

Abstract
The majority of ovarian cancers over-express the estrogen receptor (ERα) and grow in response to estrogens. We previously demonstrated that ER induction of the chemokine CXCL12 (stromal cell-derived facor-1) is required for estradiol (E2)-stimulated proliferation of human ovarian carcinoma cells. In the current study we report that known ‘endocrine disrupting chemicals’ (EDCs) display mitogenic activities in ovarian cancer cells via their ability to activate the ER and upregulate CXCL12 expression. Notably, the EDCs genistein, bisphenol A and HPTE stimulated both cell proliferation and induction of CXCL12 mRNA and protein in a manner comparable to estradiol. The effects were completely attenuated by the ER antagonist ICI 182,780, revealing that observed activities of these agents were receptor-mediated. In cell proliferation assays the mitogenic effects of estradiol and EDCs were obviated by siRNAs targeting CXCL12 and restored upon addition of exogenous CXCL12. Furthermore, an inhibitor to the CXCL12 receptor CXCR4 completely attenuated growth-stimulatory effects of E2 and EDCs. These studies highlight a potential role of EDCs possessing estrogenic activities in the etiology of ovarian cancer. Moreover, they suggest that the ER-CXCL12-CXCR4 signaling axis may represent a promising target for development of therapeutics for ER+ ovarian cancers.
Keywords: endocrine disrupting chemicals (EDCs), ovarian cancer, estrogen receptor, cell proliferation, CXCL12, CXCR4
INTRODUCTION
The steroid hormone 17β-estradiol (E2) is a key regulator of growth, differentiation, and function in a wide array of target tissues, including the male and female reproductive tracts, mammary gland, and skeletal and cardiovascular systems (1). In addition to the physiological actions of estrogens, inappropriate responses to this hormone have been shown to underlie the pathology of most breast, ovarian and uterine cancers (2, 3). The predominant biological effects of E2 are mediated through two intracellular receptors, ERα and ERβ, members of the nuclear receptor family of ligand-inducible transcription factors (4, 5). In the absence of ligand the ERs reside in the nuclei of target cells in an inactive form associated with an inhibitory heat shock protein (hsp) complex. Upon ligand binding the receptors undergo an activating conformational change which is incompatible with hsp interactions and which facilitates receptor dimerization. The receptor dimer may interact directly with target gene promoters or indirectly through interactions with other DNA-bound proteins (4, 6, 7). It is also now clear that estrogens may display rapid actions through membrane-bound ERs (8) or GPR30 (9); however, the contributions of nongenomic signaling to estrogen physiology remains to be fully understood.
In addition to endogenous estrogens, the ERs are activated by an array of natural and man-made chemicals that bind the receptors in target tissues and may mimic or block the effects of physiological hormones (10). In the past decade, a compelling body of evidence linking exposure of environmental endocrine disrupting chemicals (EDCs) to the etiology of cancer has emerged (11–13). Notably, EDCs such as the isoflavone genistein (found in soy products) has been linked to proliferative effects in ER-positive cancer cells at concentrations consistent with those detected in humans ingesting soy-rich beverages (14, 15). Paradoxically, growth inhibitory activities of genistein have also been noted due to effects on tyrosine kinase activity that manifest at higher (µM-mM) concentrations (16). Much recent attention has focused on impact of exposure to the plasticizer bisphenol A (BPA), which is used widely in consumer products, including food and water containers, soda cans, baby bottles, and medical tubing. BPA has been liberated into food, beverages and the environments for decades and accumulates to substantial levels humans; (~ 0.5–40 nM) in adult and fetal serum (17). Notably, in animals early-life exposure to BPA causes increased susceptibility to mammary and prostate tumorigenesis (18, 19). While epidemiological data awaits, in situ studies showed BPA induces neoplastic transformation in human breast epithelial cells (20). Another EDC of concern with regards to reproductive malignancies is the methoxychlor metabolite, 2,2-bis(p-hydroxyphenyl)-1,1,1-trichloroethane (HPTE). Notably, this agent mimics the proliferative effects of E2 in the uterus by augmenting ERα-IGF-I signaling (21) and displays an extensive uterine genomic profile that correlates with that of E2 (22).
The majority of human breast and ovarian tumors overexpress ERα and grow in response to estrogens, making this signaling system sensitive to the effects of EDCs possessing estrogenic activities. Surprisingly, although several decades of research in this regard, it remains to be determined how this receptor is involved in cell proliferation when activated by either native hormones or EDCs. Confusing this issue even further is the demonstration that in addition to transcriptional regulation both estrogens and EDCs exhibit activities that occur in a non-genomic manner (23). The ability to link proliferation to specific gene changes has also been difficult as several groups have demonstrated that there are over 200 primary responses to estrogens in breast cancer cells treated with estradiol and over 700 binding sites for the ER in the human genome (24–26). The inability to satisfactorily annotate the gene expression patterns identified has necessitated a candidate gene approach in defining the key genes required for proliferation. It was in this manner that we recently identified stromal cell derived factor-1 (CXCL12) as a key target of estrogens in ER-positive breast and ovarian cells (27). Specifically, CXCL12 was shown to be a primary target of ER and that upon estradiol treatment both the CXCL12 mRNA and secretion of its corresponding chemokine was increased. Neutralizing antibodies to CXCL12 blocked the mitogenic actions of estradiol whereas activation of the CXCL12 receptor CXCR4 obviated the need for estradiol supplementation. The role of CXCL12 as an ER target and mitogen in human breast and ovarian cancer cells has since been described by others (28–32). Importantly, these collective data have defined at least one genomic response and signaling pathway that is required for estrogen-stimulated cell proliferation. It is likely, however, that additional genomic and non-genomic actions of estrogens may also be required for maximal proliferative responses.
The discovery of the ER-CXCL12-CXCR4 axis, coupled with the known estrogenic effects of some EDCs prompted a closer look at the relationship between the two in cancer cell growth. The current study examined the ability of EDCs genistein, BPA and HPTE to display mitogenic effects through the ER in BG-1 ovarian carcinoma cells. BG-1 cells express physiologically relevant levels of ERα and progesterone receptors, but not ERβ, and display proliferative effects in response to estrogenic stimuli. Furthermore, BG-1 is an established model for ERα+ ovarian epithelial cancer (27, 33, 34) and contains all functional components of the ER/CXCL12/CXCR4 regulatory pathway (27). Thus using BG-1 cells as a model, the objectives of this study were to (I) evaluate the ability of EDCs to display mitogenic effects in ovarian cancer cells, (II) characterize the ability of EDCs to activate classical ER gene expression, and (III) determine whether EDCs may alter ovarian cancer cell biology by activation and/or perturbation of the ER-CXCL12-CXCR4 signaling axis.
MATERIALS AND METHODS
Biochemicals
PCR reagents were obtained from BIO-RAD (Hercules, CA). 17β-estradiol, genistein and bisphenol A were purchased from Sigma (St. Louis, MO). 2,2-bis(p-hydroxyphenyl)-1,1,1-trichloroethane (HPTE) was a gift from Dr. Wendy Jefferson, NIEHS/NIH. ICI 182,780 was a gift from Dr. A. Wakeling, AstraZeneca Pharmaceuticals, Cheshire, UK. AMD3100 (Plerixafor) was purchased from Cayman Chemical Company (Ann Arbor, Michigan). Purified human recombinant CXCL12 and the FluoReporter Blue Fluorometric dsDNA Quantitation Kit were obtained from Invitrogen (Carlsbad, CA). The CXCL12 ELISA kit was obtained from R&D Systems (Minneapolis, MN). siRNA oligos and DharmaFECT-1 Transfection Reagent were purchased from Dharmacon (Lafayette, CO). qPCR oligos were obtained from Invitrogen.
Mammalian Cell Culture and Ligand Treatments
BG-1 (human epithelial ovarian cancer) cells were maintained in DMEM F12 medium supplemented with 10% fetal calf serum (FCS), 2 mM glutamine, 0.1 mM nonessential amino acids, and 1 mM sodium pyruvate (Invitrogen). Cell were grown in a 37°C incubator with 5% CO2. For RNA analysis, cells were plated at 60% confluency in 6-well culture dishes in 2 ml phenol red-free DMEM F12 containing 8% charcoal-stripped FCS (HyClone Laboratories, Inc., Logan, UT) for 24h prior to treatment. Media was then replaced and supplemented with ethanol (vehicle), 17β-estradiol, genistein, bisphenol A or HPTE. Note figure legends for details on concentrations of agents used in specific experiments. For some studies 1 µM ICI 182,780 (ER antagonist) or 1µg/ml AMD3100 (CXCR4 antagonist) was coadministered with vehicle or treatments. For cell proliferation assays, cells were induced with vehicle, 17β-estradiol or other agents in the absence or presence of 100 ng/ml purified human CXCL12 (SDF-1β).
RNA Isolation and Quantitative PCR
RNA was isolated from cultured BG-1 cells using the RNeasy kit (Qiagen, Valencia, CA). One µg of RNA was reverse transcribed using the BioRad iScript cDNA synthesis kit. The ABI PRISM® 7900HT Real Time PCR System was used to amplify and quantitate levels of target gene cDNA. Quantitative PCR (qPCR) reactions were performed using 0.1 µl of cDNA, 10 µM gene-specific primers, and 2× SYBR Green Mastermix (Applied Biosystems). Data are the mean +/−STDEV of three biological replicates performed in triplicate. All experiments were repeated a minimum of three times. The following primers were used: CXCL12,
5’-GTGGTCGTGCTGGTCCTC-3’ (for) and 5’-GATGCTTGACGTTGGCTCTG-3’ (rev), CXCR4, 5’-TACACCGAGGAAATGGGCTCA-3’ (for) and
5’-AGATGATGGAGTAGATGGTGGG-3’(rev), c-Myc,
5’-CCGCTTCTCTGAAAGGCTCTC-3’(for) and 5’-CTGCTGCTGCTGCTGGTAG-3’(rev), WISP2, 5’-TGAGAGGCACACCGAAGAC-3’ (for) and
5’-ACAGCCATCCAGCACCAG-3’ (rev), and
36B4 5’-GGACATGTTGCTGGCCAATAA-3’ (for) and
5’-GGGCCCGAGACCAGTGTT-3’ (rev).
Mammalian Cell siRNA Transfection
Validated small interfering RNAs (siRNAs) directed against a control non-targeting sequence or 3 different regions of human CXCL12 were obtained from Dharmacon (Catalog numbers: D-001810-01, J-007873-06, J-007873-07, J-007873-08). For transfections, BG-1 cells were plated in 6-well plates for 24 h. Prior to transfection, media was replaced with phenol red-free DMEM containing 8% charcoal/dextran (C/D) filtered serum (Hyclone). Cells were transfected 48 h with 50 nmol/L siRNA final concentration using DharmaFECT-1 transfection reagent according to the manufacturer’s protocol. CXCL12 expression in cells transfected with siRNAs was analyzed 48 h post-transfection using the reagents and methods described above for qPCR. CXCL12 protein in cells transfected with siRNAs was analyzed 48 h post-transfection using the methods below (see ELISA).
Cell Proliferation Assays
Proliferation assays were optimized for BG-1 cells as described previously (27). For standard assays, BG-1 cells were seeded and adhered in 96-well plates at 2.5 × 104 cells/well. Cells were incubated in the absence or presence of hormones, purified CXCL12, and antibodies for 96 h in phenol red-free DMEM F12 containing 8% charcoal-stripped FCS (100 µl/well). After 96 h, cell proliferation was measured using the FluoReporter® Blue Fluorometric dsDNA Quantitation Kit according to the manufacturers’ protocols (Invitrogen). All values were corrected for fluorescence values read from blank controls containing media with no cells. Sextuplicate biological replicates were used for each treatment condition in each experiment, and data are the mean +/−STDEV of the 6 replicates. All experiments were repeated a minimum of three independent times.
ELISA
BG-1 cell media were analyzed for secreted CXCL12 protein as follows: BG-1 cells were seeded in 96-well plates in phenol-red free DMEMF12 supplemented with 8% charcoal/dextran filtered FBS. After 24 h cells were treated with vehicle or ligands. After 24 h and 48 h, media were collected from individual wells and flash frozen until analysis. The CXCL12 ELISA was performed in a 96-well format on triplicate samples of spent media, as according to the manufacturer’s protocol (R&D Systems). Data are the mean +/− STDEV of two replicates.
Statistical Analysis
All experiments in this study were repeated a minimum of 3 independent times. Each Figure shows a representative experiment. For proliferation assays, data points represent the mean ± STDEV for of 6 biological replicates in a single experiment; for ELISAs, data points represent the mean ± STDEV of 2 biological replicates in a single experiment; for RT-PCR data points represent the mean ± STDEV of 3 replicates. In all experiments statistical comparisons were made between control and treatments using a two-sample t-test.
RESULTS
EDCs Promote the Growth of Ovarian Cancer Cells in an ER-Dependent Manner
These studies were initiated by examining the ability of EDCs to function as estrogens in regulating proliferation of human ovarian cancer cells. BG-1 cells were plated in 96-dishes, treated with various ligands for 96 h and assayed for changes in cell growth using a quantitative fluorescent assay (see methods). Estradiol was a potent mitogen in BG-1 cells (27), reaching maximal efficacy at physically relevant doses of 1–10 nM (Fig. 1). Not surprisingly EDCs genistein, BPA and HPTE were less potent in the assay, likely reflecting their lower binding affinities for the ER. However, at 100 nM genistein and BPA were almost as efficacious as estradiol, promoting a 250% increase in cell growth; likewise, the lower affinity ligand HPTE reached maximal efficacy at 1 µM. Interestingly, at µM doses genistein was less effective at promoting proliferation, consistent with previous reports that the growth-inhibitory activities tyrosine kinase of the compound are manifest in the µM range (16). Together these data indicate that EDCs can effectively mimic the effects of estradiol in promoting the proliferation of ovarian cancer cells.
Fig. 1. Estradiol and EDCs Promote the Growth of Ovarian Cancer Cells in an ER-Dependent Manner.
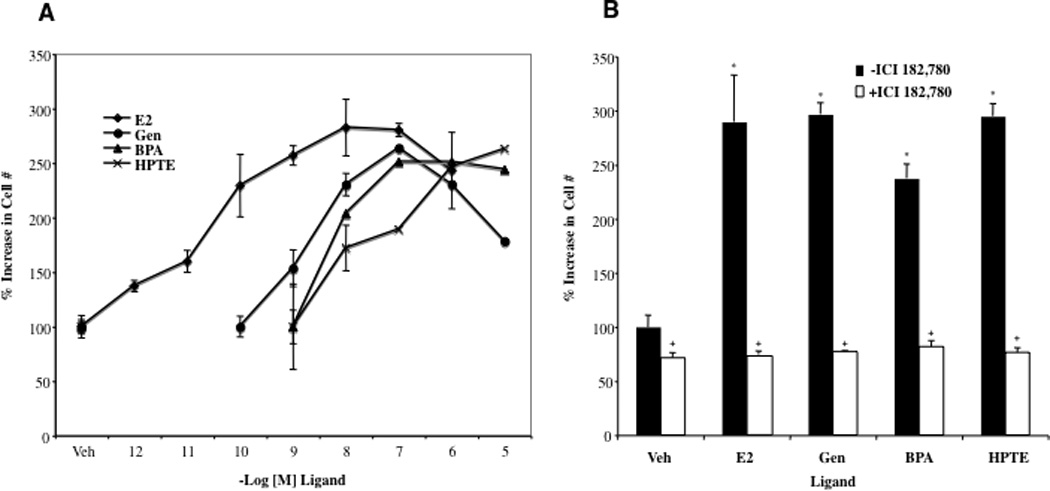
A, BG-1 cells were seeded in 96-well plates and treated with vehicle (Veh) (ethanol), 1 pM–1 µM 17β-estradiol (E2), 10 pM–10 µM Genistein (Gen), 1 nM–10 µM Bisphenol A (BPA), or 1 nM –10 µM HPTE for 92 h and then assayed for cell growth. Graphical data are represented as % increase in cell number over vehicle (set at 100%). Data points represent the average of 6 samples +/− STDEV for each condition in a representative experiment. Proliferation assays were optimized for BG-1 cells as described previously (27). B, BG-1 cells were seeded in 96-well plates and treated with Veh, 1 nM E2, 100 nM Gen, 100 nM BPA, or 1 µM HPTE as above in the absence or presence of 1 µM ICI 182,780 for 92 h. Cell proliferation (sextuplicate samples per treatment) was assayed as described above. *p < 0.05 for comparison between Veh and each treatment. + p < 0.05 for comparisons between each treatment alone and each treatment + ICI 182,780.
Previous studies have shown that the EDCs utilized here, genistein, BPA and HPTE, are capable of altering ER signaling in a variety of tissues including the breast, uterus, prostate, and heart (14, 18,19, 21, 22, 35). The impact of EDCs on ovarian function is less well characterized, however, and it was not clear whether the observed mitogenic effects in ovarian cells were due to ER-dependent mechanisms. To examine this more closely, EDCs were administered to BG-1 cells at the dose which yielded maximal efficacy in Fig. 1A (100 nM for genistein and BPA; 1 µM for HPTE). To assess ERα-dependence, additional experiments were carried out with cells coadministered EDCs together with the pure ER antagonist ICI 182,780 (Fig. 1B). Strikingly, ICI 182,780 completely attenuated the mitogenic effects of estradiol and all EDCs, and consistent with the known inverse agonist activities, decreased basal levels of proliferation to approximately 75%. These results indicate the proliferative effects of EDCs in ovarian cancer cells are mediated through ERα.
EDCs Enhance Expression of ER-Regulated Genes in Ovarian Cancer Cells
Traditionally the biological actions of estrogens, antiestrogens and EDCs have been ascribed to their ability to regulate ER transcriptional activity and impact subsequent gene expression. There are various reports that EDCs may manifest their estrogenic effects through both genomic and nongenomic signaling pathways (10, 22, 23). Previously, using microarray experiments in BG-1 cells a series of estrogen-regulated genes were identified that serve as markers for classical pathways of ER action in ovarian cancer cells (27). In the current study quantitative real-time PCR (qPCR) was used to examine the effects of EDCs on the expression of several genes that exhibited differential expression profiles in the microarrays. Dose response studies determined the concentration at which ligand displayed maximal efficacy for the selected genes (Supplemental Fig. 1). Figure 2 shows the qPCR results from BG-1 cells administered estradiol or EDCs at doses yielding maximal responses (see Supplemental data). Strikingly, EDCs were as effective in inducing expression of WISP2 and cMyc as estradiol, and coadministration of the ER antagonist ICI 182,780 diminished all responses. Thus, it appears that EDCs are capable of activating both proliferation and gene expression in ovarian cancer cells through direct actions on ERα.
Fig. 2. EDCs Enhance Expression of Classical ER-Regulated Genes in Ovarian Cancer Cells.
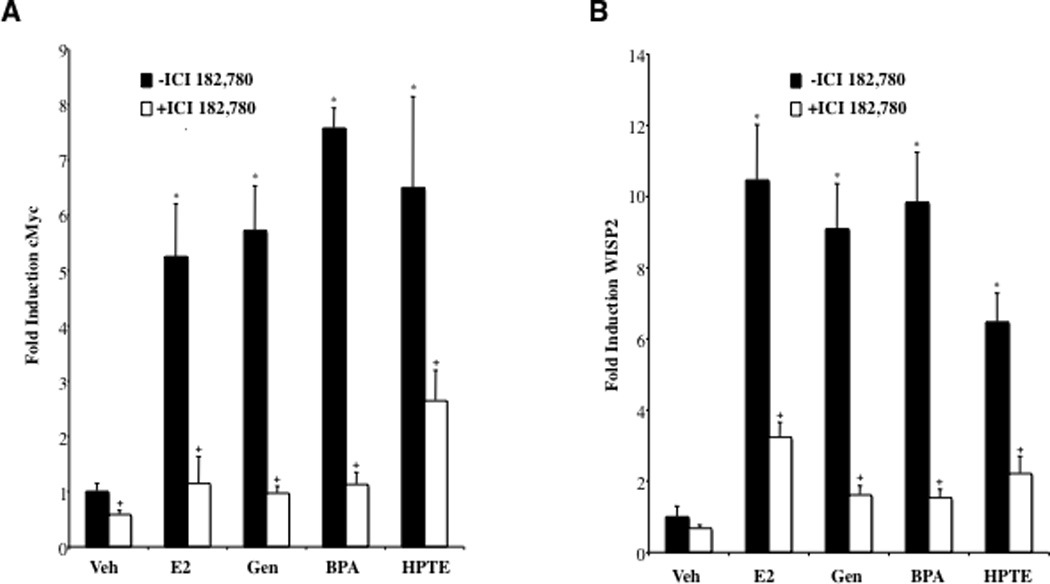
Changes in A, c-Myc and B, WISP2 RNA levels in BG-1 cells treated with EDCs were measured by real-time PCR. BG-1 cells were grown for 24 h in phenol-red free DMEM + 10% stripped FCS and then treated for an additional 24 h with vehicle (Veh), 1 nM Estradiol (E2), 100 nM Genistein (Gen), 100 nM Bisphenol A (BPA), or 1 µM HPTE in the absence or presence of 1µM ICI 182,780. Dose-response analysis revealed that these doses yielded maximal gene induction (see Suppl. Figure 1). Total RNA was harvested, and cDNA was prepared and used as a template for gene expression analysis. All values were normalized to a 36B4 control. Graphical data are represented as fold induction over vehicle (set at 1). Data points represent the average of triplicate amplification reactions +/− STDEV for each condition in a representative experiment. *p < 0.05 for comparison between Veh and each treatment. + p < 0.05 for comparisons between each treatment alone and each treatment + ICI 182,780.
With the abundance of ER regulated genes that have been annotated in cancer cells, it has been a challenge to identify those that mediate the growth-promoting effects of estrogens. Previously we characterized one such gene in demonstrating that the mitogenic effects of estradiol in breast and ovarian cancer cells require ERα-mediated induction of the growth-stimulatory chemokine CXCL12. Thus, the next series of studies addressed the question of whether like estradiol the proliferative effects of EDCs in human ovarian cancer cells are mediated through the ER-CXCL12 signaling axis.
EDCs Enhance Expression and Production of CXCL12 in Ovarian Cancer Cells
CXCL12 expression in response to estradiol and EDCs was next assessed. BG-1 cells were treated for 24 h with various doses of ligands, and RNA was harvested and processed for use in a qPCR analysis of CXCL12 expression (Fig. 3A). While estradiol was the most potent agent in the assay, genistein and BPA displayed similar efficacy to the hormone at 100 nM, the dose that yielded maximal responses to these agents in the proliferation analysis (Fig. 1). Genistein showed a decreased induction of CXCL12 at higher doses, consistent with the decreased proliferation observed in the µM range. HPTE, while a moderately weaker agonist, produced a 6-fold increase in CXCL12 levels at 1 µM, also a dose that yielded maximal cell proliferation (Fig 1). Coadministation of ICI 182,780 attenuated the responses, demonstrating that observed effects were ER-dependent.
Fig. 3. EDCs Enhance Expression and Production of CXCL12 in Ovarian Cancer Cells.
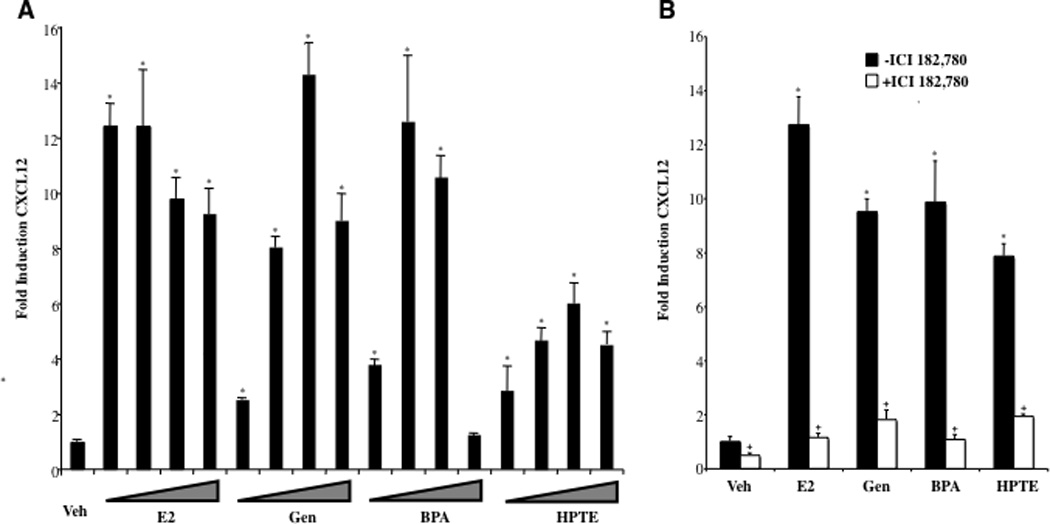
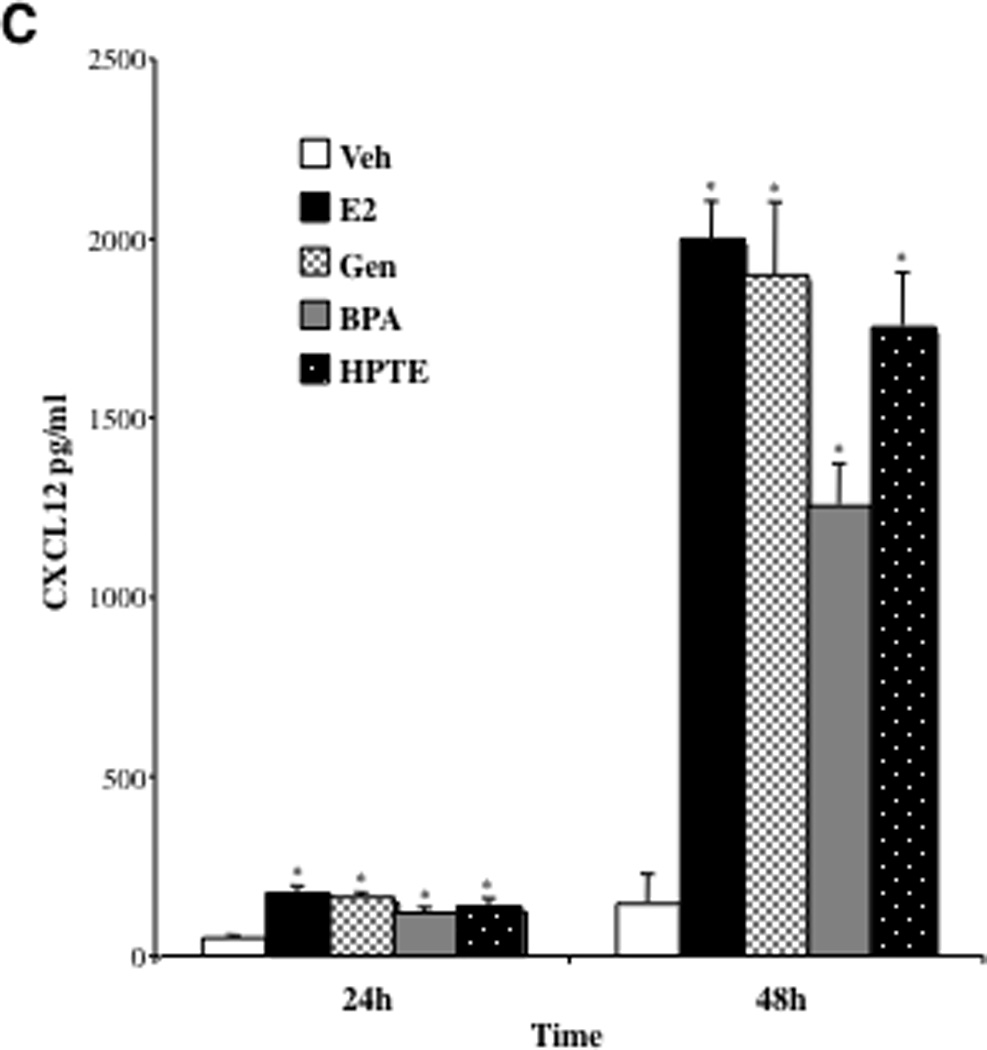
A, Changes in CXCL12 RNA levels in BG-1 cells treated with EDCs were measured by real-time PCR. BG-1 cells were grown for 24 h in phenol-red free DMEM + 10% stripped FCS and then treated for an additional 24 h with vehicle (Veh), or increasing concentrations of ligands; 100 pmol, 1 nM, 10 nM and 100 nM Estradiol (E2), 1 nM, 10 nM, 100 nM and 1 µM Genistein (Gen), 1 nM, 10 nM, 100 nM and 1 µM Bisphenol A (BPA), or 10 nM, 100 nM, 1 µM and 10 µM HPTE. Total RNA was harvested, and cDNA was prepared and used as a template for gene expression analysis. All values were normalized to a 36B4 control. Graphical data are represented as fold induction over vehicle (set at 1). Data points represent the average of triplicate amplification reactions for each condition in a representative experiment +/− STDEV. B, BG-1 cells were grown for 24 h in phenol-red free DMEM + 10% stripped FCS and then treated for an additional 24 h with Veh, 1 nM E2, 100 nM Gen, 100 nM BPA, or 1 µM HPTE in the absence or presence of 1µM ICI 182,780. Gene expression analysis was carried out as above. C, BG-1 cells were seeded in 96-well plates and treated with Veh, 1 nM E2, 100 nM Gen, 100 nM BPA, or 1 µM HPTE for 24 or 48 h. Spent media was removed from individual wells and assayed for CXCL12 protein by ELISA. Data points represent the average of duplicate assays for each condition. (A–C) *p < 0.05 for comparison between Veh and each treatment. + p < 0.05 for comparisons between each treatment alone and each treatment + ICI 182,780.
We next probed the biological significance of CXCL12 induction by the various ER ligands in ovarian cancer cells. CXCL12 protein is functional after it is secreted from cells, an event that enables it to signal through its cell surface receptor CXCR4 and trigger downstream growth-stimulatory pathways (36). Thus, the effect of estradiol and EDCs on extracellular CXCL12 production was next assessed. For these studies BG-1 cells were plated in 6-well plates and treated with various ligands for 24 or 48 h. Spent media was removed from individual cells and CXCL12 protein content was quantitated by ELISA. A 3–4 fold increase in CXCL12 was apparent with all treatments after 24 h (Fig. 3C). After 48 h, a 10-fold increase was observed for E2, Gen and HPTE (from 200 pg/ml in Veh treated cells compared with 2000 pg/ml in treated cells) and 7-fold increase with BPA. Previously it was shown that addition of 500, 1000 and 2000 pg/ml exogenous CXCL12 caused maximal growth of cultured BG-1 cells (27). Thus, EDCs, like estradiol, appear to be capable of inducing production of sufficient extracellular levels of CXCL12 for stimulating growth of ovarian cancer cells.
siRNA Knockdown of CXCL12 Attenuates the Proliferative Effects of Estradiol and EDCs in Ovarian Cancer Cells
The next objective was to determine whether if like estradiol, induction of CXCL12 is important for the proliferative actions of the various EDC ligands in the study. The growth of BG-1 cells was evaluated following treatment with either of three siRNA duplexes directed against independent regions of CXCL12. A reduction in CXCL12 mRNA was confirmed using qPCR analysis. Importantly each siRNA was effective in completely suppressing CXCL12 even in the presence of respective ligands (Fig. 4A). Likewise, quantitation of CXCL12 protein levels in the spent media of transfected cells revealed that siRNAs suppressed extracellular CXCL12 levels to that of the Veh treated control and in some cases below basal levels (Fig. 4B).
Fig. 4. siRNA-Mediated Knockdown of CXCL12 Attenuates the Proliferative Effects of Estradiol and EDCs.
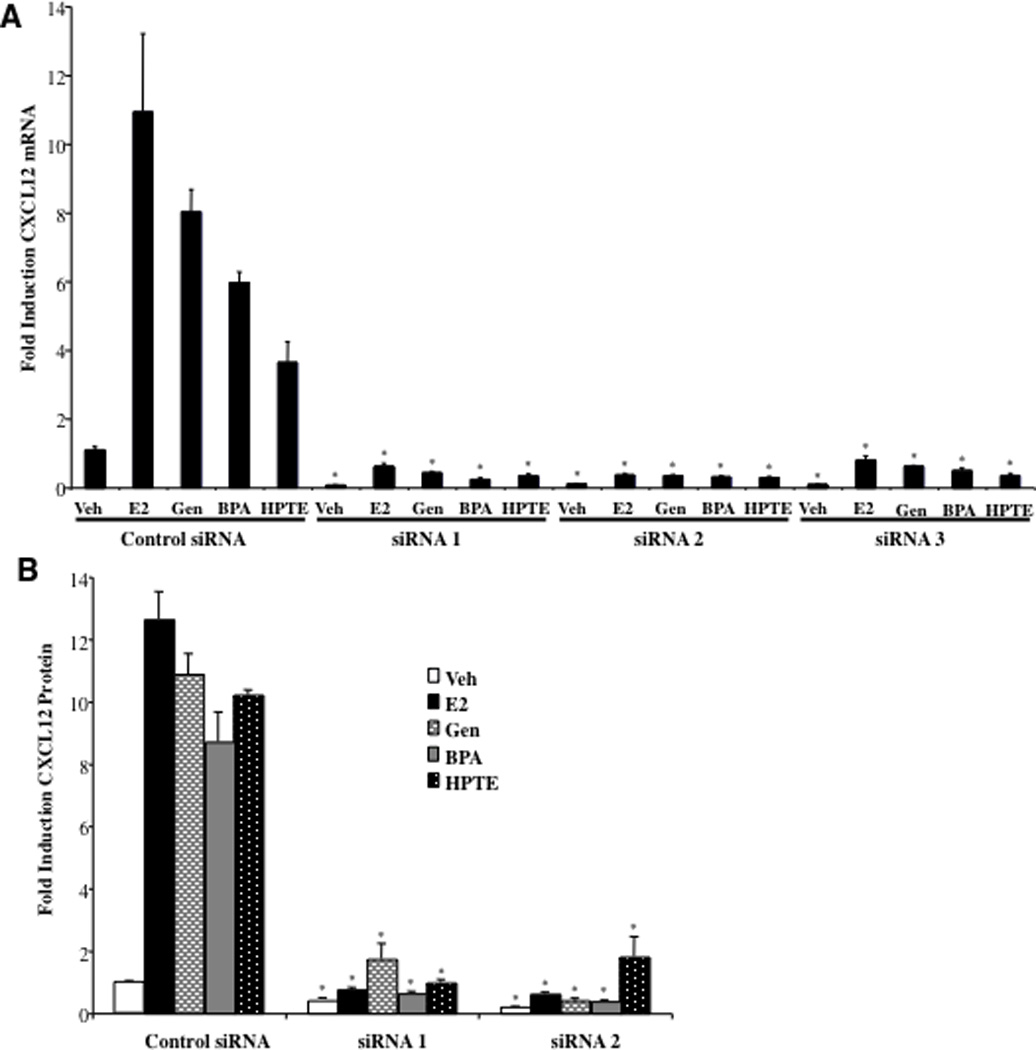
Real-time PCR analysis of endogenous CXCL12 expression. A, BG-1 cells were transfected with either a nontargeting siRNA duplex (Control siRNA) or one of three independent CXCL12 siRNAs (siRNA 1–3). After 24 h cells were treated with vehicle (Veh), 1 nM Estradiol (E2), 100 nM Genistein (Gen), 100 nM Bisphenol A (BPA), or 1 µM HPTE for an additional 24 h. 48 h post-transfection cells were harvested for total RNA, which was used for real-time PCR. Graphical data are represented as fold induction over vehicle (set at 1). Data points represent the average of triplicate amplification reactions for each condition in a representative experiment. B, CXCL12 protein production in siRNA-transfected cells. BG-1 cells with transfected with either a Control siRNA, or siRNA1 or siRNA2 targeting CXCL12. After 24 h cells were counted, seeded in 96-well plates at equal densities per siRNA treatment, and administered ligands at the concentrations above for an additional 48 h. Spent media was removed from individual wells and assayed for CXCL12 protein by ELISA. Data points represent the average of duplicate assays for each condition in a representative experiment. C, Cells with transfected with either a Control siRNA or one of three independent siRNAs targeting CXCL12. After 24 h cells were trypsinized, seeded in 96-well plates at equal densities, and administered ligands as above for 92 h and then assayed for cell growth. Graphical data are represented as % increase in cell number over vehicle (set at 100%). Data points represent the average of 6 samples for each condition in a representative experiment. For (A–C) *p < 0.05 for comparison between Control siRNA and CXCL12 siRNA 1, Control siRNA and CXCL12 siRNA 2, and Control siRNA and CXCL12 siRNA 3 for each separate ligand (Veh, E2, Gen, BPA or HPTE).
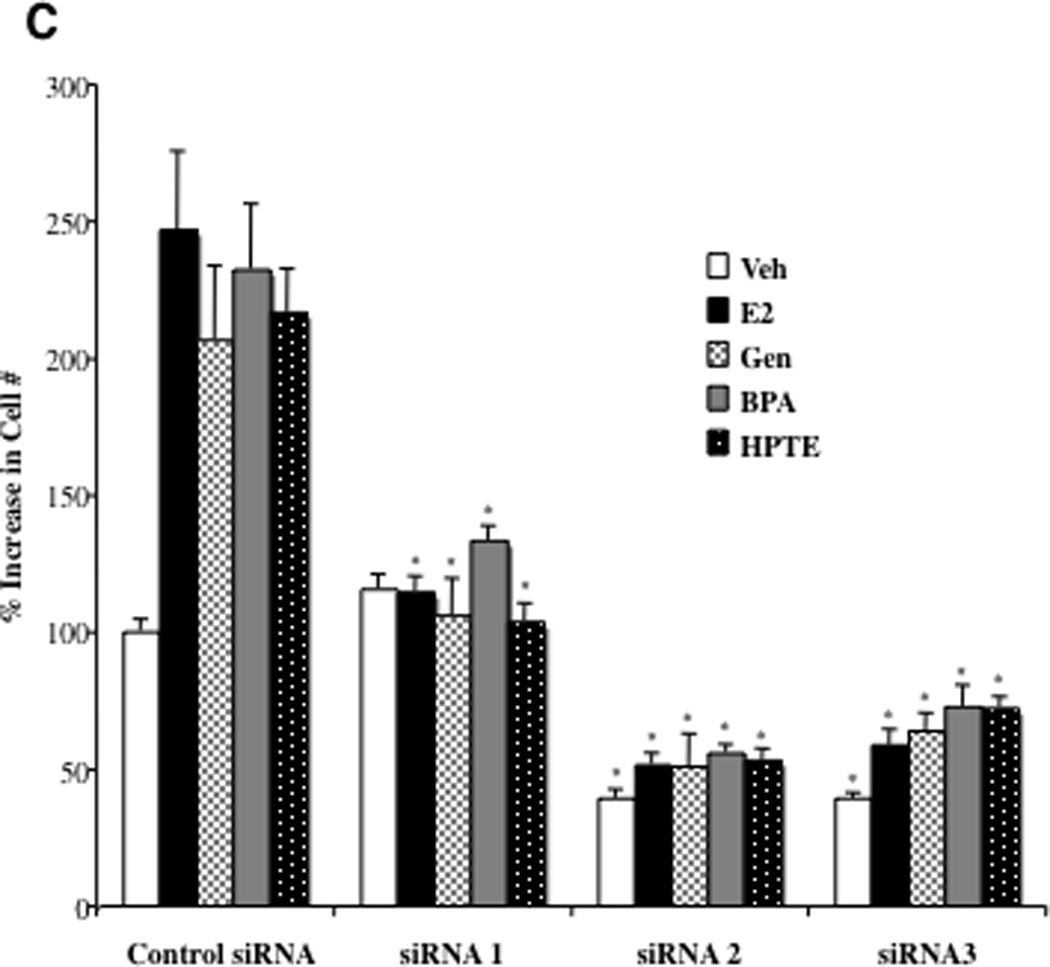
When assayed for growth, in BG-1 cells transfected with a control, non-targeting siRNA duplex, estradiol and EDCs significantly increased proliferation (Fig. 4C). Strikingly, the mitogenic activity of all ligands was completely attenuated in cells treated with each of the siRNA duplexes targeting CXCL12. Furthermore, siRNA2 and siRNA3 treatments decreased growth below that of the control, perhaps reflecting the decrease in basal CXCL12 expression observed in Fig. 4A and B. Cell viability assays confirmed that the inhibitory effects of the siRNAs were not due to cell toxicity or cell death (not shown).
The next objective was to determine whether CXCL12 is indeed a limiting factor for ovarian cancer growth stimulated by EDCs. BG-1 cells were transfected with control or siRNAs targeting CXCL12, treated with various ligands and assayed for cell proliferation. As seen before, in cells administered siRNAs to CXCL12, the mitogenic effect of estradiol and EDCs were suppressed in a similar manner (Fig. 5). Addition of exogenous CXCL12 protein to the media effectively restored the proliferative effects of all ligands. Of further note, CXCL12 treatment augmented the basal growth rate (Veh) to that of ligand-treated cells. These studies suggest that at least some of mitogenic effects of EDCs in BG-1 ovarian cancer cells require activation of classical ER pathways, and thus share a common mechanism with estradiol that involves induction of CXCL12.
Fig. 5. Exogenous CXCL12 Restores the Proliferative Effects of EDCs in Ovarian Cancer Cells.
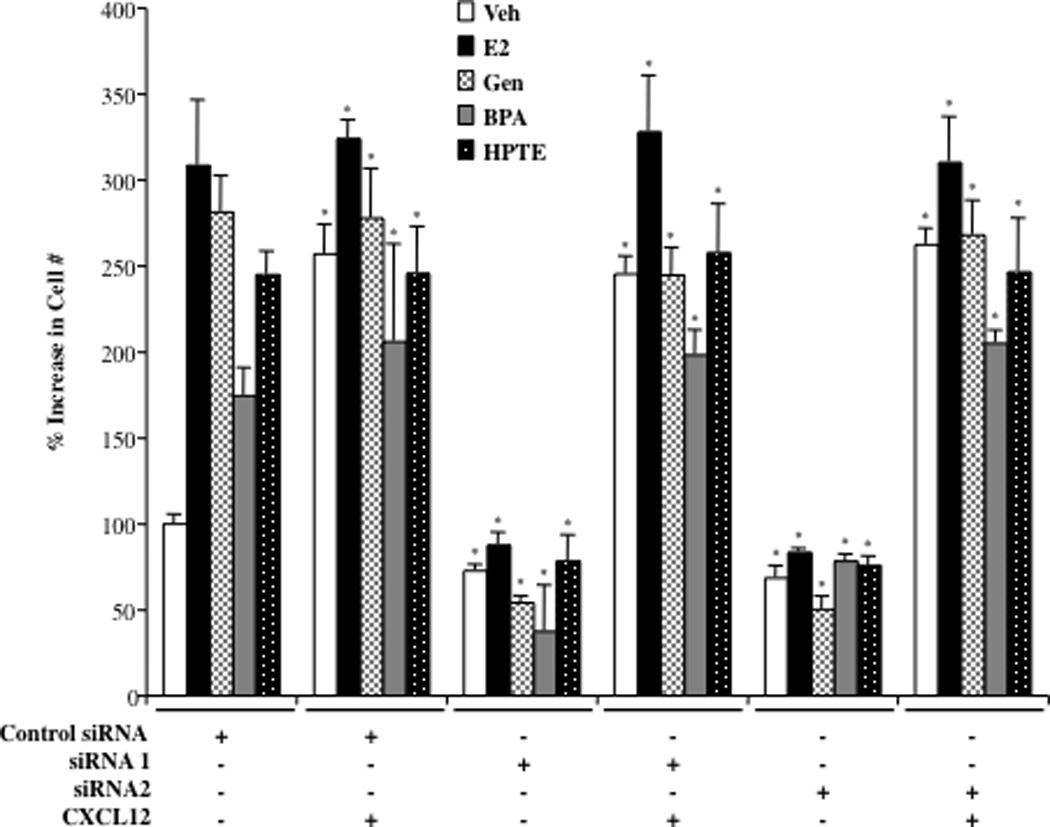
Proliferation of siRNA transfected BG-1 cells. Cells with transfected with either a nontargeting siRNA duplex (Control siRNA) or one of two independent CXCL12 siRNAs. After 24 h cells were trypsinized, seeded in 96-well plates at equal densities. Cells were administered treatments as indicated for 96 h and then assayed for cell growth. Treatments were as follows: vehicle (Veh), 1 nM Estradiol (E2), 100 nM Genistein (Gen), 100 nM Bisphenol A (BPA), 1 µM HPTE, and 100 ng/ml purified CXCL12 protein were indicated. Graphical data are represented as % increase in cell number over vehicle (set at 100%). Data points represent the average of 6 samples (+/− STDEV) for each condition in a representative experiment. *p < 0.05 for comparison between Control siRNA for each separate ligand (Veh, E2, Gen, BPA or HPTE; 1st cluster of bars) and each ligand under the other treatment conditions (+ siRNA 1, SiRNA 2 or CXCL12).
CXCR4 Antagonist AMD3100 Attenuates the Proliferative Effects of Estradiol and EDCs in Ovarian Cancer Cells
The biological effects of CXCL12 are mediated through its receptor CXCR4, which is expressed on the surface of ovarian epithelial cancer cells (28). We demonstrated previously that the CXCL12-CXCR4 signaling pathway is functional in BG-1 cells and when activated induces cell proliferation (27). qPCR analysis revealed that the CXCR4 expression is unchanged in BG-1 cells treated with E2 or EDCs (not shown). However whether the CXCL12 produced by these ligands was sufficient to induce CXCR4 activation was next assessed in the study. AMD3100 is a CXCR4 receptor antagonist that was shown previously to be effective in inhibiting the growth and invasiveness of ER+ ovarian cancer cells (32). In the current study, when coadministered with estradiol or EDCs, AMD3100 completely attenuated the proliferative effects of these ligands (Fig. 6). Taken together, these studies indicate that the mitogenic effects of estrogen and EDCs in human ovarian cells can occur through activation of the ER and elevation of extracellular CXCL12 levels, which in turn enhances activation of the CXCR4 signaling pathway, resulting in cell proliferation.
Fig. 6. CXCR4 Antagonist AMD3100 Attenuates the Proliferative Effects of Estradiol and EDCs in Ovarian Cancer Cells.
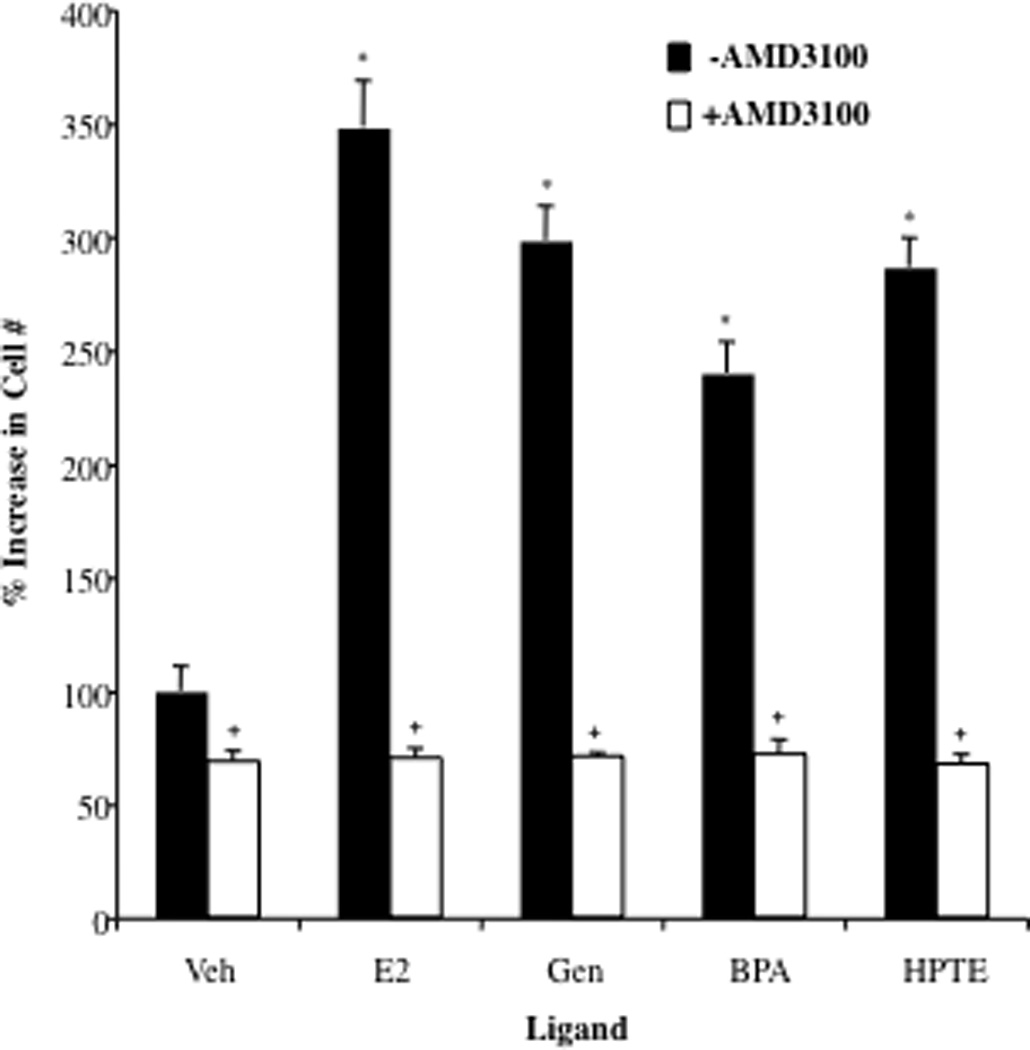
BG-1 cells were seeded in 96-well plates and treated with vehicle (Veh), 1 nM 17β-estradiol (E2), 100 nM Genistein (Gen), 100 nM Bisphenol A (BPA), or 1 µM HPTE in the absence or presence of 1 µg/ml AMD3100 for 92 h and then assayed for cell growth. Graphical data are represented as % increase in cell number over vehicle (set at 100%). Data points represent the average of 6 samples for each condition in a representative experiment +/− STDEV. *p < 0.05 for comparison between Veh and each treatment. + p < 0.05 for comparisons between each treatment alone and each treatment + AMD3100.
DISCUSSION
Endocrine Disrupting Chemicals and Ovarian Cancer
The persistent industrialization of modern society has raised increasing concern over the impact of industrial byproducts on human health. Indeed, many common organic pollutants function as endocrine disrupting chemicals (EDCs) in humans and animals, as they interfere with sex hormone signaling and reproductive function (37). In recent years there is emerging evidence that exposure to EDCs may be a risk factor for reproductive cancers. Several cohort studies demonstrate that the presence of detectable levels of polychlorinated biphenyls (PCBs), pesticides, phthalates and other toxicants in women is strongly correlated with incidence of breast cancer (38). While, epidemiological data linking EDC exposure to ovarian cancer has lagged behind, evidence from cell-based and animal studies is beginning to emerge as a cause for concern. Notably, BPA and HPTE were shown to enhance ovarian epithelial cancer cell proliferation (39, 40) and accelerate the formation of ovarian cysts and adenomas in mice (41, 42).
In the current study we have expanded these observations by showing that exposure to genistein, BPA and HPTE, 3 structurally and pharmacologically distinct EDCs, markedly accelerates the growth of human ovarian cancer cells in a manner comparable to estradiol. Notably, the proliferative effects of these agents were manifest even at relatively low (nM) concentrations, those that are found in humans with average/moderate exposure (17). Although there exists evidence that genistein may offer protection against some types of cancers, it is suggested that these effects require high (µM) concentrations. Likewise, we found that the proliferative effects were less pronounced in ovarian cells at µM levels, which may be explained by the tyrosine kinase inhibitor properties apparent at these doses. Collectively, these findings further raise the concern over the impact of exposure to these agents on reproductive health and suggest that the relationship between xenoestrogen exposure and ovarian cancer should be further explored.
Endocrine Disrupting Chemicals Activate ER-CXCL12-CXCR4 Signaling in Ovarian Cancer Cells
A significant outcome of this study was the observation that the growth-promoting effects of EDCs in ovarian cancer cells are mediated via activation of the ER and downstream target gene expression. This is notable given that both endogenous estrogens and xenoestrogens are known to activate rapid, nongenomic pathways initiated at the membrane via membrane-associated estrogen receptors or GPR30 (43–44). Genistein was shown to induce proliferation of MCF-7 breast cancer cells via activation of GPR30 (45). However, Park et al. (2009; 39) recently noted although EDCs like BPA could rapidly induce activation of extracellular signal-regulated kinase (ERK) 1/2 and p38 MAPK in ovarian cancer cells, the growth of ovarian cancer cells stimulated by xenoestrogens involved an estrogen-ER dependent genomic pathway independent of activation of cellular kinases (39).
Here, we advanced these findings by delineating an ER genomic pathway by which xenoestrogens may display proliferative effects in ovarian cancer cells. Xenoestrogens were shown to activate the ER, upregulate CXCR12 gene and protein expression, leading to an augmentation of CXCR4 signaling. Knock-down of CXCL12 attenuated the proliferative effects of EDCs, and the observation that this was reversed by exogenous CXCL12 addition indicated ER-induction of the cytokine is required for the proliferative effects of EDCs in the ovary. Thus, while it is likely that other genomic and non-genomic pathways may also contribute to maximal proliferative responses, we have identified one genomic response and signaling pathway that is required for both estrogen and xenoestrogen-stimulated growth of ovarian cancer cells.
Targeting the ER-CXCL12-CXCR4 Signaling Axis in Ovarian Cancer
The majority of ovarian cancers are epithelial adenocarcinomas that include a spectrum of disease ranging from low-grade cancers to grade I, II, III and IV. These cancers are often cystic and spread easily throughout the abdominal cavity and peritoneal surfaces. Currently the standard initial treatment for advanced staged cancers is surgical removal of the uterus, fallopian tubes and ovaries as well as any large nodules of cancer, followed by adjuvant chemotherapy. Despite the aggressive mode of treatment ovarian cancer has remained the most deadly of all gynecological cancer with a five-year survival rate for Stage III–IV cancers less than 30% following treatment (46). The relatively high recurrence rate together with the poor survival for aggressive ovarian cancer suggests that additional therapeutic approaches are necessary for the treatment of this disease.
Most ovarian cancers overexpress the ER and grow in response to hormones, suggesting that agents that target estrogen-ER pathways should be effective therapeutics. While the efficacy of antiestrogens in treatment of some ER-positive breast cancers is established, surprisingly, these agents have been widely ineffective in ovarian cancer. From a physiological standpoint it is possible that exogenous ER ligands may not accumulate in sufficient levels to block the high levels of local production of estrogens in the ovary at least in pre-menopausal women. Further complicating this issue is the evidence that ER can be activated by a number of alternative signaling pathways (cAMP, Cyclin D1, calveolin, leptin) in the absence of hormone (47). This ligand-independent activation is associated with nuclear localization of the ER and transcriptional activation, indicating that the genomic actions of the receptor are enhanced. Collectively, these observations promoted us to propose that targeting specific downstream effectors of estrogen signaling may constitute a rational approach to developing selectivity in cancer drug design and addressing the difficulties in treating ovarian malignancies. Thus, notwithstanding the limitations of the in vitro system used to study this regulatory axis, our data, taken together with that of others, indicates that the ER-CXCL12-CXCR4 signaling axis is a key player in the mitogenic effect of estrogens (and EDCs) in ovarian epithelial cancer cells. Furthermore, clinical data revealed that CXCL12 and CXCR4 were overexpressed in primary ovarian epithelial carcinomas, and CXCR4 expression correlated with metastatic spread and predicted a significantly poorer survival rate (48, 49). It is feasible therefore that agents that either block CXCL12 expression and/or CXCR4 activation could be therapeutic. Interestingly, there is emerging evidence that such an approach might be successful. Treatment of ovarian cancer cells inhibited ovarian cancer cell migration to CXCL12, but on longer incubation, caused cell death in CXCR4-positive cells in an additive manner with the drug paclitaxel (50). In mouse models of ovarian cancer, use of the selective CXCR4 antagonist AMD 3100 caused tumor cell apoptosis and significantly reduced the spread of cancer throughout the peritoneal cavity (48, 51).
In summary, the present studies demonstrate that EDCs at physiological levels enhance the growth of ovarian cancer cells by activating the ER and augmenting the CXCL12-CXCR4 signaling axis. These results support previous in situ and rodent studies showing an increase in ovarian cancer growth following exposure to EDCs with estrogenic activities. Given the current challenges in treating aggressive ovarian cancers, the clinical development of CXCL12 and/or CXCR4 inhibitors may provide an effective alternative to current primary or adjuvant therapies. Further, these studies suggest that there may exist an unappreciated relationship between xenoestrogen exposure and ovarian cancer, and further investigation into this area is warranted.
Supplementary Material
Changes in A, c-Myc and B, WISP2 RNA levels in BG-1 cells treated with EDCs were measured by real-time PCR. BG-1 cells were grown for 24 h in phenol-red free DMEM + 10% stripped FCS and then treated for an additional 24 h with vehicle (Veh), or increasing concentrations of ligands; 100 pmol–100 nM Estradiol (E2), 1 nM–1 µM Genistein (Gen), 1 nM–1 µM Bisphenol A (BPA), or 10nM–10µM HPTE. Total RNA was harvested, and cDNA was prepared and used as a template for gene expression analysis. All values were normalized to a 36B4 control. Graphical data are represented as fold induction over vehicle (set at 1). Data points represent the average of triplicate amplification reactions +/− STDEV for each condition in a representative experiment. *p < 0.05 for comparison between Veh and each treatment.
Acknowledgement
Funding: This work was supported by the Division of Intramural Research, National Institutes of Environmental Health Sciences, National Institutes of Health to KSK (Z01ES70065).
ABBREVIATIONS
- BPA
Bisphenol A
- CXCL12
chemokine (C-X-C motif) ligand 12
- CXCR4
C-X-C chemokine receptor type 4
- E2
17β-estradiol
- EDCs
Endocrine Disrupting Chemicals
- ER
Estrogen Receptor
- Hsp
heat shock protein
- HPTE
2,2-bis(p-hydroxyphenyl)-1,1,1-trichloroethane
- IGF-1
Insulin-like Growth Factor-1
- qPCR
Quantitative PCR
REFERENCES
- 1.Boon WC, Chow JD, Simpson ER. The multiple roles of estrogens and the enzyme aromatase. Prog Brain Res. 2010;181:209–232. doi: 10.1016/S0079-6123(08)81012-6. [DOI] [PubMed] [Google Scholar]
- 2.Conzen SD. Minireview: nuclear receptors and breast cancer. Mol Endocrinol. 2008;22:2215–2228. doi: 10.1210/me.2007-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Folkerd EJ, Dowsett M. Influence of sex hormones on cancer progression. J Clin Oncol. 2010;28:4038–4044. doi: 10.1200/JCO.2009.27.4290. [DOI] [PubMed] [Google Scholar]
- 4.Hall JM, Couse JF, Korach KS. The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem. 2001;276:36869–36872. doi: 10.1074/jbc.R100029200. [DOI] [PubMed] [Google Scholar]
- 5.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Safe S, Kim K. Non-classical genomic estrogen receptor (ER)/specificity protein and ER/activating protein-1 signaling pathways. J Mol Endocrinol. 2008;41:263–275. doi: 10.1677/JME-08-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stender JD, Kim K, Charn TH, Komm B, Chang KC, Kraus WL, Benner C, Glass CK, Katzenellenbogen BS. Genome-wide analysis of estrogen receptor alpha DNA binding and tethering mechanisms identifies Runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Mol Cell Biol. 2010;30:3943–3955. doi: 10.1128/MCB.00118-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levin ER. Membrane oestrogen receptor alpha signalling to cell functions. J Physiol. 2009;587:5019–5023. doi: 10.1113/jphysiol.2009.177097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maggiolini M, Picard D. The unfolding stories of GPR30, a new membrane-bound estrogen receptor. J Endocrinol. 2010;204:105–114. doi: 10.1677/JOE-09-0242. [DOI] [PubMed] [Google Scholar]
- 10.Henley DV, Korach KS. Physiological effects and mechanisms of action of endocrine disrupting chemicals that alter estrogen signaling. Hormones. 2010;9:191–205. doi: 10.14310/horm.2002.1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, Zoeller RT, Gore AC. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev. 2009;30:293–342. doi: 10.1210/er.2009-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Safe S. Clinical correlates of environmental endocrine disruptors. Trends Endocrinol Metab. 2005;16:139–144. doi: 10.1016/j.tem.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 13.Soto AM, Sonnenschein C. Environmental causes of cancer: endocrine disruptors as carcinogens. Nat Rev Endocrinol. 2010;6:363–370. doi: 10.1038/nrendo.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang X, Yang S, McKimmey C, Liu B, Edgerton SM, Bales W, Archer LT, Thor AD. Genistein induces enhanced growth promotion in ER-positive/erbB-2-overexpressing breast cancers by ER-erbB-2 cross talk and p27/kip1 downregulation. Carcinogenesis. 2010;31:695–702. doi: 10.1093/carcin/bgq007. [DOI] [PubMed] [Google Scholar]
- 15.Fanti P, Sawaya BP, Custer LJ, Franke AA. Serum levels and metabolic clearance of the isoflavones genistein and daidzein in hemodialysis patients. J Am Soc Nephrol. 1999;10:864–871. doi: 10.1681/ASN.V104864. [DOI] [PubMed] [Google Scholar]
- 16.Peterson G, Barnes S. Genistein inhibits both estrogen and growth factor-stimulated proliferation of human breast cancer cells. Cell Growth & Differentiation. 1996;7:1345–1351. [PubMed] [Google Scholar]
- 17.Welshons WV, Nagel SC, vom Saal FS. Large effects from small exposures. III. Endocrine mechanisms mediating effects of bisphenol A at levels of human exposure. Endocrinology. 2006;147:S56–S69. doi: 10.1210/en.2005-1159. [DOI] [PubMed] [Google Scholar]
- 18.Prins GS, Birch L, Tang WY, Ho SM. Developmental estrogen exposures predispose to prostate carcinogenesis with aging. Reprod Toxicol. 2007;23:374–382. doi: 10.1016/j.reprotox.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soto AM, Vandenberg LN, Maffini MV, Sonnenschein C. Does breast cancer start in the womb? Basic Clin Pharmacol Toxicol. 2008;102:125–133. doi: 10.1111/j.1742-7843.2007.00165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fernandez SV, Russo J. Estrogen and xenoestrogens in breast cancer. Toxicol Pathol. 2010;38:110–122. doi: 10.1177/0192623309354108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klotz DM, Hewitt SC, Korach KS, Diaugustine RP. Activation of a uterine insulin-like growth factor I signaling pathway by clinical and environmental estrogens: requirement of estrogen receptor-alpha. Endocrinology. 2000;141:3430–3439. doi: 10.1210/endo.141.9.7649. [DOI] [PubMed] [Google Scholar]
- 22.Hewitt SC, Korach KS. Estrogenic Activity of Bisphenol A and 2,2-bis(p-hydroxyphenyl)-1,1,1-trichloroethane (HPTE) Demonstrated in Mouse Uterine Gene Profiles. Environ Health Perspect. 2010;119:63–70. doi: 10.1289/ehp.1002347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watson CS, Jeng YJ, Kochukov MY. Nongenomic signaling pathways of estrogen toxicity. Toxicol Sci. 2010;115:1–11. doi: 10.1093/toxsci/kfp288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, Wang Q, Bekiranov S, Sementchenko V, Fox EA, Silver PA, Gingeras TR, Liu XS, Brown M. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 25.Frasor J, Danes JM, Komm B, Chang KC, Lyttle CR, Katzenellenbogen BS. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology. 2003;144:4562–4574. doi: 10.1210/en.2003-0567. [DOI] [PubMed] [Google Scholar]
- 26.Laganiere J, Deblois G, Lefebvre C, Bataille AR, Robert F, Giguere V. From the Cover: Location analysis of estrogen receptor alpha target promoters reveals that FOXA1 defines a domain of the estrogen response. Proc Natl Acad Sci U S A. 1005;102:11651–11656. doi: 10.1073/pnas.0505575102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hall JM, Korach KS. Stromal cell-derived factor 1, a novel target of estrogen receptor action, mediates the mitogenic effects of estradiol in ovarian and breast cancer cells. Mol Endocrinol. 2003;17:792–803. doi: 10.1210/me.2002-0438. [DOI] [PubMed] [Google Scholar]
- 28.Kryczek I, Wei S, Keller E, Liu R, Zou W. Stroma-derived factor (SDF-1/CXCL12) and human tumor pathogenesis. Am J Physiol Cell Physiol. 2007;292:C987–C995. doi: 10.1152/ajpcell.00406.2006. [DOI] [PubMed] [Google Scholar]
- 29.Rhodes LV, Antoon JW, Muir SE, Elliott S, Beckman BS, Burow ME. Effects of human mesenchymal stem cells on ER-positive human breast carcinoma cells mediated through ER-SDF-1/CXCR4 crosstalk. Mol Cancer. 2010;9:295. doi: 10.1186/1476-4598-9-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rhodes LV, Short SP, Neel N, Salvo VA, Zhu Y, Elliott S, Wei Y, Yu D, Sun M, Muir SE, Fonseca JP, Bratton MR, Segar C, Tilghman SL, Sobolik-Delmaire T, Horton LW, Zaja-Milatovic S, Collins-Burow BM, Wadsworth S, Beckman BS, Wood CE, Fuqua SA, Nephew KP, Dent P, Worthylake RA, Curiel TJ, Hung MC, Richmond A, Burow ME. Cytokine receptor CXCR4 mediates estrogen-independent tumorigenesis, metastasis, and resistance to endocrine therapy in human breast cancer. Cancer Res. 2011;71:603–613. doi: 10.1158/0008-5472.CAN-10-3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sauve K, Lepage J, Sanchez M, Heveker N, Tremblay A. Positive feedback activation of estrogen receptors by the CXCL12-CXCR4 pathway. Cancer Res. 2009;69:5793–5800. doi: 10.1158/0008-5472.CAN-08-4924. [DOI] [PubMed] [Google Scholar]
- 32.Scotton CJ, Wilson JL, Scott K, Stamp G, Wilbanks GD, Fricker S, Bridger G, Balkwill FR. Multiple actions of the chemokine CXCL12 on epithelial tumor cells in human ovarian cancer. Cancer Res. 2002;62:5930–5938. [PubMed] [Google Scholar]
- 33.Geisinger KR, Kute TE, Pettenati MJ, Welander CE, Dennard Y, Collins LA, Berens ME. Characterization of a human ovarian carcinoma cell line with estrogen and progesterone receptors. Cancer. 1989;63:280–288. doi: 10.1002/1097-0142(19890115)63:2<280::aid-cncr2820630213>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 34.Schmitt E, Dekant W, Stopper H. Assaying the estrogenicity of phytoestrogens in cells of different estrogen sensitive tissues. Toxicol In Vitro. 2001;15:433–439. doi: 10.1016/s0887-2333(01)00048-0. [DOI] [PubMed] [Google Scholar]
- 35.Melzer D, Rice NE, Lewis C, Henley WE, Galloway TS. Association of urinary bisphenol a concentration with heart disease: evidence from NHANES 2003/06. PLoS One. 2010;5:e8673. doi: 10.1371/journal.pone.0008673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hinton CV, Avraham S, Avraham HK. Role of the CXCR4/CXCL12 signaling axis in breast cancer metastasis to the brain. Clin Exp Metastasis. 2010;27:97–105. doi: 10.1007/s10585-008-9210-2. [DOI] [PubMed] [Google Scholar]
- 37.McLachlan JA. Environmental signaling: what embryos and evolution teach us about endocrine disrupting chemicals. Endocr Rev. 2001;22:319–341. doi: 10.1210/edrv.22.3.0432. [DOI] [PubMed] [Google Scholar]
- 38.Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, Zoeller RT, Gore AC. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev. 2009;30:293–342. doi: 10.1210/er.2009-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park SH, Kim KY, An BS, Choi JH, Jeung EB, Leung PC, Choi KC. Cell growth of ovarian cancer cells is stimulated by xenoestrogens through an estrogen-dependent pathway, but their stimulation of cell growth appears not to be involved in the activation of the mitogen-activated protein kinases ERK-1 and p38. J Reprod Dev. 2009;55:23–29. doi: 10.1262/jrd.20094. [DOI] [PubMed] [Google Scholar]
- 40.Ptak A, Wróbel A, Gregoraszczuk EL. Effect of bisphenol-A on the expression of selected genes involved in cell cycle and apoptosis in the OVCAR-3 cell line. Toxicol Lett. 2011;202:30–35. doi: 10.1016/j.toxlet.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 41.Adewale HB, Jefferson WN, Newbold RR, Patisaul HB. Neonatal bisphenol A exposure alters rat reproductive development and ovarian morphology without impairing activation of gonadotropin-releasing hormone neurons. Biol Reprod. 2009;81:690–699. doi: 10.1095/biolreprod.109.078261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Newbold RR, Jefferson WN, Padilla-Banks E. Prenatal exposure to bisphenol A at environmentally relevant doses adversely affects the murine female reproductive tract later in life. Environ Health Perspect. 2009;117:879–885. doi: 10.1289/ehp.0800045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prossnitz ER, Maggiolini M. Mechanisms of estrogen signaling and gene expression via GPR30. Mol Cell Endocrinol. 2009;308:32–38. doi: 10.1016/j.mce.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watson CS, Jeng YJ, Kochukov MY. Nongenomic signaling pathways of estrogen toxicity. Toxicol Sci. 2010;115:1–11. doi: 10.1093/toxsci/kfp288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lucki NC, Sewer MB. Genistein stimulates MCF-7 breast cancer cell growth by inducing acid ceramidase (ASAH1) gene expression. J Biol Chem. 2011;286:19399–19409. doi: 10.1074/jbc.M110.195826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rich WM, editor. Gynecologic oncology [Internet] California: Gynecologic Oncology Associates; [cited 2011 Aug 9]. Available from: http://www.gyncancer.com/ovarian-cancer.html. [Google Scholar]
- 47.Power RF, Mani SK, Codina J, Conneely OM, O'Malley BW. Dopaminergic and ligand-independent activation of steroid hormone receptors. Science. 1991;254:1636–1639. doi: 10.1126/science.1749936. [DOI] [PubMed] [Google Scholar]
- 48.Kajiyama H, Shibata K, Terauchi M, Ino K, Nawa A, Kikkawa F. Involvement of SDF-1α/CXCR4 axis in the enhanced peritoneal metastasis of epithelial ovarian carcinoma. Int J Cancer. 2008;122:91–99. doi: 10.1002/ijc.23083. [DOI] [PubMed] [Google Scholar]
- 49.Jiang YP, Wu XH, Shi B, Wu WX, Yin GR. Expression of chemokine CXCL12 and its receptor CXCR4 in human epithelial ovarian cancer: an independent prognostic factor for tumor progression. Gynecol Oncol. 2006;103:226–233. doi: 10.1016/j.ygyno.2006.02.036. [DOI] [PubMed] [Google Scholar]
- 50.Kwong H, Kulbe H, Wong D, Chakravarty P, Balkwill F. An antagonist of the chemokine receptor CXCR4 induces mitotic catastrophe in ovarian cancer cells. Mol Cancer Ther. 2009;8:1893–1905. doi: 10.1158/1535-7163.MCT-08-0966. [DOI] [PubMed] [Google Scholar]
- 51.Righi E, Kashiwagi S, Yuan J, Santosuosso M, Leblanc P, Ingraham R, Forbes B, Edelblute B, Collette B, Xing D, Kowalski M, Mingari MC, Vianello F, Birrer MJ, Orsulic S, Dranoff G, Poznansky M. CXCL12/CXCR4 blockade induces multimodal anti-tumor effects and prolongs survival of immunocompetent mice with ovarian cancer. Cancer Res. 2011;71:5522–5534. doi: 10.1158/0008-5472.CAN-10-3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Changes in A, c-Myc and B, WISP2 RNA levels in BG-1 cells treated with EDCs were measured by real-time PCR. BG-1 cells were grown for 24 h in phenol-red free DMEM + 10% stripped FCS and then treated for an additional 24 h with vehicle (Veh), or increasing concentrations of ligands; 100 pmol–100 nM Estradiol (E2), 1 nM–1 µM Genistein (Gen), 1 nM–1 µM Bisphenol A (BPA), or 10nM–10µM HPTE. Total RNA was harvested, and cDNA was prepared and used as a template for gene expression analysis. All values were normalized to a 36B4 control. Graphical data are represented as fold induction over vehicle (set at 1). Data points represent the average of triplicate amplification reactions +/− STDEV for each condition in a representative experiment. *p < 0.05 for comparison between Veh and each treatment.