

Abstract
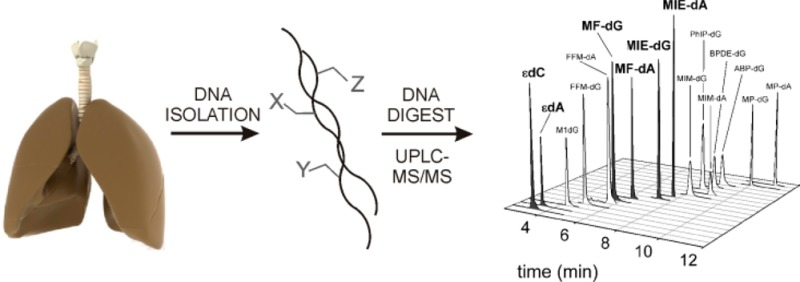
Recent studies have demonstrated that various DNA adducts can be detected in human tissues and fluids using liquid chromatography connected to tandem mass spectrometry (LC-MS/MS). However, the utility of a single DNA adduct as a biomarker in risk assessment is debatable because humans are exposed to many genotoxicants. We established a method to measure DNA adducts derived from 16 ubiquitous genotoxicants and developed an analytical technique for their simultaneous quantification by ultra performance liquid chromatography (UPLC)-MS/MS. Methods for the enrichment of the analytes from DNA hydrolysates and chromatographic separation preceding mass spectrometric analysis were optimized, and the resultant technique was used for the simultaneous analysis of the 16 DNA adducts in human lung biopsy specimens. Eleven adducts (formed by benzo[a]pyrene, 1-methylpyrene, 4-aminobiphenyl, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine, 1-methoxy-3-indolylmethylglucosinolate, 5-hydroxymethylfurfural, and malondialdehyde) were not detected in any tissue sample (limits of detection: 0.02–7.1 adducts/108 nucleosides). 3,N4-etheno-2′-deoxycytidine and 1,N6-etheno-2′-deoxyadenosine, formed from 2,3-epoxyaldehydes of endogenous lipid peroxidation products, were present in all subjects (16.9–115.3 and 27.2–179/108 nucleosides, respectively). The same was true for N2-(trans-methylisoeugenol-3′-yl)-2′-deoxyguanosine, the major adduct of methyleugenol (1.7–23.7/108 nucleosides). A minor adduct of methyleugenol and two adducts of furfuryl alcohol were detected in several pulmonary specimens. Taken together, we developed a targeted approach for the simultaneous mass spectrometric analyses of 16 DNA adducts, which can be easily extended by adducts formed from other mutagens. The method allowed one to detect adducts of furfuryl alcohol and methyleugenol in samples of human lung.
Deoxyribonucleic acid (DNA) adducts are a major cause of mutations which can lead to the development of tumors. The measurement of DNA adducts is an estimate of the exposure and the biologically effective dose and accounts for interindividual differences in absorption, metabolic activation, or detoxification and DNA repair. Thus, DNA adducts are excellent biomarkers of internal exposure to reactive metabolites and may be correlated to tumor incidences.1−3 The research on DNA adduct analysis was greatly advanced by the 32P-postlabeling method,4 one of the first techniques that provided sufficient sensitivity for the quantification of DNA adducts in human tissue samples.5−8 However, identification of the adduct remains tentative with this method, as it mainly relies on chromatographic properties of the lesion. In the last two decades, a number of techniques based on liquid chromatography combined with tandem mass spectrometry (LC-MS/MS) for DNA adduct quantification have been developed, which surpass many other analytical methods especially regarding specificity of detection.9−12 However, reports on determination of DNA adducts in human samples by LC-MS/MS-based techniques are relatively few. Some examples reported in the literature are the detection of DNA adducts of 4-aminobiphenyl in pancreas tissue from smokers,13 of acrolein14 or acetaldehyde15 in leukocytes from smokers, of estrogen adducts in breast tumor tissue,16 and of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) and 4-aminobiphenyl adducts in saliva samples.17 The 2′-deoxyguanosine (dG) adduct of PhIP (C8-PhIP-dG, C8-(2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine-N2-yl)-2′-deoxyguanosine) was detected in the normal tumor-adjacent tissue of just one of 70 breast cancer patients at a level of 3 molecules C8-PhIP-dG/109 nucleotides.18 Recently, Yun et al. reported on the quantitation of adducts from aristolochic acid in paraffin-embedded kidney samples from patients of Balkan endemic nephropathy.19
The focus of many of these studies was on the quantification of DNA adducts formed from a single genotoxicant, which restricts their applicability to molecular epidemiology studies seeking to address the role of complex chemical exposures in cancer risk. Here, a technique is in demand that allows monitoring of an individual’s internal exposure to an array of electrophilic DNA-reactive metabolites. This so-called adductome defined by measurement of multiple DNA adducts derived from common environmental and food genotoxicants may be a suitable parameter for the correlation of DNA damage with tumor incidence in epidemiological studies. A high resolution MSn technology with impressive specificity was used for the simultaneous detection of 18 adducts in hepatic DNA of mice treated with the tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and is an excellent starting point for the analyses of DNA adducts in humans.20 Chou et al. used multiple reaction monitoring (MRM) for the parallel detection of 11 oxidative stress-related DNA adducts in various human autopsy samples.21
However, the specific analysis of multiple DNA adducts is still in its early stages. There are various analytical challenges with the adductome approach, three of which are technical problems: First, the sensitivities of some LC-MS/MS methods for measurements of DNA adducts are variable and low, when compared to those of 32P-postlabeling.9 Second, the conditions of enzymatic hydrolysis and the techniques of DNA adduct enrichment preceding LC-MS/MS quantification vary considerably. For example, DNA adduct enrichment may involve extraction with 1-butanol,22 solid-phase extraction (SPE) using different types of reversed-phase-resins,23,24 precipitation of the hydrolytic enzymes from the digestion mixture with methanol21 or ethanol,25 and online column-switching chromatography.18,26 Third, a reliable quantification of DNA adducts requires isotope-labeled internal standards for each adduct, which can pose a substantial effort in chemical synthesis.
Here, we describe a targeted approach that allows for the quantification of 16 DNA adducts derived from exposure to food and environmental mutagens and carcinogens, as well as from endogenous electrophiles produced from common lipid peroxidation-related degradation products. The molecular structures of the DNA adducts are depicted in Figure S-1 of the Supporting Information. We developed a standardized method for a combined adduct enrichment following enzymatic hydrolysis of the DNA and optimized ultra performance liquid chromatography (UPLC) of the adducts for quantification by mass spectrometric MRM employing the stable isotopic dilution method. Our analytical approach was applied to the measurement of this array of DNA adducts in ten human lung samples.
Experimental Section
Chemicals
Calf intestine alkaline phosphatase, micrococcal nuclease (from Staphylococcus aureus), bovine spleen phosphodiesterase, and ribonuclease A from bovine pancreas (RNase) were purchased from Sigma-Aldrich (Steinheim, Germany). Proteinase K, HPLC-grade methanol, 2-propanol, 1-butanol, formic acid, and acetic acid were from Carl Roth GmbH (Karlsruhe, Germany). Herring sperm DNA and all other reagents and solvents (analytical grade) were from Sigma-Aldrich.
Stable Isotope-Labeled DNA Adducts
The nucleoside adducts were available as isotope-labeled standards containing 15N, 13C, or 2H. Their synthesis has been described previously: [13C10]C8-ABP-dG (C8-(4-aminobiphenyl-N4-yl)-2′-deoxyguanosine),18 adducts of furfuryl alcohol ([15N5]N6-MF-dA (N6-MF-dA, N6-((furan-2-yl)methyl)-2′-deoxyadenosine; dA, 2′-deoxyadenosine); [15N5,13C10]N2-MF-dG (N2-MF-dG, N2-((furan-2-yl)methyl)-2′-deoxyguanosine)),27 5-hydroxymethylfurfural ([15N5]N6-FFM-dA (N6-FFM-dA, N6-((2-formylfuran-5-yl)methyl)-2′-deoxyadenosine); [15N5,13C10]N2-FFM-dG (N2-FFM-dG, N2-((2-formylfuran-5-yl)methyl)-2′-deoxyguanosine)),23 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine ([15N5,13C10]C8-PhIP-dG),28 methyleugenol ([15N5]N6-MIE-dA (N6-(trans-methylisoeugenol-3′-yl)-2′-deoxyadenosine); [15N5]N2-MIE-dG (N2-MIE-dG, N2-(trans-methylisoeugenol-3′-yl)-2′-deoxyguanosine)),25 1-methylpyrene ([15N5]N6-MP-dA (N6-MP-dA, N6-(1-methylpyrenyl)-2′-deoxyadenosine); [15N5,13C10]N2-MP-dG (N2-MP-dG, N2-(1-methylpyrenyl)-2′-deoxyguanosine)),29 and 1MIM glucosinolate ([15N5]N6-1MIM-dA (N6-1MIM-dA, N6-(1-methoxy-3-indolylmethyl)-2′-deoxyadenosine); [15N5]N2-1MIM-dG (N2-1MIM-dG, N2-(1-methoxy-3-indolylmethyl)-2′-deoxyguanosine)).24 We thank Dr. Beland and Dr. Marnett for the kind contribution of [2H7]N2-BPDE-dG (N2-(7,8,9-trihydroxy-7,8,9,10-tetrahydrobenzo[a]pyrene-C10-yl)-2′-deoxyguanosine)30 and [15N3]M1dG (M1dG, 3-(2′-deoxy-β-d-erythro-pentofuranosyl)-pyrimido[1,2-a]purin-10(3H)-one), respectively, and Dr. Poulsen for [15N3]3,N4-εdC (3,N4-εdC, 3,N4-etheno-2′-deoxycytidine; dC, 2′-deoxycytidine) and [15N5]1,N6-εdA (1,N6-εdA, 1,N6-etheno-2′-deoxyadenosine).31
Human Lung Samples
Pulmonary tissue samples, from four male and six female lung tumor patients, were provided by Biopredic International (Rennes, France). Immediately after surgical intervention, the samples of nontumorous tissues were stored at −80 °C until they were homogenized for DNA isolation. Table S-1 of the Supporting Information provides an overview of the patients’ sex and age. The study was approved by the ethics committee of the University of Potsdam (Germany).
DNA Isolation
Pulmonary DNA was isolated using a Qiagen kit for the preparation of genomic DNA with slight modifications of the manufacturer’s instructions (Qiagen, Venlo, Netherlands). Briefly, 100 mg samples of frozen tissues were homogenized in 9.5 mL of the lysis buffer G2. The homogenate was digested with proteinase K (500 μL, 10 mg/mL) and DNase-free RNase A (20 μL, 10 mg/mL in water) for 2 h at 37 °C. The mixture was cooled to 4 °C and centrifuged at 10 000g for 10 min. The supernatant was loaded onto a Qiagen Genomic-tip 100/G previously equilibrated with 4 mL of QBT-buffer. The column was washed twice with a 7.5 mL QC solution. The DNA was eluted with 5 mL of QF solution that was heated to 70 °C, precipitated by adding 2.5 mL of 2-propanol and separated by centrifugation at 15 000g for 15 min. The pellet was washed with 1 mL of cold 70% ethanol, centrifuged again and dissolved in 300 μL of water. The DNA concentration was determined spectrophotometrically with a Nanodrop ND-1000 spectrometer (peqlab Biotechnologie, Erlangen, Germany).
Enzymatic Digestion of DNA
Samples containing 350 μg of DNA were dried together with fixed amounts of the isotope-labeled adduct standards in the range of 58.3 fmol (N6-MIE-dA) to 5413 fmol (C8-PhIP-dG). The conditions were adopted from optimization studies using DNA containing adducts of methyleugenol and of 1MIM glucosinolate (neoglucobrassicin).32 The efficacy of the digestion was tested via UPLC-MS/MS quantification of dG. Small portions of 6.85 μL of the digests were diluted with 130.1 μL of water containing 373 μmol of [15N5]dG. The concentration of dG in the digests was determined via the MRM peak area ratio of dG (m/z = 268.1 → 152.1) and [15N5]dG (m/z = 273.1 → 157.1). The average concentration of dG in the digests of the 20 pulmonary DNA samples was 505 μM, which corresponds to a dG content of 21.6% in the DNA samples. The actual dG content in the human genome is 20.5%.
DNA Adduct Enrichment
Four different techniques for the extraction of multiple adducts from DNA hydrolysates were tested. The extraction methods are described briefly. Chou et al. described the precipitation of the digestion enzymes with methanol.21 A DNA digest (∼440 μL) was concentrated under reduced pressure to a volume of 100 μL and diluted with 300 μL of methanol. The mixture was centrifuged, and the pellet was extracted with another 300 μL of methanol. The combined methanol fractions were evaporated to dryness. Further, we tested the efficacy of extraction with 1-butanol.22,28 The DNA digests (∼440 μL) were diluted with 560 μL of water, and 84 mg of (NH4)2SO4 was added. The samples were extracted three times with 1 mL of water-saturated 1-butanol. The combined 1-butanol phases were washed with 500 μL of water saturated with 1-butanol and evaporated to dryness. The application of Oasis columns (3 cm3, 60 mg) from Waters (Eschborn, Germany) was described.23 The columns were preconditioned with 3 mL of methanol and 3 mL of water. The DNA digestion mixtures (∼440 μL) were diluted by addition of 160 μL of water and 160 μL of methanol, vortexed thoroughly and centrifuged at 15 000g for 15 min. The supernatants were loaded onto the columns and washed with 3 mL of water/methanol (95:5). DNA adducts were eluted with 3 mL of methanol, and the solvents were evaporated. The other SPE method was based on Chromabond C18 columns (500 mg) from Macherey & Nagel (Düren, Germany). The DNA digests were diluted with 700 μL of water, thoroughly vortexed, and centrifuged at 15 000g for 15 min. The columns were prepared with 2 mL of methanol and 2 mL of water. The samples were loaded, and the columns were washed with 1 mL of water. DNA adducts were eluted with 1.5 mL of methanol, and the solvent was evaporated. For UPLC-MS/MS analyses, all samples were dissolved in 25 μL of 75% methanol.
For the recovery assessment of each extraction method, ten samples of 350 μg of herring sperm DNA were prepared, five of which were spiked with defined amounts of all isotope-labeled standards. Following enzymatic hydrolysis, the DNA adduct reference compounds were extracted and analyzed by UPLC-MS/MS. The mean peak areas A̅1 of the quantifier signals were determined. (The quantifier signals of the DNA adducts are indicated in Table S-2, Supporting Information.) The other five DNA samples were first subjected to the workup procedure, and the residuals were then spiked with defined amounts of all isotope-labeled standards. The mean peak areas of the quantifiers of the reference compounds (A̅2) were determined. The recovery value RM,Ad for the processing of a specific DNA adduct (Ad) by one of the extraction methods (M) was calculated as RM,Ad = A̅1/A̅2 × 100%.
The second parameter with a strong impact on the overall performances of the DNA adduct analyses is the ion suppression of MRM signals that is caused by residuals from DNA digestion and workup solvents (matrix effect). In order to determine the effect of the ion suppression, five samples of commercial herring sperm DNA were subjected to digestion and extraction, and the residuals were spiked with defined amounts of the isotope-labeled reference compounds. These were analyzed by UPLC-MS/MS, and the mean peak areas of the quantifier traces (A̅3) were compared with those resulting from direct injection of the reference substances (A̅4). The relative signal intensity was calculated (ISM,Ad = A̅3/A̅4 × 100%). The overall performance PM,Ad (%) was calculated from PM,Ad = RM,Ad × ISM,Ad/100%.
UPLC-MS/MS Analyses
Mixtures of DNA adducts were separated using an Acquity UPLC System (Waters) with a HSS T3 column (1.8 μm, 2.1 × 100 mm, Waters). Samples of 8 μL were injected. The chromatographic efficiencies of two eluent solvent systems were studied. The first eluent system consisted of water (solvent A) and acetonitrile (solvent B). Both solvents were acidified with 0.25% acetic acid and 0.25% formic acid. Following a washing interval of 1 min at 100% solvent A, an 11 min gradient was applied with decreases of solvent A from 100% to 70% solvent A (1 to 8 min) and then to 30% A (8 to 12 min) at a 0.35 mL/min flow rate. The second eluent system consisted of 10 mM ammonium bicarbonate buffer, pH 8.0, (A) and methanol (B). It was applied using a linear 11 min gradient from 90% solvent A to 30% solvent A (1 to 12 min) at a 0.35 mL/min flow rate.33 The UPLC was connected to a Quattro Premier XE, a linear tandem quadrupole mass spectrometer (Waters) with an electrospray interface operated in the positive ion mode. MRM was used to record 74 transitions, half of which served the detection of the analyte DNA adducts while the other half was employed to monitor the isotope-labeled internal standard reference substances. The fragmentation transitions are summarized in Table S-2 (Supporting Information) together with the transition-specific cone voltages and collision energies. Other mass spectrometric parameters were constant: temperature of the electrospray source, 110 °C; desolvation temperature, 450 °C; desolvation gas, nitrogen (850 L/h); cone gas, nitrogen (50 L/h); collision gas, argon (indicated cell pressure, ∼5 × 10–3 mbar). The dwell time was set to 50 ms, and capillary voltage was set to 1.0 kV. The RF1 lens voltage was 0.1 V.
Limits of Detection (LODs)
The LOD values were estimated from the background integrals of the quantifier-traces in 1-butanol extracted residuals of digests of 350 μg of herring sperm DNA. The LOD of a particular DNA adduct A was defined by LODA = X̅A + 3σA, with X̅A as the analyte concentration corresponding to the mean area of five measurements of the background signal and with σA as the standard deviation. The retention time and the peak width of the adduct signal were determined from the coeluting isotope-labeled reference compounds. Herring sperm DNA contained some of the adducts analyzed in this study. In particular, 3,N4-εdC, 1,N6-εdA, and both 2-methylfuranyl adducts27 were present at considerable levels and 5-methylfurfuryl adducts23 were present at moderate levels. Therefore, the LODs for nucleoside adducts of furfuryl alcohol and 5-hydroxymethylfurfural were determined with 350 μg of porcine liver DNA prepared in our laboratory, in which these adducts were below the LOD. An LOD was not determined for 3,N4-εdC and 1,N6-εdA, because each of these adducts was clearly detectable in all of the human lung samples.
Results and Discussion
Enrichment of Adducts from DNA Hydrolysates
A critical step preceding mass spectrometric DNA adduct analyses is the analyte extraction from DNA digestion mixtures. This is particularly challenging if multiple DNA adducts with differing polarity are involved. The authors of most reports on the quantification of multiple DNA adducts focused on different classes of adducts, for example, either hydrophobic “bulky adducts” derived from polycyclic aromatic hydrocarbons34 or hydrophilic adducts formed as a consequence of oxidative stress and internal exposure to degradation products of lipid peroxidation.35 We sought to develop an analytical tool for the simultaneous quantification of many DNA adducts with diverse chemical structures without losing the sensitivities of single adduct analyses. For this purpose, stable isotope-labeled internal reference substances of 16 DNA adducts were obtained (Figure S-1, Supporting Information). These DNA adducts originate from electrophilic metabolites of compounds obtained from different sources, such as food plants, environmental contamination, lipid peroxidation, or cooked meat. Molecular structures and properties of the adducts differed markedly suiting our goal to develop a versatile method for the quantification of multiple DNA adducts that may be easily extended to the quantification of other adducts in the future.
We explored four different methods of adduct extraction frequently used prior to LC-MS/MS analysis. The overall performance of these techniques is based on two parameters: the recovery of single DNA adducts and the loss of signal intensity caused by ion suppression. The parameters designated in the following with RM,Ad (recovery) and with ISM,Ad (ion suppression) were determined in independent experiments for all adduct standards (Ad) with each of the workup methods (M). Table S-3 in the Supporting Information summarizes the recovery values RM,Ad. The enrichment with methanol, 1-butanol, C18 columns, and Oasis columns yielded reasonable recoveries over 50% for 13, 10, 12, and 12 out of 16 adducts, respectively. The peak area from the analysis of a specific DNA adduct also may be reduced by ion suppression that is caused by competing ionization of constituents of DNA or of the solvents employed for sample workup. The parameter ISM,Ad was defined as the ratio from the peak areas of the quantifier signal of a DNA adduct standard (Ad) that was analyzed either together with the residual of a specific workup method (M) or by direct injection (=100%). The mass spectrometric detection was least affected by previous extraction of the DNA adducts via 1-butanol or the Oasis columns (Table S-4, Supporting Information).
The recovery of the extraction (Table S-3, Supporting Information) and the ion suppression of the MS/MS detection (Table S-4, Supporting Information) were used to determine the overall performance of the workup methods for each of the DNA adducts included in this study (Table 1). Most of the DNA adducts were analyzed with satisfactory performances using either Oasis columns or the liquid–liquid extraction with 1-butanol as an inexpensive but effective alternative. The data in Table 1 indicate that some DNA adducts were difficult to enrich by any of the methods, i.e., N6-MF-dA and N6-MP-dA. This is primarily due to the poor recovery (Table S-3, Supporting Information) and has been observed previously.27,29 It is of note that the enrichment routines using SPE columns were detrimental to the quantification of C8-PhIP-dG. Goodenough et al. previously observed this phenomenon and showed that ion suppression effects were caused by contaminants in the solvents and SPE columns.36 Considering the collected data, the liquid–liquid extraction method using 1-butanol and the SPE with Oasis columns yielded the best results. We favored the liquid–liquid extraction because it provides a satisfactory recovery of C8-PhIP-dG.
Table 1. Effectiveness of DNA Adduct Enrichment Techniques.
| performance (%)a of extraction with |
||||
|---|---|---|---|---|
| adduct | methanol | 1-butanol | C18 columns | Oasis columns |
| 1,N6-εdA | 8.5 | 24.3 | 15.2 | 90.6 |
| 3,N4-εdCb | ||||
| M1dG | 31.2 | 43.6 | 63.2 | 17.6 |
| N2-FFM-dG | 33.1 | 39.5 | 46.2 | 60.9 |
| N6-FFM-dA | 61.6 | 92.6 | 72.4 | 61.1 |
| N2-MF-dG | 21.9 | 39.3 | 41.5 | 55.2 |
| N6-MF-dA | 15.7 | 19.5 | 22.1 | 23.3 |
| N2-MIE-dG | 51.5 | 63.7 | 56.2 | 75.7 |
| N6-MIE-dA | 43.9 | 48.5 | 55.5 | 39.3 |
| N2-1MIM-dG | 43.8 | 37.4 | 30.2 | 70.3 |
| N6-1MIM-dA | 25.0 | 46.4 | 40.9 | 37.0 |
| C8-PhIP-dG | 88.7 | 38.5 | 2.0 | 14.0 |
| C8-ABP-dG | 32.6 | 31.8 | 29.7 | 46.2 |
| N2-BPDE-dG | 45.6 | 79.0 | 45.6 | 53.2 |
| N2-MP-dG | 51.1 | 54.6 | 34.5 | 54.8 |
| N6-MP-dA | 6.2 | 7.9 | 4.3 | 13.7 |
The performance PM,Ad is the residual peak area of the quantifier signal of a particular DNA adduct that remains after processing by one of the workup procedures. The performance PM,Ad (%) was calculated from PM,Ad = RM,Ad × ISM,Ad/100% with the recovery RM,Ad of one of the enrichment methods (Table S-3, Supporting Information) and the remaining signal reduced by ion suppression ISM,Ad (Table S-4, Supporting Information). Details are given in the Experimental Section.
The MRM analyses of herring sperm DNA digests showed a substantial background noise in the trace of 255.1 → 139 of the internal standard [15N3]3,N4-εdC that prohibited the evaluation.
Chromatographic Separation of DNA Adducts
The increase in simultaneously recorded mass spectrometric fragmentations goes hand in hand with a loss of chromatographic resolution. Thus, the time windows for the detection of single DNA adducts by MRM were narrowed to about 60 s. In addition, an optimal chromatographic separation of the adduct mixtures is essential. Usually, DNA adducts are separated via reversed-phase columns using acidic solvents which support the protonation of the exocyclic amino groups in adducts of dG, 2′-deoxyadenosine (dA), and 2′-deoxycytidine (dC).10 In our work group, the chromatography of DNA adducts derived from single carcinogens was conventionally performed with a UPLC system and an eluting gradient of water and acetonitrile which were acidified with acetic acid and formic acid.23−25,27,37,38 We adjusted the method successfully allowing for the separation of all DNA adducts included in this study within about 12 min. Figure S-2 in the Supporting Information shows the chromatograms of the quantifier traces from the isotope-labeled reference substances after a joint injection.
Recently, Yin et al. reported on the chromatographic separation of acrolein-derived DNA adducts 6-hydroxy-1,N2-propano-dG and 8-hydroxy-1,N2-propano-dG testing different eluents.33 The use of the ammonium bicarbonate buffer (pH 8.0) led to narrower peaks and superior sensitivity of mass spectrometric detection compared to other test eluents containing, e.g., formic acid or ammonium acetate. The ammonium bicarbonate fragments into carbon dioxide and ammonia in the heated gas phase. Yin et al. proposed that ammonium ions suppress the formation of signal-reducing metal complexes of the analytes.33 Here, we compared the LOD values of the 16 analytes using either acetic acid and formic acid or ammonium bicarbonate as eluent additives (Table 2). The application of the ammonium bicarbonate as solvent additive resulted in slightly increased sensitivities for the mass spectrometric detection of most of the analytes except N6-MIE-dA and the hydrophobic “bulky adducts” C8-PhIP-dG, N2-BPDE-dG, N2-MP-dG, and N6-MP-dA.
Table 2. Detection Limits (LOD) of DNA Adduct Analyses Using Two Different Eluent Systems for UPLC-MS/MS.
| LOD
(adducts per 108 nucleosides)a |
||
|---|---|---|
| adduct | HOAc/FAb | NH4HCO3b |
| 1,N6-εdAc | ||
| 3,N4-εdCc | ||
| M1dG | 0.63 | 0.43 |
| N2-FFM-dG | 1.6 | 0.59 |
| N6-FFM-dA | 0.62 | 0.11 |
| N2-MF-dG | 0.14 | 0.09 |
| N6-MF-dA | 0.10 | 0.04 |
| N2-MIE-dG | 1.2 | 0.55 |
| N6-MIE-dA | 0.11 | 0.31 |
| N2-1MIM-dG | 1.8 | 0.35 |
| N6-1MIM-dA | 2.2 | 0.51 |
| C8-PhIP-dG | 3.0 | 7.1 |
| C8-ABP-dG | 0.10 | 0.04 |
| N2-BPDE-dG | 1.0 | 3.0 |
| N2-MP-dG | 0.1 | 0.28 |
| N6-MP-dA | 0.01 | 0.02 |
LODA = X̅A + 3σA with σA as the mean area of the background signal at the retention time of the analyte adduct in a digest of 350 μg of unmodified DNA and σA as the standard deviation from measurements of five independent samples.
HOAc/FA: eluent solvents water (A) and acetonitrile (B), each acidified with 0.25% acetic acid (HOAc) and 0.25% formic acid (FA); NH4HCO3: eluent solvents 10 mM ammonium bicarbonate buffer, pH 8.0, (A) and methanol (B).
Sizeable peaks in the traces m/z = 276.1 → 160.0 and m/z = 252.1 → 136.0 at the retention times of [15N5]1,N6-εdA and [15N3]3,N4-εdC, respectively, indicated that the etheno adducts abound in DNA of herring sperm and porcine liver used as biological matrices.
DNA Adducts in Tissue Samples of Human Lung
The previous results showed that none of the combinations of extraction and chromatographic separation was optimal for the simultaneous detection of all DNA adducts. We chose to use the 1-butanol extraction for the adduct enrichment and ammonium bicarbonate buffer (pH 8.0) and methanol as eluents A and B, respectively, for the subsequent UPLC-MS/MS analyses. The simultaneous analysis of the 16 adducts in DNA isolated from ten human lung samples allowed one to readily detect six of the adducts in some or all of the samples. The adduct levels summarized in Table 3 were calculated from the strongest signals (“quantifier peaks”, compare Table S-2, Supporting Information) of the analytes. The validity of detection of particular adducts was checked by the observance of up to two qualifier signals. Further confirmation provided the area ratio of the quantifier peak and the most prominent qualifier peak, which usually was in the range of the corresponding values determined for the isotope-labeled reference substance in the ten samples.
Table 3. Levels of the Adducts (per 108 nucleosides) that Were Detectable in DNA Samples of Human Lunga.
| patient
number |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| adduct | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | mean |
| first analysis | |||||||||||
| 1,N6-εdA | 57.4 | 30.6 | 16.9 | 34.3 | 63.4 | 38.5 | 39.0 | 40.0 | 68.8 | 74.9 | 46.4 |
| 3,N4-εdC | 88.0 | 53.1 | 27.2 | 49.0 | 105.6 | 84.4 | 63.6 | 60.1 | 85.7 | 102.9 | 72.0 |
| N6-MF-dA | 0.88 | 1.76c | – | – | – | – | – | – | 1.30c | – | |
| N2-MF-dG | 4.6 | 6.4 | 2.9 | 1.2 | 0.3 | 5.1 | 0.2 | 9.4 | 10.4 | – | 4.1b |
| N6-MIE-dA | 0.31c | 0.60 | – | 1.18 | – | 0.55c | – | 0.39c | – | – | 0.38b |
| N2-MIE-dG | 7.5 | 18.8 | 7.8 | 23.7 | 4.7 | 21.5 | 5.6 | 8.8 | 3.0 | 1.7 | 10.3 |
| second analysis | |||||||||||
| 1,N6-εdA | 53.8 | 28.6 | 40.1 | 38.7 | 54.6 | 35.0 | 24.7 | 39.9 | 69.0 | 115.3 | 50.0 |
| 3,N4-εdC | 90.0 | 60.5 | 87.3 | 67.2 | 87.1 | 80.8 | 37.9 | 65.2 | 113.2 | 179.0 | 86.8 |
| N6-MF-dA | 0.98 | – | – | – | – | – | – | 1.21 | – | – | |
| N2-MF-dG | 4.0 | 6.1 | 2.6c | 1.3 | – | 4.8 | – | 10.1 | 10.2 | – | 3.9b |
| N6-MIE-dA | 0.42c | 0.41 | – | 0.32c | 0.45 | 0.56 | – | 0.35c | – | 0.51 | 0.35b |
| N2-MIE-dG | 8.6 | 15.5 | 7.5 | 12.6 | 11.9 | 17.6 | 6.8 | 9.4 | 3.8c | 15.8 | 11.0 |
For each of the analyses, about 500 μg of DNA was isolated from separate portions of 0.5 to 1 g of lung tissue. After enzymatic hydrolysis of 350 μg of DNA, the adducts were extracted with 1-butanol. Details are described in the Experimental Section. Hyphens (−) indicate adduct levels below the LOD.
The arithmetic mean was calculated with values of LOD/2 (Table 2) for those adduct levels < LOD.
The ratio of the peak areas from the quantifier signal and the most prominent qualifier was not in the range of the corresponding ratios calculated from the signals of the internal standard.
The etheno adducts 1,N6-εdA and 3,N4-εdC were the most abundant adducts detected in the samples of human lung. These adducts are formed from the reactions of the DNA bases with 2,3-epoxyaldehydes of 4-hydroxy-2-nonenal or croton aldehyde39 and were previously quantified in human DNA by 32P-postlabeling and immunodetection techniques.40,41 Interestingly, the adduct levels were shown to increase in affected tissues of persons with some inherited cancer risk factors, e.g., Wilson’s disease40 or familial adenomatous polyposis.42 There are only few studies of etheno adducts in human DNA samples using specific LC-MS/MS techniques and isotope-labeled reference compounds. Thus, 1,N6-εdA was found at high levels in commercial placental DNA (73.2 adducts/108 nucleosides)43 or in gastric mucosa and lung in the range of 11.9–276.3 adducts/108 nucleosides (n = 22)44 and 4.0–246.3 adducts/108 nucleosides (n = 12),21 respectively. Also in the current work, 1,N6-εdA and, in addition, 3,N4-εdC were detected in human pulmonary DNA. Representative chromatograms of the fragmentations of 1,N6-εdA and 3,N4-εdC and their reference compounds are shown in Figure 1. In the first analysis, the adduct levels were from 16.9–74.9 1,N6-εdA/108 nucleosides and 27.2–102.9 3,N4-εdC/108 nucleosides (Table 3). In contrast, we did not detect M1dG, the main adduct of the lipid peroxidation product malondialdehyde in spite of a favorable LOD. We observed that the internal reference [15N3]M1dG did not survive the conditions of the DNA hydrolysis (pH ∼ 10). This is probably due to hydrolytic ring-opening to N2-oxopropenyl-dG, the major degradation pathway of M1dG previously studied under basic conditions (pH 11.2).45
Figure 1.

UPLC-MS/MS chromatograms of digested pulmonary DNA of patient 10 containing 1,N6-εdA and 3,N4-εdC. The chromatogram of 1,N6-εdA (m/z = 276.1 → 160.0, A) is shown together with the parallel recording of [15N5]1,N6-εdA (m/z = 281.1 → 165.0, B). Panels C and D show the neutral loss of the 2′-deoxyribose from 3,N4-εdC (m/z = 252.1 → 136.0) and [15N3]3,N4-εdC (m/z = 255.1 → 139.0), respectively.
The adducts of furfuryl alcohol N2-MF-dG and N6-MF-dA were detected for the first time in human DNA. The furan derivative is a carcinogen of moderate potency;46 however, it is abundant in heat-processed foods.47 In the first analysis of this study, we identified N2-MF-dG in nine out of ten DNA samples in the range of 0.2–10.4 N2-MF-dG/108 nucleosides (Table 3). Figure 2 shows exemplary chromatograms of N2-MF-dG detected by three transitions together with the reference compound [15N5,13C10]N2-MF-dG. The MRM analysis of N6-MF-dA yielded clear quantifier signals (m/z = 332.1 → 216.1) in most samples; however, the detection of both qualifier peaks was compromised by background noise (Figure S-3, Supporting Information). Consequently, only three samples contained N6-MF-dA (0.88–1.76 adducts/108 nucleosides) at levels that allowed verification with qualifier peaks of satisfactory intensity. In previous animal experiments, N2-MF-dG and N6-MF-dA were detected in liver, kidney, and lung of mice after a single administration of furfuryl alcohol or after a four-week exposure via drinking water.27 A recent study in mice indicated that the pulmonary genotoxic effect may originate in great part from hepatic sulfo conjugation of furfuryl alcohol and transport of the reactive sulfate ester to the lung via the bloodstream.48 In addition, furfuryl alcohol is a common volatile organic compound also present in samples of indoor dust (0.4–500 μg furfuryl alcohol/g dust, n = 340 residences) and consequently may also be taken up by inhalation.49
Figure 2.

UPLC-MS/MS chromatograms of a digested DNA sample isolated from lung tissue of patient 8 containing N2-MF-dG. The left panels depict the chromatograms of N2-MF-dG with the fragmentations m/z = 348.1 → 232.0 (A), m/z = 348.1 → 164.0 (B), and m/z = 348.1 → 81.1 (C). The most intensive signal (m/z = 348.1 → 232.0) was used as quantifier signal. Simultaneously, the transitions m/z = 363.1 → 242.0 (D), m/z = 363.1 → 174.0 (E), and m/z = 363.1 → 81.1 (F) of the internal isotope-labeled standard [15N5,13C10]N2-MF-dG were monitored. Figure S-3 in the Supporting Information shows the chromatograms of the N6-MF-dA detection in the DNA of the same lung sample.
Methyleugenol is an alkenylbenzene present in herbs, spices, and essential oils and is used as a flavoring additive in the food and cosmetic industry. It is a strong hepatocarcinogen in rodents.50 The methyleugenol adducts N6-MIE-dA and N2-MIE-dG, which were recently quantified by UPLC-MS/MS in 29 out of 30 DNA samples of human liver,22 were also detected in the current study. Chromatograms of three N2-MIE-dG fragmentations and those of the reference compound [15N5]N2-MIE-dG are shown in Figure 3. Figure S-4 in the Supporting Information depicts corresponding chromatograms of N6-MIE-dA and [15N5]N6-MIE-dA. In the first analysis, N6-MIE-dA levels were determined in five of ten samples with levels from 0.31 to 1.18 adducts/108 nucleosides, whereas N2-MIE-dG was readily detectable in all samples containing 1.7–23.7 adducts/108 nucleosides. The mean N2-MIE-dG level was 27-fold higher compared to the average N6-MIE-dA level. In previous studies, the levels of N2-MIE-dG exceeded those of N6-MIE-dA by about 60-fold in the human liver22 and by 35-fold in the liver of FVB/N mice treated orally with methyleugenol.37 This is the first study, in which N6-MIE-dA and N2-MIE-dG were detected in pulmonary DNA of humans. Substantial levels, up to 126 N2-MIE-dG/108 nucleosides, were also present in the lung of FVB/N mice genetically engineered for the expression of human SULT1A1/1A2 that received a single oral dose of 50 mg methyleugenol/kg body weight (Herrmann, K. and Glatt, H. R., manuscript in preparation).
Figure 3.

UPLC-MS/MS chromatograms of a digested DNA sample isolated from patient 6 that contained the methyleugenol adduct N2-MIE-dG. Panels on the left-hand side show the chromatograms of N2-MIE-dG with the fragmentations m/z = 444.1 →328.1 (A), m/z = 444.1 → 177.1 (B), and m/z = 444.1 → 164.1 (C). The most intensive signal (m/z = 444.1 → 328.1) was used as quantifier signal. On the right-hand side are the parallel recordings of the transitions m/z = 449.1 → 333.1 (D), m/z = 449.1 → 177.1 (E), and m/z = 449.1 → 169.1 (F) of the isotope-labeled standard [15N5]N2-MIE-dG. Figure S-4 in the Supporting Information shows the chromatograms of the N6-MIE-dA detection in the DNA of the same lung sample.
The simultaneous analysis of the adducts was performed a second time using the same protocol as described above but in DNA samples isolated from distinct lung tissue specimens of the same patients (Table 3, second analysis). In most, but not in all cases, the results of the second analysis were in good agreement with the first analysis. The overall picture is described by the average values of adduct levels, which deviate only slightly between first and second analysis, and by correlations of first and second analytical results depicted in Figure S-5, Supporting Information. For example, the 1,N6-εdA levels in the lung sample patients 1, 5, 9, and 10 were the highest in both quantifications (m = 1.13, r2 = 0.632). Also, the maximum levels of N2-MF-dG were detected in samples from patients 2, 8, and 9 in both analytical runs (m = 1.04, r2 = 0.987). However, in some cases, strong deviations between pairs of results from first and second analysis were also observed. For example, the N2-MIE-dG levels in patients 4, 5, and 10 vary considerably between both analytical runs (m = 0.98, r2 = 0.304).
Conclusions
For the combined analysis of 16 structurally and chemically diverse DNA adducts, we optimized a method for the posthydrolytic enrichment and the chromatographic separation of the analytes. In the pulmonary DNA of ten lung cancer patients, we detected high levels of adducts of lipid peroxidation products (1,N6-εdA, 3,N4-εdC), of the secondary plant metabolite methyleugenol (N6-MIE-dA, N2-MIE-dG), and of the food contaminant furfuryl alcohol (N6-MF-dA, N2-MF-dG). In contrast, adducts of common environmental carcinogens that are also present in tobacco smoke, e.g., benzo[a]pyrene, 4-aminobiphenyl, and 1-methylpyrene, were not detectable. In some previous studies, adducts of benzo[a]pyrene were detected in human lung by HPLC and fluorescence detection,51 and one work reported the presence of C8-ABP-dG in 25 samples of human pulmonary tissue by immunochemistry and 32P-postlabeling.52 Recently, the data were questioned due to the low specificity of the detection methods.10,12 In agreement with the current work, no reports were published hitherto on the specific LC-MS/MS detection of DNA adducts of the common carcinogens included here, especially those of benzo[a]pyrene, 1-methylpyrene, PhIP, and 4-aminobiphenyl, in specimens of human pulmonary DNA.
In part, the results can be explained by the large variation in human intake amounts. The use of furfuryl alcohol as a flavoring agent alone leads to a possible average daily intake of 1.3 μmol/kg body weight.53 The estimated average daily intake of methyleugenol is 56.1 nmol/kg body weight,54 which exceeds the average daily doses of benzo[a]pyrene (31.7 pmol/kg body weight)55 and PhIP (26.8 pmol/kg body weight)56 by factors of 1700 and 2100, respectively. Other parameters may greatly influence the tissue concentration of a particular DNA adduct, e.g., the toxicokinetic properties of the genotoxicant, the transport of metabolites, the reactivity of the ultimate carcinogen, and the DNA repair. In view of the current results, it is interesting to note that the estimated carcinogenic hazard of methyleugenol (defined by the margin between the daily human intake and the lowest dose that induced a specific tumor in an animal model) is about 25-fold and 100-fold greater than the hazards of benzo[a]pyrene and PhIP, respectively.57 However, the mutagenic potency and the persistence of DNA adducts of different substances may vary greatly,1 and thus, the current results do not allow speculations on the carcinogenic effect of one of the substances in the lung. Future work is aimed at the DNA adduct monitoring in a larger library of specimens from healthy individuals and tumor patients, for which data on diet and smoking history are available.
Acknowledgments
The authors thank Martina Scholtyssek for her excellent technical assistance and gratefully acknowledge financial support from the German Research Foundation (MO 2520/1-1) and the German Institute of Human Nutrition (DIfE). The synthesis of C8-ABP-dG was supported by the National Cancer Institute of the National Institutes of Health under Award Number R33CA186795 (R.J.T.).
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† B.H.M.: Department Food Safety, Federal Institute for Risk Assessment (BfR), Max-Dohrn-Str. 8-10, 10589 Berlin, Germany.
Author Present Address
¶ F.S.: Department of Nutritional Toxicology, University of Potsdam, 14558 Nuthetal, Germany.
Author Present Address
# K.H.: Department Pesticide Safety, Federal Institute for Risk Assessment (BfR), Max-Dohrn-Str. 8-10, 10589 Berlin, Germany.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Otteneder M.; Lutz W. K. Mutat. Res. 1999, 424, 237–247. [DOI] [PubMed] [Google Scholar]
- Poirier M. C. Nat. Rev. Cancer 2004, 4, 630–637. [DOI] [PubMed] [Google Scholar]
- Poirier M. C.; Beland F. A. Chem. Res. Toxicol. 1992, 5, 749–755. [DOI] [PubMed] [Google Scholar]
- Randerath K.; Reddy M. V.; Gupta R. C. Proc. Natl. Acad. Sci. U.S.A. 1981, 78, 6126–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vineis P.; Husgafvel-Pursiainen K. Carcinogenesis 2005, 26, 1846–1855. [DOI] [PubMed] [Google Scholar]
- Phillips D. H. Carcinogenesis 2002, 23, 1979–2004. [DOI] [PubMed] [Google Scholar]
- Godschalk R. W.; Van Schooten F. J.; Bartsch H. J. Biochem. Mol. Biol. 2003, 36, 1–11. [DOI] [PubMed] [Google Scholar]
- Angerer J.; Ewers U.; Wilhelm M. Int. J. Hyg. Environ. Health 2007, 210, 201–228. [DOI] [PubMed] [Google Scholar]
- Klaene J. J.; Sharma V. K.; Glick J.; Vouros P. Cancer Lett. 2012, 334, 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretyakova N.; Goggin M.; Sangaraju D.; Janis G. Chem. Res. Toxicol. 2012, 25, 2007–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monien B. H. Adv. Exp. Med. Biol. 2014, 806, 383–397. [DOI] [PubMed] [Google Scholar]
- Gavina J. M.; Yao C.; Feng Y. L. Talanta 2014, 130, 475–494. [DOI] [PubMed] [Google Scholar]
- Ricicki E. M.; Soglia J. R.; Teitel C.; Kane R.; Kadlubar F.; Vouros P. Chem. Res. Toxicol. 2005, 18, 692–699. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Balbo S.; Wang M.; Hecht S. S. Chem. Res. Toxicol. 2010, 24, 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Wang M.; Villalta P. W.; Luo X.; Feuer R.; Jensen J.; Hatsukami D. K.; Hecht S. S. Chem. Res. Toxicol. 2007, 20, 108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embrechts J.; Lemiere F.; Van Dongen W.; Esmans E. L.; Buytaert P.; Van Marck E.; Kockx M.; Makar A. J. Am. Soc. Mass Spectrom. 2003, 14, 482–491. [DOI] [PubMed] [Google Scholar]
- Bessette E. E.; Spivack S. D.; Goodenough A. K.; Wang T.; Pinto S.; Kadlubar F. F.; Turesky R. J. Chem. Res. Toxicol. 2010, 23, 1234–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu D.; Turesky R. J.; Tao Y.; Langouet S. A.; Nauwelaers G. C.; Yuan J. M.; Yee D.; Yu M. C. Carcinogenesis 2012, 33, 124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun B. H.; Yao L.; Jelakovic B.; Nikolic J.; Dickman K. G.; Grollman A. P.; Rosenquist T. A.; Turesky R. J. Carcinogenesis 2014, 35, 2055–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbo S.; Hecht S. S.; Upadhyaya P.; Villalta P. W. Anal. Chem. 2014, 86, 1744–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou P. H.; Kageyama S.; Matsuda S.; Kanemoto K.; Sasada Y.; Oka M.; Shinmura K.; Mori H.; Kawai K.; Kasai H.; Sugimura H.; Matsuda T. Chem. Res. Toxicol. 2010, 23, 1442–1448. [DOI] [PubMed] [Google Scholar]
- Herrmann K.; Schumacher F.; Engst W.; Appel K. E.; Klein K.; Zanger U. M.; Glatt H. Carcinogenesis 2013, 34, 1025–1030. [DOI] [PubMed] [Google Scholar]
- Monien B. H.; Engst W.; Barknowitz G.; Seidel A.; Glatt H. R. Chem. Res. Toxicol. 2012, 25, 1484–1492. [DOI] [PubMed] [Google Scholar]
- Schumacher F.; Engst W.; Monien B. H.; Florian S.; Schnapper A.; Steinhauser L.; Albert K.; Frank H.; Seidel A.; Glatt H. Anal. Chem. 2012, 84, 6256–6262. [DOI] [PubMed] [Google Scholar]
- Herrmann K.; Engst W.; Appel K. E.; Monien B. H.; Glatt H. R. Mutagenesis 2012, 27, 453–462. [DOI] [PubMed] [Google Scholar]
- Singh R.; Arlt V. M.; Henderson C. J.; Phillips D. H.; Farmer P. B.; Gamboa da Costa G. J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2010, 878, 2155–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monien B. H.; Herrmann K.; Florian S.; Glatt H. R. Carcinogenesis 2011, 32, 1533–1539. [DOI] [PubMed] [Google Scholar]
- Lin D.; Kaderlik K. R.; Turesky R. J.; Miller D. W.; Lay J. O. Jr.; Kadlubar F. F. Chem. Res. Toxicol. 1992, 5, 691–697. [DOI] [PubMed] [Google Scholar]
- Monien B. H.; Müller C.; Engst W.; Frank H.; Seidel A.; Glatt H. R. Chem. Res. Toxicol. 2008, 21, 2017–2025. [DOI] [PubMed] [Google Scholar]
- Beland F. A.; Churchwell M. I.; Von Tungeln L. S.; Chen S.; Fu P. P.; Culp S. J.; Schoket B.; Gyorffy E.; Minarovits J.; Poirier M. C.; Bowman E. D.; Weston A.; Doerge D. R. Chem. Res. Toxicol. 2005, 18, 1306–1315. [DOI] [PubMed] [Google Scholar]
- Hillestrom P. R.; Weimann A.; Poulsen H. E. J. Am. Soc. Mass Spectrom. 2006, 17, 605–610. [DOI] [PubMed] [Google Scholar]
- Schumacher F.; Herrmann K.; Florian S.; Engst W.; Glatt H. Anal. Biochem. 2013, 434, 4–11. [DOI] [PubMed] [Google Scholar]
- Yin R.; Liu S.; Zhao C.; Lu M.; Tang M. S.; Wang H. Anal. Chem. 2013, 85, 3190–3197. [DOI] [PubMed] [Google Scholar]
- Singh R.; Teichert F.; Seidel A.; Roach J.; Cordell R.; Cheng M. K.; Frank H.; Steward W. P.; Manson M. M.; Farmer P. B. Rapid Commun. Mass Spectrom. 2010, 24, 2329–2340. [DOI] [PubMed] [Google Scholar]
- Pang B.; Zhou X.; Yu H.; Dong M.; Taghizadeh K.; Wishnok J. S.; Tannenbaum S. R.; Dedon P. C. Carcinogenesis 2007, 28, 1807–1813. [DOI] [PubMed] [Google Scholar]
- Goodenough A. K.; Schut H. A.; Turesky R. J. Chem. Res. Toxicol. 2007, 20, 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann K.; Engst W.; Meinl W.; Florian S.; Cartus A. T.; Schrenk D.; Appel K. E.; Nolden T.; Himmelbauer H.; Glatt H. Carcinogenesis 2014, 35, 935–941. [DOI] [PubMed] [Google Scholar]
- Schumacher F.; Florian S.; Schnapper A.; Monien B. H.; Mewis I.; Schreiner M.; Seidel A.; Engst W.; Glatt H. Arch. Toxicol. 2014, 88, 823–836. [DOI] [PubMed] [Google Scholar]
- De Bont R.; van Larebeke N. Mutagenesis 2004, 19, 169–185. [DOI] [PubMed] [Google Scholar]
- Bartsch H.; Nair J. Toxicology 2000, 153, 105–114. [DOI] [PubMed] [Google Scholar]
- Arab K.; Pedersen M.; Nair J.; Meerang M.; Knudsen L. E.; Bartsch H. Carcinogenesis 2009, 30, 282–285. [DOI] [PubMed] [Google Scholar]
- Schmid K.; Nair J.; Winde G.; Velic I.; Bartsch H. Int. J. Cancer 2000, 87, 1–4. [DOI] [PubMed] [Google Scholar]
- Chen H. J.; Chiang L. C.; Tseng M. C.; Zhang L. L.; Ni J.; Chung F. L. Chem. Res. Toxicol. 1999, 12, 1119–1126. [DOI] [PubMed] [Google Scholar]
- Matsuda T.; Tao H.; Goto M.; Yamada H.; Suzuki M.; Wu Y.; Xiao N.; He Q.; Guo W.; Cai Z.; Kurabe N.; Ishino K.; Matsushima Y.; Shinmura K.; Konno H.; Maekawa M.; Wang Y.; Sugimura H. Carcinogenesis 2013, 34, 121–127. [DOI] [PubMed] [Google Scholar]
- Riggins J. N.; Pratt D. A.; Voehler M.; Daniels J. S.; Marnett L. J. J. Am. Chem. Soc. 2004, 126, 10571–10581. [DOI] [PubMed] [Google Scholar]
- National Toxicology Program Toxicology and carcinogenesis studies of furfuryl alcohol (CAS No. 98-00-0) in F344/N rats and B6C3F1 mice (inhalation studies); NIH Publication No. 99-3972; NIEHS: Research Triangle Park, NC, 1999. [PubMed] [Google Scholar]
- Murkovic M.; Swasti Y. R. In Chemical Food Safety and Health; Pedreschi Plasencia F., Ciesarova Z., Eds.; Nova Publishers, Inc.: New York, 2013; p 43–55. [Google Scholar]
- Sachse B.; Meinl W.; Glatt H.; Monien B. H. Carcinogenesis 2014, 35, 2339–2345. [DOI] [PubMed] [Google Scholar]
- Nilsson A.; Lagesson V.; Bornehag C. G.; Sundell J.; Tagesson C. Environ. Int. 2005, 31, 1141–1148. [DOI] [PubMed] [Google Scholar]
- Rep. Carcinog. 2011, 12, 267–268. [PubMed] [Google Scholar]
- Boysen G.; Hecht S. S. Mutat. Res. 2003, 543, 17–30. [DOI] [PubMed] [Google Scholar]
- Culp S. J.; Roberts D. W.; Talaska G.; Lang N. P.; Fu P. P.; Lay J. O. Jr.; Teitel C. H.; Snawder J. E.; Von Tungeln L. S.; Kadlubar F. F. Mutat. Res. 1997, 378, 97–112. [DOI] [PubMed] [Google Scholar]
- Munro I. C.; Danielewska-Nikiel B. Food Chem. Toxicol. 2006, 44, 758–809. [DOI] [PubMed] [Google Scholar]
- Smith B.; Cadby P.; Leblanc J. C.; Setzer R. W. Food Chem. Toxicol. 2010, 48Suppl 1S89–97. [DOI] [PubMed] [Google Scholar]
- World Health Organization Safety evaluation of certain contaminants in food. Prepared by the Sixty-fourth meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA); WHO: Geneva (Switzerland), 2006. [PubMed] [Google Scholar]
- Bogen K. T.; Keating G. A. J. Exposure Anal. Environ. Epidemiol. 2001, 11, 155–168. [DOI] [PubMed] [Google Scholar]
- Benford D.; Bolger P. M.; Carthew P.; Coulet M.; DiNovi M.; Leblanc J. C.; Renwick A. G.; Setzer W.; Schlatter J.; Smith B.; Slob W.; Williams G.; Wildemann T. Food Chem. Toxicol. 2010, 48Suppl. 1S2–24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.