

Abstract
Oxygen therapy is known to reduce loop gain (LG) in patients with obstructive sleep apnoea (OSA), yet its effects on the other traits responsible for OSA remain unknown. Therefore, we assessed how hyperoxia and hypoxia alter four physiological traits in OSA patients. Eleven OSA subjects underwent a night of polysomnography during which the physiological traits were measured using multiple 3-min ‘drops’ from therapeutic continuous positive airway pressure (CPAP) levels. LG was defined as the ratio of the ventilatory overshoot to the preceding reduction in ventilation. Pharyngeal collapsibility was quantified as the ventilation at CPAP of 0 cmH2O. Upper airway responsiveness was defined as the ratio of the increase in ventilation to the increase in ventilatory drive across the drop. Arousal threshold was estimated as the level of ventilatory drive associated with arousal. On separate nights, subjects were submitted to hyperoxia (n = 9; FiO2 ∼0.5) or hypoxia (n = 10; FiO2 ∼0.15) and the four traits were reassessed. Hyperoxia lowered LG from a median of 3.4 [interquartile range (IQR): 2.6–4.1] to 2.1 (IQR: 1.3–2.5) (P < 0.01), but did not alter the remaining traits. By contrast, hypoxia increased LG [median: 3.3 (IQR: 2.3–4.0) vs. 6.4 (IQR: 4.5–9.7); P < 0.005]. Hypoxia additionally increased the arousal threshold (mean ± s.d. 10.9 ± 2.1 l min−1 vs. 13.3 ± 4.3 l min−1; P < 0.05) and improved pharyngeal collapsibility (mean ± s.d. 3.4 ± 1.4 l min−1 vs. 4.9 ± 1.3 l min−1; P < 0.05), but did not alter upper airway responsiveness (P = 0.7). This study demonstrates that the beneficial effect of hyperoxia on the severity of OSA is primarily based on its ability to reduce LG. The effects of hypoxia described above may explain the disappearance of OSA and the emergence of central sleep apnoea in conditions such as high altitude.
Key points
Changes in the level of inspired oxygen have dramatic effects on the pathophysiology of obstructive sleep apnoea (OSA): hyperoxia reduces the severity of OSA in some but not all patients, whereas hypoxia transforms obstructive events into central events. Given that OSA is likely to result from the interaction of key pathophysiological traits, including a compromised pharyngeal anatomy, inadequate upper airway muscle function, a large ventilatory response to a disturbance in ventilation (high loop gain) and a low arousal threshold, we examined how changes in oxygen levels alter these traits.
Our study demonstrates that the beneficial effect of hyperoxia on OSA severity is solely based on its ability to attenuate loop gain, whereas hypoxia increases loop gain and the arousal threshold in addition to improving pharyngeal collapsibility.
Such effects help to explain why oxygen therapy may not work in every patient with OSA and explain the disappearance of OSA and the emergence of central events during hypoxic conditions.
Introduction
The pathophysiology of obstructive sleep apnoea (OSA) is multi-factorial. Several key factors, known as physiological ‘traits’, have been shown to combine to cause OSA. These include: (i) poor upper airway anatomy that predisposes the airway to collapse; (ii) poor ability of the upper airway muscles to respond to a respiratory challenge and stiffen or dilate the airway; (iii) a low respiratory arousal threshold that causes an individual to arouse from sleep for very small increases in respiratory drive, and (iv) a hypersensitive ventilatory control system often referred to as a system with a high loop gain (LG) (Gold et al. 1985; Wellman et al. 2011).
Over the years, many investigators have examined the use of supplemental oxygen therapy as a therapy for OSA. However, the effects of supplemental oxygen on the severity of OSA and its consequences are highly variable (Wellman et al. 2008; Mehta et al. 2013; Xie et al. 2013). Small physiological studies indicate that oxygen therapy significantly improves the apnoea–hypopnoea index (AHI) in 36–50% of individuals, whereas OSA severity remains unchanged or worsens in other patients. For those patients in whom supplemental oxygen is beneficial, it is likely that it improves OSA by reducing the sensitivity of the ventilatory control system (i.e. by decreasing LG) (Wellman et al. 2008; Xie et al. 2013). However, like any drug, oxygen may have other important physiological effects. Although oxygen may be able to reduce the sensitivity of the ventilatory control system, the reduction in ventilatory drive may have the unwanted effect of reducing the respiratory output to the upper airway muscles (Aleksandrova, 2004), which could potentially increase upper airway collapsibility and reduce pharyngeal dilator muscle responsiveness. Such a worsening of these traits may explain why a proportion of OSA patients do not improve or actually worsen.
By contrast, exposure to hypoxaemia, such as that which may occur at altitude or in heart failure, has been clinically observed to change OSA to central sleep apnoea (CSA) (Warner et al. 1987; Burgess et al. 2004, 2006; Patz et al. 2006; Nussbaumer-Ochsner et al. 2010), which suggests that hypoxaemia may improve the upper airway anatomy or responsiveness in addition to elevating LG. It is well documented that hypoxia will raise LG (Khoo et al. 1982; Solin et al. 2000; Sands et al. 2011; Andrews et al. 2012) and that a high LG amplifies small disturbances in ventilation, yielding cyclic oscillations in ventilatory drive, as seen in CSA. However, in addition to raising LG, the conversion of OSA to CSA suggests that hypoxia might also improve the pharyngeal anatomy or responsiveness through an increased drive to the upper airway muscles (Jordan et al. 2010). However, to date there has been no systematic investigation of how either hyperoxia or hypoxia alter the underlying physiology in patients with OSA. Accordingly, the aim of this study was to assess how changes in oxygen levels alter the physiological traits responsible for OSA. The preliminary results of this analysis have been published in abstract form (Edwards et al. 2013a).
Methods
Participants
Eleven patients (five male, six female) with documented OSA defined as an AHI of >10 events h−1 (mean ± s.d. 49.9 ± 22.9 events h−1) were recruited from the sleep clinic at the Brigham and Women's Hospital. All subjects were currently treated with continuous positive airway pressure (CPAP) and had documented adherence of usage of >5 h night−1 during the month prior to enrolment. Subjects were excluded if they had any of the following conditions: concurrent sleep disorders; renal insufficiency; neuromuscular disease; uncontrolled diabetes mellitus; CSA; heart failure; uncontrolled hypertension, or a thyroid disorder. Subjects were also screened to ensure they were not taking any medications that might alter sleep or are known to affect respiration or pharyngeal muscle control. Written informed consent was obtained before subjects were enrolled in the study, which was approved by the Partners’ Human Research Committee and conformed to the standards set by the Declaration of Helsinki.
Experimental design and protocol
All subjects underwent two or three overnight studies in our laboratory. During the initial overnight study, a baseline assessment of the four physiological traits (described below) was conducted. During the following visits, the traits were reassessed while subjects breathed 15% O2 balance N2 (hypoxic condition) or 50% O2 balance N2 (hyperoxic condition). The order in which subjects were subjected to hyperoxia and hypoxia nights was randomized and at least 1 week was allowed to elapse between visits.
At each visit, subjects were instrumented with standard polysomnography apparatus that included equipment for electroencephalography (EEG), electro-oculography, sub-mental electromyography, electrocardiography and arterial oxygen saturation monitored at the finger. Subjects were also fitted with a nasal mask (Gel Mask; Respironics, Inc., Murrysville, PA, USA) through which measurements of mask pressure (Validyne Engineering Corp., Northridge, CA, USA), ventilatory flow (pneumotachometer model 3700A; Hans-Rudolph, Inc., Shawnee, KS, USA), tidal volume and end-tidal CO2 (VacuMed, Vacumetrics, Inc., Ventura, CA, USA) were obtained. The mask was connected to a positive/negative pressure source (Respironics, Inc.) to enable rapid switching between CPAP levels. During the hypoxia and hyperoxia studies, the air inlet for the pressure delivery device was connected to a large respiratory balloon that was constantly filled with the appropriate gas mixture to ensure a continuous delivery of the desired gas. All signals were sampled at 125 Hz and displayed using Nihon Kohden (Tokyo, Japan) and Spike 2 (Cambridge Electronic Design Ltd, Cambridge, UK) software.
Once all the equipment was in place, subjects were asked to sleep in the supine position. Following sleep onset, the level of CPAP was titrated to eliminate all sleep-disordered breathing. When patients were asleep in stable non-rapid eye movement (nREM) sleep, the four traits [pharyngeal anatomy/collapsibility, LG, upper airway muscle responsiveness (gain) and arousal threshold] were assessed (Fig. 1). The method for measuring these traits has been described in detail previously (Wellman et al. 2011; Edwards et al. 2012, 2014) and involves 2–3 min CPAP ‘drops’ to sub-therapeutic levels during stable nREM sleep that are repeatedly performed throughout the night. Briefly, when CPAP is dropped to a sub-therapeutic level, a reduction in ventilation is caused by the partial obstruction of the airway. The ‘passive’ anatomy (i.e. pharyngeal collapsibility) is determined by plotting mask pressure versus ventilation for the second and third breaths of all the pressure drops throughout the entire night. The data are fit with a linear regression line and ventilation at zero mask pressure (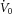 ) is used to measure pharyngeal anatomy/collapsibility. After the initial reduction in ventilation, ventilatory drive will begin to rise as a result of the accumulation of CO2 and this stimulus may activate the upper airway muscles in an attempt to reopen the airway and recover lost ventilation. Despite this partial recovery, ventilation often remains depressed or reduced below the eupnoeic level despite the increased levels of ventilatory drive. The level to which ventilatory drive has risen over the course of the drop can then be determined by abruptly returning CPAP to the therapeutic level and measuring the overshoot in ventilation. The steady-state LG is then measured as the ratio of this ventilatory overshoot (or response) to the net reduction or disturbance in ventilation from baseline. In order to be utilized in the calculation of LG, ventilation during the last 60 s of the drop must be significantly lower than eupnoeic ventilation on optimum CPAP and no arousals can occur during this interval. The components of LG, controller gain (ventilatory sensitivity to CO2) and plant gain (change in end-tidal CO2 for a corresponding change in ventilation) were also measured. Plant gain was defined as the reciprocal of the slope of the metabolic hyperbola during sleep, and controller gain as (LG)/(plant gain). The responsiveness of the upper airway muscles, which we refer to as the ‘upper airway gain’ (UAG), is measured by first calculating the difference between ventilation at the start and end of the drop, which represents how much ventilation has been recovered over the course of the drop. The ratio of this difference to the amount by which ventilation overshoots (i.e. the increase in ventilatory drive over the course of the drop) when mask pressure is returned to the holding pressure represents the ability of the airway to stiffen or dilate in response to an increase in ventilatory drive. All LG and UAG measurements were calculated from CPAP drops that did not end in arousal, and all measurements were averaged to determine a mean value for each subject.
) is used to measure pharyngeal anatomy/collapsibility. After the initial reduction in ventilation, ventilatory drive will begin to rise as a result of the accumulation of CO2 and this stimulus may activate the upper airway muscles in an attempt to reopen the airway and recover lost ventilation. Despite this partial recovery, ventilation often remains depressed or reduced below the eupnoeic level despite the increased levels of ventilatory drive. The level to which ventilatory drive has risen over the course of the drop can then be determined by abruptly returning CPAP to the therapeutic level and measuring the overshoot in ventilation. The steady-state LG is then measured as the ratio of this ventilatory overshoot (or response) to the net reduction or disturbance in ventilation from baseline. In order to be utilized in the calculation of LG, ventilation during the last 60 s of the drop must be significantly lower than eupnoeic ventilation on optimum CPAP and no arousals can occur during this interval. The components of LG, controller gain (ventilatory sensitivity to CO2) and plant gain (change in end-tidal CO2 for a corresponding change in ventilation) were also measured. Plant gain was defined as the reciprocal of the slope of the metabolic hyperbola during sleep, and controller gain as (LG)/(plant gain). The responsiveness of the upper airway muscles, which we refer to as the ‘upper airway gain’ (UAG), is measured by first calculating the difference between ventilation at the start and end of the drop, which represents how much ventilation has been recovered over the course of the drop. The ratio of this difference to the amount by which ventilation overshoots (i.e. the increase in ventilatory drive over the course of the drop) when mask pressure is returned to the holding pressure represents the ability of the airway to stiffen or dilate in response to an increase in ventilatory drive. All LG and UAG measurements were calculated from CPAP drops that did not end in arousal, and all measurements were averaged to determine a mean value for each subject.
Figure 1. Techniques for measuring the physiological traits in obstructive sleep apnoea and assessing the ventilatory response to spontaneous arousal.
A, a schematic of the ventilatory response to a continuous positive airway pressure (CPAP) drop demonstrates how all changes in ventilation were used to assess the physiological traits. Determining pharyngeal collapsibility, loop gain and upper airway gain: the drop in CPAP causes an immediate reduction in resting ventilation ( ) as a result of airway narrowing. The breaths (2–3) following the reduction in CPAP were used to calculate the pharyngeal collapsibility or
) as a result of airway narrowing. The breaths (2–3) following the reduction in CPAP were used to calculate the pharyngeal collapsibility or 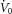 . The inset shows how the breaths from the current drop (circled) are placed on a graph of ventilation versus mask pressure in order to calculate
. The inset shows how the breaths from the current drop (circled) are placed on a graph of ventilation versus mask pressure in order to calculate 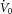 . This initial reduction in ventilation leads to an increase in respiratory drive over the course of the drop. We measure how much ventilatory drive accumulates by rapidly restoring CPAP therapy and measuring the overshoot in ventilation (x). The ratio of this ventilatory response or overshoot (x) to the net reduction in ventilation during the drop period (y) provides a measure of loop gain (x/y). A delay (δ) and time constant (τ) are then estimated from the dynamics of the ventilatory overshoot. In response to the increase in drive (x), the subject activates the upper airway muscles and partially reopens the airway, allowing ventilation to recover slightly (z). The ratio of the compensatory increase in ventilation (z) to the increase in ventilatory drive (x) across the drop provides a measure of neuromuscular compensation (z/x), to which we refer as the upper airway gain. B, determining the arousal threshold: now that we know the LG, δ and τ, a ventilatory drive signal (red line) can be calculated for each CPAP drop. In CPAP drops that cause an arousal, we quantify the arousal threshold as the level of ventilatory drive immediately preceding the arousal. C, to assess the effect of hypoxia and hyperoxia on the ventilatory response to spontaneous arousal, we calculated the ratio of the reduction in ventilation following the initial overshoot (y) and the magnitude of this overshoot (x). The solid and dashed grey lines demonstrate how a minimally and a highly underdamped system respond respectively for the same ventilatory overshoot.
. This initial reduction in ventilation leads to an increase in respiratory drive over the course of the drop. We measure how much ventilatory drive accumulates by rapidly restoring CPAP therapy and measuring the overshoot in ventilation (x). The ratio of this ventilatory response or overshoot (x) to the net reduction in ventilation during the drop period (y) provides a measure of loop gain (x/y). A delay (δ) and time constant (τ) are then estimated from the dynamics of the ventilatory overshoot. In response to the increase in drive (x), the subject activates the upper airway muscles and partially reopens the airway, allowing ventilation to recover slightly (z). The ratio of the compensatory increase in ventilation (z) to the increase in ventilatory drive (x) across the drop provides a measure of neuromuscular compensation (z/x), to which we refer as the upper airway gain. B, determining the arousal threshold: now that we know the LG, δ and τ, a ventilatory drive signal (red line) can be calculated for each CPAP drop. In CPAP drops that cause an arousal, we quantify the arousal threshold as the level of ventilatory drive immediately preceding the arousal. C, to assess the effect of hypoxia and hyperoxia on the ventilatory response to spontaneous arousal, we calculated the ratio of the reduction in ventilation following the initial overshoot (y) and the magnitude of this overshoot (x). The solid and dashed grey lines demonstrate how a minimally and a highly underdamped system respond respectively for the same ventilatory overshoot.
In addition to its use in the calculation of LG and UAG, the time course of ventilation following the return to the therapeutic pressure allows a delay and a time constant to be derived (Wellman et al. 2011). Importantly, once the LG, delay and time constant are known, the time course of the rise in ventilatory drive during each drop can be determined using a dynamic model of the ventilatory control system. Briefly, the observed changes in ventilation that occur during each CPAP drop were input into the transfer function model with the known steady-state LG, time constant and delay, which computationally transformed the changes in ventilation into a ventilatory drive signal. Once ventilatory drive is calculated, the arousal threshold can be quantified from any CPAP drop during which an arousal occurred (defined as an increase of >3 s in EEG frequency). Specifically, the arousal threshold was calculated as the level of ventilatory drive immediately preceding the arousal.
Given the importance of arousals in promoting a ventilatory overshoot (Khoo & Berry, 1996; Khoo et al. 1996) and ventilatory instability, we also examined the effects of hyperoxia and hypoxia on the magnitude and damping characteristics of the ventilatory response to spontaneous arousal (VRA). In order for a spontaneous arousal to be included in our analysis, it had to occur while the subject was on therapeutic CPAP, last 3–15 s, occur during stage 2–4 nREM sleep and be preceded and followed by ≥1 min of stable nREM sleep following pre-established guidelines (Jordan et al. 2004; Edwards et al. 2013b). Arousals were discarded if a mask leak, a change in the level of CPAP or mouth expiration occurred within 60 s before or after the arousal. Breath-by-breath measurements of inspired minute ventilation (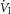 ) and end-tidal CO2 (
) and end-tidal CO2 ( ) were interpolated at 0.25 s intervals for 60 s prior to and 60 s following each arousal (start arousal = time zero), designated as time = 0. Ventilation was then normalized to the mean ventilation using the 60 s prior to the arousal. We calculated the size of the average ventilatory overshoot (defined as the peak ventilation within 15 s of time = 0), the size of the secondary undershoot (defined as the nadir ventilation within 45 s of time = 0) and the ratio of these two values (Fig. 1C) to provide another measure of the stability of the ventilatory control system. A large ratio indicates a more unstable system, whereas a low value indicates a more stable system.
) were interpolated at 0.25 s intervals for 60 s prior to and 60 s following each arousal (start arousal = time zero), designated as time = 0. Ventilation was then normalized to the mean ventilation using the 60 s prior to the arousal. We calculated the size of the average ventilatory overshoot (defined as the peak ventilation within 15 s of time = 0), the size of the secondary undershoot (defined as the nadir ventilation within 45 s of time = 0) and the ratio of these two values (Fig. 1C) to provide another measure of the stability of the ventilatory control system. A large ratio indicates a more unstable system, whereas a low value indicates a more stable system.
Statistical analysis
In order to maximize our sample size because several participants did not complete all three conditions, the effects of hyperoxia and hypoxia on OSA traits were assessed independently using either paired t tests or the signed rank test depending on whether the data were normally distributed, with Bonferroni correction for multiple comparisons (i.e. hyperoxic and hypoxic conditions). All statistical analyses were performed using SigmaPlot Version 11.0 (Systat Software, Inc., San Jose, CA, USA). A P-value of ≤0.05 was considered to indicate statistical significance. Values are presented as means ± s.e.m. or medians [interquartile range (IQR)] as appropriate.
Results
The mean ± s.d. age and body mass index of our patients were 50.4 ± 5.5 years and 36.6 ± 5.7 kg m−2, respectively. Of the 11 subjects who completed the baseline study, 10 patients provided trait measurements during hypoxia and nine provided trait measurements during hyperoxia. The effects of hyperoxia and hypoxia therapy on resting ventilatory parameters, the therapeutic CPAP level used during the study and the numbers of CPAP drops performed to assess the traits are shown in Table 1. Compared with baseline values, hyperoxia raised mean overnight oxygen saturation and hypoxia lowered it. Minute ventilation and end-tidal CO2 remained unaltered by the level of oxygen, although transient changes were observed when the patients were initially switched into hyperoxia or hypoxia. During the hypoxia night, the majority of patients (n = 8) developed short epochs of cyclic central apnoeas/hypopnoeas most commonly following either an arousal or the ventilatory overshoot consequent to the return of CPAP to therapeutic levels. When the traits were assessed under the different oxygen conditions, no differences emerged in the therapeutic CPAP level used, the number of CPAP drops performed on each night, or the number of CPAP drops used to obtain LG/upper airway gain measurements.
Table 1.
Effects of oxygen therapy on resting ventilatory and sleep parameters, continuous positive airway pressure (CPAP) drops performed and number of arousals included in the ventilatory response to spontaneous arousal (VRA) analysis
| Baseline (n = 11) | Hyperoxia (n = 9) | Hypoxia (n = 10) | |
|---|---|---|---|
| Resting ventilatory parameters | |||
| Minute ventilation (l min−1) | 7.6 ± 1.1 | 7.5 ± 0.9 | 7.6 ± 0.7 |
| End-tidal CO2 (mmHg) | 39.4 ± 2.4 | 38.2 ± 1.7 | 40.0 ± 2.9 |
| Mean overnight O2 saturation (%) | 95.0 ± 1.4 | 97.3 ± 0.9* | 84.3 ± 1.8* |
| Sleep parameters | |||
| Total recording duration (min) | 364.9 ± 59.0 | 347.9 ± 48.0 | 337.9 ± 48.0 |
| Total sleep duration (min) | 265.1 ± 31.5 | 255.3 ± 33.6 | 266.2 ± 57.1 |
| nREM duration (min) | 240.0 ± 31.2 | 229.4 ± 26.4 | 230.3 ± 58.3 |
| Stage 1 | 65 ± 38.9 | 49.1 ± 23.2 | 50.7 ± 24.5 |
| Stage 2 | 172.6 ± 35.1 | 176.5 ± 32.1 | 176.3 ± 39.2 |
| Stage 3–4 | 0 (0–1.4) | 0.5 (0–5.5) | 0.3 (0–0.5) |
| REM duration (min) | 25.1 ± 16.1 | 25.9 ± 14.4 | 36.0 ± 11.5 |
| Sleep efficiency (%) | 73.9 ± 11.0 | 74.8 ± 14.1 | 79.1 ± 13.5 |
| CPAP used and drops performed | |||
| Therapeutic pressure (cmH2O) | 11.4 ± 1.9 | 10.6 ± 2.6 | 12.0 ± 2.4 |
| Total CPAP drops (n) | 27.6 ± 7.8 | 21.9 ± 3.6 | 16.3 ± 7.6 |
| CPAP drops to assess LG/UAG (n) | 4.7 ± 2.9 | 7.4 ± 3.6 | 3.9 ± 2.1 |
| VRA analysis | |||
| Arousal number (n) | 4.8 ± 1.6 | 4.7 ± 2.6 | 6.6 ± 2.8 |
| Arousal duration (s) | 6.9 ± 1.4 | 7.4 ± 1.6 | 8.3 ± 1.7 |
Values are means ± s.d. Abbreviations: LG, loop gain; nREM, non-rapid eye movement; REM, rapid eye movement; UAG, upper airway gain.
P < 0.05 compared with data for the baseline night.
Effects of hyperoxia on OSA traits
Figure 2 demonstrates that hyperoxia lowered LG from a median of 3.4 (IQR: 2.6–4.1) to 2.1 (IQR: 1.3–2.5) (P < 0.01) as a result of a reduction in controller gain [0.47 l min−1 mmHg−1 (IQR: 0.30–0.60 l min−1 mmHg−1) vs. 0.25 l min−1 mmHg−1 (IQR: 0.19–0.34 l min−1 mmHg−1); P < 0.01] as plant gain remained unchanged (7.5 ± 0.5 mmHg l−1 min−1 vs. 7.4 ± 0.4 mmHg l−1 min−1; P = NS). There was a trend for hyperoxia to increase the circulatory delay (6.1 ± 1.1 s vs. 11.1 ± 1.6 s; P = 0.12), although this difference failed to reach statistical significance. However, hyperoxia did not alter the time constant of the ventilatory overshoot (53.6 ± 8.4 s vs. 79.3 ± 17.9 s; P = 0.6), and nor did it alter the upper airway anatomy/collapsibility, arousal threshold or UAG (Fig. 3).
Figure 2. Effects of hyperoxia and hypoxia on ventilatory control characteristics.
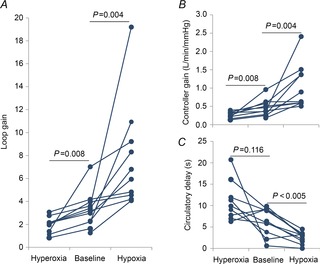
A, compared with the baseline night, hyperoxia consistently lowered loop gain in all subjects by approximately 40%, whereas hypoxia doubled loop gain (∼95%), an occurrence driven by changes in controller gain (B). C, compared with baseline, hypoxia significantly decreased the circulatory delay, whereas there was a trend for hyperoxia to raise it.
Figure 3. Effects of hyperoxia on anatomy, arousal threshold and upper airway gain.
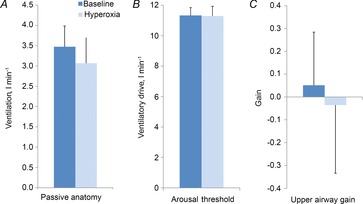
Hyperoxia did not alter the passive anatomy (A), the arousal threshold (B) or the upper airway gain (C).
Effects of hypoxia on OSA traits
Sustained overnight hypoxia increased LG [3.3 (IQR: 2.3–4.0) vs. 6.4 (IQR: 4.5–9.7); P < 0.005] via increases in controller gain [0.42 (IQR: 0.27–0.59) vs. 0.76 (IQR: 0.60–1.41); P < 0.005]. It also decreased the circulatory delay (6.2 ± 1.0 s vs. 2.5 ± 0.4 s; P < 0.005). Exposure to sustained hypoxia additionally increased the arousal threshold (10.9 ± 0.7 l min−1 vs. 13.3 ± 1.4 l min−1; P < 0.05) and improved pharyngeal collapsibility (3.4 ± 0.4 l min−1 vs. 4.9 ± 0.4 l min−1; P < 0.05), but did not alter UAG (Fig. 4).
Figure 4. Effects of hypoxia on anatomy, arousal threshold and upper airway gain.

Hypoxia significantly improved the passive anatomy (A) and increased the arousal threshold (B), but did not statistically alter the upper airway gain (C).
Effects of oxygen on VRA
The VRA could be assessed in seven of the nine patients who completed the hyperoxia nights and in all patients who completed the hypoxia nights. Compared with baseline levels, the level of oxygen did not alter the number or duration of arousals included in the analysis (Table 1). The effects of hypoxia and hyperoxia on VRA are depicted in Fig. 5. There was no difference in the magnitude of VRA with either hypoxia or hyperoxia in comparison with baseline conditions, although there was a trend for the overshoot to decrease with hyperoxia (P = 0.06). Compared with baseline, hypoxia significantly increased the magnitude of the ventilatory undershoot, whereas hyperoxia reduced it. These changes resulted in hypoxia significantly increasing the ventilatory undershoot/overshoot ratio, indicating a less stable system, whereas hyperoxia did not significantly alter this ratio.
Figure 5. Effects of oxygen on the ventilatory response to arousal.
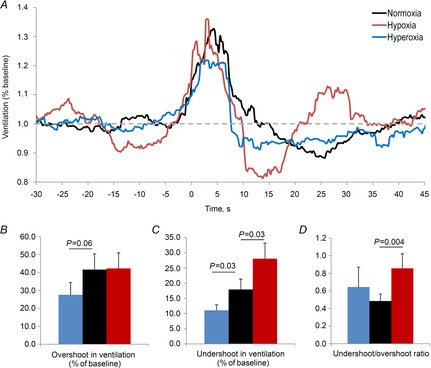
A, ensemble averaged ventilatory response to spontaneous arousal for each oxygen condition where time = 0 is the start of the scored electroencephalogram arousal. The components of the ventilatory response to arousal were also assessed, including the overshoot in ventilation (B), the undershoot in ventilation (C) and the undershoot/overshoot ratio (D).
Discussion
The major novel findings of the present study are that sustained hypoxia improved the upper airway anatomy/collapsibility, increased the arousal threshold and raised LG. Such findings may help to explain several clinical observations: the increased arousal threshold may help to explain the reduced proportion of events with arousals at altitude, and the combination of improved collapsibility and increased LG may help to explain the conversion of OSA to CSA in conditions such as altitude or congestive heart failure. By contrast with the effects of hypoxia, hyperoxia had no detrimental effects on airway anatomy or muscle responsiveness. Thus the beneficial effect of hyperoxia in the treatment of OSA is based solely on its ability to reduce LG. Such a finding highlights the need for individual trait assessment in order to individualize therapy and to better determine which OSA subjects will benefit from the lowering of LG with supplemental oxygen.
Effects of oxygen level on the four physiological traits
Effects of hyperoxia
In the present study, hyperoxia consistently lowered the steady-state LG as predicted by theory (Khoo et al. 1982) and has been previously demonstrated experimentally (Gautier et al. 1986; Chowdhuri et al. 2010a). Furthermore, the magnitude of the decrease in LG was driven solely by reductions in controller gain and is strikingly similar to the reductions in controller gain observed with the administration of sustained hyperoxia during sleep in healthy volunteers (Chowdhuri et al. 2010a). At first, our results seem inconsistent with those of our previous study, in which we reported that the ‘dynamic’ LG was lowered only in those individuals who had a high LG at baseline (Wellman et al. 2008). Although the steady-state and dynamic LGs are not directly comparable, if we estimate the ‘dynamic’ LG using our CPAP dial-down technique [see Wellman et al. (2011) and Edwards et al. (2012) for details], we see that the majority of subjects in the current study also had a somewhat high LG at baseline [median LG: 0.71 (IQR: 0.34–0.84)].
Although it is likely that the present study was statistically underpowered to detect a significant increase in the circulatory delay, we did observe a strong trend for this to increase with hyperoxia. An increase in the delay might occur because: (i) hyperoxia is able to blunt the fast responsive peripheral chemoreceptors and the changes in ventilation subsequently observed reflect the response of the more ‘sluggish’ central chemoreceptors, or (ii) hyperoxia has depressive effects on cardiac function: it has been shown to reduce cardiac output in patients with congestive heart failure in a dose-dependent manner (Haque et al. 1996), as well as to impair cardiac relaxation and increased left ventricle filling pressures (Mak et al. 2001). Nonetheless, an increase in circulatory delay may be a contributing factor to the longer respiratory events often observed in OSA patients receiving supplemental oxygen (Wellman et al. 2008; Mehta et al. 2013).
Importantly, our finding that hyperoxia did not alter any of the remaining traits suggests that the ability of oxygen therapy to improve OSA severity is driven primarily by its ability to reduce LG in normoxic individuals, specifically through reductions in the sensitivity of the carotid bodies (i.e. controller gain). Such a finding is consistent with results in animal studies that have shown that denervation of the carotid body either prevents the apnoea and periodic breathing consequent to transient ventilatory overshoots (Nakayama et al. 2003) or the unstable breathing caused in heart failure models (Marcus et al. 2014). The ubiquitous finding that oxygen therapy improves OSA severity in a proportion of individuals, whereas the remaining patients gain little or no benefit (Martin et al. 1982; Smith et al. 1984; Gold et al. 1985, 1986; Pokorski & Jernajczyk, 2000; Landsberg et al. 2001; Kumagai et al. 2008; Mehta et al. 2013), highlights the importance of understanding that OSA is caused by both anatomical and non-anatomical factors (Wellman et al. 2011; Eckert et al. 2013). If a patient has a highly collapsible airway, as recent data indicate that 23% of patients do (Eckert et al. 2013), then he or she will have OSA regardless of whether there are abnormalities in any of the other physiological traits (i.e. LG). In such patients, we expect that reducing LG with therapies such as oxygen or acetazolamide will be of little benefit in terms of reducing the AHI. However, if a patient's anatomy is of the vulnerable type found in the overwhelming majority of OSA subjects (Eckert et al. 2013), then whether or not he or she has a high LG (or defects in the other non-anatomical traits) will play a large role in whether the individual will develop OSA (i.e. LG is an effect modifier), as well as how that person will respond to treatment with oxygen. Considering an elevated LG as an effect modifier helps to explain why treatments that are intended to reduce LG often improve OSA in some but not all patients, even if they do universally lower LG as observed in the current study. Firstly, the fact that OSA is not completely resolved in most patients by such therapies suggests that an elevated LG is not the only factor causing OSA. Secondly, the reason why such therapies do not work in everyone is that these previous studies were conducted in unselected patients. If we could lower LG in patients with a mild vulnerability to upper airway collapse, who represent patients in whom an elevated LG is a large contributor to the sleep disorder (Eckert et al. 2013; Wellman et al. 2004), we predict that these patients would show dramatic improvements in the severity of their OSA. Although our hypotheses need to be tested rigorously in well-designed clinical trials, we hope that these concepts will allow clinicians to move beyond the ‘one size fits all’ treatment approach of CPAP and to begin to tailor alternative therapies to the needs of individuals based on their underlying physiology (Jordan et al. 2014; Malhotra, 2014).
Effects of hypoxia
By contrast with hyperoxia, exposure to sustained overnight hypoxia had an interesting effect on OSA traits. As expected, hypoxia raised LG via an increase in controller gain, the magnitude of which was increased by ∼80% from its baseline value. Notably, this increase is remarkably similar to the increase in controller gain (83%) observed after short periods of episodic hypoxia in healthy volunteers (Chowdhuri et al. 2010b). The improvement in pharyngeal collapsibility with hypoxia is likely to be attributable to an increase in respiratory output to the upper airway muscles providing a ‘stiffer’ and less collapsible airway. Similar improvements in upper airway collapsibility have been documented in response to sustained CO2 exposure (Jordan et al. 2010) in OSA patients. Despite the improvement in the collapsibility of the upper airway, hypoxia did not alter the responsiveness of the upper airway muscles (i.e. upper airway gain), a finding which is consistent with those of the study by Eckert et al. (2008), which demonstrated that the activation of the genioglossus muscle (a major upper airway dilator muscle) in response to brief negative pressure pulses applied in both wake and sleep was unaltered by hypoxia. Lastly, hypoxia also raised the arousal threshold by 22% in the current study. This finding is consistent with that of a previous study in healthy participants demonstrating that hypoxia increases the respiratory arousal threshold by ∼25% and the time to arousal following either resistive loading or airway occlusion (Hlavac et al. 2006). The mechanism(s) by which acute hypoxia increases the arousal threshold are unclear, but it has been proposed that hypoxia is an important neuro-inhibitory modulator that can depress respiratory afferent transmission. Taken together, these findings may help to explain the clinical observation in patients with OSA that sustained hypoxia transforms obstructive events into predominantly central events and reduces the proportion of events with arousals.
Acute sustained hypoxia during sleep not only occurs at altitude but is a key feature of many medical disorders, including congestive heart failure, chronic obstructive pulmonary disease and obesity hypoventilation syndrome, as well as moderate–severe OSA. However, the ramifications of the role of intermittent hypoxia in the pathogenesis of OSA have not been fully elucidated. Certainly, an elevated controller gain (and thus LG) in untreated OSA patients can be reversed with CPAP treatment, suggesting that an increased LG can be a consequence of OSA (Loewen et al. 2009; Salloum et al. 2009). Such a finding is consistent with the various animal studies in which exposure to intermittent hypoxia (and hypercapnia) has been shown to increase the sensitivity of the peripheral chemoreceptors. Animal studies have also shown that intermittent hypoxia may attenuate the responsiveness/recruitment of the genioglossus muscle (Edge et al. 2012), although this may be counteracted by long-term facilitation of the muscle (Tadjalli et al. 2010). Lastly, Sforza et al. (1999) reported that in OSA patients, the arousal threshold increased shortly after sleep onset, peaked between the second and third hours of the night and remained at this level for the duration of the night. Studies in sleeping neonatal animals suggest that increases in the arousal threshold can be induced by intermittent hypoxia (Johnston et al. 1998; Durand et al. 2004; Waters & Tinworth, 2005). However, whether or not such changes are driven by the sleep fragmentation associated with repetitive arousals from sleep or intermittent hypoxia per se in patients with OSA remains unclear.
Effect of oxygen level on VRA
The mechanisms that determine the magnitude of the VRA have been attributed to a combination of: (i) the sudden removal of the sleep-induced increase in upper airway resistance; (ii) a reflex ‘startle-like’ mechanism that is independent of ventilatory sensitivity during wakefulness, and (iii) the restoration of the waking chemical drive at the increased 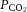 level which occurs during sleep (Phillipson, 1978; Khoo et al. 1998; Horner et al. 2001). The observation that the magnitude of the VRA is similar whether chemical drive is elevated with hypoxia or depressed with hyperoxia suggests that the overshoot in ventilation following a spontaneous arousal is chemoreceptor-independent, an observation congruent with studies suggesting its magnitude is in part similar to a ‘startle-like’ response (Horner et al. 2001; Trinder et al. 2006).
level which occurs during sleep (Phillipson, 1978; Khoo et al. 1998; Horner et al. 2001). The observation that the magnitude of the VRA is similar whether chemical drive is elevated with hypoxia or depressed with hyperoxia suggests that the overshoot in ventilation following a spontaneous arousal is chemoreceptor-independent, an observation congruent with studies suggesting its magnitude is in part similar to a ‘startle-like’ response (Horner et al. 2001; Trinder et al. 2006).
The role of arousals in the pathogenesis of OSA has been widely debated in the literature. The immediate impact of arousal is to restore pharyngeal patency and waking muscle tone in an attempt to prevent large falls in oxygen level. Furthermore, in some patients frequent recurrent arousals can act to recruit upper airway muscle activity progressively, which can lead to improved airway patency and periods of stable breathing (Jordan et al. 2011). By contrast, a large ventilatory response to arousal can also promote dynamic ventilatory instability (Khoo et al. 1996), which may contribute to OSA severity in certain individuals. To date, the effects of hyperoxia and hypoxia on VRA have not been assessed. Although there were no differences in the magnitude of the initial overshoot, we did find that the magnitude of the ventilatory undershoot following arousal was greater in hypoxia and smaller in hyperoxia, which is consistent with what we would expect physiologically. The magnitude of the ventilatory undershoot following arousal is dependent not only upon how much the  changes, but how close eupnoeic
changes, but how close eupnoeic  is from the apnoeic threshold, which is a function of controller gain (Dempsey, 2004). As the increase in ventilation following spontaneous arousal remained constant among the three conditions (and thus reduction in
is from the apnoeic threshold, which is a function of controller gain (Dempsey, 2004). As the increase in ventilation following spontaneous arousal remained constant among the three conditions (and thus reduction in  ), the changes in the undershoot with oxygen reflect the differences in controller gain. Such findings suggest that the level of oxygen may be an important contributor to whether arousals promote dynamic ventilatory instability and contribute to OSA severity.
), the changes in the undershoot with oxygen reflect the differences in controller gain. Such findings suggest that the level of oxygen may be an important contributor to whether arousals promote dynamic ventilatory instability and contribute to OSA severity.
Methodological considerations
Although our estimates of LG using our published technique often produce values similar to those obtained with other methods (Salloum et al. 2009), we have never formally validated this measurement. One way to validate our methodology for the determination of LG is to assess whether it is sensitive to the known effects of varying oxygen levels on LG: hypoxia increases LG by increasing the slope of the hypercapnic ventilatory response (i.e. controller gain), whereas hyperoxia does the opposite (Nielsen & Smith, 1951; Khoo et al. 1982; Dahan et al. 1990; Mohan & Duffin, 1997; Xie et al. 2006; Chowdhuri et al. 2010a). The observation that our measurements of LG show the expected directional changes with different levels of oxygen gives us confidence that the method of measuring LG from disturbances in ventilation provided by CPAP drops is a robust technique (Edwards et al. 2011, 2012).
Many of the previous investigations or randomized controlled trials assessing the effects of oxygen in the treatment of OSA have delivered 100% oxygen at flow rates of 2–4 l min−1, which corresponds to an FiO2 of roughly 27–33% and may be appreciably lower than the 40% administered in the current study. We elected to create hyperoxic levels which would ensure the inhibition of the peripheral chemoreceptors (i.e. controller gain) similar to those we and others have used previously (Wellman et al. 2008; Chowdhuri et al. 2010a; Xie et al. 2013). Thus there is a possibility that the reduction in LG with hyperoxia may have been greater than expected during clinical studies, although we believe that this situation is unlikely to be the case for two reasons: (i) the mean overnight oxygen saturation level in the current study is similar to that seen in previous clinical investigations, and (ii) physiological studies in OSA patients who have used 40–50% oxygen (Xie et al. 2013) have shown a reduction in AHI similar to those in the numerous clinical studies (Mehta et al. 2013). By contrast with the level of hyperoxia, the level of hypoxia administered in the current study is similar to the oxygen level at an altitude of ∼2700 m. This level was chosen in order to elucidate the physiology responsible for the switching of the OSA phenotype that has been previously reported to occur at this altitude (Burgess et al. 2004, 2006; Nussbaumer-Ochsner et al. 2010).
It was initially surprising that sustained hyperoxia and hypoxia seemingly had no effect on resting ventilation and end-tidal CO2. The finding that we did not observe a systematic change in either ventilatory characteristic may reflect the fact that the actual changes that occur in these patients are small and, because of the large individual variability, are not captured by our small sample size (i.e. the study was insufficiently powered to detect differences in resting ventilation). However, the lack of change may actually be a real phenomenon as other small studies have reported these ventilatory variables to remain unchanged during sustained hypoxic (Hlavac et al. 2006; Eckert et al. 2008) or hyperoxic (Xie et al. 2013) conditions.
We chose to study patients with OSA rather than assessing the effect that different levels of oxygen would have on the physiology of healthy participants (i.e. without OSA) for two reasons. Firstly, several previous investigations have already directly or indirectly assessed the effects of oxygen levels on several of the physiological traits measured in this study (using a variety of different techniques) in healthy participants and have been discussed above. Secondly, our main aim was to understand the mechanisms responsible for the hyperoxia-induced reduction in OSA severity, as well as the hypoxia-induced obstructive–central switch in patients with OSA. Therefore, we needed to study the relevant population (i.e. subjects with OSA). Our current work is limited by the fact that the complex nature of our study design did not allow us to assess how the changes in OSA traits during hyperoxia and hypoxia translate into alterations in the severity and pattern of sleep-disordered breathing. Nonetheless, the findings of the current study provide valuable information that helps to explain many of the clinically observed effects of different oxygen levels.
Conclusions
In summary, the major findings of our study highlight key alterations in the pathophysiology causing OSA in response to sustained exposure to both hyperoxia and hypoxia. Our study demonstrates that the beneficial effect of hyperoxia on OSA severity is based solely on its ability to attenuate LG, whereas hypoxia increased LG and the arousal threshold, in addition to improving pharyngeal collapsibility. Such effects help to explain why oxygen therapy may not work in all patients with OSA and account for the disappearance of OSA and the emergence of central events during hypoxic conditions.
Acknowledgments
The authors would like to thank Lauren Hess and Erik Smales for their laboratory assistance. The study was conducted in the Sleep Disorders Research Program Laboratory, Division of Sleep Medicine, Brigham and Women's Hospital, Boston, MA, USA.
Glossary
- AHI
apnoea–hypopnoea index
- CPAP
continuous positive airway pressure
- CSA
central sleep apnoea
- EEG
electroencephalography
- LG
loop gain
- nREM
non-rapid eye movement
- OSA
obstructive sleep apnoea
- UAG
upper airway gain
- VRA
ventilatory response to spontaneous arousal
Additional information
Competing interests
R.L.O. consults for Respironics, Inc. A.W. is a consultant for Philips Respironics, Inc., SOVA Pharmaceuticals, Inc. and Apnex Medical, Inc. A.W.'s interests were reviewed and are managed by the Brigham and Women's Hospital and Partners HealthCare in accordance with their conflict of interest policies. A.M. was a consultant for Philips Respironics, Sleep HealthCenters, Sleep Group Solutions, Apnex Medical, Inc., Pfizer, Inc. and Apnicure, Inc., but has relinquished all outside personal income since May 2012. D.P.W. was the chief medical officer for Philips Respironics until 31 December 2012 and remains a consultant for that company. He is now the chief scientific officer for Apnicure, Inc. None of the other authors have any financial relationship with a commercial entity that has an interest in the subject of this manuscript and have no other conflicts of interest.
Author contributions
B.A.E., A.W., A.M., S.A.S. and D.P.W. contributed to the conception and design of the experiments. B.A.E., S.A.S., R.L.O., P.R.G., J.P.B., D.P.W., A.M. and A.W. were involved in the collection, analysis and interpretation of data. All authors contributed to the drafting or critical revision of the article for important intellectual content. All authors approved the final version of this article.
Funding
This study was funded by the National Institutes of Health (NIH) (grant no. R01 HL102321). The authors have also received other funding support from the NIH (NIH K24 HL 093218 and NIH R01 HL090897) and the Harvard Catalyst Clinical Research Center (grant no. UL1 RR 025758-01). B.A.E. is supported by the National Health and Medical Research Council of Australia (NHMRC) C. J. Martin Overseas Biomedical Fellowship (1035115). S.A.S. is supported by an NHMRC Early Career Fellowship (1053201) and the R. G. Menzies award.
References
- Aleksandrova NP. Chemoreceptor and vagal influences on genioglossal muscle responses to inspiratory resistive load. J Physiol Pharmacol. 2004;55(Suppl. 3):7–14. [PubMed] [Google Scholar]
- Andrews G, Ainslie PN, Shepherd K, Dawson A, Swart M, Lucas S, Burgess KR. The effect of partial acclimatization to high altitude on loop gain and central sleep apnoea severity. Respirology. 2012;17:835–840. doi: 10.1111/j.1440-1843.2012.02170.x. [DOI] [PubMed] [Google Scholar]
- Burgess KR, Cooper J, Rice A, Wong K, Kinsman T, Hahn A. Effect of simulated altitude during sleep on moderate-severity OSA. Respirology. 2006;11:62–69. doi: 10.1111/j.1440-1843.2006.00785.x. [DOI] [PubMed] [Google Scholar]
- Burgess KR, Johnson PL, Edwards N. Central and obstructive sleep apnoea during ascent to high altitude. Respirology. 2004;9:222–229. doi: 10.1111/j.1440-1843.2004.00576.x. [DOI] [PubMed] [Google Scholar]
- Chowdhuri S, Shanidze I, Pierchala L, Belen D, Mateika JH, Badr MS. Effect of episodic hypoxia on the susceptibility to hypocapnic central apnea during NREM sleep. J Appl Physiol (1985) 2010b;108:369–377. doi: 10.1152/japplphysiol.00308.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhuri S, Sinha P, Pranathiageswaran S, Badr MS. Sustained hyperoxia stabilizes breathing in healthy individuals during NREM sleep. J Appl Physiol (1985) 2010a;109:1378–1383. doi: 10.1152/japplphysiol.00453.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahan A, DeGoede J, Berkenbosch A, Olievier IC. The influence of oxygen on the ventilatory response to carbon dioxide in man. J Physiol. 1990;428:485–499. doi: 10.1113/jphysiol.1990.sp018223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey JA. Crossing the apnoeic threshold: causes and consequences. Exp Physiol. 2004;90:13–24. doi: 10.1113/expphysiol.2004.028985. [DOI] [PubMed] [Google Scholar]
- Durand E, Lofaso F, Dauger S, Vardon G, Gaultier C, Gallego J. Intermittent hypoxia induces transient arousal delay in newborn mice. J Appl Physiol (1985) 2004;96:1216–1222. doi: 10.1152/japplphysiol.00802.2003. discussion 1196. [DOI] [PubMed] [Google Scholar]
- Eckert DJ, McEvoy RD, George KE, Thomson KJ, Catcheside PG. Effects of hypoxia on genioglossus and scalene reflex responses to brief pulses of negative upper-airway pressure during wakefulness and sleep in healthy men. J Appl Physiol (1985) 2008;104:1426–1435. doi: 10.1152/japplphysiol.01056.2007. [DOI] [PubMed] [Google Scholar]
- Eckert DJ, White DP, Jordan AS, Malhotra A, Wellman A. Defining phenotypic causes of obstructive sleep apnea. Identification of novel therapeutic targets. Am J Respir Crit Care Med. 2013;188:996–1004. doi: 10.1164/rccm.201303-0448OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edge D, Bradford A, Jones JF, O'Halloran KD. Chronic intermittent hypoxia alters genioglossus motor unit discharge patterns in the anaesthetized rat. Adv Exp Med Biol. 2012;758:295–300. doi: 10.1007/978-94-007-4584-1_40. [DOI] [PubMed] [Google Scholar]
- Edwards BA, Connolly JG, Campana LM, Sands SA, Trinder JA, White DP, Wellman A, Malhotra A. Acetazolamide attenuates the ventilatory response to arousal in patients with obstructive sleep apnea. Sleep. 2013b;36:281–285. doi: 10.5665/sleep.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards BA, Sands SA, Eckert DJ, White DP, Butler JP, Owens RL, Malhotra A, Wellman A. Acetazolamide improves loop gain but not the other physiological traits causing obstructive sleep apnoea. J Physiol. 2012;590:1199–1211. doi: 10.1113/jphysiol.2011.223925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards BA, Sands SA, Owens RL, Eckert DJ, Loring SH, Butler JP, Nemati S, White DP, Malhotra A, Wellman A. The use of diaphragm activity to measure the stability of the ventilatory control system (loop gain) in obstructive sleep apnea: a validation study. Am J Respir Crit Care Med. 2011;183:A5275. [Google Scholar]
- Edwards BA, Sands SA, Owens RL, Hess LB, Smales E, White DP, Malhotra A, Wellman A. D100 Ventilatory Control: From Bench to Bedside. New York, NY: American Thoracic Society; 2013a. Effects of hyperoxia and hypoxia on the physiological traits responsible for obstructive sleep apnea; pp. A5764–A5764. [Google Scholar]
- Edwards BA, Wellman A, Sands SA, Owens RL, Eckert DJ, White DP, Malhotra A. Obstructive sleep apnea in older adults is a distinctly different physiological phenotype. Sleep. 2014;37:1227–1236. doi: 10.5665/sleep.3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier H, Bonora M, Gaudy JH. Ventilatory response of the conscious or anesthetized cat to oxygen breathing. Respir Physiol. 1986;65:181–196. doi: 10.1016/0034-5687(86)90049-6. [DOI] [PubMed] [Google Scholar]
- Gold AR, Bleecker ER, Smith PL. A shift from central and mixed sleep apnea to obstructive sleep apnea resulting from low-flow oxygen. Am Rev Respir Dis. 1985;132:220–223. doi: 10.1164/arrd.1985.132.2.220. [DOI] [PubMed] [Google Scholar]
- Gold AR, Schwartz AR, Bleecker ER, Smith PL. The effect of chronic nocturnal oxygen administration upon sleep apnea. Am Rev Respir Dis. 1986;134:925–929. doi: 10.1164/arrd.1986.134.5.925. [DOI] [PubMed] [Google Scholar]
- Haque WA, Boehmer J, Clemson BS, Leuenberger UA, Silber DH, Sinoway LI. Hemodynamic effects of supplemental oxygen administration in congestive heart failure. J Am Coll Cardiol. 1996;27:353–357. doi: 10.1016/0735-1097(95)00474-2. [DOI] [PubMed] [Google Scholar]
- Hlavac MC, Catcheside PG, McDonald R, Eckert DJ, Windler S, McEvoy RD. Hypoxia impairs the arousal response to external resistive loading and airway occlusion during sleep. Sleep. 2006;29:624–631. [PubMed] [Google Scholar]
- Horner RL, Rivera MP, Kozar LF, Phillipson EA. The ventilatory response to arousal from sleep is not fully explained by differences in CO2 levels between sleep and wakefulness. J Physiol. 2001;534:881–890. doi: 10.1111/j.1469-7793.2001.00881.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston RV, Grant DA, Wilkinson MH, Walker AM. Repetitive hypoxia rapidly depresses arousal from active sleep in newborn lambs. J Physiol. 1998;510(Part 2):651–659. doi: 10.1111/j.1469-7793.1998.651bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan AS, Eckert DJ, Wellman A, Trinder JA, Malhotra A, White DP. Termination of respiratory events with and without cortical arousal in obstructive sleep apnea. Am J Respir Crit Care Med. 2011;184:1183–1191. doi: 10.1164/rccm.201106-0975OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan AS, McEvoy RD, Edwards JK, Schory K, Yang CK, Catcheside PG, Fogel RB, Malhotra A, White DP. The influence of gender and upper airway resistance on the ventilatory response to arousal in obstructive sleep apnoea in humans. J Physiol. 2004;558:993–1004. doi: 10.1113/jphysiol.2004.064238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan AS, McSharry DG, Malhotra A. Adult obstructive sleep apnoea. Lancet. 2014;383:736–747. doi: 10.1016/S0140-6736(13)60734-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan AS, White DP, Owens RL, Eckert DJ, Rahangdale S, Yim-Yeh S, Malhotra A. The effect of increased genioglossus activity and end-expiratory lung volume on pharyngeal collapse. J Appl Physiol (1985) 2010;109:469–475. doi: 10.1152/japplphysiol.00373.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoo MC, Berry RB. Modeling the interaction between arousal and chemical drive in sleep-disordered breathing. Sleep. 1996;19(Suppl):167–169. doi: 10.1093/sleep/19.suppl_10.167. [DOI] [PubMed] [Google Scholar]
- Khoo MC, Koh SS, Shin JJ, Westbrook PR, Berry RB. Ventilatory dynamics during transient arousal from NREM sleep: implications for respiratory control stability. J Appl Physiol (1985) 1996;80:1475–1484. doi: 10.1152/jappl.1996.80.5.1475. [DOI] [PubMed] [Google Scholar]
- Khoo MC, Shin JJ, Asyali MH, Kim TS, Berry RB. Ventilatory dynamics of transient arousal in patients with obstructive sleep apnea. Respir Physiol. 1998;112:291–303. doi: 10.1016/s0034-5687(98)00041-3. [DOI] [PubMed] [Google Scholar]
- Khoo MCK, Kronauer RE, Strohl KP, Slutsky AS. Factors inducing periodic breathing in humans: a general model. J Appl Physiol Respir Environ Exerc Physiol. 1982;53:644–659. doi: 10.1152/jappl.1982.53.3.644. [DOI] [PubMed] [Google Scholar]
- Kumagai T, Ishibashi Y, Kawarazaki H, Kawarazaki W, Shimizu H, Kaname S, Fujita T. Effects of nocturnal oxygen therapy on sleep apnea syndrome in peritoneal dialysis patients. Clin Nephrol. 2008;70:332–339. doi: 10.5414/cnp70332. [DOI] [PubMed] [Google Scholar]
- Landsberg R, Friedman M, Ascher-Landsberg J. Treatment of hypoxemia in obstructive sleep apnea. Am J Rhinol. 2001;15:311–313. [PubMed] [Google Scholar]
- Loewen A, Ostrowski M, Laprairie J, Atkar R, Gnitecki J, Hanly P, Younes M. Determinants of ventilatory instability in obstructive sleep apnea: inherent or acquired? Sleep. 2009;32:1355–1365. doi: 10.1093/sleep/32.10.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak S, Azevedo ER, Liu PP, Newton GE. Effect of hyperoxia on left ventricular function and filling pressures in patients with and without congestive heart failure. Chest. 2001;120:467–473. doi: 10.1378/chest.120.2.467. [DOI] [PubMed] [Google Scholar]
- Malhotra A. Hypoglossal-nerve stimulation for obstructive sleep apnea. N Engl J Med. 2014;370:170–171. doi: 10.1056/NEJMe1314084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus NJ, Del Rio R, Schultz EP, Xia XH, Schultz HD. Carotid body denervation improves autonomic and cardiac function and attenuates disordered breathing in congestive heart failure. J Physiol. 2014;592:391–408. doi: 10.1113/jphysiol.2013.266221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RJ, Sanders MH, Gray BA, Pennock BE. Acute and long-term ventilatory effects of hyperoxia in the adult sleep apnea syndrome. Am Rev Respir Dis. 1982;125:175–180. doi: 10.1164/arrd.1982.125.2.175. [DOI] [PubMed] [Google Scholar]
- Mehta V, Vasu TS, Phillips B, Chung F. Obstructive sleep apnea and oxygen therapy: a systematic review of the literature and meta-analysis. J Clin Sleep Med. 2013;9:271–279. doi: 10.5664/jcsm.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan R, Duffin J. The effect of hypoxia on the ventilatory response to carbon dioxide in man. Respir Physiol. 1997;108:101–115. doi: 10.1016/s0034-5687(97)00024-8. [DOI] [PubMed] [Google Scholar]
- Nakayama H, Smith CA, Rodman JR, Skatrud JB, Dempsey JA. Carotid body denervation eliminates apnea in response to transient hypocapnia. J Appl Physiol (1985) 2003;94:155–164. doi: 10.1152/japplphysiol.00722.2002. [DOI] [PubMed] [Google Scholar]
- Nielsen M, Smith H. Studies on the regulation of respiration in acute hypoxia; preliminary report. Acta Physiol Scand. 1951;22:44–46. doi: 10.1111/j.1748-1716.1951.tb00748.x. [DOI] [PubMed] [Google Scholar]
- Nussbaumer-Ochsner Y, Schuepfer N, Ulrich S, Bloch KE. Exacerbation of sleep apnoea by frequent central events in patients with the obstructive sleep apnoea syndrome at altitude: a randomised trial. Thorax. 2010;65:429–435. doi: 10.1136/thx.2009.125849. [DOI] [PubMed] [Google Scholar]
- Patz D, Spoon M, Corbin R, Patz M, Dover L, Swihart B, White D. The effect of altitude descent on obstructive sleep apnea. Chest. 2006;130:1744–1750. doi: 10.1378/chest.130.6.1744. [DOI] [PubMed] [Google Scholar]
- Phillipson EA. Control of breathing during sleep. Am Rev Respir Dis. 1978;118:909–939. doi: 10.1164/arrd.1978.118.5.909. [DOI] [PubMed] [Google Scholar]
- Pokorski M, Jernajczyk U. Nocturnal oxygen enrichment in sleep apnoea. J Int Med Res. 2000;28:1–8. doi: 10.1177/147323000002800101. [DOI] [PubMed] [Google Scholar]
- Salloum A, Rowley JA, Mateika JH, Chowdhuri S, Omran Q, Badr MS. Increased propensity for central apnea in patients with obstructive sleep apnea: effect of nasal continuous positive airway pressure. Am J Respir Crit Care Med. 2009;181:189–193. doi: 10.1164/rccm.200810-1658OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sands SA, Edwards BA, Berger PJ, Shepherd KL, Dawson A, Swart M, Ainslie P, Burgess K. Impact of sleep state and position on ventilatory stability in healthy adults at high altitude (5050 m) J Sleep Res. 2011;20(Suppl. 1):21. [Google Scholar]
- Sforza E, Krieger J, Petiau C. Arousal threshold to respiratory stimuli in OSA patients: evidence for a sleep-dependent temporal rhythm. Sleep. 1999;22:69–75. [PubMed] [Google Scholar]
- Smith PL, Haponik EF, Bleecker ER. The effects of oxygen in patients with sleep apnea. Am Rev Respir Dis. 1984;130:958–963. doi: 10.1164/arrd.1984.130.6.958. [DOI] [PubMed] [Google Scholar]
- Solin P, Roebuck T, Johns DP, Walters EH, Naughton MT. Peripheral and central ventilatory responses in central sleep apnea with and without congestive heart failure. Am J Respir Crit Care Med. 2000;162:2194–2200. doi: 10.1164/ajrccm.162.6.2002024. [DOI] [PubMed] [Google Scholar]
- Tadjalli A, Duffin J, Peever J. Repeated obstructive apneas induce long-term facilitation of genioglossus muscle tone. Adv Exp Med Biol. 2010;669:297–301. doi: 10.1007/978-1-4419-5692-7_61. [DOI] [PubMed] [Google Scholar]
- Trinder J, Ivens C, Kleiman J, Kleverlaan D, White DP. The cardiorespiratory activation response at an arousal from sleep is independent of the level of CO2. J Sleep Res. 2006;15:174–182. doi: 10.1111/j.1365-2869.2006.00513.x. [DOI] [PubMed] [Google Scholar]
- Warner G, Skatrud JB, Dempsey JA. Effect of hypoxia-induced periodic breathing on upper airway obstruction during sleep. J Appl Physiol (1985) 1987;62:2201–2211. doi: 10.1152/jappl.1987.62.6.2201. [DOI] [PubMed] [Google Scholar]
- Waters KA, Tinworth KD. Habituation of arousal responses after intermittent hypercapnic hypoxia in piglets. Am J Respir Crit Care Med. 2005;171:1305–1311. doi: 10.1164/rccm.200405-595OC. [DOI] [PubMed] [Google Scholar]
- Wellman A, Eckert DJ, Jordan AS, Edwards BA, Passaglia CL, Jackson AC, Gautam S, Owens RL, Malhotra A, White DP. A method for measuring and modeling the physiological traits causing obstructive sleep apnea. J Appl Physiol (1985) 2011;110:1627–1637. doi: 10.1152/japplphysiol.00972.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellman A, Jordan AS, Malhotra A, Fogel RB, Katz ES, Schory K, Edwards JK, White DP. Ventilatory control and airway anatomy in obstructive sleep apnea. Am J Respir Crit Care Med. 2004;170:1225–1232. doi: 10.1164/rccm.200404-510OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellman A, Malhotra A, Jordan AS, Stevenson KE, Gautam S, White DP. Effect of oxygen in obstructive sleep apnea: role of loop gain. Respir Physiol Neurobiol. 2008;162:144–151. doi: 10.1016/j.resp.2008.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie A, Skatrud JB, Puleo DS, Dempsey JA. Influence of arterial O2 on the susceptibility to posthyperventilation apnea during sleep. J Appl Physiol (1985) 2006;100:171–177. doi: 10.1152/japplphysiol.00440.2005. [DOI] [PubMed] [Google Scholar]
- Xie A, Teodorescu M, Pegelow DF, Teodorescu MC, Gong Y, Fedie JE, Dempsey JA. Effects of stabilizing or increasing respiratory motor outputs on obstructive sleep apnea. J Appl Physiol (1985) 2013;115:22–33. doi: 10.1152/japplphysiol.00064.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]