

Abstract
Estetrol (E4) is a natural estrogen with a long half-life produced only by the human fetal liver during pregnancy. The crystal structures of the estrogen receptor α (ERα) ligand-binding domain bound to 17β-estradiol (E2) and E4 are very similar, as well as their capacity to activate the two activation functions AF-1 and AF-2 and to recruit the coactivator SRC3. In vivo administration of high doses of E4 stimulated uterine gene expression, epithelial proliferation, and prevented atheroma, three recognized nuclear ERα actions. However, E4 failed to promote endothelial NO synthase activation and acceleration of endothelial healing, two processes clearly dependent on membrane-initiated steroid signaling (MISS). Furthermore, E4 antagonized E2 MISS-dependent effects in endothelium but also in MCF-7 breast cancer cell line. This profile of ERα activation by E4, uncoupling nuclear and membrane activation, characterizes E4 as a selective ER modulator which could have medical applications that should now be considered further.
Keywords: endothelium, estetrol, estrogen receptor, uterus
Introduction
Beside the well-characterized 17β-estradiol (E2) that is considered as the active estrogen during the estrous cycle, estriol (E3) and also estetrol (E4) are synthesized during pregnancy, but their physiological roles are essentially unknown. It is hypothesized that these two weaker estrogens could interfere with E2 and attenuate its actions in estrogen-sensitive tissues. Indeed, E3 has an affinity for estrogen receptor (ER) and a biological potency that are both tenfold lower than that of E2. When administered with E2, E3 can act as an antiestrogen and partially interfere with E2-dependent transcription (Melamed et al, 1997). E4 is viewed as a weaker estrogen, with affinity and potency 100-fold lower than those of E2 (Holinka & Gurpide, 1979), but its antagonistic actions are poorly defined. E4 shares with E2 and E3 several estrogenic activities such as uterine growth and epithelial proliferation (Holinka & Gurpide, 1979), prevention of bone demineralization (Coelingh Bennink et al, 2008b), inhibition of ovulation (Coelingh Bennink et al, 2008c), and prevention of hot flushes (Holinka et al, 2008).
E4 appears to be produced exclusively by the human fetal liver (Hagen et al, 1965). E4 also differs from E2 by having a long plasma half-life (about 28 h) (Visser & Coelingh Bennink, 2009), and it neither stimulates the production of nor binds to sex hormone binding globulin (SHBG) (Hammond et al, 2008). Because of these characteristics, E4 was evaluated, in combination with a progestin, as a new oral contraceptive in a phase II clinical trial (I. Duijkers I., C. Klipping C., Y. Zimmerman, L. Petit, M. Mawet, J-M. Foidart, H. Coelingh Bennink, in preparation). Very interestingly, E4 (up to 20 mg/day) did not elicit changes in circulating hepatic factors and thus might not increase thrombo-embolic events, which are undesirable effects of estrogen pharmaceuticals containing E2 or ethinyl-estradiol (EE) (C. Kluft Cornelis, Y. Zimmerman, M. Mawet Marie, C. Klipping, I. Duijkers Ingrid, L. Petit, J. Neuteboom, J-M Foidart, H. Coelingh Bennink, in preparation). Unfortunately, as previously reported (Valera et al, 2012), the impact of estrogen on hepatic factors is species dependent, which precludes the use of mice as an animal model to elucidate these mechanisms.
The physiological responses to estrogenic compounds are initiated by their binding to the estrogen receptors (ER), ERα and ERβ. E4 binds ERα with a modest preference over ERβ (Visser et al, 2008). ER mediates its transcriptional activity after ligand binding inducing an ordered sequence of interactions between two activation functions (AF), AF-1 and AF-2, and coactivators such as the steroid receptor coactivator (SRC) 3, a member of the p160 subfamily (McKenna & O'Malley, 2001; Metivier et al, 2003; Smith & O'Malley, 2004). In addition, estrogens can act through a distinctly different pathway by inducing rapid extra-nuclear activity via the activation of a pool of ERs localized at the plasma membrane, a process termed membrane-initiated steroid signaling (MISS) (Ascenzi et al, 2006; Wu et al, 2011). Although ERα MISS effects were initially also called ‘non-genomic’ effects, they can modulate ERα-dependent transcriptional activity in cultured cell models in vitro (La Rosa et al, 2012). However, thanks to a unique mouse model targeted for the ERα palmitoylation site membrane, we recently demonstrated a very contrasted involvement of MISS-mediated E2 action in two different tissues: the uterus in which the E2 response depends on ERα nuclear action and the arteries involving exclusively MISS of ERα to mediate E2 response (Abot et al, 2013; Adlanmerini et al, 2014).
The aim of this study was to analyze the molecular action of E4 using structural, in vitro and in vivo models. First, experiments were conducted to analyze the binding of E4 to ERα-LBD and to investigate the role of the two activation functions AF-1 and AF-2 in the transcriptional activity of E4 in comparison to E2. Second, we studied the impact of acute E4 treatment on gene expression and epithelial cell proliferation in uterus, which involved primarily genomic/transcriptional actions of ERα but not ERα MISS (Abot et al, 2013; Adlanmerini et al, 2014). Third, we analyzed the effect of chronic E4 treatment on fatty streak deposit formation at the aortic root of ovariectomized LDLr−/− (Low Density Lipoprotein receptor) mice fed with an hypercholesterolemic diet. Fourth, we evaluated the effect of E4 on endothelial functions recognized to be dependent on MISS ERα signaling, namely acceleration of endothelial healing and activation of endothelial NO synthase (Brouchet et al, 2001; Toutain et al, 2009; Chambliss et al, 2010; Wu et al, 2011; Adlanmerini et al, 2014). Finally, MISS of ERα versus nuclear action after E4 stimulation was analyzed in the breast cancer cell line, MCF-7. The present studies reveal that high doses of E4 stimulated nuclear ERα actions in the uterus but E4 failed to promote MISS in the endothelium, and a similar profile of activation was also observed in MCF-7 cells. This profile of ERα activation indicates that E4 is a selective ER modulator which could have medical applications that should now be considered further, in particular in light its lesser hepatic effects in women, which could potentially reduce venous thrombo-embolic risk.
Results
Comparison of the ERα LBD structure, of the coactivator interaction, and of the solubility/orientation in phospholipids bilayer model membranes after E2 and E4 binding
In order to gain insight into the molecular mechanism of action of E4, we first compared the crystal structures of ERα LBD complexed with E3 (3Q95) or E4 (3L03) to the published E2-ERα structure (1ERE) and we found all of them very similar in their overall conformation (Fig 1A and B). In addition, the two ligands are perfectly superimposable and interact equally with residues within the ligand-binding pocket (Fig 1B). The only significant difference between these structures is the altered orientation of helix 12 and the loop between helices 11 and 12 relative to that in the E2-ERα LBD complex (Fig 1C). However, this small difference does not prevent binding of the GRIP peptide to the E3- or E4-ERαLBD to stabilize an agonist conformation (Fig 1C). Using competitive radiometric binding assays, we found, as reported previously (Visser et al, 2008), that E4 and E3 bind to ERα with less affinity than E2 and with a small preference over ERβ (Supplementary Table S1). The binding affinity of the steroid receptor coactivator SRC3 to complexes of ligands with the ERα ligand-binding domain can be quantified by a time-resolved fluorescence resonance transfer assay (tr-FRET) (Jeyakumar et al, 2011). In this assay, E3-ERα and E2-ERα have essentially identical affinities for SRC3, and the affinity of E4-ERα, while half that of E2-ERα, is still in the low nanomolar range (Supplementary Fig S1 and Supplementary Table S2). Thus, as a hormonal ligand, while E4 has considerably lower binding affinity for ERα than E2, it forms a complex with this receptor that binds to a key coactivator protein, SRC3, almost as well as does the complex with E2.
Figure 1. Structure of E2, E3 and E4 and their respective complexed structure with ERα ligand binding domain.
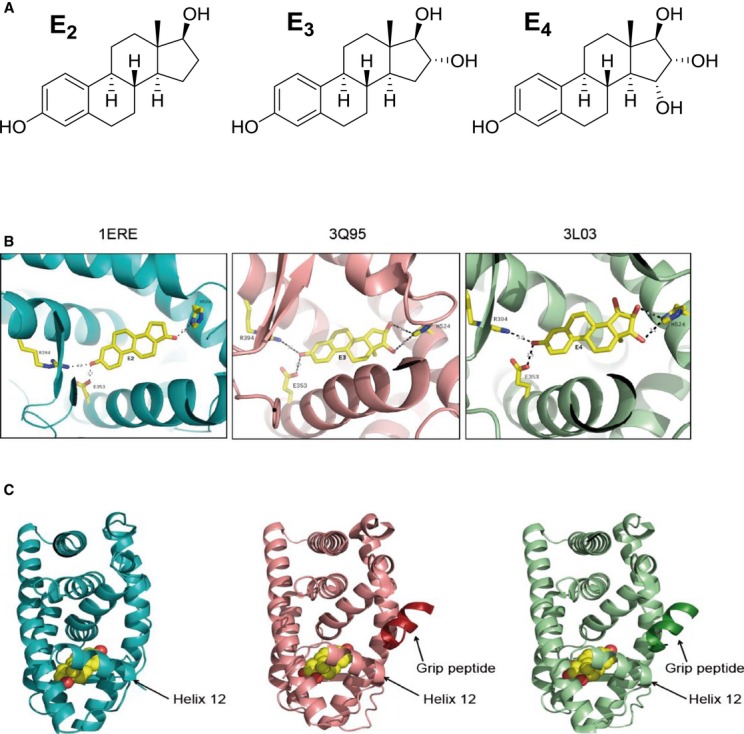
A Chemical structures of E2, E3, and E4.
B, C Structure of ERαLBD complexed with E2 (blue), E3 (red), or E4 (green). Shown are ribbon diagrams of the ERαLBD monomer. Ligand-binding site (B), shown in ball-and-stick rendering of the ligands along with their interacting residues. Hydrogen bonds are shown as dotted lines. Ligand-binding domain (C) and peptide fragment of the GRIP1 coactivator protein in complex with E3 or E4 only (darker red and darker green). Ligand is represented as a space-filled model. Position of the helix 12 is indicated by an arrow.
As a consequence of its two extra hydroxyl groups, one might expect E4 to be less hydrophobic than E2 (Fig 1A); in fact, its calculated octanol–water partition coefficient (ClogPo/w) is 2.62 versus 3.78 for E2. Thus, we hypothesized that E4 would less readily partition into the plasma membrane than E2 (Yamamoto & Liljestrand, 2004). However, we found a similar solubility for E2 (˜4 mol%) and E4 (˜2 mol%) into palmitoyl-oleoyl-phosphatidylcholine (POPC) liposomes using nuclear magnetic resonance, indicating that their uptake is equivalent (Supplementary Fig S2A). In addition, contrary to what is described by Scheidt et al (2010), we found that E2 is in an equilibrium between two orientations in the bilayer (phenol at the lipid–water interface versus phenol within the hydrophobic core), whereas the phenol of E4 is oriented more predominantly toward the lipid–water interface (Supplementary Fig S2B). While unexpected, this behavior of E4 may be a consequence of an efficient intramolecular network of hydrogen bonds, operating among the three OH groups in the D-ring that in some way effectively suppresses their polar nature, thus allowing the D-ring to reside more comfortably in the hydrophobic core of the bilayer. In contrast, the lone 17β-OH in E2, which would be fully surrounded by a hydrophobic environment when in the core of the bilayer, more effectively competes with the phenolic OH for access to the aqueous interface, resulting in the two orientations of this ligand.
Respective roles of ERα AF-1 and AF-2 in the transcription activity induced by E4
We then evaluated the ability of E4 to induce transcriptional activity of an estrogen-sensitive reporter gene (ERE-TK-Luc) in transient transfection assays in vitro. The dose–response effect of E4 was compared with that of E2 in HeLa cells transfected with an expression vector encoding the full-length ERα. E4 displayed a marked rightward dose–response shift compared to E2, requiring at least 100-fold higher hormone concentration to achieve half-maximal stimulation of the reporter gene (Fig 2A), consistent with its lower ERα binding affinity.
Figure 2. E4 induces ERE transcriptional activity in a cellular context-dependent manner in vitro in a manner similar to that of E2.

A, B HeLa (A, B) and HepG2 (B) cells were transiently transfected with the ERE-TK-Luc reporter constructs in the presence of pCR-ERα, pCR-ERαΔ79, pCR-ERαAF-10, or empty pCR vector. Cells were treated with indicated dose of E2 and E4 or vehicle (Ctrl) for 24 h. Normalized luciferase activities were expressed as fold increase above values measured with empty pCR and vehicle. Data correspond to the mean values ± SEM of at least three separate transfection experiments.
E4 modulation of activation function AF-1 and AF-2 of ERα was then evaluated in HepG2 and HeLa cell lines (Fig 2B). Whereas AF-1 is the dominant AF involved in ERα transcriptional activity in HepG2 cells, HeLa cells mediate ERα signaling mainly through AF-2 (Merot et al, 2004). Furthermore, cell permissiveness to either ERα AFs was determined by comparing the transcriptional activity of the full-length ERα with those of ERαΔ79 (deletion of only AF-1 box 1) and ERαAF-10 (additional deletion of AF-1 box 2/3). In HepG2 cells, as is the case for E2, the main region involved in E4-induced ERα transcriptional activity is the AF-1 box 1 (ERαΔ79 versus ERα, 65% decrease of the total activity, Fig 2B), the remaining activity depending upon the AF-1 box 2/3, as expected (Huet et al, 2008). In contrast, the AF-1 box 1 (ERαΔ79 versus ERα) represents < 20% of the E2- or E4-induced ERα transcriptional potency in HeLa cells. These results show that a high concentration of E4 is able to activate gene transcription through ERα via the classical ERE mechanism. In addition, as previously described for E2, both AFs are involved in this action in a cell type-dependent manner.
Impact of acute E4 treatment on uterine gene expression and epithelial proliferation
We then assessed the transcriptional activity of E4 in vivo on the uterus in C57Bl/6J mice. We selected a set of genes known to be regulated by E2 in this tissue (Hewitt et al, 2003; Watanabe et al, 2003; Abot et al, 2013) and evaluated their expression profile in ovariectomized mice after an acute dose of each estrogen alone. Dose–response studies (E2: 8, 30, 80, and 200 μg/kg and E4: 8, 30, 80, 200, 600 μg/kg, or 1 and 10 mg/kg) indicated that most of the regulated genes reached their maximum level of induction at the lowest dose of E2, that is, 8 μg/kg (Table 1), and of repression, between 8 and 30 μg/kg of E2 (Table 2). In most cases, compared to E2, E4 required a 100-fold higher dose (i.e., 1 mg/kg) to optimally activate the transcription of target genes (Table 1), although 7 of the 23 studied genes were activated at lower levels of E4. Concerning down-regulated genes, a dose of 80 μg/kg of E4 was sufficient to induce the maximal action (Table 2). Plasma analysis showed that a subcutaneous injection of 1 mg/kg E4 resulted in an E4 plasma concentration of 16,100 pg/ml after 6 h of treatment, a value close to that found for E4 in human fetal plasma (18,630 pg/ml). All E2 (8 μg/kg) target genes in the uterus were also regulated (at least twofold) by E4 (1 mg/kg) (Fig 3A, Tables 1 and 2) and have been distributed into three groups, according to the response to E2 versus E4 (Fig 3B). Cluster 1 represents genes similarly regulated by E2 at 8 μg/kg and E4 at 1 mg/kg doses; cluster 2 genes were found to be less regulated by E4 than by E2, and cluster 3 genes more regulated by E4 than by E2 at these doses. Yellow highlight is used to designate gene expression regulation by E2 that is greater than by E4 (Fig 3B, middle), whereas gene expression that is more regulated by the same dose of E4, is highlighted in blue (Fig 3B, bottom). It is noteworthy that this latter category involved mainly down-regulated genes.
Table 1.
Seven-week-old ovariectomized C57Bl/6J mice were subcutaneously injected with vehicle (Ctrl, castor oil), 17β-estradiol (E2, 1, 8, 30, 80, 200 μg/kg) or estetrol (E4, 1, 8, 30, 80, 200 μg/kg, or 1 and 10 mg/kg) and were euthanized 6 h after treatment. mRNA levels of a set of genes from uterus that were up-regulated at least twofold by E2 administration relative to placebo were measured by quantitative PCR and normalized to Hprt1 expression
| GOI | Dose E2 (µg/kg) |
Dose E4 (µg/kg) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 8 | 30 | 80 | 200 | 1 | 8 | 30 | 80 | 200 | 1,000 | 10,000 | |
| ramp3 | 0.81 ± 0.06 | 30.84 ± 1.48 P < 0.0001 |
21.71 ± 0.45 P < 0.0001 |
22.33 ± 0.42 P < 0.0001 |
29.04 ± 3.11 P < 0.0001 |
0.91 ± 0.10 | 2.01 ± 0.36 P = 0.0005 |
10.09 ± 0.13 P < 0.0001 |
16.22 ± 0.21 P < 0.0001 |
23.20 ± 0.85 P < 0.0001 |
29.87 ± 1.17 P < 0.0001 |
27.75 ± 2.34 P < 0.0001 |
| gadd45g | 1.08 ± 0.15 | 19.87 ± 0.58 P < 0.0001 |
9.05 ± 0.31 P < 0.0001 |
6.89 ± 0.44 P < 0.0001 |
11.96 ± 2.59 P < 0.0001 |
0.91 ± 0.19 | 2.31 ± 0.49 P = 0.0002 |
5.49 ± 0.89 P < 0.0001 |
6.52 ± 0.89 P < 0.0001 |
6.84 ± 0.62 P < 0.0001 |
10.96 ± 0.42 P < 0.0001 |
9.49 ± 0.69 P < 0.0001 |
| mad2l1 | 0.83 ± 0.06 | 19.63 ± 0.33 P < 0.0001 |
7.69 ± 0.54 P < 0.0001 |
5.54 ± 0.56 P < 0.0001 |
7.59 ± 0.56 P < 0.0001 |
0.79 ± 0.16 | 0.46 ± 0.17 P = 0.0014 |
1.42 ± 0.10 P = 0.0058 |
1.82 ± 0.23 P = 0.0009 |
2.80 ± 0.38 P < 0.0001 |
11.03 ± 2.03 P < 0.0001 |
15.52 ± 0.46 P < 0.0001 |
| inhbb | 0.78 ± 0.06 | 18.13 ± 1.62 P < 0.0001 |
8.93 ± 0.36 P < 0.0001 |
7.86 ± 0.41 P < 0.0001 |
10.45 ± 1.29 P < 0.0001 |
0.90 ± 0.03 | 1.44 ± 0.08 P = 0.0024 |
5.36 ± 0.23 P < 0.0001 |
5.76 ± 0.15 P < 0.0001 |
6.81 ± 0.24 P < 0.0001 |
10.55 ± 0.58 P < 0.0001 |
11.44 ± 0.99 P < 0.0001 |
| fam65b | 0.81 ± 0.04 | 12.92 ± 1.07 P < 0.0001 |
10.09 ± 0.33 P < 0.0001 |
8.03 ± 0.97 P < 0.0001 |
9.53 ± 0.86 P < 0.0001 |
0.97 ± 0.11 | 1.65 ± 0.05 P < 0.0001 |
4.42 ± 0.58 P < 0.0001 |
6.53 ± 0.31 P < 0.0001 |
8.08 ± 1.01 P < 0.0001 |
9.85 ± 0.53 P < 0.0001 |
10.77 ± 1.31 P < 0.0001 |
| fos | 0.97 ± 0.14 | 12.36 ± 2.19 P < 0.0001 |
1.42 ± 0.13 | 1.29 ± 0.21 | 3.60 ± 1.39 P = 0.075 |
0.81 ± 0.24 | 0.99 ± 0.22 | 1.64 ± 0.31 | 1.23 ± 0.10 | 3.59 ± 1.37 P = 0.0182 |
6.26 ± 1.61 P = 0.0005 |
2.91 ± 0.54 P = 0.0001 |
| aldh1a2 | 1.08 ± 0.07 | 9.94 ± 0.68 P < 0.0001 |
9.29 ± 0.36 P < 0.0001 |
9.12 ± 0.75 P < 0.0001 |
8.61 ± 0.45 P < 0.0001 |
1.25 ± 0.06 P = 0.0241 |
1.07 ± 0.13 | 5.38 ± 0.38 P < 0.0001 |
8.33 ± 0.36 P < 0.0001 |
7.62 ± 0.45 P < 0.0001 |
7.91 ± 0.39 P < 0.0001 |
7.22 ± 0.45 P < 0.0001 |
| p21 | 1.01 ± 0.06 | 9.17 ± 0.66 P < 0.0001 |
9.37 ± 0.34 P < 0.0001 |
6.49 ± 0.54 P < 0.0001 |
8.62 ± 1.04 P < 0.0001 |
1.01 ± 0.07 | 1.83 ± 0.27 P = 0.0002 |
5.57 ± 0.37 P < 0.0001 |
6.09 ± 0.60 P < 0.0001 |
6.89 ± 0.38 P < 0.0001 |
7.80 ± 0.67 P < 0.0001 |
6.33 ± 0.82 P < 0.0001 |
| aars | 0.98 ± 0.05 | 6.95 ± 0.30 P < 0.0001 |
10.50 ± 0.05 P < 0.0001 |
9.00 ± 0.75 P < 0.0001 |
9.24 ± 1.06 P < 0.0001 |
1.04 ± 0.03 | 1.33 ± 0.10 P = 0.0062 |
7.12 ± 0.04 P < 0.0001 |
7.03 ± 0.50 P < 0.0001 |
7.84 ± 0.73 P < 0.0001 |
7.94 ± 0.77 P < 0.0001 |
7.77 ± 0.62 P < 0.0001 |
| lcn2 | 1.04 ± 0.11 | 6.68 ± 0.48 P < 0.0001 |
4.93 ± 0.51 P < 0.0001 |
5.61 ± 0.96 P < 0.0001 |
9.42 ± 1.18 P < 0.0001 |
0.98 ± 0.02 | 0.81 ± 0.03 | 5.86 ± 0.56 P < 0.0001 |
8.12 ± 0.69 P < 0.0001 |
10.21 ± 0.90 P < 0.0001 |
9.04 ± 1.36 P < 0.0001 |
6.12 ± 0.42 P < 0.0001 |
| errfi1 | 1.06 ± 0.14 | 6.62 ± 0.84 P < 0.0001 |
3.41 ± 0.18 P < 0.0001 |
3.13 ± 0.12 P < 0.0001 |
4.25 ± 0.96 P < 0.0001 |
1.16 ± 0.14 | 1.94 ± 0.35 P = 0.0002 |
2.86 ± 0.56 P < 0.0001 |
2.04 ± 0.25 P < 0.0001 |
2.45 ± 0.32 P < 0.0001 |
3.10 ± 0.27 P < 0.0001 |
2.30 ± 0.16 P < 0.0001 |
| sprr2f | 0.89 ± 0.12 | 6.25 ± 1.36 P < 0.0001 |
1.37 ± 0.09 | 4.48 ± 1.35 P = 0.0002 |
4.87 ± 1.43 P = 0.0007 |
0.98 ± 0.26 | 0.41 ± 0.21 P = 0.0129 |
2.36 ± 0.55 P = 0.0007 |
3.02 ± 0.18 P < 0.0001 |
3.88 ± 0.80 P = 0.0001 |
10.70 ± 1.49 P < 0.0001 |
10.01 ± 0.75 P < 0.0001 |
| rasd1 | 1.13 ± 0.07 | 5.97 ± 0.35 P < 0.0001 |
4.18 ± 0.19 P < 0.0001 |
3.28 ± 0.23 P < 0.0001 |
3.47 ± 0.19 P < 0.0001 |
1.06 ± 0.11 | 1.42 ± 0.22 P = 0.0134 |
1.61 ± 0.32 P = 0.0035 |
1.18 ± 0.19 | 1.42 ± 0.37 | 2.49 ± 0.23 P < 0.0001 |
3.18 ± 0.13 P < 0.0001 |
| vegfa | 0.86 ± 0.10 | 5.04 ± 0.51 P < 0.0001 |
4.23 ± 0.28 P < 0.0001 |
3.06 ± 0.37 P < 0.0001 |
3.61 ± 0.29 P < 0.0001 |
0.97 ± 0.04 | 1.11 ± 0.09 | 2.13 ± 0.09 P < 0.0001 |
1.43 ± 0.12 P = 0.0037 |
1.63 ± 0.14 P < 0.0001 |
2.91 ± 0.32 P < 0.0001 |
3.60 ± 0.51 P < 0.0001 |
| cebpb | 0.92 ± 0.03 | 4.54 ± 0.23 P < 0.0001 |
2.82 ± 0.19 P < 0.0001 |
2.37 ± 0.29 P < 0.0001 |
2.72 ± 0.08 P < 0.0001 |
1.21 ± 0.04 | 0.86 ± 0.10 | 1.54 ± 0.10 P = 0.0011 |
1.08 ± 0.09 | 1.20 ± 0.13 | 1.59 ± 0.11 P < 0.0001 |
1.87 ± 0.08 P < 0.0001 |
| psat1 | 1.18 ± 0.08 | 4.31 ± 0.23 P < 0.0001 |
8.98 ± 0.45 P < 0.0001 |
9.95 ± 0.70 P < 0.0001 |
8.26 ± 1.22 P < 0.0001 |
1.15 ± 0.09 | 0.81 ± 0.05 | 5.09 ± 0.38 P < 0.0001 |
5.49 ± 0.45 P < 0.0001 |
4.96 ± 0.65 P < 0.0001 |
5.07 ± 0.51 P < 0.0001 |
5.30 ± 0.70 P < 0.0001 |
| gadd45a | 1.02 ± 0.03 | 3.70 ± 0.40 P < 0.0001 |
4.88 ± 0.35 P < 0.0001 |
4.09 ± 0.64 P < 0.0001 |
3.77 ± 0.56 P < 0.0001 |
0.81 ± 0.05 P = 0.0436 |
1.21 ± 0.12 | 3.09 ± 0.20 P < 0.0001 |
2.22 ± 0.24 P < 0.0001 |
2.54 ± 0.47 P = 0.0003 |
3.33 ± 0.40 P < 0.0001 |
3.16 ± 0.21 P < 0.0001 |
| hspa5 | 1.04 ± 0.01 | 3.28 ± 0.19 P < 0.0001 |
3.03 ± 0.18 P < 0.0001 |
3.71 ± 0.31 P < 0.0001 |
4.95 ± 0.84 P < 0.0001 |
1.01 ± 0.01 | 1.18 ± 0.04 | 2.23 ± 0.15 P < 0.0001 |
4.90 ± 0.32 P < 0.0001 |
5.17 ± 0.48 P < 0.0001 |
5.58 ± 0.54 P < 0.0001 |
5.64 ± 0.35 P < 0.0001 |
| igf1 | 1.07 ± 0.03 | 3.27 ± 0.15 P < 0.0001 |
2.82 ± 0.08 P < 0.0001 |
3.52 ± 0.23 P < 0.0001 |
3.67 ± 0.30 P < 0.0001 |
1.11 ± 0.09 | 1.07 ± 0.08 | 3.18 ± 0.15 P < 0.0001 |
4.19 ± 0.24 P < 0.0001 |
3.72 ± 0.18 P < 0.0001 |
4.01 ± 0.18 P < 0.0001 |
5.23 ± 0.89 P < 0.0001 |
| cars | 1.07 ± 0.11 | 3.16 ± 0.04 P < 0.0001 |
3.55 ± 0.27 P < 0.0001 |
3.73 ± 0.37 P < 0.0001 |
4.26 ± 0.32 P < 0.0001 |
0.96 ± 0.10 | 1.06 ± 0.08 | 3.19 ± 0.09 P < 0.0001 |
3.62 ± 0.35 P < 0.0001 |
3.47 ± 0.23 P < 0.0001 |
4.27 ± 0.54 P < 0.0001 |
3.96 ± 0.37 P < 0.0001 |
| cyr61 | 0.91 ± 0.12 | 2.73 ± 0.16 P < 0.0001 |
0.96 ± 0.07 | 1.11 ± 0.18 | 2.08 ± 0.81 P = 0.0560 |
0.73 ± 0.19 | 0.56 ± 0.07 P = 0.0442 |
0.75 ± 0.12 | 0.94 ± 0.13 | 2.65 ± 0.92 P = 0.0245 |
4.23 ± 0.57 P < 0.0001 |
3.08 ± 0.15 P < 0.0001 |
| dio2 | 0.79 ± 0.06 | 2.49 ± 0.47 P = 0.0002 |
2.05 ± 0.07 P < 0.0001 |
3.67 ± 0.33 P < 0.0001 |
4.60 ± 0.71 P < 0.0001 |
1.03 ± 0.10 | 0.73 ± 0.07 | 1.33 ± 0.08 P = 0.0036 |
2.68 ± 0.21 P < 0.0001 |
4.60 ± 0.40 P < 0.0001 |
6.30 ± 0.66 P < 0.0001 |
5.12 ± 0.38 P < 0.0001 |
| pgr | 0.87 ± 0.04 | 2.47 ± 0.17 P < 0.0001 |
1.80 ± 0.03 P < 0.0001 |
1.67 ± 0.09 P < 0.0001 |
1.90 ± 0.07 P < 0.0001 |
0.88 ± 0.05 | 1.25 ± 0.04 P = 0.0163 |
1.58 ± 0.06 P < 0.0001 |
1.55 ± 0.09 P = 0.0002 |
1.74 ± 0.11 P < 0.0001 |
2.31 ± 0.13 P < 0.0001 |
2.20 ± 0.09 P < 0.0001 |
Results were expressed as mean ± SEM (n = 4–8 mice/group). Significance of the observed effects was evaluated using Student's t-test. Gray highlight represents the maximum of regulation
Table 2.
Seven-week-old ovariectomized C57Bl/6J mice were subcutaneously injected with vehicle (Ctrl, castor oil), 17β-estradiol (E2, 1, 8, 30, 80, 200 μg/kg) or estetrol (E4, 1, 8, 30, 80, 200 μg/kg, or 1 and 10 mg/kg) and were euthanized 6 h after treatment. mRNA levels of a set of genes from uterus that were down-regulated at least twofold by E2 administration relative to placebo were measured by quantitative PCR and normalized to Hprt1 expression
| GOI | Dose E2 (µg/kg) |
Dose E4 (µg/kg) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 8 | 30 | 80 | 200 | 1 | 8 | 30 | 80 | 200 | 1,000 | 10,000 | |
| esr2 | 0.92 ± 0.09 | 0.50 ± 0.04 P = 0.0033 |
0.32 ± 0.05 P = 0.0018 |
0.34 ± 0.04 P = 0.0006 |
0.56 ± 0.08 P = 0.0077 |
0.90 ± 0.22 | 0.41 ± 0.20 P = 0.0056 |
0.32 ± 0.05 P = 0.0019 |
0.13 ± 0.04 P < 0.0001 |
0.26 ± 0.08 P < 0.0001 |
0.36 ± 0.08 P < 0.0001 |
0.34 ± 0.06 P = 0.0006 |
| fgfr1 | 0.98 ± 0.01 | 0.45 ± 0.02 P < 0.0001 |
0.40 ± 0.03 P < 0.0001 |
0.33 ± 0.04 P < 0.0001 |
0.39 ± 0.03 P < 0.0001 |
1.10 ± 0.04 | 0.95 ± 0.08 | 0.68 ± 0.05 P = 0.0020 |
0.44 ± 0.02 P < 0.0001 |
0.42 ± 0.03 P < 0.0001 |
0.40 ± 0.03 P < 0.0001 |
0.44 ± 0.05 P < 0.0001 |
| tgfbr2 | 0.90 ± 0.05 | 0.43 ± 0.01 P < 0.0001 |
0.33 ± 0.02 P < 0.0001 |
0.32 ± 0.02 P < 0.0001 |
0.39 ± 0.03 P < 0.0001 |
0.85 ± 0.04 | 0.87 ± 0.02 | 0.54 ± 0.02 P = 0.0007 |
0.47 ± 0.02 P < 0.0001 |
0.51 ± 0.03 P < 0.0001 |
0.50 ± 0.02 P < 0.0001 |
0.57 ± 0.07 P = 0.0005 |
| esr1 | 0.98 ± 0.03 | 0.41 ± 0.03 P < 0.0001 |
0.27 ± 0.01 P < 0.0001 |
0.29 ± 0.02 P < 0.0001 |
0.38 ± 0.02 P < 0.0001 |
0.90 ± 0.03 | 1.04 ± 0.04 | 0.67 ± 0.02 P = 0.0101 |
0.44 ± 0.02 P < 0.0001 |
0.44 ± 0.02 P < 0.0001 |
0.44 ± 0.02 P < 0.0001 |
0.41 ± 0.02 P < 0.0001 |
| ptov1 | 0.56 ± 0.08 P = 0.0028 |
0.40 ± 0.03 P < 0.0001 |
0.20 ± 0.02 P < 0.0001 |
0.21 ± 0.02 P < 0.0001 |
0.29 ± 0.05 P < 0.0001 |
0.65 ± 0.05 P = 0.0110 |
0.56 ± 0.03 P = 0.0019 |
0.25 ± 0.03 P < 0.0001 |
0.14 ± 0.02 P < 0.0001 |
0.24 ± 0.02 P < 0.0001 |
0.27 ± 0.03 P < 0.0001 |
0.28 ± 0.03 P < 0.0001 |
| sox17 | 1.01 ± 0.05 | 0.40 ± 0.06 P < 0.0001 |
0.43 ± 0.02 P = 0.0006 |
0.35 ± 0.04 P < 0.0001 |
0.40 ± 0.02 P < 0.0001 |
0.90 ± 0.08 | 1.02 ± 0.04 | 0.56 ± 0.04 P = 0.0044 |
0.34 ± 0.01 P < 0.0001 |
0.37 ± 0.01 P < 0.0001 |
0.27 ± 0.02 P < 0.0001 |
0.28 ± 0.03 P < 0.0001 |
| egfr | 1.19 ± 0.04 | 0.38 ± 0.02 P < 0.0001 |
0.53 ± 0.02 P = 0.0062 |
0.52 ± 0.06 P = 0.0022 |
0.63 ± 0.05 P = 0.0064 |
1.53 ± 0.08 P = 0.0011 |
0.77 ± 0.02 | 0.41 ± 0.03 P = 0.0013 |
0.29 ± 0.02 P < 0.0001 |
0.29 ± 0.03 P < 0.0001 |
0.30 ± 0.03 P < 0.0001 |
0.43 ± 0.04 P = 0.0004 |
| ptrf | 0.90 ± 0.03 | 0.34 ± 0.02 P < 0.0001 |
0.31 ± 0.02 P < 0.0001 |
0.29 ± 0.02 P < 0.0001 |
0.35 ± 0.02 P < 0.0001 |
1.13 ± 0.04 | 0.79 ± 0.01 P = 0.0258 |
0.39 ± 0.03 P < 0.0001 |
0.25 ± 0.01 P < 0.0001 |
0.25 ± 0.01 P < 0.0001 |
0.28 ± 0.02 P < 0.0001 |
0.29 ± 0.03 P < 0.0001 |
| igfbp2 | 1.07 ± 0.16 | 0.34 ± 0.06 P < 0.0001 |
0.64 ± 0.13 P = 0.0122 |
0.63 ± 0.11 P = 0.0056 |
0.58 ± 0.06 P = 0.0005 |
2.15 ± 0.42 P = 0.0002 |
2.70 ± 1.84 | 1.15 ± 0.62 | 0.38 ± 0.03 P < 0.0001 |
0.43 ± 0.11 P < 0.0001 |
0.32 ± 0.04 P < 0.0001 |
0.36 ± 0.10 P < 0.0001 |
| tgfb3 | 1.14 ± 0.07 | 0.30 ± 0.02 P < 0.0001 |
0.47 ± 0.03 P = 0.0002 |
0.41 ± 0.03 P < 0.0001 |
0.41 ± 0.04 P < 0.0001 |
1.06 ± 0.05 | 0.86 ± 0.06 | 0.31 ± 0.01 P < 0.0001 |
0.26 ± 0.01 P < 0.0001 |
0.25 ± 0.01 P < 0.0001 |
0.37 ± 0.03 P < 0.0001 |
0.53 ± 0.04 P = 0.0002 |
| vegfb | 0.93 ± 0.02 | 0.28 ± 0.02 P < 0.0001 |
0.22 ± 0.03 P < 0.0001 |
0.20 ± 0.03 P < 0.0001 |
0.27 ± 0.01 P < 0.0001 |
1.06 ± 0.06 | 0.81 ± 0.03 | 0.40 ± 0.04 P = 0.0003 |
0.23 ± 0.01 P < 0.0001 |
0.26 ± 0.01 P < 0.0001 |
0.28 ± 0.02 P < 0.0001 |
0.32 ± 0.02 P < 0.0001 |
| ar | 1.07 ± 0.04 | 0.28 ± 0.01 P < 0.0001 |
0.26 ± 0.01 P < 0.0001 |
0.31 ± 0.02 P < 0.0001 |
0.37 ± 0.02 P < 0.0001 |
0.99 ± 0.03 | 1.01 ± 0.04 | 0.51 ± 0.02 P = 0.0003 |
0.41 ± 0.01 P < 0.0001 |
0.45 ± 0.01 P < 0.0001 |
0.48 ± 0.01 P < 0.0001 |
0.51 ± 0.02 P < 0.0001 |
| igfbp6 | 0.80 ± 0.03 | 0.26 ± 0.02 P < 0.0001 |
0.19 ± 0.01 P < 0.0001 |
0.19 ± 0.02 P < 0.0001 |
0.19 ± 0.01 P < 0.0001 |
0.81 ± 0.03 | 0.75 ± 0.04 | 0.34 ± 0.01 P = 0.0003 |
0.21 ± 0.01 P < 0.0001 |
0.21 ± 0.01 P < 0.0001 |
0.18 ± 0.01 P < 0.0001 |
0.19 ± 0.02 P < 0.0001 |
| egf | 0.93 ± 0.06 | 0.26 ± 0.05 P < 0.0001 |
0.38 ± 0.02 P = 0.0003 |
0.34 ± 0.02 P < 0.0001 |
0.40 ± 0.06 P < 0.0001 |
1.20 ± 0.10 | 0.61 ± 0.04 P = 0.0036 |
0.29 ± 0.02 P < 0.0001 |
0.22 ± 0.02 P < 0.0001 |
0.33 ± 0.01 P < 0.0001 |
0.32 ± 0.03 P < 0.0001 |
0.35 ± 0.02 P < 0.0001 |
| mapk3 | 1.11 ± 0.03 | 0.24 ± 0.01 P < 0.0001 |
0.21 ± 0.02 P < 0.0001 |
0.21 ± 0.01 P < 0.0001 |
0.22 ± 0.01 P < 0.0001 |
1.13 ± 0.04 | 0.78 ± 0.02 P = 0.0497 |
0.34 ± 0.02 P < 0.0001 |
0.19 ± 0.01 P < 0.0001 |
0.19 ± 0.01 P < 0.0001 |
0.19 ± 0.01 P < 0.0001 |
0.19 ± 0.01 P < 0.0001 |
| tnxb | 0.98 ± 0.02 | 0.21 ± 0.02 P < 0.0001 |
0.30 ± 0.01 P < 0.0001 |
0.27 ± 0.03 P < 0.0001 |
0.30 ± 0.02 P < 0.0001 |
1.04 ± 0.02 | 0.89 ± 0.03 | 0.31 ± 0.01 P < 0.0001 |
0.17 ± 0.01 P < 0.0001 |
0.19 ± 0.01 P < 0.0001 |
0.23 ± 0.01 P < 0.0001 |
0.24 ± 0.03 P < 0.0001 |
| bcl2 | 1.10 ± 0.01 | 0.21 ± 0.02 P < 0.0001 |
0.24 ± 0.02 P < 0.0001 |
0.23 ± 0.01 P < 0.0001 |
0.23 ± 0.01 P < 0.0001 |
1.12 ± 0.08 | 0.59 ± 0.03 P = 0.0002 |
0.17 ± 0.02 P < 0.0001 |
0.10 ± 0.01 P < 0.0001 |
0.12 ± 0.01 P < 0.0001 |
0.16 ± 0.01 P < 0.0001 |
0.18 ± 0.01 P < 0.0001 |
| igf1r | 1.00 ± 0.04 | 0.20 ± 0.01 P < 0.0001 |
0.15 ± 0.01 P < 0.0001 |
0.11 ± 0.01 P < 0.0001 |
0.12 ± 0.01 P < 0.0001 |
1.04 ± 0.02 | 0.59 ± 0.01 P = 0.0005 |
0.21 ± 0.02 P < 0.0001 |
0.09 ± 0.01 P < 0.0001 |
0.08 ± 0.01 P < 0.0001 |
0.09 ± 0.01 P < 0.0001 |
0.09 ± 0.01 P < 0.0001 |
| pik3r2 | 0.83 ± 0.02 | 0.18 ± 0.01 P < 0.0001 |
0.14 ± 0.01 P < 0.0001 |
0.15 ± 0.02 P < 0.0001 |
0.20 ± 0.02 P < 0.0001 |
0.84 ± 0.05 | 0.82 ± 0.03 | 0.31 ± 0.02 P < 0.0001 |
0.16 ± 0.01 P < 0.0001 |
0.20 ± 0.02 P < 0.0001 |
0.21 ± 0.01 P < 0.0001 |
0.18 ± 0.02 P < 0.0001 |
| sox4 | 0.83 ± 0.03 | 0.17 ± 0.01 P < 0.0001 |
0.16 ± 0.01 P < 0.0001 |
0.16 ± 0.02 P < 0.0001 |
0.20 ± 0.01 P < 0.0001 |
0.73 ± 0.04 | 0.61 ± 0.11 P = 0.0139 |
0.19 ± 0.02 P < 0.0001 |
0.13 ± 0.01 P < 0.0001 |
0.18 ± 0.01 P < 0.0001 |
0.23 ± 0.01 P < 0.0001 |
0.22 ± 0.02 P < 0.0001 |
| lepr | 1.13 ± 0.03 | 0.16 ± 0.01 P < 0.0001 |
0.17 ± 0.01 P < 0.0001 |
0.12 ± 0.02 P < 0.0001 |
0.13 ± 0.01 P < 0.0001 |
1.26 ± 0.04 P = 0.462 |
0.79 ± 0.06 | 0.28 ± 0.03 P = 0.0001 |
0.12 ± 0.01 P < 0.0001 |
0.10 ± 0.01 P < 0.0001 |
0.07 ± 0.01 P < 0.0001 |
0.07 ± 0.01 P < 0.0001 |
| gpr30 | 1.04 ± 0.04 | 0.16 ± 0.01 P < 0.0001 |
0.20 ± 0.04 P < 0.0001 |
0.16 ± 0.01 P < 0.0001 |
0.17 ± 0.01 P < 0.0001 |
1.10 ± 0.05 | 0.74 ± 0.08 | 0.23 ± 0.03 P < 0.0001 |
0.11 ± 0.01 P < 0.0001 |
0.11 ± 0.01 P < 0.0001 |
0.11 ± 0.01 P < 0.0001 |
0.12 ± 0.02 P < 0.0001 |
| fgfr2 | 1.25 ± 0.07 | 0.15 ± 0.02 P < 0.0001 |
0.17 ± 0.01 P = 0.0001 |
0.17 ± 0.04 P < 0.0001 |
0.13 ± 0.01 P < 0.0001 |
1.03 ± 0.05 | 0.95 ± 0.02 | 0.25 ± 0.03 P = 0.0003 |
0.13 ± 0.01 P < 0.0001 |
0.12 ± 0.01 P < 0.0001 |
0.10 ± 0.01 P < 0.0001 |
0.12 ± 0.02 P < 0.0001 |
| ctsf | 1.02 ± 0.02 | 0.14 ± 0.02 P < 0.0001 |
0.11 ± 0.01 P < 0.0001 |
0.08 ± 0.01 P < 0.0001 |
0.10 ± 0.01 P < 0.0001 |
1.03 ± 0.04 | 0.71 ± 0.04 P = 0.0068 |
0.19 ± 0.02 P < 0.0001 |
0.07 ± 0.01 P < 0.0001 |
0.07 ± 0.01 P < 0.0001 |
0.06 ± 0.01 P < 0.0001 |
0.06 ± 0.01 P < 0.0001 |
| kgf | 1.28 ± 0.15 | 0.14 ± 0.01 P < 0.0001 |
0.16 ± 0.01 P < 0.0001 |
0.18 ± 0.02 P < 0.0001 |
0.24 ± 0.02 P < 0.0001 |
1.12 ± 0.07 | 1.02 ± 0.04 | 0.41 ± 0.02 P = 0.0007 |
0.31 ± 0.01 P < 0.0001 |
0.36 ± 0.03 P < 0.0001 |
0.29 ± 0.01 P < 0.0001 |
0.28 ± 0.020 P < 0.0001 |
| igfbp3 | 1.10 ± 0.06 | 0.14 ± 0.01 P < 0.0001 |
0.15 ± 0.01 P = 0.0003 |
0.13 ± 0.02 P < 0.0001 |
0.13 ± 0.02 P < 0.0001 |
1.18 ± 0.11 | 0.65 ± 0.05 P = 0.0379 |
0.21 ± 0.01 P = 0.0005 |
0.12 ± 0.01 P < 0.0001 |
0.10 ± 0.01 P < 0.0001 |
0.09 ± 0.01 P < 0.0001 |
0.11 ± 0.01 P < 0.0001 |
| hdac5 | 0.74 ± 0.06 P = 0.0438 |
0.13 ± 0.02 P < 0.0001 |
0.09 ± 0.01 P < 0.0001 |
0.08 ± 0.01 P < 0.0001 |
0.09 ± 0.01 P < 0.0001 |
0.88 ± 0.07 | 0.58 ± 0.05 P = 0.0023 |
0.19 ± 0.03 P < 0.0001 |
0.06 ± 0.01 P < 0.0001 |
0.07 ± 0.01 P < 0.0001 |
0.08 ± 0.01 P < 0.0001 |
0.06 ± 0.01 P < 0.0001 |
| vegfc | 1.06 ± 0.08 | 0.11 ± 0.01 P < 0.0001 |
0.11 ± 0.01 P < 0.0001 |
0.11 ± 0.01 P < 0.0001 |
0.13 ± 0.01 P < 0.0001 |
1.02 ± 0.05 | 0.78 ± 0.06 | 0.30 ± 0.01 P < 0.0001 |
0.18 ± 0.01 P < 0.0001 |
0.15 ± 0.01 P < 0.0001 |
0.16 ± 0.01 P < 0.0001 |
0.16 ± 0.01 P < 0.0001 |
| txnip | 1.29 ± 0.05 P = 0.0246 |
0.08 ± 0.01 P < 0.0001 |
0.09 ± 0.01 P < 0.0001 |
0.08 ± 0.01 P < 0.0001 |
0.10 ± 0.01 P < 0.0001 |
1.32 ± 0.06 P = 0.0160 |
0.68 ± 0.03 P = 0.0144 |
0.15 ± 0.01 P < 0.0001 |
0.09 ± 0.01 P < 0.0001 |
0.09 ± 0.01 P < 0.0001 |
0.07 ± 0.01 P < 0.0001 |
0.07 ± 0.01 P < 0.0001 |
| ccng2 | 0.99 ± 0.03 | 0.08 ± 0.01 P < 0.0001 |
0.08 ± 0.01 P < 0.0001 |
0.06 ± 0.01 P < 0.0001 |
0.06 ± 0.01 P < 0.0001 |
1.11 ± 0.02 | 0.58 ± 0.03 P = 0.0003 |
0.10 ± 0.02 P < 0.0001 |
0.04 ± 0.01 P < 0.0001 |
0.04 ± 0.01 P < 0.0001 |
0.05 ± 0.01 P < 0.0001 |
0.06 ± 0.01 P < 0.0001 |
Results were expressed as mean ± SEM (n = 4–8 mice/group). Significance of the observed effects was evaluated using Student's t-test. Gray highlight represents the maximum of regulation
Figure 3. Comparison of E2 and E4 on uterine gene regulation in ovariectomized mice.
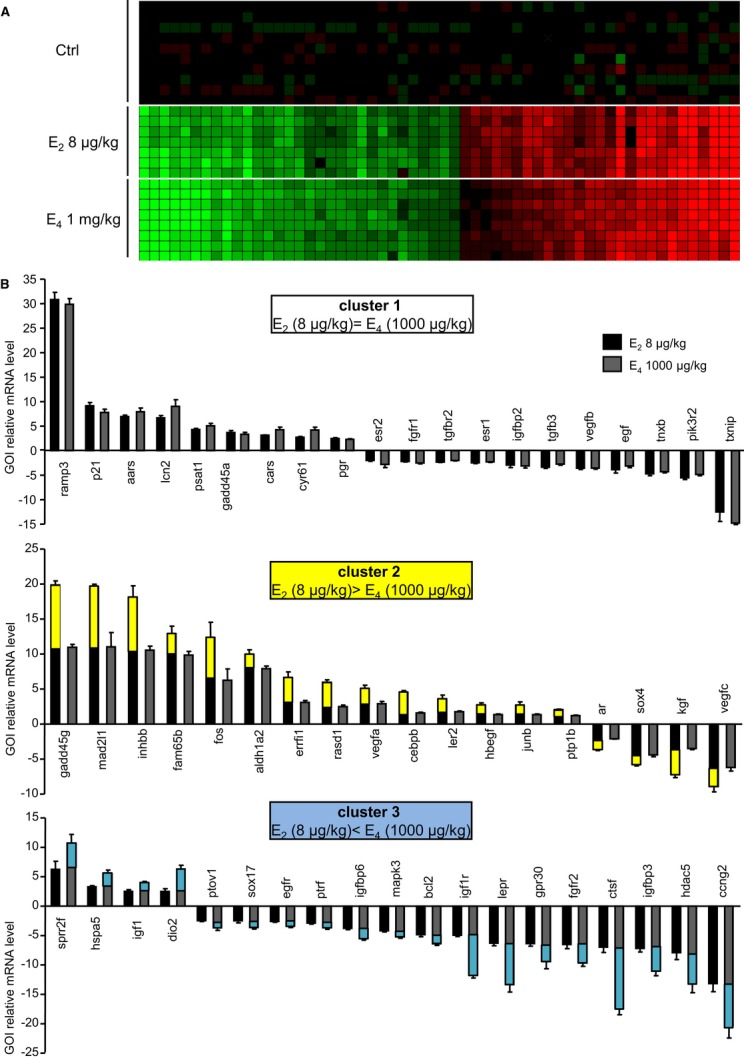
- Data obtained from 96.96 Dynamic Arrays were used to generate a cluster diagram of the significant gene expression changes. Each vertical line represents a single gene. Each horizontal line represents an individual sample. Genes that were up-regulated at least twofold following E2 administration relative to placebo are in red, whereas down-regulated genes are in green. The color intensity indicates the degree of variation in expression.
- Clustering pattern of the gene whose expression is affected by E2 and/or E4.
We next examined the relationship between gene regulation patterns and uterotrophic effects of E2 versus E4, noting histological changes and uterine epithelial cell proliferation. Luminal epithelial height (LEH) and stromal height (SH) were significantly and similarly increased with E2 (8 μg/kg) and E4 (1 mg/kg) 24 h after subcutaneous administration (Fig 4), without significant effects for doses of E4 < 1 mg/kg (Fig 4A and B, and Supplementary Fig S3A and B). Accordingly, a maximal induction of epithelial proliferation, detected by Ki-67 nuclear staining (Fig 4C and D), was observed in mice treated with either E2 8 μg/kg or E4 1 mg/kg alone. Lower doses of E4 elicited moderate to minor epithelial proliferation (Supplementary Fig S3C and D). To further analyze the interactions between E4 and E2 on ERα transcriptional activity, we then studied the effect of their combined impact on uterus. E2 (8 μg/kg) and E4 (given at either 200 μg/kg or 1 mg/kg) were co-administrated, and gene expression in the uterus was analyzed 6 h later. As shown in the Supplementary Fig S4, the gene expression profile of the E2–E4 combination was similar to that elicited by E2 alone for most of the genes (cluster 1). In some cases an intermediate response was observed using co-administration of E2–E4 compared to E2 alone (cluster 2), probably due to the lower potency of E4 (1 mg/kg) than those of E2 to induce maximal gene regulation for these genes (Fig 3, middle panel). Importantly, the histological changes and uterine epithelial cell proliferation induced by E2 (8 μg/kg) and E4 (200 μg/kg or 1 mg/kg) co-treatment did not differ from those elicited by E2 (8 μg/kg) alone (Fig 4). Taken together, these results demonstrate that E4 acts as a less potent estrogen on both gene expression and epithelial proliferation in the uterus, close to results obtained previously in rat uterus (Holinka & Gurpide, 1979).
Figure 4. Comparison of E2 and E4 on uterine histological parameters and epithelial proliferation.

Seven-week-old ovariectomized C57Bl/6J mice were injected subcutaneously with vehicle (Ctrl, castor oil), E2 (8 μg/kg), and/or E4 (200 μg/kg or 1 mg/kg) and were euthanized 24 h after treatment.
A, B Luminal epithelial height (LEH) (A) and stromal height (SH) (B) were measured.
C, D Representative (C) and quantification (D) of Ki-67 detection in transverse uterus sections (scale bar = 50 μm).
Data information: Results are expressed as mean ± SEM. To test the respective roles of each treatment, a one-way ANOVA was performed and a Bonferroni's multiple comparison test (n = 4–6 mice/group).
E4 induces an atheroprotective effect in an ERα-dependent manner
Since estrogens exert many beneficial effects on the arteries (Arnal et al, 2012), we assessed the impact of E4 on the prevention of atheroma. For this aim, we examined lipid deposition at the aortic sinus from ERα+/+LDLr−/− or ERα−/−LDLr−/− (Low Density Lipoprotein receptor) mice fed a high-cholesterol diet supplemented or not with E4 (0.6 and 6 mg/kg/day), a well-recognized model to study atheroprotective effects of estrogens (Mallat & Tedgui, 2007; Weber et al, 2008). E4 dose-dependently prevented lipid deposition in ovariectomized ERα+/+LDLr−/− mice (Fig 5A and B), decreasing the atheroma deposit by up to 80%, a level of protection similar to that obtained using a high dose of E2 (80 μg/kg/jour) (Billon-Gales et al, 2009). As previously observed with E2, this effect was completely abolished in ERα−/−LDLr−/− mice, indicating that ERα is necessary to mediate the atheroprotective effect of E4 (Fig 5A and B). Interestingly, expression of the most strongly induced gene by E2 in the aorta, Gremlin 2 (Grem2) (Schnoes et al, 2008) was found to be regulated by the highest dose of E4 in ERα+/+LDLr−/−, but not in ERα−/−LDLr−/− mice (Fig 5C), emphasizing another aspect of the ERα-dependent nuclear regulation by E4.
Figure 5. E4 prevents aortic sinus lipid deposition in hypercholesterolemic mice.
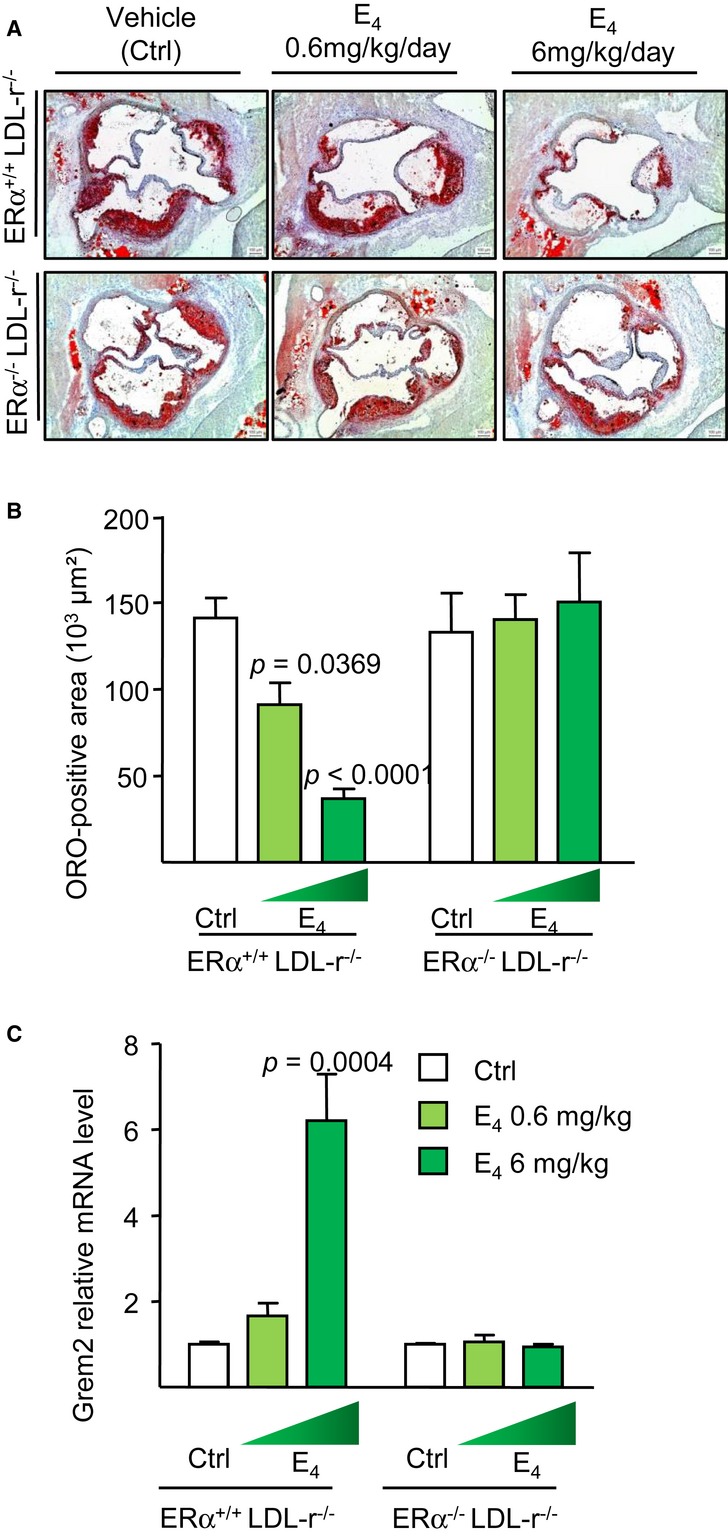
Four-week-old ovariectomized ERα+/+LDL-r−/− or ERα−/−LDL-r−/− mice were switched to atherogenic diet from the age of 6–18 weeks added with placebo (Ctrl) or E4 (0.6 or 6 mg/kg/day).
A, B Representative micrographs of Oil red-O (ORO) lipid-stained cryosections of the aortic sinus (A) and quantification of lipid deposition (B) are represented.
C Gremlin 2 (Grem2) mRNA level from aorta of these mice was quantified by qPCR and normalized to Tpt1 mRNA levels. Result was expressed according to the level in aorta from placebo set as 1.
Data information: Results are expressed as mean ± SEM. Significance of the observed effects was evaluated using one-way or two-way ANOVA followed by Bonferroni's post hoc test (n = 4–8 mice/group).
As previously observed with E2 (Billon-Gales et al, 2009), E4 (6 mg/kg/day) decreased total plasma cholesterol in ERα+/+LDLr−/− but not in ERα−/−LDLr−/− mice. However, in contrast to the action of E2, no change of HDL cholesterol level was observed in E4 treated mice (Table 3). As expected from the acute dose experiments, a dose-dependent uterine hypertrophy was observed in mice receiving E4 chronically, and this effect was totally abolished in ERα−/−LDLr−/− mice, further demonstrating the crucial role of ERα in E4 uterotrophic activity (Table 3).
Table 3.
Effect of E4 (0.6 or 6 mg/kg/day) treatment on body weight, uterine weight, plasma lipid concentrations, and Oil-red O (ORO) positive area at the aortic sinus in 18-week-old ERα+/+LDLr−/− or ERα−/−LDLr−/− mice
| ERα+/+LDLr−/− |
ERα−/− LDLr−/− |
P, two-factor ANOVA |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Ctrl (n = 10) | E4 0.6 mg/kg/day (n = 9) | E4 6 mg/kg/day (n = 7) | Ctrl (n = 7) | E4 0.6 mg/kg/day (n = 8) | E4 6 mg/kg/day (n = 4) | Genotype | E4 | Interaction | |
| Body weight (g) | 21.5 ± 0.9 | 18.7 ± 0.6 P = 0.0187 |
16.2 ± 0.3 P < 0.0001 |
20.9 ± 0.8 | 23.2 ± 0.5 | 22.0 ± 1.1 | – | – | P = 0.0004 |
| Uterine weight (mg) | 6 ± 1 | 31 ± 3 P < 0.0001 |
71 ± 7 P < 0.0001 |
3 ± 1 | 4 ± 1 | 6 ± 1 | – | – | P = 0.0001 |
| Total Chol. (mg/dl) | 1152.8 ± 142.2 | 868.4 ± 154.6 | 552.6 ± 44.0 P = 0.0065 |
1102.2 ± 205.3 | 1356.5 ± 124.5 | 1633.3 ± 276.3 | – | – | P = 0.0052 |
| HDL Chol. (mg/dl) | 62.3 ± 9.8 | 77.2 ± 15.1 | 63.7 ± 4.9 | 56.9 ± 14.9 | 61.9 ± 6.2 | 82.6 ± 24.1 | NS | NS | NS |
| ORO area (×103 µm2) | 141 ± 11 | 91 ± 13 P = 0.0369 |
37 ± 5 P < 0.0001 |
133 ± 23 | 140 ± 14 | 151 ± 28 | – | - | P = 0.0028 |
Results were expressed as mean ± SEM. Significance of the observed effects was evaluated using two-way ANOVA. When an interaction was observed between the 2 factors, effect of E4 treatment was studied in each genotype using a Bonferroni's post hoc test (n = 4–8 mice/group)
E4 fails to increase endothelial NO production and to accelerate endothelial healing
We then tested the effect of E4 on two other important vasculoprotective actions of estrogens, namely the acceleration of reendothelialization (Brouchet et al, 2001; Chambliss et al, 2010) and activation of eNOS (Wu et al, 2011), both of which are known to involve ERα MISS in the endothelium (Adlanmerini et al, 2014). First, although E2 promoted endothelial healing in the model of carotid artery electric injury, no effect was observed with E4, regardless of the dose employed (0.3, 1 or 6 mg/kg/day) (Fig 6A). Second, we tested the effect of E4 on eNOS activation in aortae by measuring eNOS phosphorylation (Fig 6B) and NO production using a NO-specific amperometric probe. Whereas E2 (10−8 M) rapidly and nicely induced eNOS phosphorylation (Fig 6B) and NO production (Fig 6C) in aortae, E4 (10−6 M) failed to produce these effects (Fig 6B and C). Together, these results suggest that E4 is not able to elicit two major endothelial actions known to be MISS ERα dependent, namely acceleration of reendothelialization and activation of eNOS.
Figure 6. E4 fails to accelerate reendothelialization and to increase NO production.

- Electric injury was applied to the distal part (3 mm precisely) of the common carotid artery, and the endothelial regeneration process was evaluated 3 days postinjury. Quantification of the reendothelialized area evaluated by Evans blue staining, and results were expressed as mean ± SEM (n = 7–23 mice per group). Significance of the observed effects was evaluated using one-way ANOVA followed by Bonferroni's post hoc test.
- Quantification expressed as mean ± SEM (n = 7 mice per group, upper panel) and representative Western blot (lower panel) of phospho-eNOS/eNOS abundance in isolated aortae treated by E2 (10−8 M), E4 (10−6 M), combination of both E2 and E4 or acetylcholine (Ach) used as a positive control during 30 min. Significance of the observed effects was evaluated using one-way ANOVA followed by Bonferroni's post hoc test (n = 8 mice/group).
- Representative trace of ex vivo amperometric measurements of NO release of aortae from 10- to 12-week-old C57Bl/6J mice exposed to E2 (10−8 M) or E4 (10−6 M) during 5 min.
- For cotreatment experiment, E4 (10−6 M) or vehicle (DMSO) was pre-incubated during 10 min prior to E2 (10−8 M) treatment. To test the respective roles of each treatment, a one-way ANOVA was performed followed by a Bonferroni's post hoc test.
Source data are available online for this figure.
The fact that E4 failed to elicit responses that are mediated via membrane ERα raises the question of whether this is due to the failure of E4 to bind to membrane ERα or the failure of membrane ERα to become activated by E4 binding, in which case E4 would be expected to have antagonist activity on this signaling pathway. To address this question, we first co-administrated E4 (6 mg/kg/day) and E2 (80 μg/kg/day), and found that this combination failed to accelerate endothelial healing (Fig 6A). Then, we tested the effect of E2 (10−8 M) on NO production by aortae ex vivo exposed to E4 (10−6 M) 10 min before, and we found that E4 inhibited the stimulatory action of E2 (Fig 6D). Accordingly, the combination of E2 (10−8 M) and E4 (10−6 M) did not stimulate eNOS phosphorylation in aortae (Fig 6B). Altogether, E4 is not only devoid of ERα MISS in the endothelium, but E4 is also able to partially antagonize these E2 MISS effects.
E4 promotes ERα-src interaction less efficiently than does E2 but induces similar ERE-dependent transcriptional activity in MCF-7
Finally, we approached the impact of E4 on ERα MISS in the breast cancer cell line, MCF-7. We failed to detect reliably the activation of MAPK by E2, in agreement with some authors (Gaben et al, 2004). We studied another well-accepted aspect of ERα MISS, that is, ERα interaction with the tyrosine kinase src using the Duolink technique (Soderberg et al, 2006). We found that E2 (10−8 M) favored this interaction, whereas a 100-fold higher dose (E4 10−6 M) was less efficient in inducing this aspect of MISS (Fig 7A). Importantly, when administrated together, the combination totally abrogated the ERα-src interaction, suggesting that, as shown above in endothelial cells, E4 was able to antagonize the action of E2 on ERα MISS. We also explored the impact of E2 10−8 M, E4 10−6 M, and their combination on the gene expression of MCF-7. As shown in Fig 7B, E2 10−8 M and E4 10−6 M similarly up-regulated the expression of genes containing ERE in their regulatory sequences, such as the gene regulated by estrogen in breast cancer 1 (GREB1) (Sun et al, 2007), the progesterone receptor (PR) (Kraus et al, 1993), and the chemokine (C-X-C motif) ligand 12 (CXCL12) (Boudot et al, 2011). Interestingly, and in striking contrast with the MISS effect, E2–E4 combination elicited the same induction than each isolated compound, showing no detectable interaction in these ERα nuclear actions.
Figure 7. E4 promotes ERα-src interaction less efficiently than does E2 but induces similar ERE-dependent transcriptional activity in MCF-7.

- MCF-7 cells were grown in medium containing 2.5% charcoal-stripped serum with vehicle or with E2 (10−8 M), E4 (10−6 M) or in combination for 5 min. After fixation, in situ PLA for ERα-Src dimers was performed with ERα- and Src-specific antibodies. The detected dimers are represented by red dots, and the nuclei were counterstained with DAPI (blue). Quantification of the number of signals per cell was performed by computer-assisted analysis as reported in the Materials and Methods section. Values correspond to the mean ± SEM of at least three separate experiments, and columns with different superscripts differ significantly using Student's t-test.
- mRNA level of the indicated gene from MCF-7 cells treated with vehicle, E2 (10−8 M), E4 (10−6 M) or combined treatment and analyzed after 24 h by qPCR. Values correspond to the mean ± SD of at least three separate experiments. To test the respective roles of each treatment, a one-way ANOVA was performed and a Bonferroni's multiple comparison test.
Discussion
Estetrol (E4), a physiological estrogen with four hydroxyl groups produced only by the fetal liver, appears to be human specific, but its physiological role is unknown. Furthermore, very few data are available concerning its molecular mechanisms of action. In this study, we demonstrate through in vitro and in vivo experiments that E4 is able to induce ERα transcriptional activity (about 100-fold above the doses of E2 required for the responses considered). Accordingly, the positioning of E4 in the ligand-binding pocket is very similar to that of E2, leading to a positioning of helix 12 and AF-2 availability that are nearly identical to that elicited by E2. Notably, although the affinity of E4 for ERα is 100-fold less than E2, the ERα complex with E4 is able to bind the important coactivator SRC3 as the complex with E2. We and others previously demonstrated that endometrial proliferation is highly dependent on the ERα nuclear actions, since this effect is abrogated in ERαAF-20 and ERαAF-10 mice (Abot et al, 2013), whereas it is fully preserved using a mouse with a point mutation of the palmitoylation site of ERα (C451A-ERα) that leads to membrane-specific loss of function of ERα (Adlanmerini et al, 2014). The potent atheroprotective effect observed in response to E4 also fits nicely not only with an ERα-dependent effect, as demonstrated by its abrogation in ERα−/− mice, but also with the nuclear action of ERα. Indeed, we previously demonstrated that E2 failed to induce its atheroprotective action using AF-20LDLR−/− mice, highlighting the importance of nuclear/transcriptional actions of ERα for atheroprotection (Billon-Gales et al, 2011).
In contrast, E4, even at high doses, is not able to elicit major endothelial actions known to be membrane ERα dependent, namely an increase in eNOS phosphorylation, in NO production, or an acceleration of reendothelialization (Chambliss et al, 2010; Adlanmerini et al, 2014). Furthermore, it antagonizes partially these MISS effects of ERα in response to E2. We also found that although E4 promotes some level of ERα-src interaction, E2/E4 combination does not promote any interaction. Already, H. Coelingh Bennink et al reported in the cancer-induced rat model that mammary tumor formation induced by DMBA treatment was stimulated by E2 and EE, but prevented by E4 (Coelingh Bennink et al, 2008a). Very recently, it has been demonstrated that E2 through a MISS effect enhanced the migration and invasiveness of human T47D breast carcinoma cells (Giretti et al, 2014). In contrast, E4 failed to stimulate and even antagonized the stimulation of T47D cells migration and invasion through matrigel by E2. According to our current understanding of MISS effects in breast cancer (Acconcia & Marino, 2011; Le Romancer et al, 2011), these data suggest that in this context E4 could have a safer profile than classic estrogens. Altogether, E4 appears to behave as a full or partial membrane ERα antagonist.
The structure as well as the conformation of ERα at the plasma membrane remains unclear, although palmitoylation appears to play an important role in its membrane localization and extranuclear-initiated actions (Acconcia et al, 2004; Adlanmerini et al, 2014). It thus appeared to us that comparing the physical interaction characteristics of these two estrogens, E2 and E4, in artificial membranes could shed some light to the lack of MISS action of E4. E4 was found to be almost as soluble as E2 in artificial membranes, ruling out the possibility that the lack of membrane signaling by E4 could be the result of its lack of availability in this cell compartment. In addition, whereas E2 was found to be in equilibrium between two orientations in the bilayer, E4 had a preferential orientation with its phenol group oriented toward interface and the three hydroxyl groups thus being at the hydrophobic core of the membrane. This orientation is rather counterintuitive, although an efficient intramolecular network of hydrogen bonds among the three D-ring OH groups might be masking their polarity more effectively than the lone 17β-OH in E2. The relationship between membrane orientation of an estrogen and its access to the ligand-binding site in membrane ERα, however, is at this point a matter of speculation, but it is clear that both E2 and E4 bind to ERα regardless of whether it is localized in the nucleus or the plasma membrane.
It is important to underline that the molecular mechanisms that mediate MISS effects of estrogen are far to be fully understood. The downstream target regulated by the ERα MISS involved various post-transcriptional modifications which probably highly differ between cell types. In endothelial cells, PI3K, Akt kinase, ERK1/2, striatin, and phosphorylation of eNOS have been described to be required for ERα MISS, whereas in vascular smooth muscle cells, expression and activity of several phosphatases such as MKP-1, SHP-1, PTEN, and PP2A mediate this pathway (Ueda & Karas, 2013). Since E4 is specific for humans and is produced only by the fetal liver, it is tempting to speculate that E4 might be conferring a very specific but important modulating effect of E2 action on fetal development, especially on brain development, as the nervous system appears to be largely influenced by MISS actions (Vasudevan & Pfaff, 2007).
Defect of E4 action via the membrane ERα pathway could also play a role on gene expression profiles and phenotypic effects of ERα action in organs that are dependent on both nuclear and membrane effects. Several authors proposed that nuclear action of ERα and of other transcription factors are regulated by MISS actions of estrogens (O'Malley & McGuire, 1968; Bjornstrom & Sjoberg, 2002; Lannigan, 2003; La Rosa et al, 2012), and the respective level of dependency of tissues on both nuclear and membrane effects could also be determined thanks to C451A-ERα and ERαAF-20 mice. Although this cross talk was not observed for cell proliferation in uterus (Adlanmerini et al, 2014), it could be important in other tissues.
This original profile of ERα activation, uncoupling nuclear and membrane activation is, to the best of our knowledge, unique and characterizes E4 as a natural endogenous selective ER modulator (Table 4), reinforcing the idea that medical applications should be pursued further. Indeed, E4, in combination with a progestin, inhibits ovulation during the reproductive life (Coelingh Bennink et al, 2008c), or alleviates the climacteric symptoms after menopause (Holinka et al, 2008). As mentioned in the introduction, two recent phase 2 clinical trials evaluated the contraceptive efficacy of 5–20 mg E4 and levonorgestrel or drospirenone as a progestin. The first study evaluated ovulation inhibition in 91 women (18–35 year old) by measuring follicular size and endometrial thickness by ultrasound and evaluating the plasma levels of FSH, LH, E2, and progesterone. No ovulation was observed during the three cycles of treatment. The second study evaluated the bleeding profile in 330 young women over six cycles. An excellent bleeding and spotting profile clearly demonstrated the capacity of E4 to maintain a stable endometrium that was superior to the control group treated with E2 and dienogest. Lack of ovulation in all women was also verified by measuring the urinary excretion of pregnanediol, a progesterone metabolite. Remarkably, changes in SHBG, corticosteroid binding globulin (CBG), angiotensinogen, triglycerides, or coagulation proteins were minimal and considerably lower than in the comparator group receiving a combination of EE and drospirenone. Altogether, these experimental and clinical studies indicate that E4 should now be considered as a natural SERM. It is able to stimulate the endometrium, but it has no or only a minimal impact on the liver function. Dedicated experimental studies and randomized clinical trials of E4 are now needed, as better therapeutic alternatives are greatly needed by physicians and patients both in the field of oral contraception and as agents to replace the loss of beneficial estrogen effects resulting from the menopause.
Table 4.
Current understanding of the impact of E2 and E4 on nuclear versus membrane initiated steroid signaling (MISS) ERα-mediated effects
| Estrogens | Cell or tissue effects |
|||
|---|---|---|---|---|
| Uterus |
MCF-7 |
Endothelial cells |
||
| Transcription/ proliferation | Transcription ERE dependent | Src-ERα interaction | Cell migration/ eNOS activation | |
| E2 | +++ | +++ | +++ | +++ |
| E4 | +++ | +++ | + | 0 |
| E2 + E4 | +++ | +++ | 0 | 0/+ |
| Prominent mechanism of action | Nuclear | Miss | ||
Materials and Methods
Expression purification and crystallization of ERα ligand-binding domain
ERα-LDB was expressed with a N-terminal Histidine tag in E. coli (BL21 DE3) and induced with isopropyl-β-d-thiogalactopyranoside (IPTG) for 16 h at 18°C. Cell pellets were lysed in 5 pellet volumes of lysis buffer [50 mM Tris pH7.6, 500 mM NaCl, 10% glycerol, 0.05% β-octyl glucoside, 10 mM imidazole, 5 mM β-mercaptoethanol, protease inhibitor (Roche) and 0.1 mg/ml lysozyme]. The lysates were centrifuged at 30,000 g for 30 min, and the supernatant was collected and loaded on a Ni-affinity resin. ERα-LDB protein was eluted with lysate buffer containing 500 mM imidazole. ERα-LDB was further purified on a size exclusion column. ERα was crystallized in complex with E2, E3 or E4, and GRIP peptide using a commercial screen formulation Index (Hampton Research) (Hsieh et al, 2006) Data collection was performed on single crystals at sector 19 (Structural Biology Center Collaborative Access Team at Agronome National Laboratory).
Cell culture and transfection assays
MCF-7 cells were maintained in DMEM (Sigma-Aldrich) supplemented with 10% fetal calf serum (FCS) (Biowest) and antibiotics (Sigma-Aldrich) at 37°C in 5% CO2. One day before treatment, cells growing in 10 cm diameter dishes were placed in phenol red-free DMEM (Sigma-Aldrich) containing 2.5% charcoal-stripped FCS (Biowest). Cells were then treated for 24 h with E2 (10−8 M), E4 (10−6 M), combined treatment or ethanol.
HepG2 and HeLa cells were maintained in DMEM (Sigma-Aldrich) supplemented with 10% fetal calf serum (FCS) (Biowest) and antibiotics (Sigma-Aldrich) at 37°C in 5% CO2. Transfections were carried out using jetPEI reagent according to manufacturer's instructions (Polyplus). One day before transfection, cells were plated in 24-well plates at 50% confluence. One hour prior to transfection, the medium was replaced with phenol red-free DMEM (Sigma-Aldrich) containing 2.5% charcoal-stripped FCS (Biowest). Transfection was carried out with 100 ng of ERE-TK promoter driven renilla luciferase (luc) reporter, 100 ng of CMV-β galactosidase (Gal) internal control, and 50 ng of pCR3.1, pCR-ERα, pCR-ERα Δ79, or pCR-ERαAF-10 expression vectors. Following an overnight incubation, cells were treated for 24 h with E2, E4, or ethanol (vehicle control). Cells were then harvested, and luciferase and β-galactosidase assays were performed as previously described (Penot et al, 2005).
Mice
All procedures involving experimental animals were performed in accordance with the principles and guidelines established by the National Institute of Medical Research (INSERM) and were approved by the local Animal Care and Use Committee. ERα-null mice (ERα−/−) were generated as previously described (Billon-Gales et al, 2009) and were kindly provided by Pr P. Chambon (Strasbourg, France). To generate the double-deficient mice, LDLr−/− female mice, purchased from Charles River (L'Arbresle, France), were crossed with ERα+/− mice. The mice were anesthetized by injection of ketamine (100 mg/kg) and xylazine (10 mg/kg) by intraperitoneal route. To analyze E4 uterine action, C57Bl/6J were ovariectomized at 4 weeks of age and were subcutaneously injected with vehicle (castor oil), E2, or E4 at different doses 3 weeks later. Mice were sacrificed 6 or 24 h after a single estrogen injection and uteri were collected.
Analysis of mRNA levels by RT-qPCR
Tissues were homogenized using a Precellys tissue homogenizer (Bertin Technol., Cedex, France), and total RNA from tissues was prepared using TRIzol (Invitrogen, Carlsbad, CA). One microgram of RNA was reverse transcribed (RT) at 25°C for 10 min and then at 37°C for 2 h in 20 μl final volume using the High Capacity cDNA reverse transcriptase kit (Applied Biosystems). For gene expression in uterus, the 96.96 Dynamic Arrays for the microfluidic BioMark system (Fluidigm Corporation, CA, USA) were used to study by high throughput qPCR the gene expression profile in 6.5 ng cDNA from each sample, as described previously (Abot et al, 2013). For gene expression in aorta, qPCR was performed using SsoFast EvaGreen Supermix (Bio-Rad) with primers validated by testing the PCR efficiency (Fontaine et al, 2013). Gene expression was quantified using the comparative Ct (threshold cycle) method.
Total RNA from MCF-7 cells was also extracted using TRIzol™ (Invitrogen) according to the manufacturer's instructions. cDNAs were generated using MMLV Reverse transcriptase (Invitrogen) and random hexamers (Promega, Madison, WI, USA). Quantitative RT-PCR was performed using the iQ SybrGreen supermix (BioRad, Hercules, CA, USA) on a BioRad MyiQ apparatus. Sequences of the primers used for cDNA amplification in the quantitative RT-PCR experiments are available upon request. Results were normalized to GAPDH expression.
Uterus immunohistochemistry
Four-micrometer paraffin-embedded transverse sections from formalin fixed uterine specimens were dewaxed in toluene and rehydrated through acetone bath to deionized water. Antigen retrieval was performed in 10 mM citrate buffer pH 6.0 for 30 min in a water bath at 95°C. Cooled sections were then incubated in peroxidase blocking solution (Dako) to quench endogenous peroxidase activity. To block non-specific binding, sections were incubated in normal goat serum (Dako) for 20 min at room temperature. Primary antibodies were all rabbit polyclonal antibodies: anti-Ki-67 antigen (Thermo-scientific). Sections were incubated 50 min at room temperature with primary antibodies. The secondary antibody, biotinylated goat anti-rabbit immunoglobulins (Thermo-Scientific), was applied for 25 min at room temperature followed by an HRP-streptavidin solution (Dako) for 25 min. Peroxidase activity was revealed by 3,3′-diaminobenzidine tetrahydrochloride substrate (Dako). Finally, sections were counterstained with Harris hematoxylin, dehydrated and coverslipped. The luminal epithelial height (LEH) and stromal height (SH) were measured from the basal membrane to the apical surface. The values are the mean of ten measurements in each transverse uterus section.
Analyses of atherosclerosis lesions
Bilateral ovariectomy was performed at 4 weeks of age. At 6 weeks of age, mice were switched to a hypercholesterolemic atherogenic diet (1.25% cholesterol, 6% fat, no cholate, TD96335, Harlan Teklad, Wisconsin) mixed with E4 (calculated to correspond to either 0.6 or 6 mg/kg/day) during 12 weeks. Over-night fasted mice were anesthetized, and blood was collected from the retro-orbital venous plexus. Lipid deposition size was evaluated at the aortic sinus as previously described (Billon-Gales et al, 2009). Briefly, each heart was frozen on a cryostat mount with OCT compound. One hundred 10-μm thick sections were prepared from the top of the left ventricle, where the aortic valves were first visible, up to a position in the aorta where the valve cusps were just disappearing from the field. After drying for 2 h, the sections were stained with oil red O and counterstained with Mayer's hematoxylin. Ten sections out of the 100, each separated by 90 μm, were used for specific morphometric evaluation of intimal lesions using a computerized Biocom morphometry system. The first and most proximal section to the heart was taken 90 μm distal to the point where the aorta first becomes rounded. The mean lesion size (expressed in μm2) in these 10 sections was used to evaluate the lesion size of each animal.
Determination of plasma lipids
Total cholesterol was assayed using the CHOD-PAD kit (Horiba ABX, Montpellier, France). The high density lipoprotein (HDL) fraction was isolated from 10 μl of serum and assayed using the ‘C-HDL + Third generation’ kit (Roche, Lyon, France).
Mouse carotid injury and quantification of reendothelialization
Bilateral ovariectomy was performed at 4 weeks of age, and concomitantly the mice received pellets implanted subcutaneously releasing either placebo, E2 (17β-estradiol 0.1 mg, 60 days release, i.e., 80 μg/kg/day, Innovative Research of America, Sarasota, FL) or an osmotic minipump releasing E4 (1 or 6 mg/kg/day). After 2 weeks treatment, carotid electric injury was performed as previously described (Brouchet et al, 2001) and reendothelialization was evaluated after 3 days. Briefly, surgery was carried out with a stereomicroscope (Nikon SMZ800), and the left common carotid artery was exposed via an anterior incision in the neck. The electric injury was applied to the distal part (3 mm precisely) of the common carotid artery with a bipolar microregulator. Three day postinjury, carotid arteries were stained with Evans blue dye and mounted with Kaiser's Glycerol gelatin (Merck). Images were acquired using DMR 300 Leica microscope using LAS V3.8 and ImageJ software. Percentage of reendothelialization was calculated relative to the initial deendothelialized area (Brouchet et al, 2001; Chambliss et al, 2010).
Western blotting
Total proteins from aortae were separated on a 10% SDS/PAGE gel and transferred to a nitrocellulose membrane. The primary antibodies used are as follows: pSer1177-eNOS (612392; BD Bioscience), eNOS (610297; BD Bioscience), and β-actin (A2066; Sigma). Revelation was performed using an HRP-conjugated secondary antibody and visualized by ECL detection according to the manufacturer's instructions (Amersham Biosciences/GE Healthcare), using ChemiDoc Imaging System (Bio-Rad). Bands were quantified using ImageJ densitometry.
Real-time NO production
Aorta from intact mice (10–12 weeks) was quickly harvested and maintained in 200 μl Krebs–Ringer oxygenated solution containing 2.5 mmol/l glucose at 37°C. A NO-specific amperometric probe [ISO-NOPF100; World Precision Instruments (WPI), Sarasota, FL] was implanted directly in the tissue, and NO release was monitored. The aorta was exposed to E2 (10−8 M) or E4 (10−6 M) during 5 min. For cotreatment experiment, E4 (10−6 M) or vehicle (DMSO) was pre-incubated during 10 min prior to E2 (10−8 M) treatment. The concentration of NO gas in the tissue was measured in real time with the data acquisition system LabTrax (WPI) connected to the free radical analyzer Apollo1000 (WPI). Data acquisition and analysis were performed with DataTrax2 software (WPI). The NO-specific amperometric probe was calibrated as previously described (Knauf et al, 2001).
Proximity Ligation Assay
The Proximity Ligation Assay (PLA) technology was developed by Olink Bioscience (Sweden) (Soderberg et al, 2006) and is commercialized by Sigma-Aldrich. For PLA, MCF-7 cells (5 × 104 cells/ml) were grown on coverslips into 24-well plates in phenol red-free DMEM/F12 containing 5% charcoal-stripped FCS and were treated or not with E2 (10 nM) or E4 (1 μM) for 5 min. Cells were then fixed in 4% paraformaldehyde for 10 min and washed in large amount of PBS, and the coverslips were treated according to manufacturer's instructions (Duolink II Fluorescence, Olink Bioscience). Then, couple of primary antibodies rabbit anti-ERα (HC20 (Santa Cruz technology) and mouse anti-Src (B12, Santa Cruz Technology) was incubated overnight at 4°C in PBS with 0.2% triton and 0.5% non-fat milk. After washes, the PLA minus and plus probes (containing the secondary antibodies conjugated with complementary oligonucleotides) were added and incubated 1 h at 37°C. The next step allows the ligation of oligonucleotides if the two proteins are in close proximity thanks to the ligase during an incubation of 30 min at 37°C. After washes, the addition of nucleotides and polymerase allows amplification by rolling-circle amplification reaction using the ligated circle as a template during an incubation of 100 min at 37°C. The amplification solution also contains fluorescently labeled oligonucleotides that hybridize to the rolling-circle amplification product. The coverslips were let drying at room temperature in the dark and were mounted with Duolink II mounting Medium containing Dapi. The hybridized fluorescent slides were viewed under a Zeiss AxioImager Z1 microscope. Images were acquired under identical conditions at objective ×40. On each samples, at least 500 cells were counted. Analyses and quantifications of these samples were performed using ImageJ software that allows counting dots on 8 bits image and the plugin ‘Counter cells’ allows analyzing cells number.
The paper explained
Problem
Estetrol (E4) is an estrogen produced by the human fetal liver only during pregnancy. A recent clinical phase II study evaluating its contraceptive properties revealed that E4 did not change the levels of hepatic-derived proteins, including coagulation factors. Thus, at variance to classically used estrogens, it might not increase thrombo-embolic events. The molecular mechanism of action of E4 is essentially unknown, and the goal of this study was to define the nuclear/transcriptional actions versus the membrane/rapid actions in comparison to E2.
Results
In this study, we show that E4 is less potent than E2 to activate estrogen receptor alpha (ERα), but a high dose is able to modulate the transcriptional activity of ERα in the uterus, the proliferation of endometrial epithelium and to prevent atheroma. In contrast, E4 was not only devoid of effects on endothelial healing and eNOS activation, but it antagonized these E2 effects that are purely membrane ERα-dependent.
Impact
Thus, E4 appears not only as less potent estrogen than E2 but behaves as a natural selective ER modulator, and its spectrum of action as safe oral contraceptive or hormonal treatment of menopause should now be considered.
Statistical analyses
Results are expressed as the mean ± SEM (Standard Error Mean). To test the effect of treatments, 1-way ANOVA was performed. To test the respective roles of treatment and genotype (ERα deficiency), a 2-way ANOVA was performed. When an interaction was observed between the two factors, the effect of treatment was studied in each genotype using a Bonferroni's post hoc test. A value of P < 0.05 was considered as statistically significant.
Acknowledgments
The staff of the animal facilities and of the ‘Plateforme d'experimentation fonctionnelle’ (A. Desquesnes) are acknowledged for skillful technical assistance. We also thank J-C.Albouys, F. Boudou, and C. Bleuart as well as J.J. Maoret and F. Martins for their excellent technical assistance and contribution to qRT-PCR experiments carried out at GeT-TQ Genopole Toulouse Facility. We thank P. Liere who performed E4 plasmatic dosage and A-L Guihot who performed experiments on phosphorylation of eNOS. The work at the INSERM unit U1048 was supported by INSERM, Université de Toulouse III and Faculté de Médecine Toulouse-Rangueil, Fondation de France, Conseil Régional Midi-Pyrénées and Fondation pour la Recherche Médicale (FRM). A. Abot was supported by a grant from the Groupe de Réflexion sur la Recherche Cardiovasculaire. The NMR facility is part of the genotoul-Ibisa PICT platform and was funded by CNRS, région Midi-Pyrénées, and European structural funds. The work at INSERM U1083-CNRS-UMR 6214 is supported by INSERM, CNRS, CHU and Université d'Angers, Fondation de France, Fondation de l'Avenir, and Conseil Régional Pays de la Loire. The work at ULg, GIGA-cancer was supported by grants from the F.R.S.-FNRS (Belgium), the DGO6 from SPW (Belgium), the IUAP (Belspo, Belgium). This work was supported by National Institutes of Health Grants PHS5R01 DK015556 to J.A.K.
Author contributions
Study was conceived by JFA and JMF. Experiments were designed by AA, CF, RS, AD, AF, SR, MCV, MB, ML, IM, AM, DH, CK, and GLF. Acquisition of all the data was realized by AG, FF, CG, AA, CF, RS, AD, AF, SR, MB, ML, IM, CP, MA, AM, DH, CK and GF, and the analysis and interpretation of data were performed by AG, AA, CF, AD, MB, IM, AM, DH, CK, GF, MM, IRL, PG, PV, FL, GLF, BSK, JAK, and JFA. The final manuscript was prepared by CF, BSK, JAK, and JFA. The whole study was supervised by JFA.
Conflict of interest
MM and JMF are associated with UTERON-A DIVISION OF ACTAVIS. This work was supported in part by a grant from UTERON.
Supporting Information
Supplementary information for this article is available online: http://embomolmed.embopress.org
References
- Abot A, Fontaine C, Raymond-Letron I, Flouriot G, Adlanmerini M, Buscato M, Otto C, Berges H, Laurell H, Gourdy P, et al. The AF-1 activation function of estrogen receptor alpha is necessary and sufficient for uterine epithelial cell proliferation in vivo. Endocrinology. 2013;154:2222–2233. doi: 10.1210/en.2012-2059. [DOI] [PubMed] [Google Scholar]
- Acconcia F, Ascenzi P, Fabozzi G, Visca P, Marino M. S-palmitoylation modulates human estrogen receptor-alpha functions. Biochem Biophys Res Commun. 2004;316:878–883. doi: 10.1016/j.bbrc.2004.02.129. [DOI] [PubMed] [Google Scholar]
- Acconcia F, Marino M. The effects of 17beta-estradiol in cancer are mediated by estrogen receptor signaling at the plasma membrane. Front Physiol. 2011;2:30. doi: 10.3389/fphys.2011.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adlanmerini M, Solinhac R, Abot A, Fabre A, Raymond-Letron I, Guihot AL, Boudou F, Sautier L, Vessieres E, Kim SH, et al. Mutation of the palmitoylation site of estrogen receptor alpha in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc Natl Acad Sci U S A. 2014;111:E283–E290. doi: 10.1073/pnas.1322057111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnal JF, Lenfant F, Flouriot G, Tremollieres F, Laurell H, Fontaine C, Krust A, Chambon P, Gourdy P. From in vivo gene targeting of estrogen receptors to optimisation of their modulation in menopause. Br J Pharmacol. 2012;165:57–66. doi: 10.1111/j.1476-5381.2011.01538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascenzi P, Bocedi A, Marino M. Structure-function relationship of estrogen receptor alpha and beta: impact on human health. Mol Aspects Med. 2006;27:299–402. doi: 10.1016/j.mam.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Billon-Gales A, Fontaine C, Douin-Echinard V, Delpy L, Berges H, Calippe B, Lenfant F, Laurell H, Guery JC, Gourdy P, et al. Endothelial estrogen receptor-alpha plays a crucial role in the atheroprotective action of 17beta-estradiol in low-density lipoprotein receptor-deficient mice. Circulation. 2009;120:2567–2576. doi: 10.1161/CIRCULATIONAHA.109.898445. [DOI] [PubMed] [Google Scholar]
- Billon-Gales A, Krust A, Fontaine C, Abot A, Flouriot G, Toutain C, Berges H, Gadeau AP, Lenfant F, Gourdy P, et al. Activation function 2 (AF2) of estrogen receptor-{alpha} is required for the atheroprotective action of estradiol but not to accelerate endothelial healing. Proc Natl Acad Sci U S A. 2011;108:13311–13316. doi: 10.1073/pnas.1105632108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornstrom L, Sjoberg M. Signal transducers and activators of transcription as downstream targets of nongenomic estrogen receptor actions. Mol Endocrinol. 2002;16:2202–2214. doi: 10.1210/me.2002-0072. [DOI] [PubMed] [Google Scholar]
- Boudot A, Kerdivel G, Habauzit D, Eeckhoute J, Le Dily F, Flouriot G, Samson M, Pakdel F. Differential estrogen-regulation of CXCL12 chemokine receptors, CXCR4 and CXCR7, contributes to the growth effect of estrogens in breast cancer cells. PLoS ONE. 2011;6:e20898. doi: 10.1371/journal.pone.0020898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouchet L, Krust A, Dupont S, Chambon P, Bayard F, Arnal JF. Estradiol accelerates reendothelialization in mouse carotid artery through estrogen receptor-alpha but not estrogen receptor-beta. Circulation. 2001;103:423–428. doi: 10.1161/01.cir.103.3.423. [DOI] [PubMed] [Google Scholar]
- Chambliss KL, Wu Q, Oltmann S, Konaniah ES, Umetani M, Korach KS, Thomas GD, Mineo C, Yuhanna IS, Kim SH, et al. Non-nuclear estrogen receptor alpha signaling promotes cardiovascular protection but not uterine or breast cancer growth in mice. J Clin Invest. 2010;120:2319–2330. doi: 10.1172/JCI38291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelingh Bennink HJ, Simoncini T, Genazzani A, Kubista E. Estetrol, a pregnancy-specific human steroid, prevents and suppresses mammary tumor growth in a rat model. Climacteric. 2008a;11(Suppl 1):29. doi: 10.1080/13697130802040325. [DOI] [PubMed] [Google Scholar]
- Coelingh Bennink HJ, Heegaard AM, Visser M, Holinka CF, Christiansen C. Oral bioavailability and bone-sparing effects of estetrol in an osteoporosis model. Climacteric. 2008b;11(Suppl 1):2–14. doi: 10.1080/13697130701798692. [DOI] [PubMed] [Google Scholar]
- Coelingh Bennink HJ, Skouby S, Bouchard P, Holinka CF. Ovulation inhibition by estetrol in an in vivo model. Contraception. 2008c;77:186–190. doi: 10.1016/j.contraception.2007.11.014. [DOI] [PubMed] [Google Scholar]
- Fontaine C, Abot A, Billon-Gales A, Flouriot G, Berges H, Grunenwald E, Vinel A, Valera MC, Gourdy P, Arnal JF. Tamoxifen elicits atheroprotection through estrogen receptor alpha AF-1 but does not accelerate reendothelialization. Am J Pathol. 2013;183:304–312. doi: 10.1016/j.ajpath.2013.03.010. [DOI] [PubMed] [Google Scholar]
- Gaben AM, Saucier C, Bedin M, Redeuilh G, Mester J. Mitogenic activity of estrogens in human breast cancer cells does not rely on direct induction of mitogen-activated protein kinase/extracellularly regulated kinase or phosphatidylinositol 3-kinase. Mol Endocrinol. 2004;18:2700–2713. doi: 10.1210/me.2003-0133. [DOI] [PubMed] [Google Scholar]
- Giretti MS, Montt Guevara MM, Cecchi E, Mannella P, Palla G, Spina S, Bernacchi G, Di Bello S, Genazzani AR, Genazzani AD, et al. Effects of estetrol on migration and invasion in T47-D breast cancer cells through the actin cytoskeleton. Front Endocrinol. 2014;5:80. doi: 10.3389/fendo.2014.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen AA, Barr M, Diczfalusy E. Metabolism of 17-beta-oestradiol-4-14-C in early infancy. Acta Endocrinol. 1965;49:207–220. [PubMed] [Google Scholar]
- Hammond GL, Hogeveen KN, Visser M, Coelingh Bennink HJ. Estetrol does not bind sex hormone binding globulin or increase its production by human HepG2 cells. Climacteric. 2008;11(Suppl 1):41–46. doi: 10.1080/13697130701851814. [DOI] [PubMed] [Google Scholar]
- Hewitt SC, Deroo BJ, Hansen K, Collins J, Grissom S, Afshari CA, Korach KS. Estrogen receptor-dependent genomic responses in the uterus mirror the biphasic physiological response to estrogen. Mol Endocrinol. 2003;17:2070–2083. doi: 10.1210/me.2003-0146. [DOI] [PubMed] [Google Scholar]
- Holinka CF, Gurpide E. In vivo effects of estetrol on the immature rat uterus. Biol Reprod. 1979;20:242–246. doi: 10.1093/biolreprod/20.2.242. [DOI] [PubMed] [Google Scholar]
- Holinka CF, Brincat M, Coelingh Bennink HJ. Preventive effect of oral estetrol in a menopausal hot flush model. Climacteric. 2008;11(Suppl 1):15–21. doi: 10.1080/13697130701822807. [DOI] [PubMed] [Google Scholar]
- Hsieh RW, Rajan SS, Sharma SK, Guo Y, DeSombre ER, Mrksich M, Greene GL. Identification of ligands with bicyclic scaffolds provides insights into mechanisms of estrogen receptor subtype selectivity. J Biol Chem. 2006;281:17909–17919. doi: 10.1074/jbc.M513684200. [DOI] [PubMed] [Google Scholar]
- Huet G, Merot Y, Le Dily F, Kern L, Ferriere F, Saligaut C, Boujrad N, Pakdel F, Metivier R, Flouriot G. Loss of E-cadherin-mediated cell contacts reduces estrogen receptor alpha (ER alpha) transcriptional efficiency by affecting the respective contribution exerted by AF1 and AF2 transactivation functions. Biochem Biophys Res Commun. 2008;365:304–309. doi: 10.1016/j.bbrc.2007.10.178. [DOI] [PubMed] [Google Scholar]
- Jeyakumar M, Carlson KE, Gunther JR, Katzenellenbogen JA. Exploration of dimensions of estrogen potency: parsing ligand binding and coactivator binding affinities. J Biol Chem. 2011;286:12971–12982. doi: 10.1074/jbc.M110.205112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauf C, Prevot V, Stefano GB, Mortreux G, Beauvillain JC, Croix D. Evidence for a spontaneous nitric oxide release from the rat median eminence: influence on gonadotropin-releasing hormone release. Endocrinology. 2001;142:2343–2350. doi: 10.1210/endo.142.6.8073. [DOI] [PubMed] [Google Scholar]
- Kraus WL, Montano MM, Katzenellenbogen BS. Cloning of the rat progesterone receptor gene 5′-region and identification of two functionally distinct promoters. Mol Endocrinol. 1993;7:1603–1616. doi: 10.1210/mend.7.12.8145766. [DOI] [PubMed] [Google Scholar]
- La Rosa P, Pesiri V, Leclercq G, Marino M, Acconcia F. Palmitoylation regulates 17beta-estradiol-induced estrogen receptor-alpha degradation and transcriptional activity. Mol Endocrinol. 2012;26:762–774. doi: 10.1210/me.2011-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannigan DA. Estrogen receptor phosphorylation. Steroids. 2003;68:1–9. doi: 10.1016/s0039-128x(02)00110-1. [DOI] [PubMed] [Google Scholar]
- Le Romancer M, Poulard C, Cohen P, Sentis S, Renoir JM, Corbo L. Cracking the estrogen receptor's posttranslational code in breast tumors. Endocr Rev. 2011;32:597–622. doi: 10.1210/er.2010-0016. [DOI] [PubMed] [Google Scholar]
- Mallat Z, Tedgui A. Cytokines as regulators of atherosclerosis in murine models. Curr Drug Targets. 2007;8:1264–1272. doi: 10.2174/138945007783220588. [DOI] [PubMed] [Google Scholar]
- McKenna NJ, O'Malley BW. Nuclear receptors, coregulators, ligands, and selective receptor modulators: making sense of the patchwork quilt. Ann N Y Acad Sci. 2001;949:3–5. doi: 10.1111/j.1749-6632.2001.tb03997.x. [DOI] [PubMed] [Google Scholar]
- Melamed M, Castano E, Notides AC, Sasson S. Molecular and kinetic basis for the mixed agonist/antagonist activity of estriol. Mol Endocrinol. 1997;11:1868–1878. doi: 10.1210/mend.11.12.0025. [DOI] [PubMed] [Google Scholar]
- Merot Y, Metivier R, Penot G, Manu D, Saligaut C, Gannon F, Pakdel F, Kah O, Flouriot G. The relative contribution exerted by AF-1 and AF-2 transactivation functions in estrogen receptor alpha transcriptional activity depends upon the differentiation stage of the cell. J Biol Chem. 2004;279:26184–26191. doi: 10.1074/jbc.M402148200. [DOI] [PubMed] [Google Scholar]
- Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, Gannon F. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- O'Malley BW, McGuire WL. Studies on the mechanism of estrogen-mediated tissue differentiation: regulation of nuclear transcription and induction of new RNA species. Proc Natl Acad Sci U S A. 1968;60:1527–1534. doi: 10.1073/pnas.60.4.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penot G, Le Peron C, Merot Y, Grimaud-Fanouillere E, Ferriere F, Boujrad N, Kah O, Saligaut C, Ducouret B, Metivier R, et al. The human estrogen receptor-alpha isoform hERalpha46 antagonizes the proliferative influence of hERalpha66 in MCF7 breast cancer cells. Endocrinology. 2005;146:5474–5484. doi: 10.1210/en.2005-0866. [DOI] [PubMed] [Google Scholar]
- Scheidt HA, Badeau RM, Huster D. Investigating the membrane orientation and transversal distribution of 17beta-estradiol in lipid membranes by solid-state NMR. Chem Phys Lipids. 2010;163:356–361. doi: 10.1016/j.chemphyslip.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Schnoes KK, Jaffe IZ, Iyer L, Dabreo A, Aronovitz M, Newfell B, Hansen U, Rosano G, Mendelsohn ME. Rapid recruitment of temporally distinct vascular gene sets by estrogen. Mol Endocrinol. 2008;22:2544–2556. doi: 10.1210/me.2008-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CL, O'Malley BW. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev. 2004;25:45–71. doi: 10.1210/er.2003-0023. [DOI] [PubMed] [Google Scholar]
- Soderberg O, Gullberg M, Jarvius M, Ridderstrale K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- Sun J, Nawaz Z, Slingerland JM. Long-range activation of GREB1 by estrogen receptor via three distal consensus estrogen-responsive elements in breast cancer cells. Mol Endocrinol. 2007;21:2651–2662. doi: 10.1210/me.2007-0082. [DOI] [PubMed] [Google Scholar]
- Toutain CE, Filipe C, Billon A, Fontaine C, Brouchet L, Guery JC, Gourdy P, Arnal JF, Lenfant F. Estrogen receptor alpha expression in both endothelium and hematopoietic cells is required for the accelerative effect of estradiol on reendothelialization. Arterioscler Thromb Vasc Biol. 2009;29:1543–1550. doi: 10.1161/ATVBAHA.109.192849. [DOI] [PubMed] [Google Scholar]
- Ueda K, Karas RH. Emerging evidence of the importance of rapid, non-nuclear estrogen receptor signaling in the cardiovascular system. Steroids. 2013;78:589–596. doi: 10.1016/j.steroids.2012.12.006. [DOI] [PubMed] [Google Scholar]
- Valera MC, Gratacap MP, Gourdy P, Lenfant F, Cabou C, Toutain CE, Marcellin M, Saint Laurent N, Sie P, Sixou M, et al. Chronic estradiol treatment reduces platelet responses and protects mice from thromboembolism through the hematopoietic estrogen receptor alpha. Blood. 2012;120:1703–1712. doi: 10.1182/blood-2012-01-405498. [DOI] [PubMed] [Google Scholar]
- Vasudevan N, Pfaff DW. Membrane-initiated actions of estrogens in neuroendocrinology: emerging principles. Endocr Rev. 2007;28:1–19. doi: 10.1210/er.2005-0021. [DOI] [PubMed] [Google Scholar]
- Visser M, Foidart JM, Coelingh Bennink HJ. In vitro effects of estetrol on receptor binding, drug targets and human liver cell metabolism. Climacteric. 2008;11(Suppl 1):64–68. doi: 10.1080/13697130802050340. [DOI] [PubMed] [Google Scholar]
- Visser M, Coelingh Bennink HJ. Clinical applications for estetrol. J Steroid Biochem Mol Biol. 2009;114:85–89. doi: 10.1016/j.jsbmb.2008.12.013. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Suzuki A, Kobayashi M, Takahashi E, Itamoto M, Lubahn DB, Handa H, Iguchi T. Analysis of temporal changes in the expression of estrogen-regulated genes in the uterus. J Mol Endocrinol. 2003;30:347–358. doi: 10.1677/jme.0.0300347. [DOI] [PubMed] [Google Scholar]
- Weber C, Zernecke A, Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol. 2008;8:802–815. doi: 10.1038/nri2415. [DOI] [PubMed] [Google Scholar]
- Wu Q, Chambliss K, Umetani M, Mineo C, Shaul PW. Non-nuclear estrogen receptor signaling in the endothelium. J Biol Chem. 2011;286:14737–14743. doi: 10.1074/jbc.R110.191791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Liljestrand HM. Partitioning of selected estrogenic compounds between synthetic membrane vesicles and water: effects of lipid components. Environ Sci Technol. 2004;38:1139–1147. doi: 10.1021/es034311w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.