

Abstract
The cellular phospholipid membrane plays an important role in cell function and cell–cell communication, but its biocomplexity and dynamic nature presents a challenge for examining cellular uptake of phospholipids and the resultant effects on cell function. Platelets, small anuclear circulating cell bodies that influence a wide variety of physiological functions through their dynamic secretory and adhesion behavior, present an ideal platform for exploring the effects of exogenous phospholipids on membrane phospholipid content and cell function. In this work, a broad range of platelet functions are quantitatively assessed by leveraging a variety of analytical chemistry techniques, including ultraperformance liquid chromatography–tandem electrospray ionization mass spectrometry (UPLC–MS/MS), vasculature-mimicking microfluidic analysis, and single cell carbon-fiber microelectrode amperometry (CFMA). The relative enrichments of phosphatidylserine (PS) and phosphatidylethanolamine (PE) were characterized with UPLC–MS/MS, and the effects of the enrichment of these two phospholipids on both platelet secretory behavior and adhesion were examined. Results show that, in fact, both PS and PE influence platelet adhesion and secretion. PS was enriched dramatically and decreased platelet adhesion as well as secretion from δ-, α-, and lysosomal granules. PE enrichment was moderate and increased secretion from platelet lysosomes. These insights illuminate the critical connection between membrane phospholipid character and platelet behavior, and both the methods and results presented herein are likely translatable to other mammalian cell systems.
The perception of the cellular phospholipid membrane as an inactive barrier between the cytosol and the extracellular space has been challenged by many recent studies. In particular, membrane-bound phospholipids have been shown to have active roles in cellular signaling and receptor expression.1,2 The membranes of mammalian cells contain phospholipids of numerous classes including phosphatidylserines (PS), phosphatidylethanolamines (PE), phosphatidylcholines (PC), and sphingomyelines (SM), as well as cholesterol and many membrane-bound peptides and proteins.3,4 Selectively examining the roles of individual membrane components is challenging because exposure to exogenous phospholipids can induce up- or down-regulation of any of the membrane components. Many studies employ model lipid bilayers, which eliminate nearly all of the biocomplexity of the cellular membrane,3 and it is unclear if studies on such model lipid bilayers translate to physiologically relevant systems. In this study, primary blood platelets are used as a platform to examine whether cellular membranes can incorporate exogenous phospholipids and if so, what effects enrichment of membrane phospholipids have on cellular function. The anuclear nature of platelets makes them an ideal platform for studies of membrane phospholipids as they have minimal capacity to up- or down-regulate protein expression in response to exposure to exogenous phospholipids.5 Additionally, platelets uniquely feature multiple types of secretory granules, each with a different type of stored cargo,6 which enables the study of phospholipid effects on different classes of granules and chemical messenger cargo.
The asymmetric distribution of phospholipids within cellular membranes has important consequences in cell–cell communication.1 Aminophospholipids, including phosphatidylserine (PS) and phosphatidylethanolamine (PE), are the abundant phospholipids in the plasma membrane, and they are localized to the inner leaflet of the plasma membrane.7−9 Upon platelet activation, both PS and PE are exposed to the outer membrane surface. It has been shown that both the asymmetric distribution at rest and scrambling of the phospholipids upon activation are critical for cellular adhesion and the chemical messenger secretion process; in fact, disruption of the phospholipid asymmetry and redistribution is known to impair these functions.10−14 Fusion between the granular membrane and the plasma membrane is a critical step of exocytosis (the secretion of preformed granule-stored chemical messenger species), and the characteristics and actions of membrane lipid species are of innate importance in these events.10,15,16 In fact, it has been shown that incubation with exogenous phospholipids can mediate both the mechanism and the kinetics of exocytotic events in model exocytotic systems such as PC12 and chromaffin cells.11−13,17 Because of their anuclear nature, platelet membranes are more stable and undergo minimal constitutive exocytosis, making it easier to draw conclusions about the direct effect of phospholipid substitution. Phospholipid content not only influences the fluidity and the curvature of the membrane but also promotes shape change and spreading of the platelets. When exposed to the outer leaflet of the platelet membrane, PS and PE serve as binding sites for circulating protein coagulation factors and also have catalytic activity in the formation of clots.
In addition to being an ideal model for studying the conserved process of exocytosis, platelets are also important players in various physiological processes, including hemostasis, inflammation, and angiogenesis, and phospholipids play a key role in these processes.6,18 As with other cells, an important regulatory component of the dynamic secretory and adhesion behavior of platelets is the phospholipid membrane, which plays an important role in influencing how platelets interact with their environment.3 While clearly important, the exact role of membrane phospholipids in platelet activation and adhesion is not well characterized, mainly due to analytical limitations in characterizing cellular uptake of phospholipids. Herein, this work provides an improved fundamental understanding of how changes in the phospholipid membrane affect platelet behavior through the measurement of numerous platelet functions on both the single-cell and ensemble levels. The methods and results presented herein give general understanding about the role of the phospholipid bilayer in cell function and can be easily adapted for use with other cell types. Additionally, the physiological relevance of studies on platelets, specifically, will lead to enhanced therapeutic approaches related to thrombosis, inflammation, and angiogenesis, among others.
Because of their small size and short lifespan (generally 1 day in vitro or 1–5 days for clinical use), platelet secretory functions are typically studied using indirect methods. In fact, platelet secretion occurs from three distinct populations of storage granules (each releasing unique messenger molecules) as well as on-demand manufacture of bioactive lipid species. To address the challenges associated with quantitative assessments of such a broad range of functions associated with a small and short-lived cell type, this work employs a variety of techniques to characterize the enrichments of PS and PE and the effects of PE or PS enrichment on both platelet secretory and adhesion behavior. In addition to measuring ensemble secretion of chemical messenger species from many platelets in suspension (platelet factor 4 (PF4) from α-granules, serotonin from δ-granules, and β-hexosaminidase from lysosomes), which reveals information about the behavior of many platelets while they are in communication with one another, single cell carbon-fiber microelectrode amperometry (CFMA) measurements enable evaluation of the phospholipid content effects on quantal release and kinetics of individual δ-granule secretion. In parallel, the PS- or PE-enriched platelet adhesion behavior was assessed within a microfluidic platform where the feature size and endothelial cell coating mimic intravenous vasculature. Results show that, in fact, both PS and PE influence platelet adhesion and secretion. PS enrichment decreased platelet adhesion and decreased the secretion of δ-, α-, and lysosomal granules while PE enrichment increased secretion from platelet lysosomes. CFMA measurements showed that PS regulates granule recruitment and influences the frequency of secretion events while PS enrichment improves the stability of granule-cell membrane fusion events. Overall, this work reveals the importance of phospholipids in regulation of platelet behavior and highlights the utility of platelets as a platform for exploring the involvement of membrane phospholipids in the functions of mammalian cells.
Materials and Methods
Safety Considerations
All manufacturer-recommended safety precautions were followed. Gloves, goggles, and labcoats should be worn when handling acetic acid, acetonitrile, methanol, acetone, or chloroform. Chloroform should be evaporated only in a hood. Animals should be handled in accordance with IACUC-approved protocols.
Platelet Isolation and Phospholipid Incubation
All reagents used were analytical grade or higher quality. KCl, MgCl2, N-2-hydroxyethylpiperazine-N′-2ethanesulfonic acid, and thrombin were purchased from Sigma-Aldrich. NaCl was purchased from BDH Chemicals. α-d-(+)-Glucose was purchased from Acros Organics. LC/MS-grade H2O and acetonitrile (ACN) were purchased from J.T. Baker. LC/MS-grade isopropyl alcohol (IPA), Na2CO3, and citric acid were purchased from Fisher Scientific. Sodium bicarbonate and sodium citrate were purchased from Mallinckrodt. Deuterated platelet activating factor (PAF-d4) was purchased from Cayman Chemical, and all phospholipids were purchased in chloroform from Avanti Polar Lipids, species no. 840032 for PS (porcine brain), no. 841118 for PE (chicken egg), no. 860061 for SM (chicken egg), and no. 840054 for PC (soy). Nine-week old C57BL/6J mice were purchased from The Jackson Laboratory.
Blood was drawn via cardiac puncture from 10 week old mice euthanized via CO2 asphyxiation (following IACUC-approved protocol no. 0806A37663) using 1 mL syringes prefilled with 200 μL of acid citrate dextrose (ACD), and washed platelets was isolated using the procedure described previously.19
The 200 μM PS, PC, PE, and SM solutions were prepared by drying appropriate volumes of phospholipids as received in chloroform in glass vials under a stream of nitrogen and sonicating in Tyrode’s buffer for 2 h, until all cloudiness had disappeared from the solutions. Phospholipid solutions were then filtered using 0.2 μm filters. Platelets were incubated with phospholipid or negative control solutions for 2 h at 37 °C.
Relative Quantification of Phospholipid Enrichment
Relative enrichments of PS, PC, PE, and SM in platelets were assessed using ultraperformance liquid chromatography coupled to tandem mass spectrometry (UPLC–MS/MS). It is known that the phospholipid standards used were not composed of pure samples of one phospholipid species, so absolute quantitation was not feasible. Rather, the primary species in each phospholipid standard was used as a reference to assess the enrichment of phospholipid-incubated platelets as compared to control, nonincubated platelets. Following incubation with PS, PC, PE, SM, or Tyrode’s buffer (control), platelets were washed twice by centrifugation at 1000g and resuspended in 6:1 Tyrode’s buffer/ACD. After the final wash, platelets were resuspended in 100 μL of Tyrode’s buffer, and phospholipids were extracted using a modified Bligh and Dyer extraction. Platelets were mixed with 400 μL of CHCl3/200 μL methanol (MeOH)/10 μL 19.1 μM PAF-d4 (internal standard) and sonicated for 20 min. A volume of 100 μL of 0.1% acetic acid in 0.1 M NaCl was then added, and samples were sonicated for an additional 10 min. Samples were centrifuged for 5 min at 1500 g, and the upper aqueous layer was removed and discarded. The lower organic layer was dried under vacuum, and phospholipids were resuspended in 200 μL of 0.1% acetic acid in 40/60 A/B (A was 20 mM ammonium acetate in water, pH 5, and organic mobile phase B was 0.1% acetic acid in 9:1 ACN/acetone) by 1 h sonication. Samples were centrifuged at 1500 g for 5 min and transferred to fresh tubes prior to UPLC–MS/MS analysis.
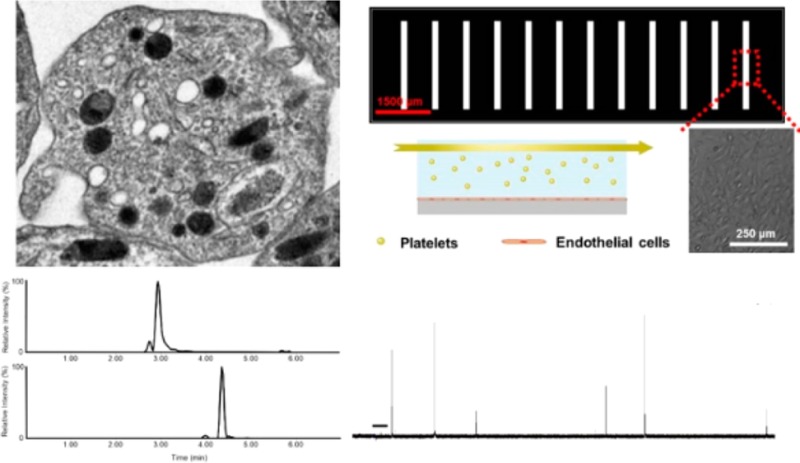
UPLC–MS/MS analysis was performed using a Waters Acquity triple quadrupole mass spectrometer using a modified version of the chromatography suggested by Rainville and Plumb with a Waters Acquity BEH C8(1.7 μm) 2.1 mm × 100 mm column.20 Deuterated platelet-activating factor (PAF-d4) was used as an internal standard. Chromatography used a flow rate of 0.6 mL/min: 60% B, 0 to 1.0 min; 60% B to 80% B, 1.0 to 2.0 min; 80% B to 84% B, 2.0 to 2.5 min; 84% B, 2.5 to 2.75 min; 84% B to 86% B, 2.75 to 3.0 min; 86% B, 3.0 to 3.25 min; 86% B to 87.7% B, 3.25 to 3.5 min; 87.7% B, 3.5 to 3.75 min; 87.7% B to 95% B, 3.75 to 4 min; 95% B, 4.0 to 5.0 min; 95% B to 60% B, 5.0 to 5.5 min; 60% B, 5.5 to 7.0 min. Electrospray ionization tandem mass spectrometry (ESI-MS/MS) was operated in positive ionization mode using the following parameters: capillary, 3.8 kV; extractor, 3.00 V; rf lens, 0.30 V; source temperature, 120 °C; desolvation temperature, 400 °C; cone gas flow, 20 L/h; desolvation gas flow, 800 L/h; collision gas flow, 0.2 mL/min; low-mass resolution (Q1), 12.00; high-mass resolution (Q1), 12.00; ion energy (Q1), 0.30; and species-specific transitions are listed in Table 1.
Table 1. Summary of the UPLC–MS/MS Analysis of Each Phospholipid.
| phospholipid incubation condition | control | PS | PE |
|---|---|---|---|
| transition used for relative quantitation | n/a | 812.5 → 208.0 | 718.3 → 577.3 |
| injection precision (RSD) | n/a | 5.91% | 15.9% |
| RSD of biological replicates | n/a | 23.3 | 13.3 |
| total protein in pelleted platelets (μg/mL ± SD) | 117 ± 17 | 113 ± 26 | 118 ± 11 |
| percent increase in platelet phospholipid upon incubation (range in 4 replicates) | n/a | 760–874 | 12.9–29.7 |
| average percent phospholipid increase (percent ± SD) | n/a | 808 ± 59 | 18.7 ± 7.5 |
Relative quantification of PS, PC, PE, and SM in each of control and phospholipid-incubated sample was accomplished using a calibration curve of phospholipid standards subjected to the same extraction procedure as the platelet samples; however, no platelets were present in the calibration solutions. To account for variation in pelleting behavior of platelets induced by phospholipid incubation, phospholipid quantification results were normalized to average protein values of control- or phospholipid-incubated nonactivated platelet pellets, as determined using a Pierce bicinchoninic acid (BCA) assay.
Adhesion of Platelets Incubated with Phospholipids
A microfluidic device, employed to monitor platelet adhesion behavior with varied phospholipid content, was fabricated as was previously described.21An endothelial cell coating of the microfluidic channel is achieved as previously described22 to simulate blood vessel architecture. Detailed descriptions of device fabrication, endothelial cell culture, and endothelial cell coating of the microfluidic device can be found in the Supporting Information. Devices with a uniform monolayer of endothelial cells were selected for experiments, and the channel was washed with fresh Tyrode’s buffer before exposure of endothelial cells to a stream of platelets. To facilitate visual distinction of platelets from endothelial cells, platelets were labeled with 5-chloromethylfluorescein diacetate (CMFDA19, Invitrogen) dye prior to phospholipid incubation. After phospholipid incubation, platelets were activated with 5 μM ADP and introduced into the microfluidic channel. Flow control was accomplished using a syringe pump, and platelet suspensions were introduced onto the device through Teflon tubing. The endothelial cell-coated channel was exposed to a stream of platelets for 20 min at a constant flow rate of 30 μL/h. This flow rate was chosen to avoid shear stress levels above 0.01 N/m2 (COMSOL simulation data not shown) to minimize shear stress-induced platelet activation. After endothelial cell exposure to a stream of platelets, the cell culture channel was washed with fresh Tyrode’s buffer and fluorescence images were obtained on an inverted microscope (Nikon, Melville, NY) equipped with a CCD camera (QuantEM, Photometrics, Tucson, AZ) using Metamorph version 7.7.5 imaging software. Platelets adhered to endothelial cells were counted, and for each experimental condition, the results from five images (450 μm × 500 μm each) were averaged; in each condition, five biological replicates were measured.
Ensemble Platelet Secretion Measurements
Platelet δ-granule secretion was measured after incubation of the washed platelets with phospholipids using a modified version of a previously published HPLC method making use of an electrochemical detector.23,24 For α-granule secretion, a PF4 Sandwich-ELISA assay kit was purchased from R&D Systems and used as directed. Lysosomal secretion was measured via a modified β-Hexosaminidase assay.25 Briefly, following incubation of platelets with phospholipid solutions, platelets were exposed to Tyrode’s buffer containing 10 U/mL thrombin (a physiological stimulant of platelet activation) or Tyrode’s buffer (as control). Platelets were centrifuged at 500 RCF to pellet. Supernatant portions were used for the analysis of the secreted species (serotonin by HPLC, PF4 with the ELISA assay, and secreted hexosaminidase with the β-hexosaminidase assay). Secreted platelet-activating factor (PAF) was measured using a previously published UPLC–MS/MS method.24 Because of the low recovery of PAF in Tyrode’s buffer, PAF secretion was measured from platelets incubated in phosphate buffered saline (free of Ca2+ and Mg2+, containing 1 g/L glucose). A BCA protein assay was used for normalization of the results to prevent any differences in the data due to pelleting differences between control and phospholipid-incubated platelets. Ensemble platelet secretion measurements are reported as secreted species per microgram of pelleted protein.
Total Protein Quantitation
Total protein in pelleted platelets was quantified with a Pierce bicinchoninic acid (BCA) assay from Thermo Scientific, used as directed. Protein was extracted from platelet pellets with mammalian protein extraction reagent from Thermo Scientific, used as directed.
Carbon-Fiber Microelectrode Amperometry Measurements
CFMA experiments were performed as previously described.26,27After incubation with the phospholipid of interest, a drop of the washed platelets was added to an experimental chamber filled with Tyrode’s buffer supplemented with the phospholipid of interest. A 7-μm-diameter carbon-fiber microelectrode (fabricated in house) was placed onto an individual platelet using piezoelectric micromanipulators, and then the platelet was activated by local application of Tyrode’s buffer containing 10 U/mL thrombin solution. With an applied potential of 700 mV versus a Ag/AgCl reference at the carbon-fiber microelectrode, serotonin secretion from δ-granules was measured as current versus time for 90 s after the stimulation. Each phospholipid condition was compared with its own control measured from the same platelet sample on the same day. Each current spike was analyzed, and the aggregate data from at least 20 individual platelets was treated statistically as has been previously described.23,28
Results and Discussion
Relative Quantitation of Phospholipids
Quantitation of lipid species presents an analytical chemistry challenge due to the limited solubility of phospholipids in solvents commonly used for liquid chromatography. Herein, relative phospholipid enrichment of platelet membranes was assessed using an UPLC–MS/MS method. Mobile phase composition and column selection were based on previously published work reported by Rainville and Plumb,20 and chromatography was modified from the same report to a 7 min separation by adjusting the chromatographic conditions to optimize elution for each compound. An absolute quantification method was not possible because the phospholipid standards contained mixtures of fatty acid tails with a common headgroup, but this relative quantitation method can be adjusted for use with many phospholipids of interest. Fragmentation transitions for the primary component of each phospholipid were monitored, and lipids extracted from phospholipid-incubated platelets were compared to those extracted from control platelets. Phospholipid concentration values were then normalized to extracted protein values from platelet pellets incubated with phospholipids and washed under the same conditions as the platelets from which the phospholipids were extracted. Calibration curves were prepared to ensure a linear detector response over the relevant phospholipid concentration range. Selected reaction monitoring (SRM) transitions that were used for phospholipid enrichment assessment are given in Table 1. All phospholipid transitions were monitored in all platelet samples. For phosphatidylserines (PS) and phosphatidylethanolamines (PE), enrichment was observed for the phospholipid in which the platelets had been incubated (Table 1). Thus, 2 h incubation was enough time for platelets to take up exogenous phospholipid. This allowed a direct comparison of the effect of enrichment of each particular phospholipid on platelet secretion and adhesion behavior. The percent increase in phospholipid content was 808 ± 59 and 18.7 ± 7.5 for PS and PE conditions, respectively (Table 1). This study is the first to demonstrate that platelets exposed to exogenous phospholipids can take them up and can do so preferentially. The aforementioned errors in phospholipid enrichment are the standard deviations of the enrichment. Surprisingly, the % enrichment for PS incubation was drastically higher than the enrichment for PE.
Platelets were incubated with each of the phospholipids for 2 h, long enough to enrich the platelets with the phospholipid of interest but short enough to minimize de novo regulation of other phospholipids. The drastically larger PS enrichment compared to PE enrichment is potentially due to a paucity of the particular PS species monitored in control platelets, which results in a much larger relative increase.
In addition to PS and PE, effects of incubation with the phospholipids sphingomyelin (SM) and phosphatidylcholine (PC) were also examined. While these two phospholipids influenced platelet secretion and adhesion behavior, incubation with PC and SM did not consistently result in enrichment of the phospholipid with which the platelets were incubated. Effects of PC and SM on platelet behavior can be found in the Supporting Information.
Phospholipids and Platelet Adhesion
The effect of plasma membrane phospholipid content on platelet adhesion was assessed on an endothelial cell-coated microfluidic platform to mimic in vivo vascular conditions. Consistent with other assessments described herein, fluorescently labeled platelets were incubated with PE or PS prior to injection through the endothelial cell-coated microfluidic channel. A short video of platelets flowing through the endothelial cell-coated channel can be found in the Supporting Information. In the microfluidic adhesion experiments, adenosine diphosphate (ADP) was used to activate platelets. The use of ADP as the stimulus makes it possible to decouple investigation of adhesion from that of secretion because ADP is known to induce platelet adhesion without initiating secretion.29 While the adhesion of ADP-activated platelets from each condition was relatively consistent, the number of platelets adhered to endothelial cells varied from day to day; as such, comparisons of platelet adhesion to endothelial cells under each phospholipid condition were calculated with respect to the number of control platelets adhered to endothelial cells.
The ratios measured for each condition were tested to see if they were statistically different from the value 1 because a ratio of 1 indicates the same number of adhered platelet in the control and phospholipid-enriched platelets. As seen in Figure 1, platelets enriched with PS showed reduced adherence to endothelial cells compared to the control condition (0.625 ± 0.075, p = 0.004). In contrast, platelets enriched with PE adhered to the endothelial layer in significantly higher numbers, with a ratio of 1.55 ± 0.02 (p = 0.04). Preincubation with either PC or SM also resulted in suppressed adhesion (Supporting Information).
Figure 1.
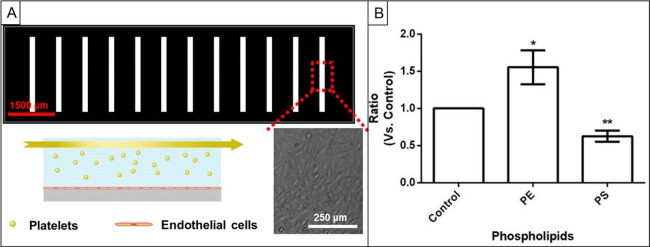
Platelet adhesion measurements. (A) Schematic of microfluidic platelet adhesion experiment with light microscopy inset showing platelets adhering to the endothelial cell layer. (B) Ratio of the number of platelets adhered to the endothelial cell layer at each phospholipid enrichment condition compared to the control.
Although PS is mostly known for its procoagulant function, PS enrichment in fact diminished the platelet adhesion compared to control platelets. Besides providing a negatively charged surface to facilitate the binding of the coagulant factors, flipping PS from the inner leaflet of the plasma membrane to the outer leaflet provides reorganization of the coagulation proteins in the plasma membrane. Upon PS incubation, it is likely that the PS concentration on the outer leaflet of the platelet membrane increased and impaired the translocation of the endogenous PS from the inner leaflet to the outer leaflet, thus diminishing the reorganization of the coagulation proteins that would facilitate platelet adhesion to endothelial cells. Moreover, since PS is negatively charged, PS enrichment of the platelet membrane will increase the anionic character of the membrane and prevent its interactions with other cells due to electrostatic repulsion. This observation is in agreement with previous findings in which the higher plasma level of PS decreased platelet coagulation and thrombus formation.7,30 Unlike PS, PE enrichment increased the positive charge on the platelet membrane and boosted platelet adhesion by causing 50% more platelet adhesion than control platelets. Previous work published by Zieseniss et al. showed that oxidized PE is one of the major components of low density lipoproteins that cause very strong thrombotic function.31 A similar effect explains the observations of this work and reveals that PE synthesis and oxidation may be a good target for thrombosis prevention.
Assessment of α-Granule, δ-Granule, Lysosomal Release and PAF Secretion at Altered Phospholipid Levels with Ensemble Secretion Assays
Bulk secretion assays were performed under conditions with phospholipid enrichment. In all cases, the amount of secreted molecules from each type of granule was normalized according to platelet protein content. Platelet factor 4 (PF4) was measured from α-granules, serotonin was measured from δ-granules, β-hexosaminidase was measured from lysosomes, and platelet-activating factor (PAF) was measured as it was manufactured from platelet lipid membranes.
Each of the measured species was selected because of their important physiological functions, both in platelets and across other immune cell classes. The protein content of α-granules includes soluble proteins that are secreted to promote coagulation as well as proteins that are bound to the external platelet surface during activation.32−34 PF4, a soluble secreted protein, promotes blood clotting through binding to circulating anticoagulation molecules such as heparin.35 δ-granules influence coagulation through storage and secretion of small biogenic amines, polyphosphate, Ca2+, ADP, and ATP. Because of its electroactive nature, serotonin can be detected at the single-platelet and bulk-suspension level, making it an ideal a marker of δ-granule secretion.
Lysosomes contain acid hydrolases, which are a class of enzymes found in other secretory cells including mast cells and polymorphonuclear leukocytes.25,36 While the secretion of δ-granules and α-granules from platelets has been studied extensively, relatively little is known about platelet lysosomal secretion. β-Hexosaminidase (β-Hex) is easily assayed using an absorbance measurement and is widely used as a marker of acid hydrolase activity. As its name suggests, PAF is a secreted phospholipid that is widely regarded as a potent contributor to the platelet activation cascade. Additionally, PAF is secreted from and acts on many other cell types, including macrophages, mast cells, and polymorphonuclear leukocytes.37
Ensemble assay results show that an increase in the PS content impairs both α- and δ-granule secretion as well as lysosomal secretion. While control platelets secreted 0.043 ± 0.011 μmol of serotonin/μg of protein upon stimulation with thrombin, PS incubation led to a significant decrease in the average amount of δ-granule-localized serotonin secreted to 0.015 ± 0.010 μmol serotonin/μg protein (Figure 2D; p = 0.008). A more drastic effect was observed in α-granule secretion; upon PS enrichment, PF4 secretion was completely attenuated, as PF4 values were not significantly different (p = 0.2) from those of the unactivated platelets (Figure 2B). PS also impairs lysosomal secretion and PAF secretion, though to a lesser extent than α- and δ-granule secretion. However, unlike α- and δ-granular release, lysosomal secretion and PAF secretion were unchanged by PE enrichment compared to control platelets.
Figure 2.

Assessment of phospholipid effects on ensemble platelet granule secretion. (A) Transmission electron micrograph of a platelet and a representative figure illustrating three different platelet granule types; δ-granule, bull’s eye shape; lysosome, pink shape; and α-granule, uniformly filled irregular shapes. Phospholipids are asymmetrically distributed in the platelet membrane. (B) PF4 release from α-granules decreased with enrichment of each of the phospholipids studied. (C) Lysosomal release decreased with PS enrichment. (D) δ-granule secretion was also suppressed upon incubation with phospholipids. (E) PAF secretion was suppressed upon PS enrichment. *p < 0.05 and **p < 0.01 compared to thrombin-stimulated control platelets.
During exocytosis, phospholipids not only serve as a matrix, accommodating essential proteins for membrane fusion but, by controlling the fluidity and curvature of the membrane, they influence the energy required for each step of exocytosis, especially the formation and stability of the granule-cell membrane fusion pore.17,38−41 It is clear from literature precedent that phospholipids interact with the exocytotic machinery and, moreover, that there are specific subunits of proteins involved in recruitment and secretion of exocytotic granules.42,43 A similar scenario may be true for lysosomal release where specific interactions of PE are required. Although phospholipid regulation of lysosomal function has been reported in other cell types44 there is not any precedent work either with evaluation of increased cellular phospholipid content or on platelets; thus, future work will explore these data further.
Platelets, like many other eukaryotic cell types, secrete bioactive lipids that are enzymatically synthesized from phospholipids, and it is interesting to consider that changes in the composition of the phospholipid bilayer may influence de novo-generated bioactive lipids. The interactions between phospholipids and circulating or membrane-bound enzymes are clearly necessary for bioactive lipid generation. Herein, PS enrichment suppressed PAF secretion, although similar effects were not observed with PE enrichment. This could be a result of the different degrees of enrichment of PS and PE or different action of the two species. For example, suppression of PAF secretion upon enrichment with PS could be a result of increased interactions between PS and the enzymes responsible for PAF synthesis (phospholipase A2 and lyso-PAF acetyltransferase) that result in suppression of enzyme activity or suppression of enzyme interaction with phosphatidylcholine.
Effect of Phospholipids on Single Platelet δ-Granule Secretion
While the ensemble assays clearly indicate that phospholipid content influences platelet secretion, mechanistic insight about why or how these changes occur would be helpful. In fact, because δ-granules contain electroactive serotonin, CFMA can be employed to achieve biophysical understanding of the phospholipid influence on platelet secretion. The submillisecond time resolution of the CFMA technique enables a detailed characterization and comparison of the δ-granule secretion in control and phospholipid-enriched platelets.26 Parameters analyzed for each δ-granule, manifested in amperometric traces as individual current spikes, include the total secreted serotonin (Q), kinetics of secretion (t1/2), total number of granule fusion events per platelet (N), and the percent of fusion events that exhibit a foot feature. In amperometric analysis of exocytosis, the presence of a foot feature (a small increase in measured current that is immediately adjacent to a large “full fusion” current spike) reveals information about the stability of the membrane fusion pore that forms prior to dilation of the pore for more complete chemical messenger secretion. The numbers of individual platelets analyzed using CFMA per phospholipid condition were (control = 47, PE = 27); (control = 20, PS = 18); (control = 20, PC = 21); and (control = 58, SM = 45).
Although incubation of the platelets with PE resulted in enrichment of this phospholipid, PE-enriched platelets did not show a significant change in δ-granule secretion (Figure 3). The number of events with a foot feature was also not significantly changed with PE incubation compared to the control condition (p = 0.08).
Figure 3.

Effect of PE on δ-granule quantal release and release kinetics. (A) PE did not influence the amount of serotonin released from single δ-granules. Kinetics of the release (B and C) were also unchanged. (D) Similar numbers of δ-granules were exocytosed from individual platelets with comparable fusion pore stability (E). Cumulative frequencies of the release are similar for each condition (F). (G) Representative amperometric traces from a control platelet (top) and PE-enriched platelet (bottom). Thrombin was applied for 3 s, as indicated by the black bar on traces in part G.
In contrast, PS, the most significantly enriched phospholipid of those considered, did induce platelet δ-granule secretion alterations (Figure 4). Upon PS enrichment, the amount of chemical messenger secreted per granule (the quantal secretion) did not change (Q = 263 ± 36 fC vs 318 ± 29 fC for control and PS conditions, respectively; p = 0.2). Since the average number of granules secreted from individual platelets also did not change between these conditions, it can be concluded that the total amount of serotonin secretion per platelet does not change with PS enrichment. This contradicts the bulk serotonin secretion measurement done using HPLC where enriched PS lowered the amount of secreted serotonin. Since single cell measurements are performed by measuring the action of one cell at a time in a relatively isolated space, it is unlikely that the activated secretion of one platelet can have a downstream effect of activation on another one. In a bulk suspension of platelets, however, activated platelets can secrete chemical messenger species that have the downstream effect of activating other platelets. By combining both single cell and ensemble measurements of platelet function, this study reveals insight into both the platelet behavior at the cellular level as well as the behavior of platelets in a suspension where they can influence one another. Ensemble measurements yield insight into the downstream effects of platelet activation and the feedback loops involved in platelet function. The discrepancy between the single cell and ensemble measurement indicates that the downstream effect of activated platelets on the larger platelet population is important. However, without single cell measurements, it is not possible to observe the actual response to the stimulation nor the heterogeneity in the response among a population of platelets.
Figure 4.

Effect of PS on δ-granule quantal release and release kinetics. (A) PS did not influence the amount of serotonin released from single granules. Similar Trise (B) but higher T1/2, **p = 0.005 (C) values were obtained, indicating slower chemical messenger secretion with enriched PS. (D) The number of granules exocytosed from single platelets did not change. (E) PS increased fusion pore stability, *p = 0.05. (F) Cumulative frequency analysis shows initially fast but later slowed granule trafficking in PS-enriched platelets. (G) Representative amperometric traces from a control platelet (top) and PE-enriched platelet (bottom). Thrombin was applied for 3 s, as indicated by the black bar on traces in part G.
Although CFMA reveals a similar amount of serotonin is extruded from platelet granules in both control and PS-enriched conditions, the secretion kinetics slow at higher membrane PS concentrations (t1/2 values are 12.4 ± 3.0 and 21.7 ± 2.4 ms for control and PS conditions, respectively; p = 0.005). While the number of granules secreted from individual platelets (N values) did not change, the platelets enriched with PS appear to have two different phases in terms of time evolution of exocytosis (Figure 4F). In the early phase, the granules that were docked before activation are released with a comparable frequency to the control condition. However, the frequency of the granular secretion decreases as the cell secretion continues, showing slower granule trafficking and docking compared to the control condition. This implies a regulatory effect of PS on granular recruitment but not docking. Finally, fusion pore analysis showed that PS enrichment increases the stability of the fusion pore, with foot events occurring in 23.6 ± 4.6% of the total secretion events, compared to 12.4 ± 3.0% foot events observed for the control.
Although PE supports negative membrane curvature, PS packs better on positively curved membranes. The role of phospholipid curvature in exocytosis becomes more prominent during fusion pore formation.17,38−41 Fusion pores are highly curved membrane structures that require phospholipids with positive curvature to pack in the outer leaflet and the phospholipids with negative curvature to pack on the inner leaflet for stabilization. To achieve a stable fusion pore, the phospholipid with appropriate curvature should be on the appropriate side of the membrane. Although PE incubation slightly increased the % of foot events, the changes were not significant (p = 0.08). While it is known that the lipids with negative curvature stabilize the fusion pore better, PS (with positive curvature) provided more stable fusion pores compared to untreated platelets. This can be attributed to the exposed PS on the surface of the membrane upon activation which stabilizes the positive curvature of the fusion pore. Previous work has reported that the time of PS transport from the inner leaflet to the outer leaflet of the membrane is 3 min.7 During 90 s of amperometric recordings, it is likely that more than half of the PS was exposed to the plasma outer surface. Moreover, previous work demonstrated that PC12 cells exposed to PS also displayed more stable fusion pore events; this was attributed to the interaction of PS with synaptotagmin, giving rise to a high negative curvature membrane.17,38−41,45 Platelets have synaptotagmin-like protein 1, and its interaction with PS is not known but certainly may follow a similar behavior.46
Conclusions
Herein, this study has demonstrated the utility of platelets as a tool for understanding how different phospholipids can act on different aspects of platelet function. Platelets were exposed to two of the most common inner-leaflet phospholipids, PS and PE, and this study demonstrates that platelets can incorporate exogenous phospholipids into their membranes. Results indicated that enrichment of PS generally suppresses platelet function, and although PE at the enriched level does not affect platelet α- and δ-granule secretion, it overactivates the thrombotic function of platelets. Moreover, this work highlights the possibility of interaction between phospholipids and important proteins involved in the secretion process, and the results and methods herein can be easily applied to other mammalian cell types using analogous cellular machinery. Elucidating the function of lipid–protein interactions will improve our current understanding of platelet secretion since it is obvious that the platelet secretion is regulated synergistically by both protein and lipid species.
Acknowledgments
The authors would like to acknowledge Sarah M. Gruba for assistance with preparing figures and Dr. Melissa A. Maurer-Jones for acquiring transmission electron micrographs of platelets. The authors would also like to acknowledge the University of Minnesota Nanofabrication Center, and this work was supported by the National Institutes of Health New Innovator Award 1 DP2 OD004258-01 to C.L.H. and a University of Minnesota Doctoral Dissertation Fellowship to A.F.M.
Supporting Information Available
Effects of PC and SM incubation on platelet secretion and adhesion; additional details regarding platelet isolation from whole blood, microfluidic device fabrication and coating with endothelial cells, and UPLC–MS/MS measurement of secreted PAF. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
S.K. and A.F.M. contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Zwaal R. F. A.; Schroit A. J. Blood 1997, 89, 1121–1132. [PubMed] [Google Scholar]
- Dykstra M.; Cherukuri A.; Sohn H. W.; Tzeng S. J.; Pierce S. K. Annu. Rev. Immunol. 2003, 21, 457–481. [DOI] [PubMed] [Google Scholar]
- Bach D.; Wachtel E. Biochim. Biophys. Acta: Biomembranes 2003, 1610, 187–197. [DOI] [PubMed] [Google Scholar]
- Fadeel B.; Xue D. Crit. Rev. Biochem. Mol. 2009, 44, 264–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert P.; Devine D. V. Vox Sanguinis 2010, 99, 112–122. [DOI] [PubMed] [Google Scholar]
- Ge S.; White J. G.; Haynes C. L. ACS Chem. Biol. 2010, 5, 819–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemskerk J.; Bevers E.; Lindhout T. Thromb. Haemostasis 2002, 88, 186–193. [PubMed] [Google Scholar]
- Zwaal R.; Comfurius P.; Bevers E. Biochim. Biophys. Acta: Rev. Biomembranes 1998, 1376, 433–453. [DOI] [PubMed] [Google Scholar]
- Zwaal R. F.; Comfurius P.; van Deenen L. L. Nature 1977, 268, 358–360. [DOI] [PubMed] [Google Scholar]
- Chasserot-Golaz S.; Coorssen J. R.; Meunier F. A.; Vitale N. Cell. Mol. Neurobiol. 2010, 30, 1335–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama Y.; Maxson M. M.; Sawada T.; Nakano A.; Ewing A. G. Brain Res. 2007, 1151, 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Hui E.; Chapman E. R.; Jackson M. B. Mol. Biol. Cell 2009, 20, 5086–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amatore C.; Arbault S.; Bouret Y.; Guille M.; Lemaître F.; Verchier Y. ChemBioChem 2006, 7, 1998–2003. [DOI] [PubMed] [Google Scholar]
- Kato N.; Nakanishi M.; Hirashima N. Biochemistry 2002, 41, 8068–8074. [DOI] [PubMed] [Google Scholar]
- Chernomordik L. V.; Kozlov M. M. Nat. Struct. Mol. Biol. 2008, 15, 675–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik L.; Kozlov M. M.; Zimmerberg J. J. Membr. Biol. 1995, 146, 1–14. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Jackson M. B. Biophys. J. 2010, 98, 2524–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaumenhaft R. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1152–1160. [DOI] [PubMed] [Google Scholar]
- Peters C. G.; Michelson A. D.; Flaumenhaft R. Blood 2012, 120, 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainville P.; Plumb R.. Separating Phospholipids with UPLC/MS; Waters: Milford, MA, 2007.
- Kim D.; Haynes C. L. Anal. Chem. 2012, 84, 6070–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.; Lin Y.-S.; Haynes C. L. Anal. Chem. 2011, 83, 8377–8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge S.; Woo E.; Haynes C. L. Biophys. J. 2011, 101, 2351–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer A. F.; Thompson J. T.; Wang Y.; Koseoglu S.; Haynes C. L.; Dalluge J. J. Analyst 2013, 138, 5697–5705. [DOI] [PubMed] [Google Scholar]
- Blank U.; Rivera J. Assays for Regulated Exocytosis of Mast Cells. In Current Protocols in Cell Biology; John Wiley & Sons, Inc.: New York, 2006. [DOI] [PubMed] [Google Scholar]
- Kim D.; Koseoglu S.; Manning B. M.; Meyer A. F.; Haynes C. L. Anal. Chem. 2011, 83, 7242–7249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge S.; Woo E.; White J. G.; Haynes C. L. Anal. Chem. 2011, 83, 2598–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge S.; White J. G.; Haynes C. L. Anal. Chem. 2009, 81, 2935–2943. [DOI] [PubMed] [Google Scholar]
- Platelets and Megakaryocytes: Functional Assays; Gibbins J. M., Mahaut-Smith M. P., Eds.; Methods in Molecular Biology, Vol. 1; Humana Press, Inc.: Totowa, NJ, 2004; p 272. [Google Scholar]
- Silver M. J. Am. J. Physiol. 1965, 209, 1128–1136. [DOI] [PubMed] [Google Scholar]
- Zieseniss S.; Zahler S.; Muller I.; Hermetter A.; Engelmann B. J. Biol. Chem. 2001, 276, 19828–19835. [DOI] [PubMed] [Google Scholar]
- Blair P.; Flaumenhaft R. Blood Rev. 2009, 23, 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger G.; Caen J. P.; Berndt M. C.; Cramer E. M. Blood 1993, 82, 3034–3044. [PubMed] [Google Scholar]
- Suzuki H.; Murasaki K.; Kodama K.; Takayama H. Br. J. Hamaetol. 2003, 121, 904–912. [DOI] [PubMed] [Google Scholar]
- Briquet-Laugier V.; Lavenu-Bombled C.; Schmitt A.; Leboeuf M.; Uzan G.; Dubart-Kupperschmitt A.; Rosa J. P. J. Thromb. Haemost. 2004, 2, 2231–2240. [DOI] [PubMed] [Google Scholar]
- Emiliani C.; Ciferri S.; Mencarelli S.; Mezzasoma A. M.; Momi S.; Orlacchio A.; Gresele P. Platelets 2006, 17, 20–29. [DOI] [PubMed] [Google Scholar]
- Prescott S. M.; Zimmerman G. A.; Stafforini D. M.; McIntyre T. M. Annu. Rev. Biochem. 2000, 69, 419–445. [DOI] [PubMed] [Google Scholar]
- Lai A. L.; Tamm L. K.; Ellena J. F.; Cafiso D. S. J. Biol. Chem. 2011, 286, 25291–25300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon H. T.; Kozlov M. M.; Martens S. Cell 2010, 140, 601–605. [DOI] [PubMed] [Google Scholar]
- Churchward M. A.; Rogasevskaia T.; Brandman D. M.; Khosravani H.; Nava P.; Atkinson J. K.; Coorssen J. R. Biophys. J. 2008, 94, 3976–3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller N.; Benatti C. R.; Rand R. P. Biophys. J. 2003, 85, 1667–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed G. L.; Fitzgerald M. L.; Polgár J. Blood 2000, 96, 3334–3342. [PubMed] [Google Scholar]
- Orita S.; Naito A.; Sakaguchi G.; Maeda M.; Igarashi H.; Sasaki T.; Takai Y. J. Biol. Chem. 1997, 272, 16081–16084. [DOI] [PubMed] [Google Scholar]
- Clement A. B.; Gamerdinger M.; Tamboli I. Y.; Lütjohann D.; Walter J.; Greeve I.; Gimpl G.; Behl C. J. Neurochem. 2009, 111, 669–682. [DOI] [PubMed] [Google Scholar]
- Richard J. P.; Leikina E.; Langen R.; Henne W. M.; Popova M.; Balla T.; McMahon H. T.; Kozlov M. M.; Chernomordik L. V. Biochem. J. 2011, 440, 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumueller O.; Hoffmeister M.; Babica J.; Prelle C.; Gegenbauer K.; Smolenski A. P. Blood 2009, 114, 1396–1404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.