

Abstract
A current challenge in metabolomics is the reliable quantitation of many metabolites. Limited resolution and sensitivity combined with the challenges associated with unknown metabolite identification have restricted both the number and the quantitative accuracy of blood metabolites. Focused on alleviating this bottleneck in NMR-based metabolomics, investigations of pooled human serum combining an array of 1D/2D NMR experiments at 800 MHz, database searches, and spiking with authentic compounds enabled the identification of 67 blood metabolites. Many of these (∼1/3) are new compared with those reported previously as a part of the Human Serum Metabolome Database. In addition, considering both the high reproducibility and quantitative nature of NMR as well as the sensitivity of NMR chemical shifts to altered sample conditions, experimental protocols and comprehensive peak annotations are provided here as a guide for identification and quantitation of the new pool of blood metabolites for routine applications. Further, investigations focused on the evaluation of quantitation using organic solvents revealed a surprisingly poor performance for protein precipitation using acetonitrile. One-third of the detected metabolites were attenuated by 10–67% compared with methanol precipitation at the same solvent-to-serum ratio of 2:1 (v/v). Nearly 2/3 of the metabolites were further attenuated by up to 65% upon increasing the acetonitrile-to-serum ratio to 4:1 (v/v). These results, combined with the newly established identity for many unknown metabolites in the NMR spectrum, offer new avenues for human serum/plasma-based metabolomics. Further, the ability to quantitatively evaluate nearly 70 blood metabolites that represent numerous classes, including amino acids, organic acids, carbohydrates, and heterocyclic compounds, using a simple and highly reproducible analytical method such as NMR may potentially guide the evaluation of samples for analysis using mass spectrometry.
Metabolite profiling of human serum/plasma is of major interest for the investigation of virtually all human diseases.1−3 This interest stems from the clinical relevance of blood arising from its close association (directly or indirectly) with essentially every living cell in the human body combined with its relatively easy access for investigations. Nuclear magnetic resonance (NMR) spectroscopy is one of the two most widely used analytical techniques to analyze blood metabolites, the other being mass spectrometry (MS). Metabolite profiling of serum/plasma is generally met with two major challenges: first, the need to alleviate interference from the massive amount of serum/plasma proteins (6-8 g/dL); and, second, the need to unravel the inherent complexity of the mixture of compounds in the biofluid to reliably detect, identify and quantitate metabolites individually. To attempt to alleviate these challenges, MS employs protein precipitation and liquid or gas chromatography prior to detection. However, neither protein precipitation nor chromatography is generally deployed in NMR analysis; traditionally, intact serum/plasma is used in the analysis using 1D NMR, in which protein signals are suppressed using the CPMG sequence.1 Although analysis of intact serum/plasma is attractive, numerous limitations increasingly make this approach less suitable for metabolomics studies. In particular, (1) the number of metabolites detected using intact serum/plasma is restricted to ∼30 or less, which is far fewer compared with the actual number of blood metabolites;2 (2) concentrations of many detected metabolites are grossly underestimated as a result of attenuation caused by metabolite binding to serum/plasma proteins;4−7 (3) residual macromolecule signals in the CPMG spectra often cause a distorted spectral baseline, which deleteriously affects metabolite quantitation; and (4) the copious proteins present in serum/plasma cause reduced transverse relaxation (T2) times for metabolite signals and facilitate exchange between protein-bound and free metabolites, which together result in significantly broadened NMR peaks and poor quantitative accuracy.
Efforts focused on alleviating these bottlenecks have included physically removing serum/plasma proteins using ultrafiltration; solid phase extraction; or protein precipitation using an organic solvent, such as methanol, acetonitrile, acetone, perchloric acid or trichloroacetic acid.8−11 These approaches have been shown to achieve significant improvements in the number of metabolites identified in blood. Notably, a comprehensive analysis of ultrafiltered serum, as a part of investigations of the human serum metabolome, resulted in identification of 49 metabolites, the number believed to be an upper limit for human serum, especially using 1H 1D NMR.2,12 More recently, another exhaustive study of ultrafiltered human plasma focused on the global characterization of a NIST SRM (National Institute of Standards and Technology Standard Reference Material) identified a total of 39 metabolites.13 Incidentally, two studies from the same group involving applications to colorectal cancer14 and pancreatic cancer,15 presumably using ultrafiltered serum, report identification of 55 and 58 metabolites, respectively; however, the lack of pertinent experimental and NMR spectral analysis details makes these studies difficult for translation to more routine applications. Further, both studies utilized Chenomx software,16 which often provides numerous hits in the identification of unknown metabolites and, hence, can lead to incorrect metabolite identification, especially for low-concentration (∼1 μM) metabolites.
To make further progress in NMR-based metabolomics, there is a strong need to enhance the number of identified metabolites, provide experimental details to allow easy reproduction of spectra, and establish spectral assignment/analysis protocols for unambiguous metabolite identification. Establishment of a robust workflow will facilitate translation of the approach for routine use, particularly given the highly reproducible and quantitative nature of NMR. The current study is focused on enhancing the pool of quantifiable metabolites in blood using a protein precipitation approach that we recently showed has superior performance over ultrafiltration for blood metabolite quantitation.7 In the current work, comprehensive analyses of a pooled human serum were made combining an array of 1D and 2D NMR experiments, database searches, and spiking experiments using authentic compounds to achieve this goal, resulting in 67 quantified metabolites (including two beta sugars). Importantly, apart from identifying a new pool of blood metabolites, detailed experimental protocols and exhaustive peak labeling, especially for characteristic metabolite peaks, are provided to enable easy reproduction of serum spectra, reliable metabolite identification and quantitation. Further, investigations based on the absolute concentrations of identified metabolites reveal a surprisingly poor performance for protein precipitation using acetonitrile compared with methanol, an important outcome that contradicts commonly held opinion that acetonitrile performs better as a serum protein precipitation solvent in metabolomics studies.
Materials and Methods
Methanol, acetonitrile, sodium phosphate, monobasic (NaH2PO4), sodium phosphate, dibasic (Na2HPO4), and 3-(trimethylsilyl)propionic acid-2,2,3,3-d4 sodium salt (TSP) were obtained from Sigma-Aldrich (St. Louis, MO). Sixty-five standard compounds used for spiking to confirm peak assignments were obtained from Sigma-Aldrich, except for 3-hydroxyisovaleric acid, which was obtained from Fisher Scientific (Waltham, MA) (Supporting Information (SI) Table S1). Deuterium oxide (D2O) was obtained from Cambridge Isotope laboratories, Inc. (Andover, MA). Pooled human serum was obtained from Innovative Research, Inc. (Novi, MI). Deionized (DI) water was purified using an in-house Synergy Ultrapure Water System from Millipore (Billerica, MA). All chemicals were used with no further purification.
Preparation of Phosphate Buffer
Buffer solution was prepared by dissolving 928.6 mg of anhydrous NaH2PO4 and 320.9 mg of anhydrous Na2HPO4 in 100 g of D2O and used without further pH correction.
Solutions of Authentic Compounds for Spiking Experiments
Stock solutions (1 mL; 1 mM) for all 65 compounds (see SI Table S1) were prepared, separately, in D2O by diluting their 50 mM stock solutions, which were first prepared by weighing the compounds and dissolving in D2O solvent.
Serum Protein Precipitation Using Methanol
Sixteen 300 μL serum samples were mixed with methanol in 1:1, 1:2, 1:3 or 1:4 ratios (v/v) (see SI Table S2), vortexed, and incubated at −20 °C for 20 min. The mixtures were centrifuged at 13 400 rcf for 30 min to pellet proteins. Supernatants were decanted to fresh vials and dried. The dried samples were mixed with 100 μL of phosphate buffer in D2O containing 66.17 μM TSP, made up to 600 μL with phosphate buffer in D2O, and transferred to 5 mm NMR tubes.
Serum Protein Precipitation Using Acetonitrile
Sixteen 300 μL serum samples were mixed with acetonitrile in 1:1, 1:2, 1:3 or 1:4 ratios (v/v) (see SI Table S2), vortexed, and incubated at −20 °C for 20 min. The mixtures were centrifuged at 13 400 rcf for 30 min to pellet proteins. Supernatants were decanted to fresh vials and dried. The dried samples were mixed with 100 μL of a solution of phosphate buffer in D2O containing 66.17 μM TSP, made up to 600 μL with phosphate buffer in D2O, and transferred to 5 mm NMR tubes.
Ultrafiltration
Centrifugal filters (3 kDa cutoff; Amicon Microcon, YM-3; Sigma-Aldrich) were washed with water and centrifuged thrice with 300 μL of water at 13 400 rcf for 20 min, each time. Two 300 μL serum samples were then transferred to filter tubes and centrifuged for 20 min at 13 400 rcf. The filtrates were mixed separately with a 100 μL solution of phosphate buffer in D2O containing 66.17 μM TSP. The solutions were made up to 600 μL with the phosphate buffer in D2O and transferred to 5 mm NMR tubes.
NMR Spectroscopy
All NMR experiments were performed at 298 K on a Bruker Avance III 800 MHz spectrometer equipped with a cryogenically cooled probe and Z-gradients suitable for inverse detection. A few spiking experiments were performed on a Bruker 700 MHz spectrometer equipped with a room temperature probe and Z-gradients suitable for inverse detection. The one-pulse or NOESY pulse sequence along with the CPMG (Carr–Purcell–Meiboom–Gill) pulse sequence, all with water suppression using presaturation, were used for 1H 1D NMR experiments. To confirm unknown metabolite identification, spectra were obtained after each addition of 5–10 μL of stock solution (1 mM) of the authentic compounds to the methanol-precipitated (2:1 v/v) serum samples (see SI Table S1); in the case of volatile compounds, such as acetone, ethanol, 2-butanol, dimethylamine, and urea, spiking experiments were performed using ultrafiltered serum samples. To enable comparison of metabolite concentrations from various protein precipitation methods (SI Table S2), the CPMG experiments were performed with 128 transients and a sufficiently long recycle delay (D1 = 15 s). To aid unknown metabolite identification, homonuclear two-dimensional (2D) experiments, such as 1H–1H double quantum filtered correlation spectroscopy (DQF-COSY) and 1H–1H total correlation spectroscopy (TOCSY) experiments, were performed on serum samples after protein precipitation using methanol (2:1 v/v). The 2D experiments were performed with suppression of the residual water signal by presaturation during the relaxation delay. For DQF-COSY and TOCSY experiments, sweep widths of 9600 Hz were used in both dimensions; 512 or 400 FIDs were obtained with t1 increments for DQF-COSY or TOCSY, respectively, each with 2048 complex data points. The number of transients used was 16, and the relaxation delay was 2.0 s for DQF-COSY and 1.5 s for TOCSY. The resulting 2D data were zero-filled to 1024 points in the t1 dimension. A 90° shifted squared sine-bell window function was applied to both dimensions before Fourier transformation. Chemical shifts were referenced to the internal TSP signal for 1H 1D or 2D spectra. Bruker Topspin versions 3.0 or 3.1 software packages were used for NMR data acquisition, processing, and analyses.
Peak Assignment, Unknown Metabolite Identification and Metabolite Quantitation
Initial peak assignments relied on established literature values, specifically, the human metabolome database,17 the biological magnetic resonance data bank,18 and publications from our laboratory on the serum metabolome.7,19 Unknown metabolite identification involved a combination of literature/database searches,17 chemical shift, peak multiplicity, and J couplings measurements, and comprehensive 2D DQF-COSY and TOCSY spectral analyses. The putative new compounds were finally confirmed by spiking with authentic compounds (see SI Table S1). Chenomx NMR Suite Professional Software package (version 5.1; Chenomx Inc., Edmonton, Alberta, Canada) was used to quantitate the metabolites. This software allows fitting spectral lines using the standard metabolite library for 800 MHz 1H NMR spectra and, in particular, the determination of concentrations in complicated, overlapped spectral regions. One complication that arises is that the proximity of chemical shift values for multiple metabolite signals often result in the software providing multiple library hits for the same metabolite peak; the correct metabolite identification therefore relied on the newly established metabolite identification as annotated for a typical 1H NMR spectrum (vide infra). Peak-fitting with reference to the internal TSP signal enabled the determination of absolute concentrations for identified metabolites in protein-precipitated serum except for 2-oxoisovaleric, which was absent in the Chenomx library and was therefore quantitated by manual integration using the Bruker Topspin versions 3.0 or 3.1 software package.
Results and Discussion
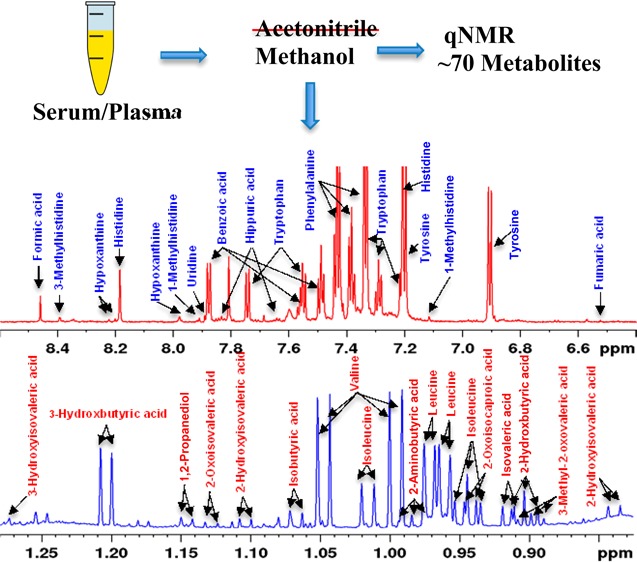
Both protein-precipitated and ultrafiltered serum provided highly resolved 1H NMR spectra for quantitative analysis of metabolites. A total of 67 blood metabolites were identified on the basis of comprehensive analyses of the NMR spectra. Importantly, more than 1/3 of the identified metabolites are new compared with the previous comprehensive investigation, made as a part of an analysis of human serum metabolome.2 Table 1 lists all 67 identified metabolites with the newly identified NMR-detectable metabolites highlighted in bold. Figures 1 and 2 show typical spectra for protein precipitated serum and ultrafiltered serum along with the annotations for characteristic peaks for all 67 metabolites. Both anomers (α and β) for glucose as well as mannose were distinctly identified. Six of the metabolites, namely, acetone, ethanol, methanol, dimethylamine, 2-propanol, and urea (see SI Table S3), were not observed in protein precipitated serum because they are lost due to sample drying after protein precipitation. Further, 1,2-propanediol, despite being a liquid at room temperature with a boiling point of 187 °C, is detected in protein precipitated serum; however, its NMR peak intensity is attenuated due to sample drying (see Figures 1 and 2), and hence, under such circumstances, its peak intensity underestimates its concentration in blood. In principle, deuterated methanol could be used without drying, but this approach would add considerable expense, and the chemical shifts would, of course, be different.
Table 1. Chemical Shifts (in ppm), J Couplings (in Hz) and Multiplicities for the Pool of 67 Metabolites Identified in Human Serum by NMRa,b.
| metabolite | human serum | authentic compounds2,30,31 |
|---|---|---|
| 1-methylhistidine | 7.116 (s), 7.908 (s) | 3.07 (dd), 3.16 (dd), 3.68 (s), 3.96 (dd), 7.0 (s), 7.67 (s) |
| 1,2-propanediol | 1.146 (d; J = 6.461), 3.457 (dd), 3.545(dd), 3.894(m) | 1.130 (d), 3.434 (dd), 3.537 (dd), 3.870 (m) |
| 2-hydroxybutyric acid | 0.903 (t; J = 7.459), 4.001 (m) | 0.886 (t), 1.641 (m), 1.734 (m), 3.990 (dd) |
| 2-hydroxyisovaleric acid | 0.839 (d; J = 6.868) | 0.82 (d), 0.95 (d), 2.01 (m), 3.84 (d) |
| 2-aminobutyric acid | 0.984 (t; J = 7.606) | 0.982 (t), 1.906 (m), 3.718 (dd) |
| 2-propanol | 1.177 (d; J = 6.160) | 1.162 (d), 4.012 (m) |
| 2-oxoisocaproic acid | 0.941 (d; J = 6.633), 2.097 (m), 2.616 (d; J = 7.021) | 0.93 (d), 2.09 (m), 2.65 (d) |
| 2-oxoisovaleric acid | 1.128 (d; J = 7.058) | 1.11 (d), 3.01 (m) |
| 3-hydroxybutyric acid | 1.204 (d; J = 6.233 Hz), 2.311 (m), 2.414 (m), 4.156 (m) | 1.204 (d), 2.314 (m), 2.414 (m), 4.160 (m) |
| 3-hydroxyisovaleric acid | 1.274 (s) | 1.26 (s), 2.35 (s) |
| 3-methylhistidine | 8.391 (s) | 3.24 (m), 3.70 (s), 3.93 (dd), 7.05 (s), 7.92 (s) |
| 3-methyl-2-oxovaleric acid | 0.899 (t; J = 7.518), 1.104 (d; J = 6.736) | 0.90 (t), 1.10 (d), 1.46 (m), 1.70 (m), 2.93 (m) |
| acetic acid | 1.924 (s) | 1.91 (s) |
| acetone | 2.236 (s) | 2.22 (s) |
| acetylcarnitine | 3.201 (s) | 2.13 (s), 2.48 (dd), 2.61 (dd), 3.18 (s), 3.61 (d), 3.82 (dd). 5.57 (q) |
| N-acetylglycine | 3.751 (s) | 2.05 (s), 3.76 (d), 8.0 (br.s) |
| alanine | 1.485 (d; J = 7.296), 3.805 (q) | 1.46 (d), 3.76 (q) |
| arginine | 1.664 (m), 1.731 (m), 1.918 (m), 3.255 (t), 3.775 (t) | 1.68 (m), 1.90 (m), 3.23 (t), 3.76 (t) |
| asparagine | 2.861 (d; J = 7.687), 2.883 (d; J = 7.687) | 2.84 (m), 2.94 (m), 4.00 (dd) |
| 2.949 (d; J = 4.294), 2.970 (d; J = 4.294), 4.015 (dd) | ||
| aspartic acid | 2.675 (d; J = 8.823), 2.697 (d; J = 8.823), | 2.66 (dd), 2.80 (dd), 3.89 (dd) |
| 2.807 (d; J = 3.713), 2.829 (d; J = 3.713), 3.912 (dd) | ||
| benzoic acid | 7.487 (t), 7.559 (t), 7.875 (d; J = 8.041) | 7.473 (dd), 7.544 (t), 7.864 (d) |
| betaine | 3.272 (s) | 3.25 (s), 3.89 (s) |
| carnitine | 3.233 (s) | 2.425 (m), 3.215 (s), 3.419 (m), 4.555 (m) |
| choline | 3.209 (s), 3.526 (m), 4.070 (m) | 3.189 (s), 3.507 (m), 4.058 (m) |
| citric acid | 2.553 (d; J = 15.124), 2.676 (d; J = 15.124) | 2.53 (d), 2.65 (d) |
| creatine | 3.040 (s), 3.935 (s) | 3.02 (s), 3.92 (s) |
| creatinine | 3.051 (s), 4.066 (s) | 3.03 (s), 4.05 (s) |
| dimethylamine | 2.726 (s) | 2.50 (s) |
| dimethylglycine | 2.934 (s) | 2.91 (s), 3.71 (s) |
| ethanol | 1.188 (t; J = 7.106), 3.664 (q) | 1.17 (t), 3.65 (q) |
| formic acid | 8.459 (s) | 8.44 (s) |
| fumaric acid | 6.524 (s) | 6.51 (s) |
| α-glucose | 3.416 (m), 3.539 (m), 3.715 (m), 3.771 (m), 3.842 (m), 5.238 (d; J = 3.798) | 3.391, 3.524, 3.701, 3.762, 3.821, 3.831, 5.223 (d) |
| β-glucose | 3.251 (dd), 3.414 (m), 3.490 (m), 3.729 (m), 3.902 (m), 4.652 (d; J = 7.989) | 3.232, 3.391, 3.461, 3.478, 3.707, 3.883, 4.634 (d) |
| glutamic acid | 2.064 (m), 2.130 (m), 2.357 (m), 3.766 (dd) | 2.040 (m), 2.119 (m), 2.341 (m), 3.748 (dd) |
| glutamine | 2.145 (m), 2.459 (m), 3.787 (t) | 2.125 (m), 2.446 (m), 3.766 (t) |
| glycerol | 3.555 (d; J = 6.636), 3.570 (d; J = 6.636), | 3.551 (m), 3.644 (m), 3.775 (tt) |
| 3.647 (d; J = 4.341), 3.662 (d; J = 4.341) | ||
| glycine | 3.564 (s) | 3.54 (s) |
| hippuric acid | 7.641 (t), 7.837 (d) | 3.96 (d), 7.54 (m), 7.62 (tt), 7.82 (dd) |
| histidine | 7.205 (s), 8.183 (s) | 3.16 (dd), 3.23 (dd), 3.98 (dd), 7.09 (d), 7.90 (d) |
| hypoxanthine | 8.199 (s), 8.221 (s) | 8.17 (s), 8.20 (s) |
| isobutyric acid | 1.067 (d; J = 6.823) | 1.21 (d), 2.59 (m) |
| isoleucine | 0.944 (t; J = 7.485), 1.015 (d; J = 6.994), 3.678 (d; J = 4.173) | 0.926 (t), 0.997 (d), 1.248 (m), 1.457 (m), 1.968 (m), 3.661 (d) |
| isovaleric acid | 0.915 (d; J = 6.645) | 0.90 (d), 1.94 (dq), 2.05 (d) |
| lactic acid | 1.332 (d; J = 6.976), 4.115 (q; J = 6.976) | 1.32 (d), 4.10 (q) |
| leucine | 0.961 (d; J = 6.227), 0.972 (d; J = 6.050), 1.718 (m), 3.740 (m) | 0.948 (t), 1.700 (m), 3.722 (m) |
| lysine | 1.452 (m), 1.512 (m), 1.733 (m), 1.913 (m), 3.030 (t; J = 7.627) | 1.46 (m), 1.71 (m), 1.89 (m), 3.02 (t), 3.74 (t) |
| α-mannose | 5.186 (d; J = 1.708) | 3.66, 3.75, 3.80, 3.83, 3.85, 3.91, 5.17 |
| β-mannose | 4.907 (d; J = 0.949) | 3.37, 3.56, 3.63, 3.72, 3.88, 3.92, 4.89 |
| methanol | 3.362 (s) | 3.341 (s) |
| methionine | 2.140 (s), 2.648 (t), 3.872 (m) | 2.157 (m), 2.631 (t), 3.851 (dd) |
| myoinositol | 3.625 (t) | 3.268 (t), 3.524 (dd), 3.613 (t), 4.053 (t) |
| ornithine | 3.060 (t; J = 7.489) | 1.727 (m), 1.826 (m), 1.933 (m), 3.046 (t), 3.774 (t) |
| phenylalanine | 7.333 (d; J = 7.466), 7.381 (t; J = 7.356), 7.432 (t) | 3.19 (m), 3.98 (dd), 7.32 (d), 7.36 (m), 7.42 (m) |
| proline | 4.138 (dd; J = 6.372; J = 8.716) | 1.99 (m), 2.06 (m), 2.34 (m), 3.33 (dt), 3.41 (dt), 4.12 (dd) |
| pyroglutamic acid | 2.407 (m), 2.507 (m), 4.182 (dd; J = 5.904; J = 9.081) | 2.02 (m), 2.39 (m), 2.50 (m), 4.17 (dd) |
| sarcosine | 2.742 (s) | 2.73 (s), 3.60 (s) |
| serine | 3.945 (d; J = 5.903), 3.960 (d; J = 5.903), 3.986 (d; J = 3.542), 4.000 (d; J = 3.542) | 3.832 (dd), 3.958 (m) |
| succinic acid | 2.415 (s) | 2.393 (s) |
| sucrose | 5.419 (d) | 3.46 (t), 3.55 (dd), 3.57 (s), 3.75 (t), 3.82 (m), 3.87–3.89 (m), 4.04 (t), 4.21 (d), 5.40 (d) |
| threonine | 1.337 (d), 3.596 (d; J = 4.931), 4.261 (dq; J = 4.918; J = 6.536) | 1.316 (d), 3.575 (d), 4.244 (m) |
| tryptophan | 7.208 (t), 7.289 (t), 7.329 (s), 7.546 (d), 7.741 (d; J = 8.017) | 3.292 (dd), 3.472 (dd), 4.046 (dd), 7.194 (m), 7.274 (m), 7.310 (s), 7.531 (d), 7.723 (d) |
| tyrosine | 6.906 (d; J = 8.503), 7.199 (d; J = 8.503) | 3.024 (dd), 3.170 (dd), 3.921 (dd), 6.877 (m), 7.170 (m) |
| urea | 5.787 (br.s) | 5.78 (br. s) |
| uridine | 4.250 (m), 4.366 (m), 5.906 (d; J = 8.010), 5.923 (d; J = 4.532), 7.886 (d; J = 8.010) | 3.801 (dd), 3.907 (dd), 4.121 (m), 4.220 (dd), 4.344 (dd), 5.882 (d), 5.902 (d), 7.864 (d) |
| valine | 0.996 (d; J = 7.061), 1.047 (d; J = 7.061), 2.281 (m), 3.617 (d; J = 4.408) | 0.976 (d), 1.029 (d), 2.261 (m), 3.601 (d) |
| xanthine | 7.977 (s) | 7.892 (s) |
Chemical shifts for characteristic peaks of metabolites that provide unambiguous information for identification and quantitation using 1D 1H NMR are shown in bold. Chemical shifts for authentic compounds are also shown separately for comparison. Newly identified metabolites are shown in bold.
s, singlet; br. s, broad singlet; d, doublet; dd, doublet of doublets; t, triplet; dt, doublet of triplets; q, quartet; dq, doublet of quartets; m, multiplet.
Figure 1.


(a) A typical 800 MHz (cryo-probe) 1D CPMG 1H NMR spectrum of a pooled human serum after protein precipitation using methanol with expanded regions (b–h) and annotations for all identified metabolites.
Figure 2.
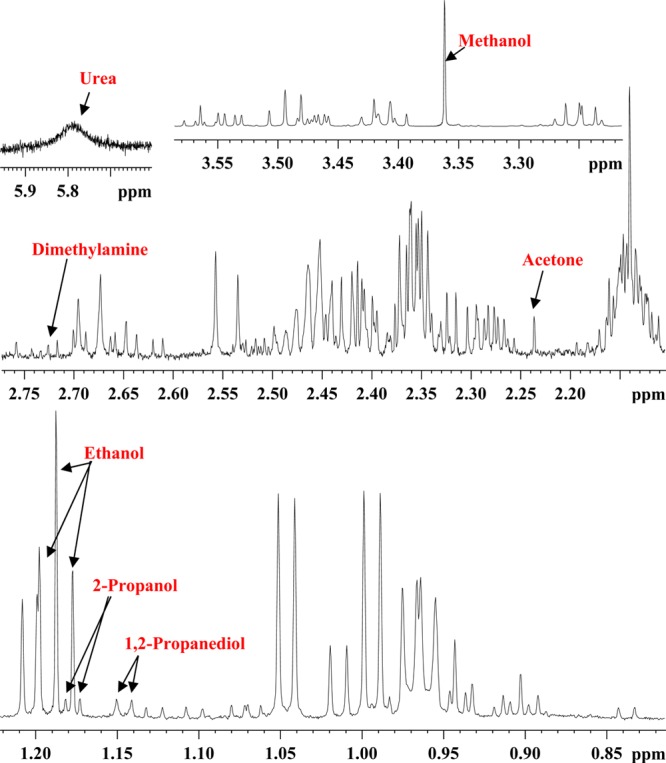
Parts of the 700 MHz 1D 1H NMR spectrum of a pooled human serum sample obtained after ultrafiltration using a 3 kDa filter with expanded regions highlighting volatile metabolites that were not detected in protein-precipitated serum because of sample drying (see Figure 1).
Despite the potential of NMR-based metabolomics of blood for the study of human diseases, advances that rely on intact serum/plasma analysis using CPMG sequence are met with numerous challenges, including the limited resolution, unknown metabolite identification, and the deleterious effects of copious proteins.20 In particular, the small number of quantifiable metabolites (∼30) combined with the grossly attenuated metabolite concentrations7 has limited the utility of this popular NMR analysis method and yielded more limited data compared with that obtained from mass spectrometry. Several efforts that involve removal of serum/plasma proteins by ultrafiltration or precipitation that show significant improvements in the number of quantifiable metabolites have been made;2,9−11 however, these approaches lack comprehensive evaluation for routine quantitation of blood metabolites. Thus far, many NMR studies have used ultrafiltration because of its efficient protein removal under the assumption that metabolites are recovered optimally.2,13−15 Only recently, on the basis of comprehensive analysis of 44 blood metabolites utilizing a variety of protein removal methods, we showed that protein precipitation exhibits superior performance over ultrafiltration for quantitating blood metabolites.7 In the current study, we have used the protein precipitation approach and identified an additional set of quantifiable blood metabolites.
Two important aspects of the analysis of blood metabolites by NMR are unknown metabolite identification and unambiguous peak assignment for routine applications. The large chemical shift databases currently available help immensely to narrow peak assignments to a relatively small list of metabolites. However, what remains challenging is the unambiguous peak assignment to specific metabolites, especially for low concentration metabolites, most of whose peaks are often buried underneath abundant metabolite signals. Thus, although a rich metabolite library consisting of nearly 300 compounds is available in the Chenomx library, for example, and is helpful for unknown metabolite identification,16 the numerous hits that it gives often lead to ambiguous or incorrect identification. The remedy for this bottleneck is a one-time establishment of the peak identities for human blood serum/plasma NMR spectra.
An advantage of using serum or plasma as the sample matrix results in stable chemical shift values that are very similar to those of other serum/plasma samples. Since, under the given experimental conditions, virtually identical spectra for blood serum/plasma can be obtained and because of the highly reproducible nature of NMR, a one-time establishment of peak assignments enables translation for routine applications. Therefore, in addition to the information provided in Table 1, Figures 1 and 2, with annotations of the characteristic peaks useful for integration for all 67 identified metabolites, serve as important visual guides to identify blood metabolites routinely. Once the unambiguous identification of blood metabolites is achieved, they can be reliably quantitated using any quantitation software package, such as Chenomx, Mnova (http://mestrelab.com), or the one available with any NMR instrument vendor.
Utilizing the new and enhanced pool of blood metabolites, a comprehensive assessment of the performance of two commonly used protein precipitation solvents, methanol and acetonitrile, was made. The results reveal a surprisingly poor performance for protein precipitation using acetonitrile: nearly 1/3 of the metabolites were attenuated by 10 to 67% compared with methanol precipitation at the same solvent-to-serum ratio of 2:1. In addition, nearly 2/3 of the metabolites were attenuated further by up to 65% upon increasing the acetonitrile-to-serum ratio to 4:1 (Figure 3, SI Tables S4 and S5). For a 3:1 acetonitrile-to-serum ratio, nearly 50% of the metabolites were attenuated by up to 53%. In contrast, only formic acid was attenuated by 10% upon increasing the methanol-to-serum ratio from 2:1 to 3:1, although 26% of the metabolites were attenuated by up to 33% upon increasing the methanol-to-serum ratio from 2:1 to 4:1.
Figure 3.

Comparison of absolute concentrations (in μM) of metabolites detected in pooled human blood serum and quantitated using 800 MHz NMR spectroscopy after protein precipitation using methanol (MeOH) (a, b, c, d) or acetonitrile (ACN) (e, f, g, h) at solvent-to-serum ratios of 2:1, 3:1, and 4:1. Methanol performs most optimally over a wide range and methanol-to-serum ratio of 2:1 provides the best performance.
As an example, SI Figure S1 illustrates the reduction in the NMR peak intensities for two typical metabolites, glutamic acid (50%) and threonine (39%), upon increasing the proportion of acetonitrile; under identical conditions, no appreciable changes in peak intensities were observed for methanol precipitation. These results indicate that the extraction of metabolites by methanol precipitation, apart from being more quantitative, is less susceptible to variation caused by different amounts of methanol. Acetonitrile, on the other hand, deleteriously affects metabolite quantities and, hence, is likely unsuitable for quantitative analysis of blood metabolites without employing recovery or internal standards. This is particularly important because, due to the lack of a comprehensive evaluation of blood metabolite quantitation, acetonitrile is still incorrectly (in our opinion) considered to be a better solvent for serum protein precipitation in metabolomics.7 It may be noted that protein precipitation using a 1:1 solvent-to-serum ratio retained a high level of residual proteins compared with the 2:1 ratio for both methanol and acetonitrile, and hence, the 1:1 ratio was not considered for further quantitative evaluation (see SI Figures S2 and S3).
The vast differences in metabolite recovery between methanol and acetonitrile solvents observed in the present study may be understood on the basis of differences in solubility of metabolites dissolved in these two solvents. Virtually all identified and quantified metabolites in this study are hydrophilic in nature, and hence, all are soluble in water. However, these metabolites have more limited solubility in organic solvents such as acetonitrile and, to a much lesser extent, methanol. In the aqueous mixture of serum, methanol is quite hydrophilic and recovers all water-soluble metabolites, although at higher methanol concentrations one can see some reduced recovery. Hydrophilic metabolites are less soluble in acetonitrile, and therefore, at the higher proportions of this organic solvent, the metabolite recovery decreases substantially. Studies have shown that solubility of polar metabolites decreases significantly as the solvent is switched from water to alcohol. For example, it has been shown that the relative solubility of amino acids such as glycine, alanine, isoleucine, phenylalanine and asparagine decreases as much as 3 orders of magnitude from water to pure methanol.21 The solubility for highly polar metabolites such as histidine, glutamic acid, aspartic acid, and asparagine could not even be measured in acetonitrile at a proportion greater than ∼1:2 (w/v).22 In accordance with these results, the recovery of a number of highly polar metabolites, including citric acid, succinic acid, lysine, aspartic acid, asparagine, glutamine, glutamic acid, threonine, and histidine, is reduced drastically as the proportion of acetonitrile increases (Figure 3 and SI Figure S1).
MS plays an important role in the metabolomics analysis of blood owing to its inherently high sensitivity and complementarity to NMR. In MS analysis, prior protein removal from blood serum/plasma is an important prerequisite. Considering the challenges involved in alleviating the interference of residual proteins in MS analysis, sample processing has been the subject of numerous investigations.20,23−29 The performance of sample processing in such MS-focused investigations is typically evaluated using the total number of ions detected and not on the comprehensive quantitative analysis of metabolites. Such an evaluation limits the quantitative accuracy, as identities for a majority of the ions thus detected remain unknown. In the current study, we have shown that absolute concentrations for numerous classes of metabolites, which represent amino acids, organic acids, carbohydrates, and heterocyclic compounds, can be obtained from a simple NMR method. Beyond metabolomics studies and applications, this ability of NMR to quantitatively evaluate a relatively large number of metabolites representing many important classes may also offer new avenues to assess sample processing methods for analysis using MS.
In conclusion, we describe the NMR identification of numerous unknown metabolites in blood and provide a simple optimized NMR approach to quantitate the enhanced pool of blood metabolites on a routine basis. A one-time establishment of the identity for 67 metabolites, a significant portion of which is newly established in this study, is made by a comprehensive analysis of pooled human blood serum using an array of 1D and 2D NMR techniques, database searches, and spiking experiments using authentic compounds. An important aspect of this study is that the characteristic peaks for all identified metabolites are marked for easy identification and quantitation of metabolites in the NMR spectrum. This, we believe, is critical for widespread use of enhanced and NMR-based blood metabolite profiling because the chemical shift databases alone are often insufficient for unambiguous assignment. This issue is particularly acute for low-concentration metabolites, given the peak overlap and chemical shift sensitivity to sample conditions such as pH, salt, and temperature. The high reproducibility of NMR combined with sample processing and analysis protocols provided here enable obtaining identical spectra for blood serum/plasma on a routine basis. Hence, even nonexpert NMR users can easily identify and quantify blood metabolites following the described approach, which is important for effective use of this tool. In addition, based on the comprehensive analysis of the pool of metabolites established in this study, the performance of serum protein precipitation using different proportions of methanol or acetonitrile was evaluated. Methanol performs most optimally over a wide range of concentrations, whereas acetonitrile shows a surprisingly poor performance. Finally, the ability to quantitatively detect a large number of metabolites, which represent many classes, including amino acids, organic acids, carbohydrates, and heterocyclic compounds, using a simple NMR method described in this study potentially offers new avenues to assess sample processing for analysis using MS. The raw and annotated data from this study will be made available to the metabolomics community through major metabolomics resources, including the Metabolomics Workbench (http://www.metabolomicsworkbench.org/), MetaboLights (http://www.ebi.ac.uk/metabolights/), and the Northwest Metabolomics Research Center (http://depts.washington.edu/nwmrc/).
Acknowledgments
We gratefully acknowledge financial support from the NIH (National Institute of General Medical Sciences 2R01GM085291).
Supporting Information Available
Five tables and three figures detailing metabolite data. This material is available free of charge via the Internet at http://pubs.acs.org/.
The authors declare the following competing financial interest(s): Daniel Raftery reports holding equity and an executive position at Matrix Bio, Inc.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Beckonert O.; Keun H. C.; Ebbels T. M.; Bundy J.; Holmes E.; Lindon J. C.; Nicholson J. K. Nat. Protoc. 2007, 2112692–2703. [DOI] [PubMed] [Google Scholar]
- Psychogios N.; Hau D. D.; Peng J.; Guo A. C.; Mandal R.; Bouatra S.; Sinelnikov I.; Krishnamurthy R.; Eisner R.; Gautam B.; Young N.; Xia J.; Knox C.; Dong E.; Huang P.; Hollander Z.; Pedersen T. L.; Smith S. R.; Bamforth F.; Greiner R.; McManus B.; Newman J. W.; Goodfriend T.; Wishart D. S. PLoS One 2011, 62e16957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagana Gowda G. A.; Raftery D. Curr. Metabolomics 2013, 13227–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson J. K.; Gartland K. P. NMR Biomed. 1989, 2277–82. [DOI] [PubMed] [Google Scholar]
- Chatham J. C.; Forder J. R. Biochim. Biophys. Acta 1999, 14261177–184. [DOI] [PubMed] [Google Scholar]
- Bell J. D.; Brown J. C.; Kubal G.; Sadler P. J. FEBS Lett. 1988, 235, 81–86. [DOI] [PubMed] [Google Scholar]
- Nagana Gowda G. A.; Raftery D. Anal. Chem. 2014, 86115433–5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wevers R. A.; Engelke U.; Heerschap A. Clin. Chem. 1994, 407 Pt 11245–1250. [PubMed] [Google Scholar]
- Daykin C. A.; Foxall P. J.; Connor S. C.; Lindon J. C.; Nicholson J. K. Anal. Biochem. 2002, 3042220–230. [DOI] [PubMed] [Google Scholar]
- Tiziani S.; Emwas A. H.; Lodi A.; Ludwig C.; Bunce C. M.; Viant M. R.; Günther U. L. Anal. Biochem. 2008, 377116–23. [DOI] [PubMed] [Google Scholar]
- Fan T. W. In The Handbook of Metabolomics, Methods in Pharmacology and Toxicology; Fan T. W., Higashi R. M., Lane A. N., Eds.; Springer: New York, 2012; pp 7–27. [Google Scholar]
- Bouatra S.; Aziat F.; Mandal R.; Guo A. C.; Wilson M. R.; Knox C.; Bjorndahl T. C.; Krishnamurthy R.; Saleem F.; Liu P.; Dame Z. T.; Poelzer J.; Huynh J.; Yallou F. S.; Psychogios N.; Dong E.; Bogumil R.; Roehring C.; Wishart D. S. PLoS One 2013, 89e73076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simón-Manso Y.; Lowenthal M. S.; Kilpatrick L. E.; Sampson M. L.; Telu K. H.; Rudnick P. A.; Mallard W. G.; Bearden D. W.; Schock T. B.; Tchekhovskoi D. V.; Blonder N.; Yan X.; Liang Y.; Zheng Y.; Wallace W. E.; Neta P.; Phinney K. W.; Remaley A. T.; Stein S. E. Anal. Chem. 2013, 852411725–11731. [DOI] [PubMed] [Google Scholar]
- Farshidfar F.; Weljie A. M.; Kopciuk K.; Buie W. D.; Maclean A.; Dixon E.; Sutherland F. R.; Molckovsky A.; Vogel H. J.; Bathe O. F. Genome Med. 2012, 4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bathe O. F.; Shaykhutdinov R.; Kopciuk K.; Weljie A. M.; McKay A.; Sutherland F. R.; Dixon E.; Dunse N.; Sotiropoulos D.; Vogel H. J. Cancer Epidemiol. Biomarkers Prev. 2011, 201140–147. [DOI] [PubMed] [Google Scholar]
- Weljie A. M.; Newton J.; Mercier P.; Carlson E.; Slupsky C. M. Anal. Chem. 2006, 78, 4430–4442. [DOI] [PubMed] [Google Scholar]
- Wishart D. S.; Jewison T.; Guo A. C.; Wilson M.; Knox C.; Liu Y.; Djoumbou Y.; Mandal R.; Aziat F.; Dong E.; Bouatra S.; Sinelnikov I.; Arndt D.; Xia J.; Liu P.; Yallou F.; Bjorndahl T.; Perez-Pineiro R.; Eisner R.; Allen F.; Neveu V.; Greiner R.; Scalbert A. Nucleic Acids Res. 2013, 41Database issueD801–D807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich E. L.; Akutsu H.; Doreleijers J. F.; Harano Y.; Ioannidis Y. E.; Lin J.; Livny M.; Mading S.; Maziuk D.; Miller Z.; Nakatani E.; Schulte C. F.; Tolmie D. E.; Wenger R. K.; Yao H.; Markley J. L. Nucleic Acids Res. 2008, 36Database issueD402–D408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagana Gowda G. A.; Tayyari F.; Ye T.; Suryani Y.; Wei S.; Shanaiah N.; Raftery D. Anal. Chem. 2010, 82218983–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuckovic D. Anal. Bioanal. Chem. 2012, 40361523–1548. [DOI] [PubMed] [Google Scholar]
- Orella C. J.; Kirwan D. J. Ind. Eng. Chem. Res. 1991, 30, 1040–1045. [Google Scholar]
- Gekko K.; Ohmae E.; Kameyama K.; Takagi T. Biochim. Biophys. Acta 1998, 13871–2195–205. [DOI] [PubMed] [Google Scholar]
- Dunn W. B.; Broadhurst D.; Begley P.; Zelena E.; Francis-McIntyre S.; Anderson N.; Brown M.; Knowles J. D.; Halsall A.; Haselden J. N.; Nicholls A. W.; Wilson I. D.; Kell D. B.; Goodacre R. Human Serum Metabolome (HUSERMET) Consortium. Nat. Protoc. 2011, 671060–1083. [DOI] [PubMed] [Google Scholar]
- Ivanisevic J.; Zhu Z. J.; Plate L.; Tautenhahn R.; Chen S.; O’Brien P. J.; Johnson C. H.; Marletta M. A.; Patti G. J.; Siuzdak G. Anal. Chem. 2013, 85146876–6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce S. J.; Tavazzi I.; Parisod V.; Rezzi S.; Kochhar S.; Guy P. A. Anal. Chem. 2009, 8193285–3296. [DOI] [PubMed] [Google Scholar]
- Jiye A.; Trygg J.; Gullberg J.; Johansson A. I.; Jonsson P.; Antti H.; Marklund S. L.; Moritz T. Anal. Chem. 2005, 77248086–8094. [DOI] [PubMed] [Google Scholar]
- Zelena E.; Dunn W. B.; Broadhurst D.; Francis-McIntyre S.; Carroll K. M.; Begley P.; O’Hagan S.; Knowles J. D.; Halsall A.; Wilson I. D.; Kell D. B. Anal. Chem. 2009, 8141357–1364. [DOI] [PubMed] [Google Scholar]
- Tulipani S.; Llorach R.; Urpi-Sarda M.; Andres-Lacueva C. Anal. Chem. 2013, 851341–348. [DOI] [PubMed] [Google Scholar]
- Want E. J.; O’Maille G.; Smith C. A.; Brandon T. R.; Uritboonthai W.; Qin C.; Trauger S. A.; Siuzdak G. Anal. Chem. 2006, 783743–752. [DOI] [PubMed] [Google Scholar]
- Cui Q.; Lewis I. A.; Hegeman A. D.; Anderson M. E.; Li J.; Schulte C. F.; Westler W. M.; Eghbalnia H. R.; Sussman M. R.; Markley J. L. Nat. Biotechnol. 2008, 262162–164. [DOI] [PubMed] [Google Scholar]
- Prestegard J. H.; Miner V. W.; Tyrell P. M. Proc. Natl. Acad. Sci. U.S.A. 1983, 80237192–7196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.