging. Soleus had an increased expression of NRF2 mRNA at 3 days of HU only, SOD1 mRNA expression was significantly higher at both 3 and 7 days of HU compared to control, while catalase mRNA level was significantly up-regulated at 3 days of HU only and underwent a significant drastic reduction at 7 days of HU. The protein levels (Fig. 2B) of SOD1 were significantly higher at both 3 and 7 days of HU, reflecting the gene expression levels, while no change of catalase content at both 3 and 7 days of HU in comparison to control was found.
Figure 2. Redox imbalance occurs in soleus muscle following HU.
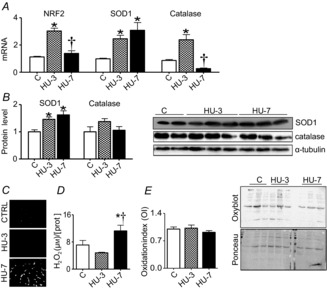
A, quantification of mRNA levels of NRF2, SOD1 and catalase by real-time PCR. B, quantification of protein levels of SOD1 and catalase by Western blot. C, superoxides accumulation on muscle cryosections stained with fluorescent dye DHE. D, H2O2 concentration. E, level of protein carbonylation in muscle by oxidation index and relative oxyblot. C, control; HU-3, 3 days of hindlimb unloading; HU-7, 7 days of hindlimb unloading. In A, B, D and E: *significantly different from C (P < 0.05); †significantly different from HU-3 (P < 0.05). Data are presented as means ± SEM.
Figure 2C shows representative muscle cryosections stained with fluorescence probe DHE to visualize accumulation of superoxides. A higher superoxide content was observed at 7 days of HU, but not at 3 days, compared to control muscle. H2O2 concentration was determined by a fluorimetric method (Fig. 2D). A significantly higher H2O2 concentration was found following 7 days of HU than in controls. Protein carbonylation was determined by Oxyblot analysis by calculating the protein OI. As shown in Fig. 2E the protein OI was not different at 3 and 7 days of HU compared to controls.
Disuse induces catabolic pathways and reduces protein synthesis
The adaptations to disuse of the two major catabolic systems, the ubiquitin proteasome and the autophagy systems, were studied. Figure 3A shows the mRNA level of MuRF1 (muscle-specific ring finger protein-1) and atrogin-1 ubiquitin ligases (ubiquitin proteasome) and of Beclin1 and p62 (autophagy system). A significant MuRF1 and atrogin-1 mRNA up-regulation was observed at 3 days of HU. At 7 days of HU, there was a lower but still significant MuRF1 up-regulation compared to control muscle, whereas atrogin-1 expression was not different from the control level. As regards autophagy, p62 mRNA relative expression was significantly higher at 3 days of HU compared to control. Beclin1 did not change at both times considered.
Figure 3. Catabolic pathways are induced and protein synthesis is reduced in soleus muscle in the early phases of HU.
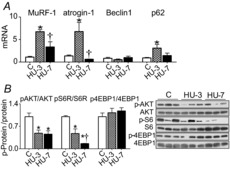
A, quantification of mRNA levels of MuRF-1 and atrogin-1 (ubiquitin proteasome system) and of Beclin1 and p62 (autophagy system) by real-time PCR. B, activity levels of AKT, S6R and 4EBP1 by Western blot analysis of the ratio between the content in the phosphorylated (p) and total forms. C, control; HU-3, 3 days of hindlimb unloading; HU-7, 7 days of hindlimb unloading. *Significantly different from C (P < 0.05); †significantly different from HU-3 (P < 0.05). Data are presented as means ± SEM.
In addition to the increase of protein degradation, muscle atrophy can result from a decrease in protein synthesis. To study the effect of disuse on the protein synthesis pathway, phosphorylation levels of key components of the PI3K-AKT pathway were analysed. Figure 3B shows representative Western blots and relative contents of phosphorylated/whole expression level of AKT kinase, 4EBP1 (eukaryotic translation initiation factor 4E-binding protein 1) and S6 ribosomal protein. A significantly lower phosphorylated AKT was found at 3 and 7 days of HU compared to control. The phosphorylated S6Rp was significantly lower at 3 days of HU in comparison to control and further decreased at 7 days of HU. The phosphorylation level of 4EBP1 did not change at any of the experimental times analysed.
Antioxidant treatment does not prevent induction of catabolic systems following HU
To clarify whether the early redox imbalance was responsible for the early activation of catabolic systems, we unloaded mice treated with the antioxidant trolox, a vitamin E analogue, for 3 days, i.e. the time point at which we found both atrophy (Fig. 1) and activation of catabolic systems (Fig. 3).
First we checked the effectiveness of the antioxidant treatment through NRF2, SOD1 and catalase expression. The data in Fig. 4A show significantly higher NRF2, SOD1 and catalase expression in HU-3 placebo mice in comparison to control placebo mice. Trolox completely prevented the induction of NRF2, SOD1 and catalase mRNAs in unloaded mice, indicating that the treatment was able to blunt oxidative stress.
Figure 4. Antioxidant treatment does not prevent catabolic systems induction and atrophy in the early phases of HU.
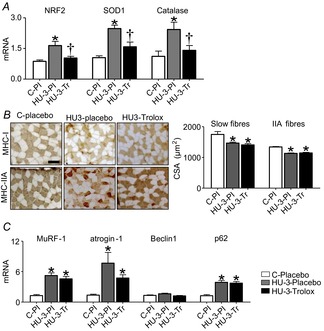
A, quantification of mRNA levels of NRF2, SOD1 and catalase by real-time PCR. B, Cross-cryosections of soleus muscles using anti-MHC-I (BA-F8) and anti-MHC-IIA (SC-71) monoclonal antibodies and relative CSA measurements. C, quantification of mRNA of MuRF-1, atrogin-1 (ubiquitin proteasome system), Beclin1 and p62 (autophagy system) by real-time PCR. C-Pl, placebo control; HU-3-Pl, placebo 3 days of hindlimb unloading; HU-3-Tr, Trolox 3 days of hindlimb unloading. *Significantly different from C-placebo (P < 0.05); †significantly different from HU-3-Pl (P < 0.05). Data are presented as means ± SEM.
CSA of individual muscle fibres was determined in soleus of control placebo, HU-3 placebo and HU-3 trolox-treated mice. Trolox did not prevent muscle fibre atrophy in slow or fast fibres (Fig. 4B). Type I and IIA fibres of unloaded trolox mice showed a degree of atrophy similar to that of placebo-treated animals (type I 19 and 16%, respectively; type IIA 14.5 and 15.5%, respectively).
To test the role of oxidative stress in triggering muscle protein breakdown in disuse atrophy, gene expression of MuRF1, atrogin-1, Beclin1 and p62 was analysed following trolox treatment. mRNA levels of MuRF1 and atrogin-1 were significantly higher both in HU-3 placebo and in HU-3 trolox in comparison to control placebo. As regards autophagy, p62 mRNA level was significantly higher in HU-3 placebo and HU-3 trolox groups than in control placebo. No difference of Beclin1 expression among the different groups was found (Fig. 4C).
Oxidative metabolism is impaired in the early phases of HU
A metabolic programme has been suggested to play a role in controlling muscle mass (Sandri et al. 2006). Metabolic adaptations occur in disuse. The latter observations prompted the hypothesis that metabolic adaptations might not just be a consequence of disuse, but could be involved in the pathogenesis of muscle atrophy (Brocca et al. 2010). Therefore, we analysed expression of (i) PGC-1α, a transcription factor involved in mitochondria homeostasis; (ii) mitochondrial complexes (OXPHOS); and (iii) DRP1 (dynamin-related protein 1), an important factor involved in mitochondrial fission machinery. PGC-1α mRNA (Fig. 5A) and protein levels (Fig. 5B) were significantly lower at 3 and 7 days of HU compared to control. As regards protein levels in mitochondrial complexes, a significantly reduced expression of Complex I, Complex III and Complex V was found at both 3 and 7 days of HU (Fig. 5D). DRP1 level was determined by Western blot and a significant up-regulation at 3 days of HU was observed (Fig. 5C).
Figure 5. Mitochondrial dysfunction is established early during HU in soleus muscle.
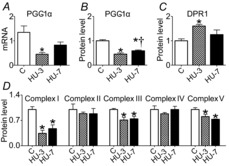
A, quantification of mRNA levels of PGC-1α by real-time PCR. B, quantification of protein levels of PGC-1α by Western blot. C, quantification of protein levels of DRP1 involved in fission machinery by Western blot. D, quantification of protein levels of mitochondrial complexes by Western blot. C, control; HU-3, 3 days of hindlimb unloading; HU-7, 7 days of hindlimb unloading. *Significantly different from control (P < 0.05); †significantly different from HU-3 (P < 0.05). Data are presented as means ± SEM.
Elevated PGC-1α levels preserve CSA in soleus unloaded muscle
We tested the hypothesis that a metabolic programme could trigger muscle atrophy by assessing whether its overexpression would lead to beneficial effects in HU. Hind-limb suspended muscle-specific PGC-1α transgenic (TgPGC-1α) and wild-type mice were studied at 3 days of HU.
As shown in Fig. 6, maintenance of PGC-1α expression in hind limb suspended soleus was sufficient to mitigate muscle atrophy. In fact, slow and fast fibres showed only 8 and 7% of atrophy, respectively.
Figure 6. Increased PGC-1α expression in muscle prevents muscle atrophy during HU.
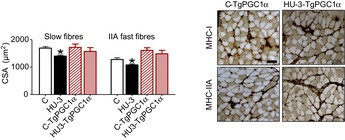
CSA of slow and fast fibres of soleus stained anti-MHC-I (BA-F8) and anti-MHC-IIA (SC-71) monoclonal antibodies and relative measurements of CSA. Scale bar: 100 μm. C, control, wild type; HU-3, 3 days of hindlimb unloading, wild type; C-TgPGC-1α, control, transgenic PGC-1α; HU3-TgPGC-1α, 3 days of hindlimb unloading, transgenic PGC-1α. *Significantly different from control (P < 0.05). Data are presented as means ± SEM.
Elevated PGC-1α levels prevent early catabolic pathways induction in soleus unloaded muscle without affecting protein synthesis
As the peak of expression of catabolic systems was reached at 3 days of HU, we monitored whether PGC-1α expression elicited any effect on atrogenes induction early in HU. As shown in Fig. 7A, high levels of PGC-1α in HU muscles significantly blunted the up-regulation of MuRF-1 and atrogin-1 genes and completely prevented p62 induction.
Figure 7. Increased PGC-1α expression in muscle prevents the activation of catabolic systems and mitigates decrease of protein synthesis.
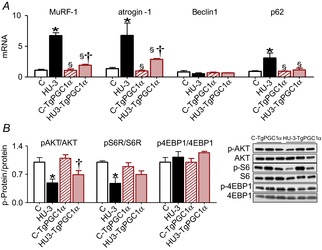
A, quantification of mRNA levels of MuRF-1 and atrogin-1 (ubiquitin proteasome system) and of Beclin1 and p62 (autophagy system) by real-time PCR. B, activity levels of AKT, S6R and 4EBP1 by Western blot analysis of the ratio between the content in the phosphorylated (p) and total forms. C, control, wild type; HU-3, 3 days of hindlimb unloading, wild type; C-TgPGC-1α, control, transgenic PGC-1α; HU3-TgPGC-1α, 3 days of hindlimb unloading, transgenic PGC-1α. *Significantly different from control (P < 0.05); §significantly different from HU-3 (P < 0.05); †significantly different from C-TgPGC-1α (P < 0.05). Data are presented as means ± SEM.
To test whether PGC-1α elicited an effect on the protein synthesis pathway we studied AKT, S6 and 4EBP1 mRNA expression. As shown in Fig. 7B, PGC-1α transgenic mice showed an attenuation of S6 dephosphorylation and no significant protection of AKT inhibition, which was significantly decreased as in unloaded wild-type mice. The phosphorylation level of 4EBP1 remained similar at both experimental time points. Therefore, PGC-1α had minor effects on pathways related to protein synthesis, as recently described (Bonaldo & Sandri, 2013).
Discussion
It is widely believed that disuse skeletal muscle atrophy is caused by an imbalance between muscle protein synthesis and muscle protein break-down. The general aim of this study was to clarify the adaptations of the intracellular signalling pathways involved in muscle protein synthesis and muscle protein break-down and to determine the underlying triggering phenomena. HU, in which the hindlimbs are suspended off the ground to remove the normal gravitational load on the muscles, is a commonly used model to study disuse muscle atrophy. The present work confirms the widely accepted view that HU is able to induce significant atrophy of the antigravitary soleus (Fitts et al. 2001; Hurst & Fitts, 2003) and extend it to early times, i.e. 3 days following HU (Fig. 1).
Redox imbalance has been considered one of the major triggers of skeletal muscle disuse atrophy (Powers et al. 2005, 2010; Moylan & Reid, 2007). The role of oxidative stress has been elegantly demonstrated in respiratory muscles following mechanical ventilation (Zergeroglu et al. 2003; Shanely et al. 2004; Falk et al. 2006). However, notwithstanding the large amount of work devoted to the issue, it is still unclear whether redox imbalance is actually the major cause of disuse atrophy of limb muscles in a model which mimics moderate decrease in neuromuscular activity (Brocca et al. 2010; Pellegrino et al. 2011; Powers et al. 2012). Here we show that redox imbalance is not a major trigger of muscle atrophy in limb muscles following HU, whereas a major role is played by a metabolic programme controlling muscle mass.
Catabolic pathways are induced and protein synthesis is decreased in soleus in the early phases of HU
The ubiquitin proteasome and the autophagy lysosome systems play a critical role during myofibre shrinkage. The activation of these pathways requires a transcriptional-dependent up-regulation of a subset of genes named atrophy-related genes or atrogenes. The role of MuRF-1 and atrogin-1 in breaking down myofibrillar proteins in disuse in small mammals is well established (Ikemoto et al. 2001). The induction of these atrogenes precedes muscle atrophy (Sandri, 2008). Consistently we found an early increase of MuRF1 and atrogin-1 mRNA (Fig. 3A). The activation was transient, fading away at 7 days of HU, in agreement with other studies on HU (Bodine et al. 2001; Haddad et al. 2006; Kline et al. 2007), cast immobilization (Krawiec et al. 2005; Caron et al. 2009), denervation (Bodine et al. 2001; Kline et al. 2007; Sacheck et al. 2007; Bertaggia et al. 2012) and several other atrophic conditions such as corticosteroid administration (Cho et al. 2010), nerve crash (Caron et al. 2009) and spinal cord isolation (Sacheck et al. 2007).
Autophagy is known to play a crucial role in the turnover of cell components both in constitutive conditions and in response to various stimuli, such as cellular stress, nutrient deprivation, amino acid starvation and cytokines (Mizushima et al. 2008). However, excessive stimulation of the autophagy machinery is documented to be deleterious and could lead to cell death (Levine & Yuan, 2005). It has been recently understood that an increase in activity of the autophagy system can play a relevant role in muscle atrophy (Sandri, 2010a,b). In other words, too much autophagy impairs myofibre homeostasis, causing excessive removal of cellular components and leading to muscle atrophy when excessive catabolic activity is sustained for long periods. We found a slight activation of autophagy in the early phase of disuse atrophy suggested by the increased expression of p62, the major protein involved in delivering ubiquitinated proteins to the autophagosome (Fig. 3A). To our knowledge there are few and conflicting data concerning autophagy activity in HU. They have all been obtained after chronic disuse, showing induction (Maki et al. 2012), suppression (Liu et al. 2012) and no modification of autophagy (Andrianjafiniony et al. 2010). Collectively, the results concerning the activation of catabolic systems show that, in the early stages of disuse, soleus atrophy is mainly supported by the ubiquitin proteasome system, whereas autophagy does not seem to be a major phenomenon.
The PI3K-AKT pathway is known to play a key role in activating muscle protein synthesis (Rommel et al. 2001; Sandri, 2008). We found a down-regulation of the anabolic PI3K-AKT pathway through the reduced phosphorylation of AKT and S6Rp (Fig. 3B) indicating that a decrease in muscle protein synthesis plays a role in both the early and the later phases of disuse. This is consistent with earlier studies showing a decrease in the PI3K-AKT pathway following acute (12 h) (Hornberger et al. 2001), short (7 days) (Hornberger et al. 2001; Dupont et al. 2011) and chronic disuse (Sugiura et al. 2005; Dupont et al. 2011) in soleus muscle. As found in other conditions (Magne et al. 2013) 4E-BP1 phosphorylation level following HU was unchanged. This was unexpected because a decrease in AKT phosphorylation is generally able to decrease phosphorylation of its downstream targets, including 4E-BP1. One possible explanation is that the very fast kinetics of phosphorylation and dephosphorylation of the different kinases of the PI3K-AKT pathway could make their concomitant activation an unlikely event.
Redox imbalance does not trigger disuse muscle atrophy
The adaptations in the expression of NRF2, SOD1 and catalase (Fig. 2) indicate that redox imbalance occurred early into HU. NRF2 is, in fact, a constitutively active transcription factor sensing redox balance and controlling genes of antioxidant defence systems (Baird & Dinkova-Kostova, 2011; Hur & Gray, 2011). The lack of accumulation of superoxides and H2O2 at 3 days of HU indicates that, in the early phase of disuse, the antioxidant defence system efficiently reacts to the initial ROS increase. A prompt response to redox imbalance in soleus is expected due to its high oxidative metabolism and therefore to its natural exposure to superoxides.
SOD1 catalyses the reduction of superoxide anions to hydrogen peroxide (H2O2) preventing superoxide accumulation and catalase converts H2O2 into H2O and O2 preventing H2O2 accumulation. The mismatch between SOD1 and catalase expression and H2O2 accumulation found at 7 days of HU indicates a clear alteration of the antioxidant defence system, providing a likely basis for protein carbonylation (Lawler et al. 2003). However, no signs of carbonylation were detected with Oxyblot (Fig. 2) probably because protein carbonylation is a massive and late phenomenon, which is unlikely to be a sensitive index of redox imbalance. Consistent with an early alteration of antioxidant defence systems and with protein carbonylation being its late consequence, protein carbonylation was found following 15 days of HU in our previous study (Brocca et al. 2010). Regardless of the lack of protein carbonylation, redox imbalance could still be responsible for the early activation of catabolic systems (Fig. 3). Therefore, the data discussed so far could still fit with the hypothesis that oxidative stress plays a major role in disuse atrophy.
However, antioxidant treatment successfully counteracted redox imbalance (Fig. 4A), but did not prevent activation of catabolic systems and muscle atrophy (Fig. 4C), indicating that activation of catabolic systems is not causally linked to redox imbalance. The observation of no impact of antioxidant treatment on muscle atrophy is in agreement with several studies (Ikemoto et al. 2002; Koesterer et al. 2002; Servais et al. 2007; Brocca et al. 2010; Desaphy et al. 2010; Glover et al. 2010). Recently, a partial prevention of muscle atrophy after inhibition of xanthine oxidase by allopurinol during hindlimb unloading was shown, suggesting a causal role of oxidative stress in determining muscle atrophy (Derbre et al. 2012). By contrast, Matuszczak et al. (2004) found no muscle atrophy mitigation after allopurinol treatment in the same disuse model.
The lack of a causal link with muscle atrophy in HU does not necessarily imply a minor role of redox imbalance in all models of disuse. The role of an alteration of redox homeostasis in disuse atrophy has been shown, in fact, to vary through muscles, models and species (Pellegrino et al. 2011). The causal role of oxidative stress has been clearly demonstrated in mechanical ventilation (Shanely et al. 2002, 2004; Zergeroglu et al. 2003; Falk et al. 2006) and immobilization in humans and rodents (Kondo et al. 1992, 1993) also by the use of a mitochondrially targeted antioxidant that prevented myofibre atrophy (Min et al. 2011). One of the possible reasons for a differential role of oxidative stress may be that mechanical ventilation, limb immobilization and HU could decrease neuromuscular activity and muscle size to different extents (Fischbach & Robbins, 1969; Froese & Bryan, 1974; Alford et al. 1987; De-Doncker et al. 2005). Moreover, redox imbalance could be responsible of other disuse-induced phenomena. To our knowledge, this is the first report in which superoxide and H2O2 have been measured at different experimental times in the early stages of disuse and correlated with the expression of the major endogenous antioxidant systems and catabolic systems.
Oxidative metabolism is impaired in soleus in the early phases of HU
Alternatively to redox imbalance, we considered metabolic alterations as potential triggers of disuse atrophy. We found an early impairment of oxidative metabolism and signs of mitochondrial dysfunction, suggesting that a metabolic programme could be responsible for disuse atrophy.
PGC-1α is one of the master regulators of mitochondrial biogenesis and oxidative metabolism (Puigserver & Spiegelman, 2003) and is known to inhibit FoxO3 (Sandri et al. 2006). The observed PGC-1α down-regulation (Fig. 5A and B) could therefore account for the reduced expression levels of the respiratory chain complexes I, III and V (Fig. 5D). The latter results are in agreement with the observation that a variety of genes related to glycolysis and oxidative phosphorylation are coordinately suppressed in atrophying muscles (Lecker et al. 2004). Furthermore, a down-regulation of metabolic enzymes (Brocca et al. 2010) and a decrease of mitochondrial oxidative capacity (Momken et al. 2011) have been shown in the later phases of muscle disuse. As the early phases of disuse did not change MHC isoform composition (Table 1), the observed adaptations in oxidative metabolism could not be dependent on a shift of muscle phenotype.
A decrease in PGC-1α mRNA expression has been found in the acute phase (Mazzatti et al. 2008) and chronic phase (Momken et al. 2011; Liu et al. 2012) of hindlimb unloading, in denervation atrophy (Sandri et al. 2006) and in several catabolic states such as diabetes (Patti et al. 2003) and ageing (Anderson & Prolla, 2009). PGC-1α down-regulation (Fig. 5) could be responsible for the activation of the catabolic systems (Fig. 3A) through FoxO3 disinhibition and the subsequent enhancement of ubiquitine ligase and autophagy gene expression, even in the absence of ROS accumulation (Fig. 2).
Moreover, it has been recently understood that expression of the mitochondrial fission machinery is sufficient to cause muscle wasting in mice, whereas inhibition of mitochondrial fission prevents muscle loss during denervation, indicating that disruption of the mitochondrial network is a crucial amplificatory loop of the muscle atrophy programme (Romanello et al. 2010; Romanello & Sandri, 2010). Therefore, the early DRP1 overexpression in soleus could play a role in disuse atrophy (Fig. 3).
Collectively, the results indicate that mitochondrial alterations could sustain muscle protein breakdown in soleus muscle disuse.
Elevated PGC-1α levels preserve CSA and prevent the early catabolic pathways induction in soleus unloaded muscle
To test the hypothesis of a causative role of a metabolic programme in disuse muscle atrophy, we subjected HU to Tg-mice overexpressing PGC-1α (TgPGC-1α). The maintained high levels of PGC-1α prevented MuRF-1 and atrogin-1 induction as well as autophagy activation, protecting muscle from proteolysis. Consistently, atrophy following 3 days of HU was mitigated in soleus (Fig. 6C). The latter findings are consistent with the observations that PGC-1α overexpression prevented muscle atrophy induced by denervation (Sandri et al. 2006) and amyotrophic lateral sclerosis (Da Cruz et al. 2012) through the inhibition of FoxO3 transcriptional activity and catabolic pathway activation.
CSA of muscle fibres of unloaded transgenic mice was lower than CSA of muscle fibres of control transgenic mice (7% vs. 16.3%), although the difference did not reach statistical significance. The lack of complete recovery of mass could depend on the protein levels of PGC-1α, which in transgenic mice were similar to and not higher than those observed in control wild-type mice, probably due to the promoter driving expression of the PGC-1α transgene, MCK, which is less expressed in slow than in fast muscles (Lin et al. 2002). Transgene PGC-1α expression may not be sufficient to prevent the complete activation of catabolic systems and consequently the reduction of protein synthesis (Bonaldo & Sandri, 2013). The latter hypothesis would explain why the administration of resveratrol in unloaded rats slightly blunts soleus mass loss, despite maintenance of PGC-1α expression at normal levels (Momken et al. 2011). It can be hypothesized that, to completely prevent disuse muscle atrophy, it is not sufficient to prevent the disuse-induced drop in PGC-1α, but it is essential to raise PGC-1α levels above basal levels. Moreover, the lack of complete recovery of atrophy in soleus of TgPGC-1α mice could also be accounted for by inhibition of the AKT/mTOR pathway and therefore of protein synthesis playing a role in disuse atrophy, consistent with several previous observations (Thomason & Booth, 1990). Interestingly, PGC-1α has been shown not to affect the AKT/mTOR pathway (Bonaldo & Sandri, 2013). Consistently, PGC-1α overexpression did not counteract the disuse-induced lower activation of such pathway (Fig. 7B) failing to counteract the loss of mass due to lower protein synthesis.
Note that mitochondrial dysfunction could not only trigger muscle atrophy but could also cause ROS production and redox imbalance. Disuse-induced redox imbalance (Fig. 2) could therefore be a secondary phenomenon to a decrease in PGC-1α. It could not play a major role at least in the early adaptations leading to muscle atrophy, but still cause relevant phenomena, i.e. protein carbonylation and altered protein function. In fact, it has been suggested that oxidative stress in the myoplasm plays a pivotal role in altering muscle function rather than in triggering muscle atrophy (Kuwahara et al. 2010).
The almost complete recovery of muscle mass in unloaded TgPGC-1α mice indicates that mitochondrial alteration plays a major role in disuse atrophy and that compounds inducing PGC-1α expression could be useful to treat or prevent muscle atrophy.
Conclusions
Oxidative stress is widely considered a major cause of muscle loss not only in disuse, but in most chronic diseases, triggering carbonylation of proteins and activation of catabolic pathways involved in their degradation. Here we show that administration of an antioxidant prevents redox imbalance, but does not prevent activation of catabolic pathways and muscle atrophy. We indicate that alterations of oxidative metabolism and mitochondrial dynamics, occurring in soleus muscle, are not just a consequence of disuse, but a major cause of activation of catabolic pathways and loss of mass. This conclusion is confirmed by the observation that muscle-specific overexpression of PGC-1α, a master regulator of mitochondrial biogenesis, prevents activation of catabolic systems and disuse muscle atrophy.
Acknowledgments
We thank Professor Francesco Zorzato who kindly donated the TgPGC-1α mice and Mr Luigi Guidotti for excellent technical help during all experiments related to muscle disuse.
Glossary
- CSA
cross-sectional area
- CuZnSOD1
superoxide dismutase1
- DHE
dihydroethidium
- DNP
dinitrophenylhydrazone
- DRPI
dynamin-related protein 1
- 4EBP1
eukaryotic translation initiation factor 4E-binding protein 1
- HU
hindlimb unloading
- MHC
myosin heavy chain
- MuRF-1
muscle-specific ring finger protein-1
- NRF2
nuclear factor erythroid derived 2 like 2 (Nfe2l2)
- OI
oxidative index
- PGC-1α
peroxisome proliferative activated receptor-γ coactivator 1α
- ROS
reactive oxygen species
Additional information
Competing interests
The authors declare no conflict of interest.
Author contributions
R.B. and M.A.P.: conception and design of the experiments; J.C. and L.B.: collection, analysis and interpretation of data; M.S., R.B. and M.A.P.: drafting the article or revising it critically for important intellectual content. All authors made comments on the manuscript and read and approved the final version. All experiments were performed at the Department of Molecular Medicine, University of Pavia, Italy.
Funding
This study was supported by Cariplo Foundation, Italy (grant no. 2010.0764), and the European Commission for the MYOAGE grant (no. 223576) funded under FP7. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- Alford EK, Roy RR, Hodgson JA, Edgerton VR. Electromyography of rat soleus, medial gastrocnemius, and tibialis anterior during hind limb suspension. Exp Neurol. 1987;96:635–649. doi: 10.1016/0014-4886(87)90225-1. [DOI] [PubMed] [Google Scholar]
- Anderson R, Prolla T. PGC-1α in aging and anti-aging interventions. Biochim Biophys Acta. 2009;1790:1059–1066. doi: 10.1016/j.bbagen.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrianjafiniony T, Dupre-Aucouturier S, Letexier D, Couchoux H, Desplanches D. Oxidative stress, apoptosis, and proteolysis in skeletal muscle repair after unloading. Am J Physiol Cell Physiol. 2010;299:C307–C315. doi: 10.1152/ajpcell.00069.2010. [DOI] [PubMed] [Google Scholar]
- Baird L, Dinkova-Kostova AT. The cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol. 2011;85:241–272. doi: 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]
- Balasubramanian VP, Varkey B. Chronic obstructive pulmonary disease: effects beyond the lungs. Curr Opin Pulm Med. 2006;12:106–112. doi: 10.1097/01.mcp.0000208449.73101.ac. [DOI] [PubMed] [Google Scholar]
- Benov L, Sztejnberg L, Fridovich I. Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic Biol Med. 1998;25:826–831. doi: 10.1016/s0891-5849(98)00163-4. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Bonaldo P. Dysfunction of mitochondria and sarcoplasmic reticulum in the pathogenesis of collagen VI muscular dystrophies. Ann N Y Acad Sci. 2008;1147:303–311. doi: 10.1196/annals.1427.009. [DOI] [PubMed] [Google Scholar]
- Bertaggia E, Coletto L, Sandri M. Posttranslational modifications control FoxO3 activity during denervation. Am J Physiol Cell Physiol. 2012;302:C587–C596. doi: 10.1152/ajpcell.00142.2011. [DOI] [PubMed] [Google Scholar]
- Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- Bonaldo P, Sandri M. Cellular and molecular mechanisms of muscle atrophy. Dis Model Mech. 2013;6:25–39. doi: 10.1242/dmm.010389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault JJ, Jespersen JG, Goldberg AL. Peroxisome proliferator-activated receptor gamma coactivator 1α or 1β overexpression inhibits muscle protein degradation, induction of ubiquitin ligases, and disuse atrophy. J Biol Chem. 2010;285:19460–19471. doi: 10.1074/jbc.M110.113092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocca L, Cannavino J, Coletto L, Biolo G, Sandri M, Bottinelli R, Pellegrino MA. The time course of the adaptations of human muscle proteome to bed rest and the underlying mechanisms. J Physiol. 2012;590:5211–5230. doi: 10.1113/jphysiol.2012.240267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocca L, Pellegrino MA, Desaphy JF, Pierno S, Camerino DC, Bottinelli R. Is oxidative stress a cause or consequence of disuse muscle atrophy in mice? A proteomic approach in hindlimb-unloaded mice. Exp Physiol. 2010;95:331–350. doi: 10.1113/expphysiol.2009.050245. [DOI] [PubMed] [Google Scholar]
- Caron AZ, Drouin G, Desrosiers J, Trensz F, Grenier G. A novel hindlimb immobilization procedure for studying skeletal muscle atrophy and recovery in mouse. J Appl Physiol. 2009;106:2049–2059. doi: 10.1152/japplphysiol.91505.2008. [DOI] [PubMed] [Google Scholar]
- Cho JE, Fournier M, Da X, Lewis MI. Time course expression of Foxo transcription factors in skeletal muscle following corticosteroid administration. J Appl Physiol. 2010;108:137–145. doi: 10.1152/japplphysiol.00704.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Cruz S, Parone PA, Lopes VS, Lillo C, McAlonis-Downes M, Lee SK, Vetto AP, Petrosyan S, Marsala M, Murphy AN, Williams DS, Spiegelman BM, Cleveland DW. Elevated PGC-1α activity sustains mitochondrial biogenesis and muscle function without extending survival in a mouse model of inherited ALS. Cell Metab. 2012;15:778–786. doi: 10.1016/j.cmet.2012.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De-Doncker L, Kasri M, Picquet F, Falempin M. Physiologically adaptive changes of the L5 afferent neurogram and of the rat soleus EMG activity during 14 days of hindlimb unloading and recovery. J Exp Biol. 2005;208:4585–4592. doi: 10.1242/jeb.01931. [DOI] [PubMed] [Google Scholar]
- Derbre F, Ferrando B, Gomez-Cabrera MC, Sanchis-Gomar F, Martinez-Bello VE, Olaso-Gonzalez G, Diaz A, Gratas-Delamarche A, Cerda M, Vina J. Inhibition of xanthine oxidase by allopurinol prevents skeletal muscle atrophy: role of p38 MAPKinase and E3 ubiquitin ligases. PLoS One. 2012;7:e46668. doi: 10.1371/journal.pone.0046668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desaphy JF, Pierno S, Liantonio A, Giannuzzi V, Digennaro C, Dinardo MM, Camerino GM, Ricciuti P, Brocca L, Pellegrino MA, Bottinelli R, Camerino DC. Antioxidant treatment of hindlimb-unloaded mouse counteracts fiber type transition but not atrophy of disused muscles. Pharmacol Res. 2010;61:553–563. doi: 10.1016/j.phrs.2010.01.012. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont E, Cieniewski-Bernard C, Bastide B, Stevens L. Electrostimulation during hindlimb unloading modulates PI3K-AKT downstream targets without preventing soleus atrophy and restores slow phenotype through ERK. Am J Physiol Regul Integr Comp Physiol. 2011;300:R408–417. doi: 10.1152/ajpregu.00793.2009. [DOI] [PubMed] [Google Scholar]
- Falk DJ, Deruisseau KC, Van Gammeren DL, Deering MA, Kavazis AN, Powers SK. Mechanical ventilation promotes redox status alterations in the diaphragm. J Appl Physiol (1985) 2006;101:1017–1024. doi: 10.1152/japplphysiol.00104.2006. [DOI] [PubMed] [Google Scholar]
- Fischbach GD, Robbins N. Changes in contractile properties of disused soleus muscles. J Physiol. 1969;201:305–320. doi: 10.1113/jphysiol.1969.sp008757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitts RH, Riley DR, Widrick JJ. Functional and structural adaptations of skeletal muscle to microgravity. J Exp Biol. 2001;204:3201–3208. doi: 10.1242/jeb.204.18.3201. [DOI] [PubMed] [Google Scholar]
- Froese AB, Bryan AC. Effects of anesthesia and paralysis on diaphragmatic mechanics in man. Anesthesiology. 1974;41:242–255. doi: 10.1097/00000542-197409000-00006. [DOI] [PubMed] [Google Scholar]
- Glover EI, Yasuda N, Tarnopolsky MA, Abadi A, Phillips SM. Little change in markers of protein breakdown and oxidative stress in humans in immobilization-induced skeletal muscle atrophy. Appl Physiol Nutr Metab. 2010;35:125–133. doi: 10.1139/H09-137. [DOI] [PubMed] [Google Scholar]
- Gondin J, Brocca L, Bellinzona E, D'Antona G, Maffiuletti NA, Miotti D, Pellegrino MA, Bottinelli R. Neuromuscular electrical stimulation training induces atypical adaptations of the human skeletal muscle phenotype: a functional and proteomic analysis. J Appl Physiol. 2011;110:433–450. doi: 10.1152/japplphysiol.00914.2010. [DOI] [PubMed] [Google Scholar]
- Haddad F, Adams GR, Bodell PW, Baldwin KM. Isometric resistance exercise fails to counteract skeletal muscle atrophy processes during the initial stages of unloading. J Appl Physiol. 2006;100:433–441. doi: 10.1152/japplphysiol.01203.2005. [DOI] [PubMed] [Google Scholar]
- Hornberger TA, Hunter RB, Kandarian SC, Esser KA. Regulation of translation factors during hindlimb unloading and denervation of skeletal muscle in rats. Am J Physiol Cell Physiol. 2001;281:C179–C187. doi: 10.1152/ajpcell.2001.281.1.C179. [DOI] [PubMed] [Google Scholar]
- Hur W, Gray NS. Small molecule modulators of antioxidant response pathway. Curr Opin Chem Biol. 2011;15:162–173. doi: 10.1016/j.cbpa.2010.12.009. [DOI] [PubMed] [Google Scholar]
- Hurst JE, Fitts RH. Hindlimb unloading-induced muscle atrophy and loss of function: protective effect of isometric exercise. J Appl Physiol. 2003;95:1405–1417. doi: 10.1152/japplphysiol.00516.2002. [DOI] [PubMed] [Google Scholar]
- Ikemoto M, Nikawa T, Takeda S, Watanabe C, Kitano T, Baldwin KM, Izumi R, Nonaka I, Towatari T, Teshima S, Rokutan K, Kishi K. Space shuttle flight (STS-90) enhances degradation of rat myosin heavy chain in association with activation of ubiquitin-proteasome pathway. FASEB J. 2001;15:1279–1281. doi: 10.1096/fj.00-0629fje. [DOI] [PubMed] [Google Scholar]
- Ikemoto M, Okamura Y, Kano M, Hirasaka K, Tanaka R, Yamamoto T, Sasa T, Ogawa T, Sairyo K, Kishi K, Nikawa T. A relative high dose of vitamin E does not attenuate unweighting-induced oxidative stress and ubiquitination in rat skeletal muscle. J Physiol Anthropol Appl Human Sci. 2002;21:257–263. doi: 10.2114/jpa.21.257. [DOI] [PubMed] [Google Scholar]
- Kline WO, Panaro FJ, Yang H, Bodine SC. Rapamycin inhibits the growth and muscle-sparing effects of clenbuterol. J Appl Physiol. 2007;102:740–747. doi: 10.1152/japplphysiol.00873.2006. [DOI] [PubMed] [Google Scholar]
- Koesterer TJ, Dodd SL, Powers S. Increased antioxidant capacity does not attenuate muscle atrophy caused by unweighting. J Appl Physiol. 2002;93:1959–1965. doi: 10.1152/japplphysiol.00511.2002. [DOI] [PubMed] [Google Scholar]
- Kondo H, Miura M, Itokawa Y. Antioxidant enzyme systems in skeletal muscle atrophied by immobilization. Pflugers Arch. 1993;422:404–406. doi: 10.1007/BF00374299. [DOI] [PubMed] [Google Scholar]
- Kondo H, Miura M, Nakagaki I, Sasaki S, Itokawa Y. Trace element movement and oxidative stress in skeletal muscle atrophied by immobilization. Am J Physiol. 1992;262:E583–E590. doi: 10.1152/ajpendo.1992.262.5.E583. [DOI] [PubMed] [Google Scholar]
- Krawiec BJ, Frost RA, Vary TC, Jefferson LS, Lang CH. Hindlimb casting decreases muscle mass in part by proteasome-dependent proteolysis but independent of protein synthesis. Am J Physiol Endocrinol Metab. 2005;289:E969–E980. doi: 10.1152/ajpendo.00126.2005. [DOI] [PubMed] [Google Scholar]
- Kuwahara H, Horie T, Ishikawa S, Tsuda C, Kawakami S, Noda Y, Kaneko T, Tahara S, Tachibana T, Okabe M, Melki J, Takano R, Toda T, Morikawa D, Nojiri H, Kurosawa H, Shirasawa T, Shimizu T. Oxidative stress in skeletal muscle causes severe disturbance of exercise activity without muscle atrophy. Free Radic Biol Med. 2010;48:1252–1262. doi: 10.1016/j.freeradbiomed.2010.02.011. [DOI] [PubMed] [Google Scholar]
- Lawler JM, Song W, Demaree SR. Hindlimb unloading increases oxidative stress and disrupts antioxidant capacity in skeletal muscle. Free Radic Biol Med. 2003;35:9–16. doi: 10.1016/s0891-5849(03)00186-2. [DOI] [PubMed] [Google Scholar]
- Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004;18:39–51. doi: 10.1096/fj.03-0610com. [DOI] [PubMed] [Google Scholar]
- Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Liu H, Zhou JS, Cao JF, Zhou XB, Choi AM, Chen ZH, Shen HH. Caveolin-1 inhibits expression of antioxidant enzymes through direct interaction with nuclear erythroid 2 p45-related factor-2 (Nrf2) J Biol Chem. 2012;287:20922–20930. doi: 10.1074/jbc.M112.352336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, Lowell BB, Bassel-Duby R, Spiegelman BM. Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- Liu J, Peng Y, Cui Z, Wu Z, Qian A, Shang P, Qu L, Li Y, Long J. Depressed mitochondrial biogenesis and dynamic remodeling in mouse tibialis anterior and gastrocnemius induced by 4-week hindlimb unloading. IUBMB Life. 2012;64:901–910. doi: 10.1002/iub.1087. [DOI] [PubMed] [Google Scholar]
- Magne H, Savary-Auzeloux I, Migne C, Peyron MA, Combaret L, Remond D, Dardevet D. Unilateral hindlimb casting induced a delayed generalized muscle atrophy during rehabilitation that is prevented by a whey or a high protein diet but not a free leucine-enriched diet. PLoS One. 2013;8:e70130. doi: 10.1371/journal.pone.0070130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki T, Yamamoto D, Nakanishi S, Iida K, Iguchi G, Takahashi Y, Kaji H, Chihara K, Okimura Y. Branched-chain amino acids reduce hindlimb suspension-induced muscle atrophy and protein levels of atrogin-1 and MuRF1 in rats. Nutr Res. 2012;32:676–683. doi: 10.1016/j.nutres.2012.07.005. [DOI] [PubMed] [Google Scholar]
- Matuszczak Y, Arbogast S, Reid MB. Allopurinol mitigates muscle contractile dysfunction caused by hindlimb unloading in mice. Aviat Space Environ Med. 2004;75:581–588. [PubMed] [Google Scholar]
- Mazzatti DJ, Smith MA, Oita RC, Lim FL, White AJ, Reid MB. Muscle unloading-induced metabolic remodeling is associated with acute alterations in PPARδ and UCP-3 expression. Physiol Genomics. 2008;34:149–161. doi: 10.1152/physiolgenomics.00281.2007. [DOI] [PubMed] [Google Scholar]
- Min K, Smuder AJ, Kwon OS, Kavazis AN, Szeto HH, Powers SK. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J Appl Physiol (1985) 2011;111:1459–1466. doi: 10.1152/japplphysiol.00591.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra SK, Misra V. Muscle sarcopenia: an overview. Acta Myol. 2003;22:43–47. [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momken I, Stevens L, Bergouignan A, Desplanches D, Rudwill F, Chery I, Zahariev A, Zahn S, Stein TP, Sebedio JL, Pujos-Guillot E, Falempin M, Simon C, Coxam V, Andrianjafiniony T, Gauquelin-Koch G, Picquet F, Blanc S. Resveratrol prevents the wasting disorders of mechanical unloading by acting as a physical exercise mimetic in the rat. FASEB J. 2011;25:3646–3660. doi: 10.1096/fj.10-177295. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Handschin C, Arlow D, Xie X, St Pierre J, Sihag S, Yang W, Altshuler D, Puigserver P, Patterson N, Willy PJ, Schulman IG, Heyman RA, Lander ES, Spiegelman BM. Errα and Gabpa/b specify PGC-1α-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc Natl Acad Sci U S A. 2004;101:6570–6575. doi: 10.1073/pnas.0401401101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moylan JS, Reid MB. Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve. 2007;35:411–429. doi: 10.1002/mus.20743. [DOI] [PubMed] [Google Scholar]
- Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, Kashyap S, Miyazaki Y, Kohane I, Costello M, Saccone R, Landaker EJ, Goldfine AB, Mun E, DeFronzo R, Finlayson J, Kahn CR, Mandarino LJ. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A. 2003;100:8466–8471. doi: 10.1073/pnas.1032913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino MA, Canepari M, Rossi R, D'Antona G, Reggiani C, Bottinelli R. Orthologous myosin isoforms and scaling of shortening velocity with body size in mouse, rat, rabbit and human muscles. J Physiol. 2003;546:677–689. doi: 10.1113/jphysiol.2002.027375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino MA, Desaphy JF, Brocca L, Pierno S, Camerino DC, Bottinelli R. Redox homeostasis, oxidative stress and disuse muscle atrophy. J Physiol. 2011;589:2147–2160. doi: 10.1113/jphysiol.2010.203232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK, Duarte J, Kavazis AN, Talbert EE. Reactive oxygen species are signalling molecules for skeletal muscle adaptation. Exp Physiol. 2010;95:1–9. doi: 10.1113/expphysiol.2009.050526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK, Kavazis AN, DeRuisseau KC. Mechanisms of disuse muscle atrophy: role of oxidative stress. Am J Physiol Regul Integr Comp Physiol. 2005;288:R337–R344. doi: 10.1152/ajpregu.00469.2004. [DOI] [PubMed] [Google Scholar]
- Powers SK, Smuder AJ, Judge AR. Oxidative stress and disuse muscle atrophy: cause or consequence? Curr Opin Clin Nutr Metab Care. 2012;15:240–245. doi: 10.1097/MCO.0b013e328352b4c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- Roberts-Wilson TK, Reddy RN, Bailey JL, Zheng B, Ordas R, Gooch JL, Price SR. Calcineurin signaling and PGC-1α expression are suppressed during muscle atrophy due to diabetes. Biochim Biophys Acta. 2010;1803:960–967. doi: 10.1016/j.bbamcr.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, Murphy MP, Beckman JS. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci U S A. 2006;103:15038–15043. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanello V, Guadagnin E, Gomes L, Roder I, Sandri C, Petersen Y, Milan G, Masiero E, Del Piccolo P, Foretz M, Scorrano L, Rudolf R, Sandri M. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010;29:1774–1785. doi: 10.1038/emboj.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanello V, Sandri M. Mitochondrial biogenesis and fragmentation as regulators of muscle protein degradation. Curr Hypertens Rep. 2010;12:433–439. doi: 10.1007/s11906-010-0157-8. [DOI] [PubMed] [Google Scholar]
- Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, Yancopoulos GD, Glass DJ. Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol. 2001;3:1009–1013. doi: 10.1038/ncb1101-1009. [DOI] [PubMed] [Google Scholar]
- Sacheck JM, Hyatt JP, Raffaello A, Jagoe RT, Roy RR, Edgerton VR, Lecker SH, Goldberg AL. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. 2007;21:140–155. doi: 10.1096/fj.06-6604com. [DOI] [PubMed] [Google Scholar]
- Sandri M. Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda) 2008;23:160–170. doi: 10.1152/physiol.00041.2007. [DOI] [PubMed] [Google Scholar]
- Sandri M. Autophagy in health and disease. 3. Involvement of autophagy in muscle atrophy. Am J Physiol Cell Physiol. 2010a;298:C1291–C1297. doi: 10.1152/ajpcell.00531.2009. [DOI] [PubMed] [Google Scholar]
- Sandri M. Autophagy in skeletal muscle. FEBS Lett. 2010b;584:1411–1416. doi: 10.1016/j.febslet.2010.01.056. [DOI] [PubMed] [Google Scholar]
- Sandri M, Lin J, Handschin C, Yang W, Arany ZP, Lecker SH, Goldberg AL, Spiegelman BM. PGC-1α protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc Natl Acad Sci USA. 2006;103:16260–16265. doi: 10.1073/pnas.0607795103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servais S, Letexier D, Favier R, Duchamp C, Desplanches D. Prevention of unloading-induced atrophy by vitamin E supplementation: links between oxidative stress and soleus muscle proteolysis? Free Radic Biol Med. 2007;42:627–635. doi: 10.1016/j.freeradbiomed.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanely RA, Van Gammeren D, Deruisseau KC, Zergeroglu AM, McKenzie MJ, Yarasheski KE, Powers SK. Mechanical ventilation depresses protein synthesis in the rat diaphragm. Am J Respir Crit Care Med. 2004;170:994–999. doi: 10.1164/rccm.200304-575OC. [DOI] [PubMed] [Google Scholar]
- Shanely RA, Zergeroglu MA, Lennon SL, Sugiura T, Yimlamai T, Enns D, Belcastro A, Powers SK. Mechanical ventilation-induced diaphragmatic atrophy is associated with oxidative injury and increased proteolytic activity. Am J Respir Crit Care Med. 2002;166:1369–1374. doi: 10.1164/rccm.200202-088OC. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Abe N, Nagano M, Goto K, Sakuma K, Naito H, Yoshioka T, Powers SK. Changes in PKB/Akt and calcineurin signaling during recovery in atrophied soleus muscle induced by unloading. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1273–R1278. doi: 10.1152/ajpregu.00688.2004. [DOI] [PubMed] [Google Scholar]
- Thomason DB, Booth FW. Atrophy of the soleus muscle by hindlimb unweighting. J Appl Physiol (1985) 1990;68:1–12. doi: 10.1152/jappl.1990.68.1.1. [DOI] [PubMed] [Google Scholar]
- Zergeroglu MA, McKenzie MJ, Shanely RA, Van Gammeren D, DeRuisseau KC, Powers SK. Mechanical ventilation-induced oxidative stress in the diaphragm. J Appl Physiol (1985) 2003;95:1116–1124. doi: 10.1152/japplphysiol.00824.2002. [DOI] [PubMed] [Google Scholar]