

Abstract

Heparan sulfate (HS) 3-O-sulfation determines the binding specificity of HS/heparin for antithrombin III and plays a key role in herpes simplex virus (HSV) infection. However, the low natural abundance of HS 3-O-sulfation poses a serious challenge for functional studies other than the two cases mentioned above. By contrast, multiple distinct isoforms of 3-O-sulfotranserases exist in mammals (up to seven isoenzymes). Here we describe a novel peeling reaction that specifically degrades HS chains with 3-O-sulfated glucosamine at the reducing-end. When HS/heparin is enzymatically depolymerized for compositional analysis, 3-O-sulfated glucosamine at the reducing ends appears to be susceptible to degradation under mildly basic conditions. We propose a 3-O-desulfation initiated peeling reaction mechanism based on the intermediate and side-reaction products observed. Our discovery calls for the re-evaluation of the natural abundance and functions of HS 3-O-sulfation by taking into consideration the negative impact of this novel peeling reaction.
Heparan sulfate (HS) is a linear sulfated polysaccharide that is covalently linked to proteoglycans core proteins on cell surfaces and in extracellular matrixes. Mostly through the sulfo groups along its chain, HS binds to a plethora of protein ligands and is involved in the regulation of a variety of biological processes including development, angiogenesis, coagulation, tumor metastasis, and viral infection.1 HS is synthesized in the Golgi apparatus as 1,4-linked repeating disaccharide units of glucuronic acid (GlcA) residues and N-actetylglucosamine (GlcNAc) residues, then undergoes a series of enzyme modifications to form the ligand-binding domains found in the mature structures. Specifically, the GlcNAc residues can be selectively de-N-acetylated and N-sulfated; the GlcA residues can be epimerized to iduronic acid (IdoA); most importantly, sulfo groups can be added by a series of O-sulfotransferases to the 2-O position of uronic acid residues and the 6-O and 3-O position of the glucosamine residues. The activities of HS modifying enzymes are controlled by numerous factors and the biosynthetic reactions are typically incomplete, thus giving rise to mature polysaccharides consisting of domains of high, low, and intermediate sulfation, overlaid by heterogeneity of modifications and chain lengths.2
Among all the HS modifications, 3-O-sulfation is considered to be the last step and comprises only 0.5% of the total sulfation.3−6 However, the 3-O-sulfo groups are essential modifications that determine the activity of HS/heparin in a number of biological and pharmacological processes, including the anticoagulant activation of antithrombin by heparin7−9 and HS mediated herpes simplex virus (HSV) attachment to host-cell and viral entry.10−12 It is also implicated in the binding of FGF and FGF receptors,13−15 epithelial-mesenchymal transition inducing,16 and kal-1 dependent neurite branching.17 Given the unusually low natural abundance of the HS 3-O-sulfation, it is intriguing that as many as seven 3-O-sulfotransferase (Hs3st) isoforms have been identified in mammals, making them the largest family of HS modification enzymes.18,19 Previous studies show that the Hs3st genes are regulated both spatially and temporally in different types of cells or tissues, suggesting in vivo functional differences exist between members of this family.20 It has been further hypothesized that each of Hs3sts creates HS domains with distinct functions.21 However, other than the involvement of 3-O-sulfation in the anticoagulant activation of antithrombin and HSV viral entry, the structure and function of the Hs3st isozyme specific HS domains remain largely unknown.19 One of the major roadblocks preventing detailed analysis of such HS domains is the apparent rarity of HS 3-O-sulfation.
The peeling reaction is a base-catalyzed elimination reaction, resulting in the stepwise loss of the monosaccharide residue from the reducing-end of carbohydrate polymers. Peeling reactions contribute to the degradation of oligosaccharides released from glycopeptides and glycoproteins using hydrozinolysis or β-elimination.22−25 The degradation is the most facile for reducing-end monosaccharides substituted at the 3-position, due to loss of the substituent (Scheme 1). 1,4-Linked glycans need harsh conditions to induce structural rearrangements before undergoing peeling reactions.26,27 Because the HS copolymer is comprised of 1,4-linked repeating disaccharide units, the degree of peeling under alkaline conditions used in sample processing and workup has not been a concern in the field. However, because of the complexity of HS, HS samples are often analyzed after being depolymerized by bacterial heparin lyases.28 Although the optimum digestion pH of these lyases is 7.0–7.5,28 reductive amination is often performed subsequently to increase saccharide hydrophobicity29 or charges in electrophoresis30−32 and to add a chromophore or fluorophore,33−35,71 exposing the lyase products to the alkaline conditions introduced by primary amine and reducing reagents. The susceptibility of different HS saccharides under these milder basic conditions has never been either noticed or assessed.
Scheme 1. General Peeling Reaction Mechanism of 1,3-Linked Glycans.

Here, we report that when 3-O-sulfated HS glucosamine is present at the reducing-end of HS saccharides produced by polysaccharide lyase, a peeling reaction will occur under mildly basic conditions. We propose that the reaction first causes the desulfation at the 3-position of the glucosamine followed by the loss of the glucosamine monosaccharide. The reaction is obviously accelerated by an unintended increase in either pH or temperature. Because of this 3-O-sulfation specific peeling reaction, there is concern that previously reported abundances of HS 3-O-sulfation in lyase-generated saccharides may have underestimated its biological abundance.
Materials and Methods
Materials
Porcine intestinal mucosa (PIM) heparan sulfate (HS) was purchased from Celsus Laboratories, Inc. (Cincinnati, OH). Heparin lyase I from Flavbacterium heparinum was purchased from Iduron (Manchester, U.K.). Fondaparinux (GlcNS6S-GlcA-GlcNS3S6S-IdoA2S-GlcNS6S-Me, C31H43N3Na10O49S8) was purchased from Organon Sanofi-Synthelabo LLC (West Orange, NJ). Fondaparinux was prepared by dialysis using a 100 Da molecular weight cutoff membrane filter before LC–MS analysis. Enoxaparin was from Sanofi prefilled injection syringe (30 mg/0.3 mL). PAPS was purchased from Sigma-Aldrich. Hs3st-1 was obtained from R&D Systems, Minneapolis, MN. Synthetic HS tetrasaccharide was prepared as previously described.36
Preparing Size-Defined HS Oligosaccharides
PIM HS (∼50 mg) was digested using heparin lyase I distributively in a small volume and then separated by a gel filtration column. Briefly, 50 mg of HS was dissolved in digestion buffer (100 mM NaCl, 20 mM Tris-HCl, 1 mM Ca(OAc)2, pH 7.4) and divided into five 400 μL aliquots, to each of which were added 50 mU of heparin lyase I, and digested at 37 °C for 3 h. The digestion products were then combined and applied onto a 1.5 cm × 170 cm Bio-Rad Econo Column packed with Bio-Rad P-10 gel (fine beads, particle size 45–90 μm, separation range 1 500–20 000 Da) equilibrated with 100 mM NH4HCO3. The flow rate was set at 0.05 mL/min, and the effluent was monitored by UV at 232 nm. The fractions corresponding to the same chromatographic peak from the UV profile were pooled and vacuum-dried twice to remove the remaining ammonium bicarbonate.
HS Compositional Analysis Using Hydrophilic Interaction Liquid Chromatography–mass Spectrometry (HILIC–MS)
The compositions of oligosaccharides of tetrasaccharide or greater sizes were analyzed by chip-based Amide-80 chromatography with MS detection as previously described.37−41
Separation of HS Oligosaccharides by Strong Anion Exchange (SAX) Chromatography
Approximately 30 nmol of size-fractioned HS oligosaccharides were further separated using an IonPac AS7 column (4.6 mm × 250 mm, Thermo Scientific) using a previously described method.72 The mobile phase used were A, H2O (pH 3.5 by HCl); B, 2 M NaCl (pH 3.5 by HCl). A Beckman Gold HPLC system was used to deliver a linear gradient as follows: 1–10 min, 0% B, 10–70 min, 0–100% B; 85–90 min, 100–0% B. The effluents were detected by UV at 232 nm and chromatographic peaks were collected and desalted using a PD MiniTrap G-10 desalting cartridge (GE Health, Piscataway, NJ) for further analysis.
Electron Detachment Dissociation (EDD) Analysis of HS Oligosaccharides
Desalted HS oligosaccharide was subject to electrostatic nanospray on a 12 T solariX hybrid Fourier transform ion cyclotron resonance (FTICR) mass spectrometry system (Bruker Daltonics, Bremen, Germany). The EDD experiment was carried out as previously described.42−44
Hs3st Modification of Synthetic HS Tetrasaccharide Substrates
The enzymatic modification of synthetic tetrasaccharides by Hs3st was carried out using a protocol modified from our previous publication.26 Each reaction consists of 0.01 μg/uL synthetic tetrasaccharide, 0.004 μg/uL Hs3st-1, 0.16 mM PAPS, 50 mM MOPS, pH 7.0, 2.5 mM MnCl2, 2.5 mM MgCl2, 1.25 mM CaCl2, 0.01% BSA, in ∼50 μL volume. After incubation at 37 °C for overnight (∼16 h), the modified tetrasaccharide was separated from proteins and salts on a Superdex Peptide PC 3.2/30 column (GE Biosciences, Piscataway, NJ) equilibrated with 100 mM NH4HCO3.
pH-Dependent Lyase Digestion of the Fondaparinux Pentasaccharide and the Synthetic Tetrasaccharide
A 1 nmol quantity of fondaparinux pentasaccharide was incubated with 10 mU heparin lyase I in a 50 μL reaction buffered by either 50 mM NaOAc, pH 6.0 or 50 mM Tris-HCl, pH 7.0 or 50 mM Tris-HCl, pH 8.0, in the presence of 1 mM Ca(OAc)2 and 0.1% BSA, at 37 °C overnight. A 0.2 μg quantity of the synthetic tetrasaccharides (with or without Hs3st modification) was digested by a mixture of 10 mU heparin lyase I and heparin lyase II in a similar way.
Testing the Base Susceptibility of Purified HS Oligosaccharides
A 1 nmol quantity of purified HS oligosaccharides were vacuum-dried and resuspended in either 0.1 M NH3·H2O, pH ∼11.0, or 0.1 M Tris-HCl, pH 8.0. The reactions were incubated at 37 °C overnight and vacuum-dried again to remove excess NH3·H2O.
Preparation of Lyase-Resistant Enoxaparin Tetrasaccharides
The enoxaparin was obtained from the Lovenox injection syringe. A total of ∼100 μg of enoxaparin was digested by a mixture of heparin lyase I, II, and III (10 mU each) in a digestion buffer consisting of 50 mM NaOAc, pH 6.0, 1 mM Ca(OAc)2, and 0.1% BSA. After incubation at 37 °C overnight, the digestion products were fractionated using a Superdex Peptide PC 3.2/30 column (GE Biosciences, Piscataway, NJ) equilibrated with 100 mM NH4HCO3. The chromatographic peak corresponding to tetrasaccharide was collected as the lyase-resistant tetrasaccharides and vacuum-dried immediately to avoid unwanted degradation.
Analysis of HS Oligosaccharides Using Size Exclusion Chromatography–Mass Spectrometry (SEC–MS)
The digestion products of HS tetrasaccharides and pentasaccharides by heparin lyases were analyzed using size exclusion chromatography (SEC)–mass spectrometry as previously described.45
Reductive-Amination Labeling of HS Oligosaccharides
A 1 nmol quantity of 3-O-sulfate containing hexasaccharide [1,2,3,1,7] was vacuum-dried and resuspended in 10 μL of labeling reagent mixture consisting of 0.8 M sodium cyanoborohydride and 0.5 M n-butylamine dissolved in a varying ratio of AcOH and DMSO (v/v = 1:1, 3:7, and 1:9). The reactions were incubated at 65 °C for 3 h. The reaction products were then desalted by PD MiniTrap G-10 cartridges and vacuum-dried.
Nomenclature and Data Analysis
HS oligosaccharide compositions are given as [ΔHexA,HexA,GlcN,Ac,SO3] (ΔHexA, 4,5-unsaturated hexuronic acid; HexA, hexuronic acid; GlcN, glucosamine; Ac, acetate; SO3, sulfate), denoting the number of the corresponding residues. Fragment ions from tandem MS are labeled using the conventional carbohydrate fragmentation nomenclature46 with HS specific modifications. Specifically, HS structures without modification of sulfation (S) and/or acetylation (Ac) are named as the backbone followed by the modifications in parentheses, such as 0,2X2 (1Ac, 2S) and Y1 (3S).
Results and Discussion
Discovery of a Base-Sensitive HS Oligosaccharide
As we were profiling the compositions of a size-fractioned HS hexasaccharide sample prepared by depolymerizing the commercial porcine intestine mucosa HS (HSPIM) using heparin lyase I, we identified abundant HS pentasaccharides with the composition of [1,2,2,1,4] (HS oligosaccharide compositions are given as [ΔHexA,HexA,GlcN,Ac,SO3], denoting the number of the corresponding residues and modifications) (Figure 1). Because of the repeating disaccharide units in the HS chains, the finding of such pentasaccharides with one glucosamine residue missing is unusual. Although odd-numbered oligosaccharide may result from the endogenous activity of mammalian heparanase,47,48 we did not detect a comparable amount of odd-numbered oligosaccharide with the composition of [0,2,3,X,Y], which would be simultaneously produced on the reducing-end side of the heparanase cleavage sites. We therefore suspected an unnatural source of the [1,2,2,1,4] pentasaccharides. Since the mobile phase used in size-fractioning the HS oligosaccharides consists of 100 mM ammonium bicarbonate, which had a slightly alkaline pH (∼8.0), we hypothesized that the [1,2,2,1,4] pentasaccharide could be a base-catalyzed degradation product from a [1,2,3,X,Y] hexasaccharide. To test this hypothesis, we incubated the HS oligosaccharide fraction with 0.1 M ammonium hydroxide (pH ∼11.0) and profiled the HS compositions before and after the incubation (Figure 1B). The results demonstrated that the elevated pH increased the abundance of the [1,2,2,1,4] pentasaccharide in the fraction. The results further revealed that the increase of the pentasaccharide was accompanied by a sharp decrease in the abundance of highly sulfated hexasaccharides, predominately in the composition of [1,2,3,1,7], suggesting that the unusual pentasaccharide [1,2,2,1,4] might be a base-catalyzed degradation product of the [1,2,3,1,7] hexasaccharide.
Figure 1.

Discovery of the base-susceptible hexasaccharide [1,2,3,1,7] in the MS profiling of the size-fractioned HS oligosaccharide profiles: (A) SEC fractionation of HS oligosaccharides obtained from limited lyase digestion of HSPIM and (B) composition profiles of the hexasaccharide fraction with and without NH3·H2O treatment. An unusual HS pentasaccharide with the composition of [1,2,2,1,4] was identified and found to be a base-catalyzed degradation product of a hexasaccharide with the composition of [1,2,3,1,7].
In order to purify base-sensitive hexasaccharide [1,2,3,1,7], we subjected the hexasaccharide fraction to further separation by strong anion exchange chromatography (SAX) as shown in Figure 2A. Individual chromatographic peaks corresponding to highly sulfated saccharides were desalted separately and analyzed by HILIC–MS. We observed the composition [1,2,3,1,7] in both peaks I and II. However, treatment of fractions I and II with basic conditions showed that only the [1,2,3,1,7] from the peak II was degraded into [1,2,2,1,4] in a pH-dependent manner (Figure 2B). In the absence of the additional base treatment, peak II underwent degradation to a small extent. When treated with pH 8.0 tris buffer, more than half of the [1,2,3,1,7] was converted into [1,2,2,1,4]. When treated with 0.1 M ammonium hydroxide (pH ∼11.0), nearly all [1,2,3,1,7] was degraded to [1,2,2,1,4], indicating the [1,2,3,1,7] isomer in peak II underwent the observed degradation described in Figure 1.
Figure 2.

Isolation of the base-susceptible hexasaccharide [1,2,3,1,7] from size-fractioned HS oligosaccharides: (A) SAX chromatogram of the size-fractioned HS hexasaccharides. The composition [1,2,3,1,7] was found in peaks I and II. (B) Degradation of the hexasaccharide [1,2,3,1,7] from peak II under basic conditions. The hexasaccharide [1,2,3,1,7] from peak II to either control (ddH2O) or basic conditions (Tris-HCl, pH8.0, or 0.1 M NH3·H2O) treatment. The products were analyzed on HILIC–MS. The base peak chromatograms (BPCs) showed that the hexasaccharide [1,2,3,1,7] (peak b) was converted into pentasaccharide [1,2,2,1,4] (peak a) under basic conditions.
We next characterized the structure of the [1,2,3,1,7] isomer in the SAX fractions using tandem mass spectrometry and discovered that it contains a N,3,6-sulfated glucosamine (GlcNS3S6S) at its reducing-end (Supplemental Figure 1 in the Supporting Information). As the degradation of the hexasaccharide [1,2,3,1,7] to the pentasaccharide [1,2,2,1,4] resulted in the loss of a glucosamine residue together with three sulfo groups, it appears that the GlcNS3S6S residue at the reducing-end of the hexasaccharide [1,2,3,1,7] was lost in this degradation reaction.
Reducing-End 3-O-Sulfation Required for the Peeling Reaction
Because the degradation reaction is apparently catalyzed by mildly basic conditions and specific to the structure of the hexasaccharide [1,2,3,1,7] we characterized, we first hypothesized that this is a novel peeling reaction that is specific to the GlcNS3S6S residues at the reducing-end. In order to test this hypothesis, we took advantage of the synthetic heparin pentasaccharide fondaparinux, which has the GlcNS3S6S residue in its sequence (GlcNS6S-GlcA-GlcNS3S6S-IdoA2S-GlcNS6S-Me). Digesting this synthetic pentasaccharide with heparin lyase I releases the GlcNS3S6S residue to the reducing-end of the trisaccharide product GlcNS6S-GlcA-GlcNS3S6S (Figure 3). We then tested if lyase I treatment of fondaparinux at various pH conditions would cause the degradation of the particular trisaccharide. As expected, we observed two chromatographic products at pH 6.0 and 7.0, corresponding to the intact disaccharide product (peak b) and the trisaccharide product (peak a). However, at pH 8.0, an additional product (peak c) with the mass corresponding to GlcNS6SHexA appeared as the abundance of the trisaccharide (peak a) decreased at the same time, consistent with the degradation of the trisaccharide GlcNS6S-GlcA-GlcNS3S6S into disaccharide GlcNS6S-GlcA. This result suggests that the peeling reaction we discovered is not a unique reaction specific to the hexasaccharide [1,2,3,1,7] and applies to other HS oligosaccharides with GlcNS3S6S residues at the reducing-end.
Figure 3.

Digestion of heparin pentasaccharide fondaparinux by heparin lyase I at different pH conditions. The lyase products were analyzed using SEC–MS, and the total ion chromatograms (TICs) demonstrated the conversion of the trisaccharide digestion product (peak a) into disaccharide degradation product (peak c) at pH 8.0. The structures of peak a, b, and c were determined by their masses.
We next tested the necessity of the 3-O-sulfation for the proposed peeling reaction. A tetrasaccharide with the sequence of GlcA-GlcNS-IdoA2S-GlcNS6S-R (R = (CH2)5NH2) was chosen from a previously published synthetic HS oligosaccharide library36 and modified at the GlcNS residue with recombinant Hs3st-1 (Supplemental Figure 2 in the Supporting Information). After the enzymatic reaction, both the synthetic tetrasaccharide substrate and the Hs3st product were digested with heparin lyase I at different pHs. As shown in Figure 4, lyase I digestion of the control tetrasaccharide produced GlcA-GlcNS and IdoA2S-GlcNS6S-R as identified using SEC–MS, both of which remained stable at elevated pH. Similarly, the Hs3st-1 modified tetrasaccharide produced the 3-O-sulfation containing GlcA-GlcNS3S and IdoA2S-GlcNS6S-R after lyase I digestion at pH 6.0. However, when the Hs3st-1 modified tetrasaccharide was digested at pH 8.0, the 3-O-sulfated product diminished on the chromatograph, demonstrating that the newly formed GlcA-GlcNS3S was indeed susceptible to degradation under mildly basic conditions. Therefore, by directly comparing the susceptibility of GlcA-GlcNS and GlcA-GlcNS3S, we proved that 3-O-sulfation is necessary for the novel peeling reaction we discovered. Since the disaccharide GlcA-GlcNS3S does not contain 6-O-sulfation, 6-O-sulfation is apparently not necessary for the degradation to occur.
Figure 4.

Digestion of synthetic (A) and Hs3st-1 modified (B) tetrasaccharide at different pH conditions. The lyase products were analyzed using SEC–MS, and the total ion chromatograms (TICs) demonstrated the 3-O-sulfation dependent degradation of the disaccharide product (peak b) at pH 8.0.
Proposed Mechanism
As this reducing-end 3-O-sulfation specific peeling reaction is both unprecedented and in sharp contrast to the traditional notion about the stability of 1,4 linked reducing end monosaccharides against peeling reactions, we investigated the mechanism of this reaction. Two new ions appeared in the extracted mass spectra of the peeling reaction products of the Hs3st-1 modified tetrasaccharide: m/z 416.05 and 434.06 (Supplemental Figure 3 in the Supporting Information). The ion at m/z 416.05 drew our attention because (1) this mass matched with that of GlcA-GlcNS3S with a loss of one water molecule (H2O) and one sulfo group (−SO3), (2) The SEC elution time of this ion was greater than that of GlcA-GlcNS3S, indicating that m/z 416.05 did not result from loss of SO3 from GlcA-GlcNS3S during ionization (Supplemental Figure 3 in the Supporting Information). We reasoned that loss of 3-O-sulfation could have occurred in a manner similar to that of peeling observed for mucin-type GalNAc saccharides with 3-substituents (Scheme 1). This elimination would create a double bond at the C2–C3 position of the glucosamine, which further set the stage for the elimination (peeling) at the C-4 position as the C-2 -NH- group gets deprotonated at basic conditions (Scheme 2A). This proposed two-step mechanism would explain the observed specificity of this novel peeling reaction to 3-O-sulfated glucosamine residues, as the elimination of the 3-O-sulfation is required for the peeling step.
Scheme 2. (A) Proposed Mechanism for the 3-O-Desulfation Initiated Peeling Reaction and (B) Proposed Michael Addition Mechanism on the Intermediate Product 3 to Yield the Side Product 5.

R′ = H or SO3H; R″ = COCH3 or SO3H.
In the proposed mechanism (Scheme 2A), the newly formed ion at m/z of 434.06 had the mass of the 3-O-desulfated intermediate product 3 plus one water molecule (H2O). We reasoned that this product could be formed by Michael addition of a hydroxyl group to the 3 position under basic conditions (Scheme 2B). Therefore, the observation of the side product 5 supported the two-step peeling reaction proposed in Scheme 2A. Importantly, a similar side product was also observed in the peeling reaction of the hexasaccharide [1,2,3,1,7] (Supplemental Figure 4 in the Supporting Information).
Impact of the Peeling Reaction on the Analysis of HS 3-O-Sulfation
We tested the extent to which this novel peeling reaction affects the determination of the abundance of 3-O-sulfation under routine analysis conditions for HS oligosaccharides. It has been established that the glycosidic bond to the nonreducing side of the 3-O-sulfation containing disaccharide unit is resistant to the depolymerization by heparin lyases.21,49−56 Therefore, exhaustive depolymerization of 3-O-sulfation containing HS/heparin by heparin lyases will generate lyase-resistant HS tetrasaccharides that have GlcNS3S6S at the reducing-ends. As the 3-O-sulfation content is necessary for anticoagulation activity of low molecular weight heparin (LMWH), manufacturers of LMWH use the abundances of the lyase-resistant, 3-O-sulfation containing tetrasaccharides to gauge product quality.57 We therefore tested the extent to which lyase-resistant LMWH tetrasaccharides are prone to the 3-O-sulfation specific peeling reaction. We digested the LMWH enoxaparin exhaustively using heparin lyases, isolated the resistant tetrasaccharides from the products, and treated them with pH 8.0 Tris-HCl buffer. As shown in Figure 5, the abundances of the tetrasaccharide compositions [1,1,2,1,4] and [1,1,2,1,3], which were previously shown to be the major 3-O-sulfated lyase-resistant tetrasaccharides from enoxaparin,58 decreased significantly after the treatment. In addition, a new peak was observed at m/z 634.0, corresponding to the trisaccharide composition [1,1,1,1,1]. The results showed that the lyase-resistant tetrasaccharide compositions [1,1,2,1,4] and [1,1,2,1,3] were converted to the trisaccharide composition [1,1,1,1,1] at slightly basic conditions, losing one glucosamine residue with three and two sulfo groups, respectively. Since these lyase-resistant tetrasaccharide from enoxaparin had 3-O-sulfated glucosamine residues at the reducing-ends, these results suggested they were prone to the 3-O-sulfation specific peeling reaction. Our discovery therefore raised concern about the degradation of the lyase-resistant tetrasaccharides from LMWH due to alkaline storage/analysis conditions.
Figure 5.
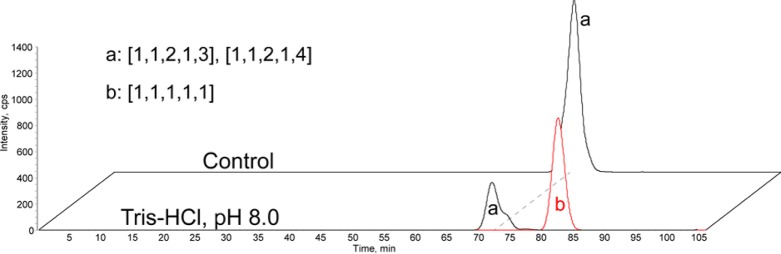
Susceptibility of lyase-resistant tetrasaccharides from enoxaparin to the 3-O-sulfation specific peeling reaction. Purified enoxaparin tetrasaccharides were subjected to either control (ddH2O) or basic condition (Tris-HCl, pH 8.0) treatment. The products were analyzed using SEC–MS. Peak a, the combined extracted ion chromatograms (EICs) of previously reported lyase-resistant tetrasaccharides from enoxaparin; peak b, the EIC of a trisaccharide resulted from the 3-O-sulfation specific peeling reaction of the tetrasaccharides.
We then considered widely used analytical methods that might cause this unwanted reaction. One common practice in analyzing HS/heparin oligosaccharides is reductive-amination to add chromophore groups for optical detection for chromatography33−35,71 or capillary electrophoresis29−32 or to add stable isotope labels for MS studies.33,59−62 For analysis of HS, reductive amination using anthranilic acid analogues,59,60 2-aminoacridone,35 aniline33 entail use of acetic acid, a large excess of the primary amine, and the reducing reagent NaBH3CN.63,64 Hyadrazide labeling using BODIPY uses similar conditions without the reducing agent.34,65,66 In order to evaluate the extent to which typical reducing end labeling conditions induce the 3-O-sulfation specific peeling reaction, we used the 3-O-sulfation containing hexasaccharide [1,2,3,1,7] under a series of solution conditions with respect to concentration of acetic acid. As expected, at commonly used labeling conditions (AcOH/DMSO = 3:7), we found that ∼36% of starting saccharide was converted to the peeling reaction product [1,2,2,1,4] with loss of the reducing end GlcNS3S6S residue (Figure 6). The degree of peeling scaled inversely with the acetic acid concentration. Using AcOH/DMSO = 1:1, we observed >90% of the labeled 3-O-sulfation containing hexasaccharide (Figure 6). In conclusion, the results indicated that the 3-O-sulfation specific peeling reaction takes place under commonly used reductive-amination conditions and raising the acetic acid concentration in the reaction would help to maintain the 3-O-sulfated substrate labeled in its intact form.
Figure 6.

Labeling the 3-O-sulfation containing hexasaccharide [1,2,3,1,7] at different acetic acid concentrations. The desalted products were analyzed on HILIC–MS. Peak a, EIC of pentasaccharide degradation product labeled with butylamine; peak b, the EIC of the intact hexasaccharide [1,2,3,1,7] labeled with butylamine; BA, butylamine.
Our results suggest that effort should be made to maintain pH conditions below neutral during purification and workup of HS saccharides from polysaccharide lyase digests. One way to avoid problems is to use methods that do not require reducing end derivatization. Our group has used HILIC–MS in the present work and elsewhere. Other groups have used reversed phase ion paring.67,68 In recent work, a reversed phase ion pairing mobile phase pH of 6.5 was used for the analysis of lyase-resistant, 3-O-sulfated tetrasaccharides in heparins using LC–MS.69
Conclusions
In this study, we discovered a novel peeling reaction that specifically degrades HS oligosaccharides that contain 3-O-sulfated glucosamine residue at the reducing-ends. We proposed that this specificity likely resulted from a de-3-O-sulfation initiated, two-step peeling reaction mechanism. Because of the high degree of heterogeneity, naturally existing HS/heparin are often analyzed after exhaustive or partial depolymerization by bacterial heparin lyases. Consequently, the 3-O-sulfated glucosamine residues are present at the reducing-end of lyase product saccharides during the analysis and become susceptible to this peeling reaction. As demonstrated in the paper, the 3-O-sulfation specific peeling reaction occurs under much milder conditions than the traditional types of peeling reactions that occur to 4-linked reducing terminal residues.26,27 This raises the possibility that unintentional peeling reactions occur during chemical and/or enzymatic reactions, chromatography, and sample storage. In our tests, factors such as unintentional pH and temperature increases could very easily accelerate the peeling reaction (Figures 3 and 4 and Supplemental Figure 5 in the Supporting Information). For example, a boiling procedure was carried out to inactivate the lyases in one of the previous studies assessing the 3-O-sulfation content in human follicular fluid HS, which could have potentially degraded some of the 3-O-sulfated saccharides.70 Another concern is the reductive-amination based labeling, as it is widely used in a variety of analytical methods for HS di- and oligo-saccharides. Acetic acid is often used in these reactions to open the reducing-end sugar ring, but an excess amount of acetic acid is also avoided both to keep some of the reacting amine in unprotonated form and to prevent the loss of sulfate groups. In our study, the amount of acetic acid used in common reductive amination reactions (AcOH/DMSO = 3:7) are often not enough to counterbalance the alkaline conditions introduced by primary amine and reducing reagents, which leads to the loss of the reducing-end 3-O-sulfated glucosamine through the peeling reaction mechanism we discovered. It has been previously reported that 3-O-sulfation changes the susceptibility of glucosamine residues to nitrous acid, another popular method to depolymerize HS chains for compositional analysis.19,54 Taken together, our results suggest that the natural abundances of 3-O-sulfation might need to be reassessed.
Acknowledgments
This work was supported by NIH Grants P41GM104603 (J.Z.), R01HL098950 (J.Z.), and P41GM103390 (G.-J.B.). The authors thank Prof. Jian Liu (University of North Carolina, Chapel Hill) and Prof. Jeffrey D. Esko (University of California, San Diego) for critical reading of the manuscript and Prof. Yitzhak Tor (University of California, San Diego) for insightful suggestions on the proposed mechanism.
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ Y.H. and Y.M. contributed equally to this work.
The authors declare no competing financial interest.
This paper published ASAP on December 22, 2014. A new reference was added for an incorrect reference cited in the MATERIALS AND METHODS section. The revised version was reposted on December 23, 2014.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Bishop J. R.; Schuksz M.; Esko J. D. Nature 2007, 446, 1030. [DOI] [PubMed] [Google Scholar]
- Esko J. D.; Selleck S. B. Annu. Rev. Biochem. 2002, 71, 435. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Beeler D. L.; Lawrence R.; Lech M.; Liu J.; Davis J. C.; Shriver Z.; Sasisekharan R.; Rosenberg R. D. J. Biol. Chem. 2001, 276, 42311. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Lawrence R.; Schwartz J. J.; Bai X.; Wei G.; Esko J. D.; Rosenberg R. D. J. Biol. Chem. 2001, 276, 28806. [DOI] [PubMed] [Google Scholar]
- Colliec-Jouault S.; Shworak N. W.; Liu J.; de Agostini A. I.; Rosenberg R. D. J. Biol. Chem. 1994, 269, 24953. [PubMed] [Google Scholar]
- Liu J.; Shworak N. W.; Fritze L. M. S.; Edelberg J. M.; Rosenberg R. D. J. Biol. Chem. 1996, 271, 27072. [DOI] [PubMed] [Google Scholar]
- Atha D. H.; Lormeau J. C.; Petitou M.; Rosenberg R. D.; Choay J. Biochemistry 1985, 24, 6723. [DOI] [PubMed] [Google Scholar]
- Atha D. H.; Lormeau J. C.; Petitou M.; Rosenberg R. D.; Choay J. Biochemistry 1987, 26, 6454. [DOI] [PubMed] [Google Scholar]
- Lindahl U.; Backstrom G.; Thunberg L.; Leder I. G. Proc. Natl. Acad. Sci. U.S.A. 1980, 77, 6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla D.; Liu J.; Blaiklock P.; Shworak N. W.; Bai X.; Esko J. D.; Cohen G. H.; Eisenberg R. J.; Rosenberg R. D.; Spear P. G. Cell 1999, 99, 13. [DOI] [PubMed] [Google Scholar]
- Tiwari V.; O’Donnell C. D.; Oh M. J.; Valyi-Nagy T.; Shukla D. Biochem. Biophys. Res. Commun. 2005, 338, 930. [DOI] [PubMed] [Google Scholar]
- Tiwari V.; Clement C.; Xu D.; Valyi-Nagy T.; Yue B. Y.; Liu J.; Shukla D. J. Virol. 2006, 80, 8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeehan W. L.; Wu X.; Kan M. J. Biol. Chem. 1999, 274, 21511. [DOI] [PubMed] [Google Scholar]
- Ye S.; Luo Y.; Lu W.; Jones R. B.; Linhardt R. J.; Capila I.; Toida T.; Kan M.; Pelletier H.; McKeehan W. L. Biochemistry 2001, 40, 14429. [DOI] [PubMed] [Google Scholar]
- Patel V. N.; Lombaert I. M.; Cowherd S. N.; Shworak N. W.; Xu Y.; Liu J.; Hoffman M. P. Dev. Cell 2014, 29, 662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song K.; Li Q.; Jiang Z. Z.; Guo C. W.; Li P. Cancer Biol. Ther. 2011, 12, 388. [DOI] [PubMed] [Google Scholar]
- Tecle E.; Diaz-Balzac C. A.; Bulow H. E. G3 2013, 3, 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Pedersen L. C. Appl. Microbiol. Biotechnol. 2007, 74, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker B. E.; Xu D.; Lawrence R.; Esko J. D. Matrix Biol. 2014, 35, 60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwallader A. B.; Yost H. J. Dev. Dyn. 2006, 235, 3423. [DOI] [PubMed] [Google Scholar]
- Xiao Z.; Zhao W.; Yang B.; Zhang Z.; Guan H.; Linhardt R. J. Glycobiology 2011, 21, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter S. R.; Williams J. M.; Clamp J. R. Carbohydr. Res. 1990, 205, 181. [DOI] [PubMed] [Google Scholar]
- Patel T.; Bruce J.; Merry A.; Bigge C.; Wormald M.; Jaques A.; Parekh R. Biochemistry 1993, 32, 679. [DOI] [PubMed] [Google Scholar]
- Merry A. H.; Neville D. C.; Royle L.; Matthews B.; Harvey D. J.; Dwek R. A.; Rudd P. M. Anal. Biochem. 2002, 304, 91. [DOI] [PubMed] [Google Scholar]
- Suzen S.; Williams M. J. Pept. Sci. 1999, 5, 283. [DOI] [PubMed] [Google Scholar]
- Murase T.; Kajihara Y. Carbohydr. Res. 2010, 345, 1702. [DOI] [PubMed] [Google Scholar]
- Totani K.; Matsuo I.; Ihara Y.; Ito Y. Bioorg. Med. Chem. 2006, 14, 5220. [DOI] [PubMed] [Google Scholar]
- Ernst S.; Langer R.; Cooney C. L.; Sasisekharan R. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 387. [DOI] [PubMed] [Google Scholar]
- Kitagawa H.; Kinoshita A.; Sugahara K. Anal. Biochem. 1995, 232, 114. [DOI] [PubMed] [Google Scholar]
- Hitchcock A.; Bowman M.; Staples G.; Zaia J. Electrophoresis 2008, 29, 4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamari F. N.; Kuhn R.; Karamanos N. K. J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2003, 793, 15. [DOI] [PubMed] [Google Scholar]
- Militsopoulou M.; Lamari F. N.; Hjerpe A.; Karamanos N. K. Electrophoresis 2002, 23, 1104. [DOI] [PubMed] [Google Scholar]
- Lawrence R.; Olson S. K.; Steele R. E.; Wang L.; Warrior R.; Cummings R. D.; Esko J. D. J. Biol. Chem. 2008, 283, 33674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skidmore M.; Atrih A.; Yates E.; Turnbull J. E. Methods Mol. Biol. 2009, 534, 157. [DOI] [PubMed] [Google Scholar]
- Yang B.; Chang Y.; Weyers A. M.; Sterner E.; Linhardt R. J. J. Chromatogr., A 2012, 1225, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arungundram S.; Al-Mafraji K.; Asong J.; Leach F. E. 3rd; Amster I. J.; Venot A.; Turnbull J. E.; Boons G. J. J. Am. Chem. Soc. 2009, 131, 17394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Shi X.; Yu X.; Leymarie N.; Staples G. O.; Yin H.; Killeen K.; Zaia J. Anal. Chem. 2011, 83, 8222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staples G. O.; Naimy H.; Yin H.; Kileen K.; Kraiczek K.; Costello C. E.; Zaia J. Anal. Chem. 2010, 82, 516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naimy H.; Leymarie N.; Zaia J. Biochemistry 2010, 49, 3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staples G.; Bowman M.; Costello C. E.; Hitchcock A.; Lau J.; Leymarie N.; Miller C.; Naimy H.; Shi X.; Zaia J. Proteomics 2009, 9, 686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchcock A.; Yates K. E.; Costello C.; Zaia J. Proteomics 2008, 8, 1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff J. J.; Laremore T. N.; Busch A. M.; Linhardt R. J.; Amster I. J. J. Am. Soc. Mass Spectrom. 2008, 19, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H. B.; Leach F. E. 3rd; Arungundram S.; Al-Mafraji K.; Venot A.; Boons G. J.; Amster I. J. J. Am. Soc. Mass Spectrom. 2011, 22, 582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Yu X.; Mao Y.; Costello C. E.; Zaia J.; Lin C. Anal. Chem. 2013, 85, 11979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X.; Zaia J. J. Biol. Chem. 2009, 284, 11806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domon B.; Costello C. E. Glycoconjugate J. 1988, 5, 397. [Google Scholar]
- Peterson S. B.; Liu J. Matrix Biol. 2013, 32, 223–227. [DOI] [PubMed] [Google Scholar]
- Peterson S.; Liu J. J. Biol. Chem. 2012, 287, 34836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S.; Yoshida K.; Sugiura M.; Sugahara K.; Khoo K. H.; Morris H. R.; Dell A. J. Biol. Chem. 1993, 268, 4780. [PubMed] [Google Scholar]
- Yamada S.; Sakamoto K.; Tsuda H.; Yoshida K.; Sugahara K.; Khoo K. H.; Morris H. R.; Dell A. Glycobiology 1994, 4, 69. [DOI] [PubMed] [Google Scholar]
- Sugahara K.; Tohno-oka R.; Yamada S.; Khoo K. H.; Morris H. R.; Dell A. Glycobiology 1994, 4, 535. [DOI] [PubMed] [Google Scholar]
- Yamada S.; Murakami T.; Tsuda H.; Yoshida K.; Sugahara K. J. Biol. Chem. 1995, 270, 8696. [PubMed] [Google Scholar]
- Tsuda H.; Yamada S.; Yamane Y.; Yoshida K.; Hopwood J. J.; Sugahara K. J. Biol. Chem. 1996, 271, 10495. [DOI] [PubMed] [Google Scholar]
- Liu J.; Shriver Z.; Blaiklock P.; Yoshida K.; Sasisekharan R.; Rosenberg R. D. J. Biol. Chem. 1999, 274, 38155. [DOI] [PubMed] [Google Scholar]
- Sundaram M.; Qi Y.; Shriver Z.; Liu D.; Zhao G.; Venkataraman G.; Langer R.; Sasisekharan R. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriver Z.; Raman R.; Venkataraman G.; Drummond K.; Turnbull J.; Toida T.; Linhardt R.; Biemann K.; Sasisekharan R. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 10359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozug J.; Wudyka S.; Gunay N. S.; Beccati D.; Lansing J.; Wang J.; Capila I.; Shriver Z.; Kaundinya G. V. Anal. Bioanal. Chem. 2012, 403, 2733. [DOI] [PubMed] [Google Scholar]
- Zhang Q. Q.; Chen X.; Zhu Z. J.; Zhan X. Q.; Wu Y. F.; Song L. K.; Kang J. W. Anal. Chem. 2013, 85, 1819. [DOI] [PubMed] [Google Scholar]
- Bowman M. J.; Zaia J. Anal. Chem. 2010, 82, 3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman M.; Zaia J. Anal. Chem. 2007, 76, 5777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchcock A. M.; Yates K. E.; Shortkroff S.; Costello C. E.; Zaia J. Glycobiology 2006, 17, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchcock A. M.; Costello C. E.; Zaia J. Biochemistry 2006, 45, 2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anumula K. R. Anal. Biochem. 2006, 350, 1. [DOI] [PubMed] [Google Scholar]
- Anumula K. R. Anal. Biochem. 2000, 283, 17. [DOI] [PubMed] [Google Scholar]
- Guimond S. E.; Puvirajesinghe T. M.; Skidmore M. A.; Kalus I.; Dierks T.; Yates E. A.; Turnbull J. E. J. Biol. Chem. 2009, 284, 25714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skidmore M. A.; Guimond S. E.; Dumax-Vorzet A. F.; Atrih A.; Yates E. A.; Turnbull J. E. J. Chromatogr., A 2006, 1135, 52. [DOI] [PubMed] [Google Scholar]
- Thanawiroon C.; Rice K. G.; Toida T.; Linhardt R. J. J. Biol. Chem. 2004, 279, 2608. [DOI] [PubMed] [Google Scholar]
- Kuberan B.; Lech M.; Zhang L.; Wu Z. L.; Beeler D. L.; Rosenberg R. J. Am. Chem. Soc. 2002, 124, 8707. [DOI] [PubMed] [Google Scholar]
- Li G.; Yang B.; Li L.; Zhang F.; Xue C.; Linhardt R. J. Anal. Biochem. 2014, 455, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Agostini A. I.; Dong J. C.; de Vantery Arrighi C.; Ramus M. A.; Dentand-Quadri I.; Thalmann S.; Ventura P.; Ibecheole V.; Monge F.; Fischer A. M.; HajMohammadi S.; Shworak N. W.; Zhang L.; Zhang Z.; Linhardt R. J. J. Biol. Chem. 2008, 283, 28115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnis P.; Coppi A.; Toida T.; Toyoda H.; Kinoshita-Toyoda A.; Xie J.; Kemp M. M.; Linhardt R. J. J. Biol. Chem. 2007, 282, 25376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenauer M. R.; Meissen J. K.; Seo Y.; Ames J. B.; Leary J. A. Anal. Chem. 2009, 81, 10179–10185. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.