

Abstract
MYC family oncoproteins (MYC, N-MYC and L-MYC) function as basic helix-loop-helix-leucine zipper (bHLH-Zip) transcription factors that are activated (i.e., overexpressed) in well over half of all human malignancies (Boxer & Dang, 2001; Beroukhim et al, 2010). In this issue of EMBO Molecular Medicine, Eilers and colleagues (Peter et al, 2014) describe a novel approach to disable MYC, whereby inhibition of the ubiquitin ligase HUWE1 stabilizes MIZ1 and leads to the selective repression of MYC-activated target genes.
See also: S Peter et al (December 2014)
Targeting MYC is a high priority for cancer therapeutics. This is no easy task, as transcription factors are notoriously difficult to inhibit with small molecules, and selectivity is also a hurdle, as MYC oncoproteins heterodimerize with MAX, a requisite and related bHLH-Zip protein that dimerizes with other bHLH-Zip proteins (Blackwood & Eisenman, 1991). Finally, achieving a suitable therapeutic window is also a concern, as MYC proteins are required for development and for the growth of normal cell types, where the MYC:MAX complex binds to and directly induces or represses the transcription of a large cast of target genes that harbor CAC/AGTG E-box elements, which in turn then provoke a proliferative state that amplifies global RNA production (Lin et al, 2012; Nie et al, 2012; Sabo et al, 2014; Walz et al, 2014). Regardless, hope for targeting MYC has recently come from the development of compounds that disrupt the MYC–MAX interaction (Hart et al, 2014), that provoke MYC destruction (Brockmann et al, 2013) or that block the transcription of MYC itself, by targeting the bromodomain protein BRD4 (Delmore et al, 2011). Eilers and colleagues (Peter et al, 2014) attempt novel approach to disable MYC, by inhibiting the ubiquitin ligase HUWE1 to stabilize MIZ1, thus leading to the selective repression of MYC-activated target genes.
Over the past 2 years, the concept that MYC oncoproteins function as selective transcription factors was challenged by studies, suggesting that MYC was a non-specific amplifier of all active genes (Lin et al, 2012; Nie et al, 2012). More recent findings from the Amati and Eilers laboratories have, however, shown that: (i) transcription regulation by MYC is indeed selective; (ii) transcriptional amplification reflects a secondary response to the proliferative state that is provoked by the expression of direct MYC targets; and (iii) the ratio of MYC and MIZ1 bound to each promoter controls if a given target gene is activated or repressed (Sabo et al, 2014; Walz et al, 2014). Peter et al (2014) now exploit this MYC-MIZ1 balancing act, where they show that inhibiting the HUWE1 ubiquitin ligase in colon cancer cells tips the response in favor of MIZ1.
Heretofore, HUWE1 was known to function as an E3 ligase that ubiquitylates and directs the destruction of N-MYC and MIZ1 (Zhao et al, 2009; Yang et al, 2010) and to add K63-linked ubiquitin chains to MYC without affecting its turnover (Adhikary et al, 2005). Based on the newly recognized importance of the MYC to MIZ1 ratio at a given promoter, Peter et al reasoned that HUWEI1 might be a target that could be exploited to override MYC transcriptional programs. Specifically, the authors hypothesized that blocking HUWE1 expression or function would stabilize MIZ1 and lead to binding of MIZ1 to MYC:MAX complexes at key target genes, to switch transcription into an off state and disable cancer cell growth.
The authors used an array of approaches to test this hypothesis. First, as predicted, knockdown of HUWE1 effectively blocked colorectal cancer cell growth ex vivo and, importantly, blocked the progression of tumor xenografts in vivo. These responses were not due to direct effects on MYC levels but were rather squarely pinned on MYC transactivation functions, where a large cast of genes normally induced by MYC was now repressed. Further, the effects of HUWE1 depletion on MYC-activated genes were not due to indirect effects on cell cycle arrest, as many of these targets are not cell cycle regulated, and they were also independent of possible effects on the p53 circuit. Most intriguingly, the effects of HUWE1 knockdown on MYC-activated genes were selective, as there were no effects of genes normally repressed by MYC.
These findings motivated an in vitro ubiquitin-based screen of a large library of compounds (> 840K), to identify small-molecule probes that selectively blocked the auto-ubiquitination of the HECT domain by HUWE1 in the presence of the E1 UBA1 and the E2 UbcH5b. Top hits from the screen were then counter-screened for activity against UBA1, UbcH5b and the ubiquitin ligase NEDD4, and the top two passing muster were shown to block the ubiquitination of validated targets of HUWE1 in cells, including that of the anti-apoptotic protein MCL1 and the checkpoint protein TopBP1. Notably, the genetic studies provided suggest that the top two hits identified, which have rather modest potency (IC50 of 0.9–3 μM), do indeed target HUWE1. Most importantly, treatment of colorectal cancer cells with these agents, but not treatment of normal colonic epithelial cells or embryonic stem cells, triggered cell growth arrest and, again, blocked the expression of target genes that are activated by MYC, without affecting those that are repressed by MYC. Finally, the HUWE1 inhibitors had little-to-no effects on MYC target genes expression in cells already depleted of HUWE1.
Proof of the relevance to the HUWE1-to-MIZ1 circuit came from a series of convincing studies that established that: (i) inhibition or knockdown of HUWE1 induced stabilization of MIZ1 and triggered MIZ1 binding on target genes normally activated by MYC; (ii) inhibition of HUWE1 has no effect on the formation of MYC:MAX complexes nor upon the expression of MXD proteins that also dimerize with MAX; and (iii) knockdown of MIZ1 reversed most of the effects of HUWE1 inhibition or silencing.
Collectively, these findings suggest that MYC can selectively be targeted in cancer by disabling the HUWE1 ubiquitin ligase that normally controls MIZ1 protein levels (Fig1). In tumors where there is a preponderance of MYC oncoproteins, the balance is in favor of transcription activating MYC:MAX complexes, which induce the expression of their direct targets that then in turn provoke a hyperproliferative state that amplifies transcription. Inhibition of HUWE1 and elevated levels of MIZ1 then restores this balance, as MIZ1 binds to MYC:MAX complexes to form ternary MIZ1:MYC:MAX complexes that repress genes that are activated by MYC, thus abolishing the hyperproliferative response (Fig1).
Figure 1. MIZ1–MYC equilibrium controls cell fate.
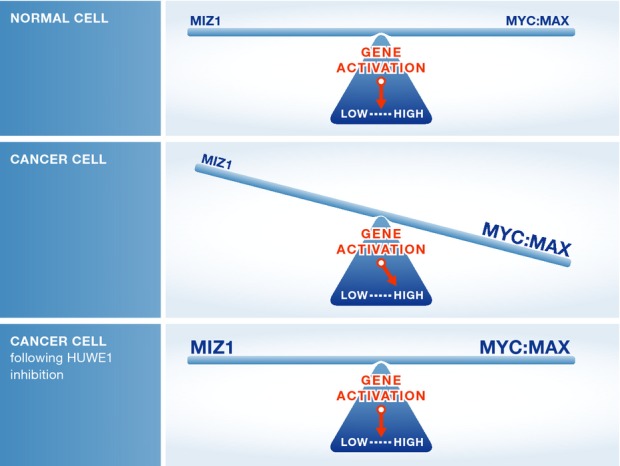
In normal cells, HUWE1-directed ubiquitylation of MIZ1 controls its levels to balance the control of MYC transcription targets. In cancer, MYC oncoproteins are overexpressed, which tips the balance to activating MYC:MAX complexes that activate direct targets, which in turn lead to a hyperproliferative state that includes an amplification of transcription (Lin et al, 2012; Nie et al, 2012; Sabo et al, 2014; Walz et al, 2014). Inhibition or silencing of HUWE1 in cancer cells re-establishes a proper equilibrium, by provoking increases in the levels of MIZ1 that forms repressive MIZ1:MYC:MAX ternary complexes on growth-associated target genes that are activated by MYC.
The small molecules identified thus far are, however, only the first steps toward targeting this circuit in the oncology clinic. Indeed, the current tool compounds lack the potency and proper drug-like properties needed for in vivo testing of safety and efficacy. Moreover, once developed, such HUWE1-targeting agents may have to be used in combination with other drugs, as knockdown of HUWE1 alone is not sufficient to induce tumor regression. Finally, other important studies need to be performed before attempting to translate these findings and include those confirming the role of this circuit in additional MYC-driven malignancies and those that interrogate possible mechanisms of resistance to such agents, which, for example, could include silencing of MIZ1 or gain-of-function somatic mutations in HUWE1 that block the function of these small molecules. Nonetheless, the facts that HUWE1 is synthetically lethal for MYC-expressing tumor cells and that this is a tractable enzyme amenable to therapeutics raises hope that drugs that target this ubiquitin ligase can ultimately be added to our armament of agents to treat the broad spectrum of aggressive malignancies that have MYC involvement.
References
- Adhikary S, Marinoni F, Hock A, Hulleman E, Popov N, Beier R, Bernard S, Quarto M, Capra M, Goettig S, et al. The ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell. 2005;123:409–421. doi: 10.1016/j.cell.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwood EM, Eisenman RN. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science. 1991;251:1211–1217. doi: 10.1126/science.2006410. [DOI] [PubMed] [Google Scholar]
- Boxer LM, Dang CV. Translocations involving c-myc and c-myc function. Oncogene. 2001;20:5595–5610. doi: 10.1038/sj.onc.1204595. [DOI] [PubMed] [Google Scholar]
- Brockmann M, Poon E, Berry T, Carstensen A, Deubzer HE, Rycak L, Jamin Y, Thway K, Robinson SP, Roels F, et al. Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell. 2013;24:75–89. doi: 10.1016/j.ccr.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart JR, Garner AL, Yu J, Ito Y, Sun M, Ueno L, Rhee JK, Baksh MM, Stefan E, Hartl M, et al. Inhibitor of MYC identified in a Krohnke pyridine library. Proc Natl Acad Sci USA. 2014;111:12556–12561. doi: 10.1073/pnas.1319488111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA. Transcriptional amplification in tumor cells with elevated c-Myc. Cell. 2012;151:56–67. doi: 10.1016/j.cell.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell. 2012;151:68–79. doi: 10.1016/j.cell.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter S, Bultinck J, Myant K, Jaenicke LA, Walz S, Muller J, Gmachl M, Treu M, Boehmelt G, Ade CP, et al. Tumor cell-specific inhibition of MYC function using small molecule inhibitors of the HUWE1 ubiquitin ligase. EMBO Mol Med. 2014;6:1525–1541. doi: 10.15252/emmm.201403927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, Morelli MJ, Bora P, Doni M, Verrecchia A, et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature. 2014;511:488–492. doi: 10.1038/nature13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, Rycak L, Dumay-Odelot H, Karim S, Bartkuhn M, et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature. 2014;511:483–487. doi: 10.1038/nature13473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Do H, Tian X, Zhang C, Liu X, Dada LA, Sznajder JI, Liu J. E3 ubiquitin ligase Mule ubiquitinates Miz1 and is required for TNFalpha-induced JNK activation. Proc Natl Acad Sci USA. 2010;107:13444–13449. doi: 10.1073/pnas.0913690107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, D'Arca D, Lim WK, Brahmachary M, Carro MS, Ludwig T, Cardo CC, Guillemot F, Aldape K, Califano A, et al. The N-Myc-DLL3 cascade is suppressed by the ubiquitin ligase Huwe1 to inhibit proliferation and promote neurogenesis in the developing brain. Dev Cell. 2009;17:210–221. doi: 10.1016/j.devcel.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]