

Abstract
Similarities between tumors and the inflammatory response associated with wound healing have been recognized for more than 150 years and continue to intrigue. Some years ago, based on our then recent discovery of vascular permeability factor (VPF)/vascular endothelial growth factor (VEGF), I suggested that tumors behaved as wounds that do not heal. More particularly, I proposed that tumors co-opted the wound healing response in order to induce the stroma they required for maintenance and growth. Work over the past few decades has supported this hypothesis and has put it on a firmer molecular basis. In outline, VPF/VEGF initiates a sequence of events in both tumors and wounds that includes the following: increased vascular permeability; extravasation of plasma, fibrinogen and other plasma proteins; activation of the clotting system outside the vascular system; deposition of an extravascular fibrin gel which serves as a provisional stroma and a favorable matrix for cell migration; induction of angiogenesis and arterio-venogenesis; subsequent degradation of fibrin and its replacement by “granulation tissue” (highly vascular connective tissue); and, finally, vascular resorption and collagen synthesis, resulting in the formation of dense fibrous connective tissue (called “scar tissue” in wounds and “desmoplasia” in cancer). A similar sequence of events also takes place in a variety of important inflammatory diseases that involve cellular immunity.
Keywords: Tumors, Wounds, VEGF, Vascular Permeability, Fibrin
Introduction
Back in 1986 I published an essay in the New England Journal of Medicine entitled “Tumors: Wounds that do not heal” (1). That essay called attention to many of the similarities that existed between solid tumors and wound healing. It also made a somewhat “tongue in cheek” proposition; namely, that tumors are parasites that invoke the wound healing response to acquire the stroma they need for survival and growth. That article attracted considerable interest, and I was pleased that the editor of Cancer Immunology Research invited me to prepare an update that would evaluate the relationships between tumors and wound healing in the light of subsequent research. Happily, the concept of tumors as healing wounds continues to have resonance (2-14). In fact, upon reviewing more recent literature, I have come to advocate it more strongly today than I did in 1986. Further, I suggest that the concept can be extended beyond tumors to a variety of chronic inflammatory diseases that are mediated by cellular immunity; for example, delayed hypersensitivity reactions and important human illnesses such as rheumatoid arthritis and psoriasis, have many features of aberrant wound healing (15-18).
But first, a disclaimer. I was certainly not the first to point out that tumors share properties with healing wounds. Threads of that general way of thinking go at least as far back as Virchow (reviewed in (2)) and there are many examples since. In the early 1970s Haddow proposed that tumor formation might represent an “overhealing” (19). Dolberg and colleagues (20) noted the proclivity of Rous sarcoma virus to induce tumors in virus injection sites (a kind of minor wounding), and, despite subsequent viremia, found that tumors developed elsewhere only at sites where a wound had been inflicted. Surgeons have long recognized the tendency of tumors to recur in healing resection margins, and many investigators have reported that the wound healing environment provides an opportunistic matrix for tumor growth (13,14).
The novelty of my 1986 report was based on two at the time recent findings, namely, the discovery of vascular permeability factor (VPF, subsequently renamed vascular endothelial growth factor (VEGF)) as a tumor product (21-23) and the recognition that the chronic vascular hyperpermeability induced by VPF/VEGF likely accounted for the fibrin deposited in solid tumors and in early-stages of wound healing (24). Together these findings anticipated that tumors and wound healing could be linked together in a fundamental way at the molecular level. In the intervening years, there has been increasing evidence for this possibility.
VPF/VEGF (hereafter VEGF) was of interest, first, because of its immediate, short term (minutes) activity, that of increasing microvascular permeability to plasma and plasma proteins with a potency some 50,000 times that of histamine (23). In addition, mid to longer term (days, weeks), VEGF not only induced chronic vascular permeability but also reprogrammed the gene expression profile of endothelial cells, leading to endothelial-cell activation, proliferation and survival (protection from apoptosis); angiogenesis; and arteriovenogenesis (25-31). Together these activities can account for much of what happens in tumor stroma generation and wound healing.
Much has been learned in the past 29 years about the relationships between tumors and wound healing. A recent Internet search listed more than 9,000 references when these terms were queried together, and a comprehensive review of the subject would require a book-length tome. This essay is necessarily quite limited in scope and intends to provide a personal perspective, dealing primarily with selected topics with which I have had experience. I hope that these topics will be of general interest and regret the omission of others that are equally important.
Tumors require stroma and depend on the host for its provision
Normal tissues comprise two interdependent compartments, the parenchyma (generally epithelium) and the stroma, a complex mixture of fixed tissue cells (fibroblasts, histiocytes, mast cells), inflammatory cells (myeloid cells, lymphocytes, macrophages), blood vessels of multiple types, matrix proteins (e.g., collagens I and III, fibronectin) and proteoglycans (e.g., versican). Stroma provides support for the parenchyma and its vascular component is necessary for providing oxygen and nutrients and for clearing waste products such as carbon dioxide and other metabolites. Tumors, regardless of their site of origin, have the same needs as normal tissues and are organized into these same two compartments, i.e., parenchyma (malignant cells) and stroma. The composition of tumor stroma mimics that of normal tissues. It is generally thought to be non-malignant, though there is evidence that, at the very least, tumor stromal fibroblasts and endothelial cells can share some of the functional properties of malignant cells such as increased proliferation and migration (32-35). It seems strange and paradoxical that the host provides tumors with the stroma they need to grow, spread and ultimately kill it. In this respect tumors behave like other, more traditional parasites, acting in a suicidal manner that makes no long-term sense either for the tumor or for the host on which it preys.
The host provides stroma for different tumors in different ways that require greater or lesser amounts of malignant cell participation. In the case of leukemias, for example, the blood plasma in which the tumor cells circulate affords an ideal, “free of charge” medium that supplies malignant cells with nutrition and clears their waste metabolic products in the same manner and with the same efficiency as it does for normal tissues.
Other tumor cells grow in a different sort of plasma protein-rich liquid. Ovarian cancers, for example, extend into the peritoneal cavity where they grow suspended in the ascites fluid they induce; likewise, lung and metastatic breast cancers that invade the pleural cavity induce extensive outpourings of fluid in which they grow in suspension. The mechanisms of this fluid accumulation were not understood until the discovery of VEGF (21-23,36). VEGF is highly expressed by cancer cells (25,26) and is responsible for plasma leakage and so for fluid (e.g., ascites) accumulation; indeed, antibodies against VEGF can prevent or reverse plasma accretion (22).
Finally, solid tumors, whether carcinomas or sarcomas, induce the vascularized connective tissue stroma that they need in order to survive, grow and metastasize. VEGF plays an essential role in this process as well, increasing vascular permeability to plasma and so initiating a chain of events that eventuates in the formation of a vascularized connective tissue stroma that closely resembles that found in healing wounds (25,26,37-39).
Normal wound healing
Whether a mosquito bite, a cut finger, or a myocardial infarct, wound healing proceeds through a series of steps that, broadly considered, may be summarized as hemostasis, humoral inflammation, cellular inflammation, angiogenesis, and generation of mature connective tissue stroma. These steps can be used as a framework for comparison with tumor stroma generation. I argue that tumors invent nothing new and generate stroma by activating the wound healing response as outlined in Fig. 1. In short, tumors disguise themselves as wounds and call upon the host to “heal” them.
Figure 1.

Schematic diagram of stroma formation in tumors and wounds. Stroma formation is initiated by tumor or wound cell expression of VEGF that leads to vascular hyperpermeability and consequent plasma protein extravasation, fibrin deposition and generation of highly vascular immature and then mature connective tissue stroma.
Hemostasis
Vascular damage with resultant local hemorrhage is a nearly universal feature of tissue injury. To avoid catastrophe, hemorrhage must be contained without delay, and this is accomplished by a combination of arterial contraction (reducing blood flow to the injured site) and blood clotting. Gross hemorrhage, of the type associated with acute tissue injury, is uncommon in cancer, though it can be dramatic, as when tumors erode a major blood vessel and generate bleeding in amounts that preclude hemostasis. However, microhemorrhages are common in both solid tumors and healing wounds, emanating from newly formed, fragile blood vessels (see below). Abnormal intravascular hemostasis has a long historical association with cancer (reviewed in (40-42)). More than a century ago Trousseau recognized that migratory thrombophlebitis (Trousseau's sign) was a harbinger of underlying cancer, particularly of adenocarcinomas that express procoagulant activities that seep into the circulation and induce intravascular clotting. Many leukemias also express procoagulant activities and not uncommonly induce disseminated intravascular coagulation (reviewed in (42)).
Humoral Inflammation: Increased microvascular permeability and extravascular clotting
Vascular permeability in normal tissues, wounds and cancer
The endothelial cells lining the vasculature provide the ultimate barrier to the passage of solutes. Normal tissues require a “basal” level of endothelial cell permeability for provision of nutrients and clearance of waste products (18,43). Basal vascular permeability (BVP) takes place in capillaries and involves the two-way exchange of gases, water, and other solutes such as salts, sugars and metabolites. Passage of these small molecules takes place primarily by a paracellular (i.e., intercellular) route in the space between adjacent capillary endothelial cells (Fig. 2). Normally, however, the inter-endothelial cell space is too small to accommodate the passage of large molecules such as plasma proteins. Nonetheless, small amounts of albumin, immunoglobulins and other plasma proteins do enter the tissues; they have been thought to do so by way of caveolae, small cytoplasmic vesicles, which shuttle across capillary endothelium from lumen to ablumen or interconnect to form transient transendothelial cell channels (Fig. 2). Recently, however, this hypothesis has been challenged by the finding that caveolin knockout mice that lack caveolae nonetheless have not only normal but slightly elevated permeability to plasma proteins (reviewed in (44)). Further work will be required to resolve this conundrum. In acute inflammation, such as that occurring at wound sites, vascular permeability is greatly increased as can be observed, for example, by the local swelling associated with a mosquito bite. Increased vascular permeability results primarily from histamine released from tissue mast cells which degranulate as a direct consequence of injury or in response to IgE-mediated immunity (45). Unlike the largely small molecule plasma filtrate characteristic of BVP, filtrate of acute vascular hyperpermeability (AVH) consists of a plasma protein-rich exudate whose content approximates that of plasma. In the case of trivial wounds, such as a mosquito bite, humoral inflammation is limited after a few minutes by cessation of mast cell degranulation and histamine release. However, increased vascular permeability becomes chronic and persists for days or weeks in wounds of greater magnitude as will be discussed below.
Figure 2.
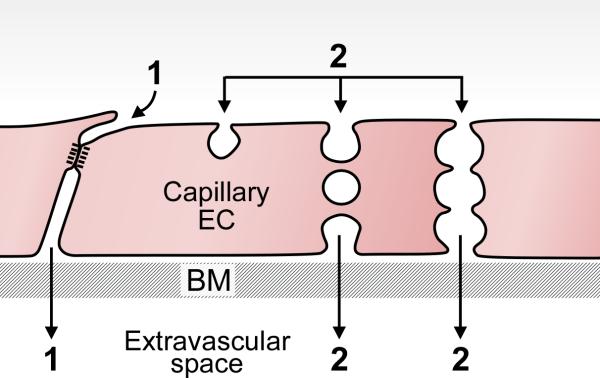
Schematic diagram illustrating pathways by which molecules of varying sizes cross the normal capillary barrier. Path 1, paracellular pathway through inter-endothelial cell cleft for small molecule extravasation. This pathway is closed to large molecules such as plasma proteins by specialized adherens and occludens junctions. Path 2, caveolae may shuttle across the capillary endothelium or form a chain of vesicles that connect the lumen and albumen to provide a pathway for plasma protein extravasation. BM, basement membrane. Modified from (43).
Whereas BVP involves capillaries, the AVH of inflammation takes place primarily from postcapillary venules, microvessels which are situated immediately downstream of capillaries. In contrast to capillary endothelial cells, those of venules are cuboidal and their cytoplasm is characterized by clusters of interconnected vesicles and vacuoles that together form an organelle termed the vesiculo-vacuolar organelle (VVO) (46-49) (Figs. 3A, 4A-C). VVOs traverse endothelial cells from lumen to ablumen and additionally open to the interendothelial cell cleft, either below or above sites of specialized junctional (adherens or tight junction) attachments.
Figure 3.

(A) Schematic diagram of a normal venule comprising cuboidal endothelial cells with prominent VVOs and a zone of tight apposition representing occludens and adherens junctions as in Fig. 2. Some VVO vesicles attach to the intercellular cleft, above or below (arrow) specialized junctions. Paths 1 and 2 indicate potential pathways for transcellular (VVO) and paracellular (intercellular) plasma extravasation, respectively. Basement membrane (BM) is intact and the endothelium is covered by pericytes (not shown). (B) AVH. Acute exposure to VEGF-A causes VVOs to open, allowing transcellular passage of plasma proteins (Path 3), possibly by mechanical pulling apart of stomatal diaphragms. Plasma extravasation may also take place through opened intercellular junctions (Path 4). (C) CVH. Prolonged VEGF-A stimulation causes venules to transform into mother vessels. Plasma may extravasate either through residual VVO vesicles (Path 5) or through fenestrae (Path 6). Modified from (43).
Figure 4.

A. Electron micrograph illustrating a cross section of a normal venule with typical prominent VVOs in the cytoplasm. B. An ultrathin section of a single VVO, typically located adjacent to the intercellular cleft that separates adjacent endothelial cells. Three sequences of interconnecting vesicles-vacuoles (a,b,c) form transendothelial chains as observed in this and subsequent serial sections. C. Computer-generated three-dimensional reconstruction of portion of a VVO, extending from the lumen (bottom, with two openings indicated by arrow) to the ablumen (top, four openings were identified but are not shown in this projection). D. Electron micrograph of a venule in skin following local injection of VEGF and iv injection of colloidal carbon. Open arrowheads indicate five separate carbon-filled, trans-endothelial cell pores that pass through adjacent endothelial cells. The intercellular cleft and occludens-type junctions (solid arrows) between these two apposed cells remain intact. E. Typical, mother vessel (MV) with thinned cytoplasm, enlarged lumen filled with red blood cells, and detached pericytes. Arrow points to a mitotic figure. Inset: the normal venule in A is reproduced at the same magnification as the MV to illustrate differences in relative size of normal venules and MV. F. VVO of a MV supplying a mouse tumor is filled with reaction product 10 seconds after iv injection of tracer horseradish peroxidase. Reaction product is confined to VVO vesicles that extend from lumen (L) to ablumen (open arrowhead). Intercellular cleft (solid arrowhead) contains no peroxidase reaction product, providing definitive evidence that protein tracer passed preferentially through the cell via VVOs rather than by a paracellular route between endothelial cells. G. MV endothelium is extremely thinned and spanned by few residual vesicles, one of which (arrowhead) traverses cytoplasm to touch both luminal and abluminal plasma membrane. Another (solid arrow) forms deep abluminal invagination. Open arrows indicate intercellular cleft which is closed and not able to accommodate circulating ferritin tracer. H. Fenestrated portion of MV endothelium in a mouse tumor following iv injection of 150kDa fluoresceinated dextran. Dextran particles are visualized in vascular lumen and immediately above and below fenestrae with diaphragms (solid arrows), and abundantly in the underlying basal lamina. One fenestra (open arrow) contains dextran particles and lacks a visible diaphragm. R, red blood cell; L, vascular lumen. Bar, 200 nm. Panels were produced from references, as follows: A, E, G, (67). B, C, (47). D, (50). F, (46). H, (49).
The pathways by which large molecules such as plasma proteins extravasate from venules in AVH are the subject of some controversy. As in capillaries, the space between adjacent venular endothelial cells is too small to admit the passage of large molecules such as plasma proteins. However, short-lived gaps or pores that traverse venular endothelium can be observed following acute exposure to histamine or other vascular permeabilizing agents (Fig. 4D) (48,50-52). Majno proposed that these agents caused adjacent endothelial cells to contract and pull apart, forming intercellular (paracellular) openings of sufficient size to permit extravasation of large molecules such as plasma proteins. More recently, however, VVOs have been proposed as an alternative transendothelial cell route for plasma extravasation (46-50). The vesicles and vacuoles comprising VVOs are linked to each other and to the plasma membrane by stomata that are normally closed by thin diaphragms that appear similar to those found in caveolae (Fig. 3A,B; 4B). Vascular permeability inducing agents such as histamine and VEGF cause the diaphragms connecting vesicles and vacuoles to open, thereby providing a transcellular pathway for plasma and plasma protein extravasation (Fig. 3B). The underlying mechanism could be mechanical, as was Majno's endothelial cell contraction model. If so, endothelial cell-cell junctions would serve as intact anchors and the actin-myosin contractions induced by permeability factors would pull apart the diaphragms linking adjacent VVO vesicles and vacuoles, resulting in a transcellular pore for plasma extravasation. The relative importance of the paracellular and VVO transcellular pathways in AVH remains to be determined, and likely varies in different tissues and in response to different stimuli.
Extravascular clotting follows plasma extravasation
Unlike the plasma protein-poor filtrate of BVP, the fluid that leaks in AVH is a plasma protein-rich exudate that includes fibrinogen and other clotting proteins. An important consequence of plasma protein extravasation is activation of clotting in the extravascular space (Fig. 1) (53). Extravascular clotting is mediated by fibroblasts and other fixed tissue cells, which, like the platelets responsible for intravascular hemostasis, express tissue factor and provide a phospholipid surface that favors prothrombinase activity. Extravascular clotting leads to deposition of fibrin in the form of a gel that retains leaked plasma and delays its clearance via lymphatics; consequently, the swelling induced by a mosquito bite does not resolve immediately. However, after a few minutes, in the absence of further plasma leakage swelling dissipates. This results from fibrin degradation by plasmin, a protease that is generated from plasminogen, a plasma protein that also extravasates in AVH (Fig. 1).
In more substantial wounds, such as a cut finger or myocardial infarct, increased microvascular permeability persists for days or weeks, long after bleeding is staunched. The chronic vascular hyperpermeability (CVH) that accompanies the wound healing response results from prolonged expression of VEGF (mast cell histamine and serotonin may also play lesser roles (54)). As in AVH, the fluid that extravasates in CVH is a plasma protein-rich exudate that approaches the overall composition of plasma. Prolonged VEGF expression is induced in wounds by ischemia that results from locally compromised blood flow. Low oxygen tension activates the transcription factor HIF-1, which, in turn, upregulates VEGF expression (55). As examples, VEGF expression is greatly increased in cardiac myocytes within 6 hours of myocardial infarction (56), and, in the skin, keratinocytes overexpress VEGF within 24h of wounding (38). Other sources of VEGF in both tumors and wounds include stromal cells, particularly macrophages (15,38). VEGF, of course, also reprograms endothelial cell gene expression to induce angiogenesis; thus, as a new vasculature is induced, local oxygen tension is restored, and expression of both HIF-1 and VEGF dial down.
Increased vascular permeability, extravascular clotting and fibrin deposition are also features of solid tumors (Fig. 1). In fact, my greatest moment in science (21,24) was the observation of fibrin deposits in solid tumors. I deduced from that observation that extravascular deposition of fibrin required two preceding events; namely, 1. Tumor blood vessels needed to become hyperpermeable to plasma proteins such as fibrinogen and other clotting factors, and 2. Clotting followed with deposition of an extravascular fibrin gel. Clotting in malignancy was of interest because we found that not only did tumor cells express tissue factor on their plasma membranes, but also that their plasma membranes provided a surface for prothrombinase activation; thus, in extravascular clotting, tumor cells were able to provide the functions played by platelets in intravascular clotting (41) (Fig. 1). In addition, we also found that tumor cells shed portions of their plasma membranes in the form of vesicles, now called exosomes, which possessed pro-coagulant activity, thus allowing clotting to extend well beyond the tumor cell locale (40,41).
I was particularly captivated by the thought that tumor cells were secreting a unique vascular permeabilizing factor or VPF that accounted for the permeability of tumor blood vessels. We pursued this line of investigation and determined the responsible factor to be a protein present both in tumor cell culture supernatants (21) and in tumor ascites fluid (36,57). Donald Senger and I purified VEGF to homogeneity in 1983 (22). Ironically, as we subsequently learned, VEGF is not unique to tumors and plays a central role in wound healing and in chronic inflammatory diseases (15-17,38).
Nearly all malignant tumor cells overexpress VEGF and they do so for a variety of reasons (reviewed in (25,26)). As in wounds, tumors are often hypoxic and may remain so because the new blood vessels that they induce provide at best a marginal supply of oxygen. However, tumors often overexpress VEGF for additional reasons that are unrelated to oxygen tension (25,26). These include the activity of oncogenes, loss of tumor suppresser genes, and soluble factors such as hormones, cytokines, and several growth factors, all of which can upregulate VEGF expression. A crucial difference between tumors and wounds is that, in tumors, VEGF expression continues indefinitely as the multiple factors that induce it persist; in contrast, in wounds, VEGF expression falls with healing as new blood vessels form and oxygen tensions normalize.
Cellular Inflammation
Inflammatory cells of multiple types play important roles in tumors and wounds (2-5,45,58,59). The first step in cellular inflammation is of course entry of inflammatory cells into the tissues, and, as with plasma and its solutes, vascular endothelium provides the primary barrier to inflammatory cell diapedesis. Inflammatory cells enter wound sites by way of venules, but it has not been determined which tumor vessels afford inflammatory cell entry, or, for that matter, tumor cell intravasation, the first step in metastatic spread. As with plasma extravasation, the pathways by which inflammatory cells cross the endothelial cell barrier to enter tumors and healing wounds have been long debated. It is now well established that inflammatory cells, including polymorphonuclear leukocytes, lymphocytes and monocytes, traverse endothelium and enter tissues by both paracellular and transendothelial cell routes, but the circumstances favoring either pathway are poorly understood (60-62).
Neutrophils are the first inflammatory cell type to respond to cell injury, and, as recent studies have shown, they act not only to ingest bacteria but also to extrude DNA nets that, like fibrin gels, serve as barriers to bacterial spread (63). Neutrophils are also common in tumors, where they are attracted to foci of necrosis. Macrophages succeed neutrophils in wounds and are prominent in tumors as well where they play a variety of roles (2-5,58,59). One of these roles is to express VEGF and so to supplement the VEGF secreted by parenchymal cells (15-17,38). Of course, macrophages also secrete many other cytokines and growth factors besides VEGF. The contribution of bone marrow-derived cells to tumor stroma is the subject of considerable recent investigation (reviewed in (64)). Finally, as readers of this Journal are well aware, there has been a tremendous resurgence of interest in immunotherapy as an approach to treating cancer and the role that different classes of lymphocytes play in favoring or inhibiting that process. Lymphocytes also participate in wound healing but their role is not well defined.
Angiogenesis
Single injections of histamine or VEGF induce a profound but short-lived and completely reversible increase in the permeability of normal venules. A similar burst of AVH follows immediately after wounding or implantation of tumor cells. However, chronic exposure to VEGF, as occurs in tumors and wounds, induces chronic vascular hyperpermeability (CVH), accompanied by profound changes in vascular structure and endothelial cell gene expression patterns. In contrast to the capillaries and venules involved in BVP and AVH, the blood vessels that leak in tumor and wound CVH do not correspond to any type of normal blood vessel. Instead, preexisting normal venules and capillaries evolve over the course of a few days into highly abnormal, greatly enlarged “mother” vessels (MV) (Figs. 3c, 4F-H) (54,65-68). Identical MVs can be induced in mouse tissues with an adenovirus expressing VEGF-A (Fig. 5). MV formation requires degradation of venular and capillary basement membranes (BM). BMs are rigid, noncompliant (nonelastic) structures composed of type IV collagen, laminin and proteoglycans that limit the expansion of normal microvessels to about 30% (69). Therefore, BMs must be degraded if MVs are to acquire cross-sectional lumens that are typically 4-5-fold larger than those of the normal microvessels from which they arose. Recent work has shown that BM degradation results from the increased expression and activation of pericyte cathepsin proteases, accompanied by decreased expression of a family of high affinity, competitive cysteine protease inhibitors that normally limit cathepsin activity (69). As a result of BM degradation, pericytes detach and formerly normal microvessels become fragile structures that are lined only by endothelium and are susceptible to microhemorrhages.
Figure 5.

Schematic diagram of the angiogenic and arterio-venogenic responses induced by VEGF in mouse tissues.
MV luminal enlargement follows BM degradation. It is likely a passive event that is driven by centripetal intravascular pressure on endothelial cells that have lost the constraints normally imposed by the BM and attached pericytes. To accommodate this increase in luminal size, endothelial cells thin and expand to cover a greatly enlarged surface area. This expansion requires a substantial increase in plasma membrane. While some membrane may result from de novo synthesis, a significant amount comes from the transfer to the cell surface of membrane that is stored within the VVOs of normal venules. VVOs contain sufficient membrane to permit a more than three-fold expansion of vessel surface area (48). Thus, in addition to providing a transvenular endothelial cell pathway for plasma extravasation in AVH, VVOs have an additional function in MV generation; they provide an intracellular store of membrane that can be translated to the cell surface to provide the additional plasma membrane required for covering a greatly enlarged surface.
MVs continue to form for as long as VEGF is highly expressed. Once formed, however, they are unstable and evolve over days to weeks into several types of “daughter” vessels (Fig. 5) (65-68). Some MVs maintain their large size and stability by acquiring a supporting coat of pericytes or smooth muscle cells. The resulting vessels closely resemble the vascular malformations (VM) that develop, for example, in the brain, skin and other tissues in other circumstances, suggesting a common mechanism by which such malformations may form in a variety of disease states (65).
Tumors express high amounts of VEGF that typically spill into plasma and ascites fluid in the high picograms to low nanograms per ml range (36,57,70). However, in solid tumors, VEGF expression is not distributed evenly. Furthermore, with declining local VEGF levels, MVs evolve into another vessel subtype, glomeruloid microvascular proliferations (GMP). GMPs are poorly organized vascular structures that are so named because of their macroscopic resemblance to the glomeruli of normal kidneys. GMPs are common in glioblastoma multiforme and are also found in other human cancers (71). Recent studies indicate that GMPs result from the collapse of MVs associated with declining VEGF levels. GMPs subsequently devolve into normal-appearing capillaries and venules, completing a cycle from normal to abnormal and then back to structurally normal microvesssels (Fig. 5). The mechanisms involved in GMP devolution are not well understood.
Macromolecular tracers extravasate from MVs to a large extent by a transcellular route (Figs. 3C; 4F-H) (18,43,46,48-50,67). It will be remembered that, in the course of MV formation substantial amounts of venular endothelial cell VVO membranes are transferred to the plasma membrane to accommodate the required increase in surface area. As a result, MVs have many fewer and less complex VVOs than the normal venular endothelium from which they were derived. However, because of endothelial cell thinning, the path length for molecular extravasation across MV endothelium is greatly shortened, such that tracers need to pass through only a few, often only one or two, vesicles or vacuoles to reach the ablumen. Macromolecules also extravasate through a second transcellular route, fenestrae (Fig. 4H), which are specialized zones of extreme endothelial cell thinning that occur in both MVs and GMPs. Pores of the type described in the venular endothelial cells of AVH also occur in MVs. As in AVH, there is debate as to whether these pores are transcellular or paracellular (18,43,48-52,67). Resolving this question will require determining whether or not specialized junction proteins are present in the plasma membranes immediately surrounding these openings, a technical tour de force that is not likely to be accomplished any time soon.
Surprisingly, the new blood vessels supplying healing wounds and inflammatory sites have been less carefully studied than those supplying tumors. As in tumors, hyperpermeable MVs are the first angiogenic vessel type to form in skin wounds (37,38) and myocardial infarcts (72). GMPs are also present in healing myocardium (72) and have been called “neovascular tufts” in oxygen-induced retinopathy (73). Capillaries are numerous at later stages of wound healing, but it is not known whether they arise from MVs or by some other mechanism.
In addition to angiogenesis, both tumors and healing wounds exhibit prominent enlarged arteries and veins that feed and drain the angiogenic vascular bed (Fig. 5) (65,66). Very little is known about the genesis or molecular properties of these important vessels. Their formation could be driven directly by exposure to VEGF (arterial and venular endothelial cells express VEGF receptors) or they could develop secondarily, driven by the increased needs of newly formed angiogenic vessels.
Generation of mature connective tissue stroma
Increased vascular permeability and extravascular clotting have important sequelae. The stromal cells of normal tissues are bathed in a plasma protein-poor filtrate. However, in AVH and CVH the composition of the interstitial fluid undergoes dramatic change. Not only does the fluid become enriched in plasma proteins, but, with clotting, it has also changed in character from plasma to serum. Serum has components, many as yet undefined, which dramatically alter the gene expression pattern of cultured fibroblasts, changes that correlate with increased malignancy in breast and other cancers (11). Similar changes in gene expression pattern may be anticipated in the fibroblasts of healing wounds and tumors, but the consequences of these changes in vivo have yet to be demonstrated. Other plasma proteins that extravasate from leaky blood vessels include fibronectin, vitronectin and osteopontin. These proteins likely have important functions, such as contributing to cell migration by virtue of sequences that favor cell attachment and detachment (74). Also, fibronectin is crosslinked to fibrin by clotting factor XIII (74).
Insertion of fibrin into tissues has important consequences (18,24,39). In addition to trapping plasma water and solutes, fibrin provides a “provisional” stroma that imposes organization on both wounds and solid tumors (74). Different tumors deposit and lyse fibrin with different efficiencies and, as a result, exhibit greater or lesser fibrin content. The fibrin present at any one time reflects a balance between fibrin deposition (clotting) and degradation (fibrinolysis) (Fig. 1). We have proposed that the amount of net fibrin present correlates with the amount of mature, connective tissue stroma that ultimately develops and so predicts the variable amounts of desmoplasia in different solid tumors (75).
Fibrin has multiple roles in stroma generation (18). First, it is a promiscuous substrate that interacts with the integrins expressed by many different types of cancer cells. This promiscuity also favors and supports the attachment and migration of a variety of host cell types, including inflammatory, fixed tissue, and endothelial cells. The capacity of fibrin to support cell migration can be readily demonstrated in vitro (76,77). Macrophages and fibroblasts migrate without difficulty through fibrin gels, even without the addition of chemotactic factors. Endothelial cells cultured in fibrin gels rapidly form lumens and organize into vascular structures (78). Fibrin also binds to and sequesters growth factors, protecting them from degradation. It also induces the expression of proangiogenic molecules such as IL8 and tissue factor. Fragment E, a fibrin degradation product, is directly proangiogenic.
Perhaps the best evidence for the importance of fibrin in stroma formation comes from in vivo experiments in which fibrin gels were implanted in the subcutaneous space of guinea pigs (39). Over time, fibroblasts and new blood vessels migrated into these gels, degrading them and replacing them with granulation tissue (vascularized connective tissue) identical to that forming in both healing wounds and tumors. Thus, fibrin can by itself induce angiogenesis and the formation of immature, highly vascularized stroma (granulation tissue). Subsequently, blood vessels are resorbed, increased amounts of collagen are synthesized, and granulation tissue matures into poorly vascularized scar tissue. A variety of growth factors and cytokines such as TGFbeta, IL8, etc. are surely involved, but the overall scheme by which they are orchestrated, either in tumors or wounds, is poorly understood. Also, for unknown reasons the process may stall as in the case of diabetic foot ulcers which fail to heal and are characterized by persistent pericapillary fibrin cuffs (79).
It should be noted that an older literature suggested that cancers could arise in scars (80). As already noted, tumors have a proclivity for growth in the microenvironment of healing wounds, but not, as far as is known, in that of healed wounds (7,13,14,20). It is likely, therefore, that so-called “scar cancers” did not arise in preexisting scars, but instead generated the desmoplastic stroma in which they became embedded.
Finally, I would note that, contrary to the title of this essay, it is not quite true that tumors cannot heal, at least in part. Solid tumors are heterogeneous in structure, and different parts of the same tumor may exhibit different stages of healing. For example, the dense, poorly vascularized, collagenous stroma in which centrally placed tumor cells are embedded is indistinguishable from the scar tissue of healed wounds. The desmoplastic stroma surrounding such tumor cells may serve as a “cocoon” that protects them from both chemotherapy and the host's inflammatory response. It will be interesting to determine whether, with improvements in immunotherapy, cytotoxic lymphocytes can reach these cells. In contrast to centrally placed tumor cells that are embedded in a “healed” matrix, goings-on at the tumor-host interface resemble wounds at active stages of healing. Invading tumor cells express VEGF and so induce AVH, new rounds of increased vascular permeability, plasma leakage and fibrin deposition, in the surrounding normal microvessels, followed by CVH as normal venules and capillaries evolve into MVs.
Looking back—and moving forward
Tumors, of course, are much more than wounds. Unlike the epithelial cells that comprise the parenchyma of normal tissues, tumor cells undergo dramatic genetic changes that release them from regulatory signals that limit invasion and proliferation. One of the most important of these is acquisition by tumor cells of the capacity to overexpress VEGF and so engage the wound healing system to generate the stroma they need for survival and growth. Much remains to be learned, however, and it is certain that future studies of tumor stroma generation will increase our understanding of wound healing, just as, conversely, studies of wound healing will enhance our knowledge of tumor biology. Studies of both are important because there are large gaps in our knowledge that could, if filled, lead to new therapeutic approaches that prevent tumor stroma generation and so interfere with tumor growth; in fact, the anti-VEGF therapies currently in vogue are really an attempt to tackle one aspect of the wound healing response, i.e., angiogenesis (81,82). The need for improved wound healing is also very great, whether in reference to the more rapid healing of myocardial infarcts or for the healing of the intractable foot ulcers that beset innumerable patients in nursing homes. Finally, as I have only touched on, the steps and principles involved in generating tumor and wound stroma also apply to a variety of chronic inflammatory diseases which are also characterized by overexpression of VEGF, increased vascular permeability, fibrin deposition and replacement of fibrin provisional stroma with vascularized connective tissue. I expect that the next 29 years will be even more exciting than the last.
Acknowledgements
This work was supported by NIH grants P01 CA92644 and R01CA142262 and by a contract from the National Foundation for Cancer Research.
References
- 1.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–9. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 2.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–45. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 3.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 5.Sun B, Karin M. Inflammation and liver tumorigenesis. Front Med. 2013;7:242–54. doi: 10.1007/s11684-013-0256-4. [DOI] [PubMed] [Google Scholar]
- 6.Barr LC, Carter RL, Davies AJ. Encapsulation of tumours as a modified wound healing response. Lancet. 1988;2:135–7. doi: 10.1016/s0140-6736(88)90686-1. [DOI] [PubMed] [Google Scholar]
- 7.Schuh AC, Keating SJ, Monteclaro FS, Vogt PK, Breitman ML. Obligatory wounding requirement for tumorigenesis in v-jun transgenic mice. Nature. 1990;346:756–60. doi: 10.1038/346756a0. [DOI] [PubMed] [Google Scholar]
- 8.Feng Y, Santoriello C, Mione M, Hurlstone A, Martin P. Live imaging of innate immune cell sensing of transformed cells in zebrafish larvae: parallels between tumor initiation and wound inflammation. PLoS Biol. 2010;8:e1000562. doi: 10.1371/journal.pbio.1000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Byun JS, Gardner K. Wounds that will not heal: pervasive cellular reprogramming in cancer. Am J Pathol. 2013;182:1055–64. doi: 10.1016/j.ajpath.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riss J, Khanna C, Koo S, Chandramouli GV, Yang HH, Hu Y, et al. Cancers as wounds that do not heal: differences and similarities between renal regeneration/repair and renal cell carcinoma. Cancer Res. 2006;66:7216–24. doi: 10.1158/0008-5472.CAN-06-0040. [DOI] [PubMed] [Google Scholar]
- 11.Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, et al. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol. 2004;2:E7. doi: 10.1371/journal.pbio.0020007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schafer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 2008;9:628–38. doi: 10.1038/nrm2455. [DOI] [PubMed] [Google Scholar]
- 13.Abramovitch R, Marikovsky M, Meir G, Neeman M. Stimulation of tumour angiogenesis by proximal wounds: spatial and temporal analysis by MRI. Br J Cancer. 1998;77:440–7. doi: 10.1038/bjc.1998.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong SY, Reiter JF. Wounding mobilizes hair follicle stem cells to form tumors. Proc Natl Acad Sci U S A. 2011;108:4093–8. doi: 10.1073/pnas.1013098108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown LF, Olbricht SM, Berse B, Jackman RW, Matsueda G, Tognazzi KA, et al. Overexpression of vascular permeability factor (VPF/VEGF) and its endothelial cell receptors in delayed hypersensitivity skin reactions. J Immunol. 1995;154:2801–7. [PubMed] [Google Scholar]
- 16.Detmar M, Brown LF, Claffey KP, Yeo KT, Kocher O, Jackman RW, et al. Overexpression of vascular permeability factor/vascular endothelial growth factor and its receptors in psoriasis. J Exp Med. 1994;180:1141–6. doi: 10.1084/jem.180.3.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fava RA, Olsen NJ, Spencer-Green G, Yeo KT, Yeo TK, Berse B, et al. Vascular permeability factor/endothelial growth factor (VPF/VEGF): accumulation and expression in human synovial fluids and rheumatoid synovial tissue. J Exp Med. 1994;180:341–6. doi: 10.1084/jem.180.1.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nagy JA, Dvorak AM, Dvorak HF. Vascular hyperpermeability, angiogenesis, and stroma generation. Cold Spring Harbor Perspect Med. 2012;2:a006544. doi: 10.1101/cshperspect.a006544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haddow A. Addendum to “molecular repair, wound healing, and carcinogenesis: tumor production a possible overhealing”? Adv Cancer Res. 1974;20:343–66. doi: 10.1016/s0065-230x(08)60113-x. [DOI] [PubMed] [Google Scholar]
- 20.Dolberg DS, Hollingsworth R, Hertle M, Bissell MJ. Wounding and its role in RSV-mediated tumor formation. Science. 1985;230:676–8. doi: 10.1126/science.2996144. [DOI] [PubMed] [Google Scholar]
- 21.Dvorak HF, Orenstein NS, Carvalho AC, Churchill WH, Dvorak AM, Galli SJ, et al. Induction of a fibringel investment: an early event in line 10 hepatocarcinoma growth mediated by tumor-secreted products. J Immunol. 1979;122:166–74. [PubMed] [Google Scholar]
- 22.Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–5. doi: 10.1126/science.6823562. [DOI] [PubMed] [Google Scholar]
- 23.Senger DR, Perruzzi CA, Feder J, Dvorak HF. A highly conserved vascular permeability factor secreted by a variety of human and rodent tumor cell lines. Cancer Res. 1986;46:5629–32. [PubMed] [Google Scholar]
- 24.Dvorak HF, Dvorak AM, Manseau EJ, Wiberg L, Churchill WH. Fibrin gel investment associated with line 1 and line 10 solid tumor growth, angiogenesis, and fibroplasia in guinea pigs. Role of cellular immunity, myofibroblasts, microvascular damage, and infarction in line 1 tumor regression. J Natl Cancer Inst. 1979;62:1459–72. [PubMed] [Google Scholar]
- 25.Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002;20:4368–80. doi: 10.1200/JCO.2002.10.088. [DOI] [PubMed] [Google Scholar]
- 26.Dvorak HF. Rous-Whipple Award Lecture. How tumors make bad blood vessels and stroma. Am J Pathol. 2003;162:1747–57. doi: 10.1016/s0002-9440(10)64309-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Connolly DT, Heuvelman DM, Nelson R, Olander JV, Eppley BL, Delfino JJ, et al. Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J Clin Invest. 1989;84:1470–8. doi: 10.1172/JCI114322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferrara N, Keyt B. Vascular endothelial growth factor: basic biology and clinical implications. EXS. 1997;79:209–32. doi: 10.1007/978-3-0348-9006-9_9. [DOI] [PubMed] [Google Scholar]
- 29.Keck PJ, Hauser SD, Krivi G, Sanzo K, Warren T, Feder J, et al. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science. 1989;246:1309–12. doi: 10.1126/science.2479987. [DOI] [PubMed] [Google Scholar]
- 30.Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–9. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- 31.Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest. 1999;103:159–65. doi: 10.1172/JCI5028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hida K, Hida Y, Amin DN, Flint AF, Panigrahy D, Morton CC, et al. Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Res. 2004;64:8249–55. doi: 10.1158/0008-5472.CAN-04-1567. [DOI] [PubMed] [Google Scholar]
- 33.Moinfar F, Man YG, Arnould L, Bratthauer GL, Ratschek M, Tavassoli FA. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: implications for tumorigenesis. Cancer Res. 2000;60:2562–6. [PubMed] [Google Scholar]
- 34.Tlsty TD. Stromal cells can contribute oncogenic signals. Semin Cancer Biol. 2001;11:97–104. doi: 10.1006/scbi.2000.0361. [DOI] [PubMed] [Google Scholar]
- 35.Kuperwasser C, Chavarria T, Wu M, Magrane G, Gray JW, Carey L, et al. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc Natl Acad Sci U S A. 2004;101:4966–71. doi: 10.1073/pnas.0401064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagy JA, Masse EM, Herzberg KT, Meyers MS, Yeo KT, Yeo TK, et al. Pathogenesis of ascites tumor growth: vascular permeability factor, vascular hyperpermeability, and ascites fluid accumulation. Cancer Res. 1995;55:360–8. [PubMed] [Google Scholar]
- 37.Brown LF, Van de Water L, Harvey VS, Dvorak HF. Fibrinogen influx and accumulation of cross-linked fibrin in healing wounds and in tumor stroma. Am J Pathol. 1988;130:455–65. [PMC free article] [PubMed] [Google Scholar]
- 38.Brown LF, Yeo KT, Berse B, Yeo TK, Senger DR, Dvorak HF, et al. Expression of vascular permeability factor (vascular endothelial growth factor) by epidermal keratinocytes during wound healing. J Exp Med. 1992;176:1375–9. doi: 10.1084/jem.176.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dvorak HF, Harvey VS, Estrella P, Brown LF, McDonagh J, Dvorak AM. Fibrin containing gels induce angiogenesis. Implications for tumor stroma generation and wound healing. Lab Invest. 1987;57:673–86. [PubMed] [Google Scholar]
- 40.Dvorak HF, Quay SC, Orenstein NS, Dvorak AM, Hahn P, Bitzer AM, et al. Tumor shedding and coagulation. Science. 1981;212:923–4. doi: 10.1126/science.7195067. [DOI] [PubMed] [Google Scholar]
- 41.Dvorak HF, Van DeWater L, Bitzer AM, Dvorak AM, Anderson D, Harvey VS, et al. Procoagulant activity associated with plasma membrane vesicles shed by cultured tumor cells. Cancer Res. 1983;43:4434–42. [PubMed] [Google Scholar]
- 42.Carr JM, Dvorak AM, Dvorak HF. Circulating membrane vesicles in leukemic blood. Cancer Res. 1985;45:5944–51. [PubMed] [Google Scholar]
- 43.Nagy JA, Benjamin L, Zeng H, Dvorak AM, Dvorak HF. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis. 2008;11:109–19. doi: 10.1007/s10456-008-9099-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang SH, Feng D, Nagy JA, Sciuto TE, Dvorak AM, Dvorak HF. Vascular permeability and pathological angiogenesis in caveolin-1-null mice. Am J Pathol. 2009;175:1768–76. doi: 10.2353/ajpath.2009.090171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marichal T, Tsai M, Galli SJ. Mast cells: potential positive and negative roles in tumor biology. Cancer Immunol Res. 2013;1:269–79. doi: 10.1158/2326-6066.CIR-13-0119. [DOI] [PubMed] [Google Scholar]
- 46.Dvorak AM, Kohn S, Morgan ES, Fox P, Nagy JA, Dvorak HF. The vesiculo-vacuolar organelle (VVO): a distinct endothelial cell structure that provides a transcellular pathway for macromolecular extravasation. J Leukoc Biol. 1996;59:100–15. [PubMed] [Google Scholar]
- 47.Feng D, Nagy JA, Hipp J, Dvorak HF, Dvorak AM. Vesiculo-vacuolar organelles and the regulation of venule permeability to macromolecules by vascular permeability factor, histamine, and serotonin. J Exp Med. 1996;183:1981–6. doi: 10.1084/jem.183.5.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feng D, Nagy JA, Dvorak AM, Dvorak HF. Different pathways of macromolecule extravasation from hyperpermeable tumor vessels. Microvasc Res. 2000;59:24–37. doi: 10.1006/mvre.1999.2207. [DOI] [PubMed] [Google Scholar]
- 49.Feng D, Nagy JA, Pyne K, Hammel I, Dvorak HF, Dvorak AM. Pathways of macromolecular extravasation across microvascular endothelium in response to VPF/VEGF and other vasoactive mediators. Microcirculation. 1999;6:23–44. [PubMed] [Google Scholar]
- 50.Feng D, Nagy JA, Hipp J, Pyne K, Dvorak HF, Dvorak AM. Reinterpretation of endothelial cell gaps induced by vasoactive mediators in guinea-pig, mouse and rat: many are transcellular pores. J Physiol. 1997;504.3:747–61. doi: 10.1111/j.1469-7793.1997.747bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Majno G, Shea SM, Leventhal M. Endothelial contraction induced by histamine-type mediators: an electron microscopic study. J Cell Biol. 1969;42:647–72. doi: 10.1083/jcb.42.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baluk P, Hirata A, Thurston G, Fujiwara T, Neal CR, Michel CC, et al. Endothelial gaps: time course of formation and closure in inflamed venules of rats. Am J Physiol. 1997;272:L155–70. doi: 10.1152/ajplung.1997.272.1.L155. [DOI] [PubMed] [Google Scholar]
- 53.Dvorak HF, Senger DR, Dvorak AM, Harvey VS, McDonagh J. Regulation of extravascular coagulation by microvascular permeability. Science. 1985;227:1059–61. doi: 10.1126/science.3975602. [DOI] [PubMed] [Google Scholar]
- 54.Qin L, Zhao D, Xu J, Ren X, Terwilliger EF, Parangi S, et al. The vascular permeabilizing factors histamine and serotonin induce angiogenesis through TR3/Nur77 and subsequently truncate it through thrombospondin-1. Blood. 2013;121:2154–64. doi: 10.1182/blood-2012-07-443903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Semenza GL. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Ann Rev Pathol. 2014;9:47–71. doi: 10.1146/annurev-pathol-012513-104720. [DOI] [PubMed] [Google Scholar]
- 56.Li J, Brown LF, Hibberd MG, Grossman JD, Morgan JP, Simons M. VEGF, flk-1, and flt-1 expression in a rat myocardial infarction model of angiogenesis. Am J Physiol. 1996;270:H1803–11. doi: 10.1152/ajpheart.1996.270.5.H1803. [DOI] [PubMed] [Google Scholar]
- 57.Yeo KT, Wang HH, Nagy JA, Sioussat TM, Ledbetter SR, Hoogewerf AJ, et al. Vascular permeability factor (vascular endothelial growth factor) in guinea pig and human tumor and inflammatory effusions. Cancer Res. 1993;53:2912–8. [PubMed] [Google Scholar]
- 58.Allavena P, Mantovani A. Immunology in the clinic review series; focus on cancer: tumour-associated macrophages: undisputed stars of the inflammatory tumour microenvironment. Clin Exp Immunol. 2012;167:195–205. doi: 10.1111/j.1365-2249.2011.04515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Novak ML, Koh TJ. Phenotypic transitions of macrophages orchestrate tissue repair. Am J Pathol. 2013;183(5):1352–63. doi: 10.1016/j.ajpath.2013.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carman CV. Mechanisms for transcellular diapedesis: probing and pathfinding by ‘invadosome-like protrusions’. J Cell Sci. 2009;122:3025–35. doi: 10.1242/jcs.047522. [DOI] [PubMed] [Google Scholar]
- 61.Carman CV, Springer TA. Trans-cellular migration: cell-cell contacts get intimate. Curr Opin Cell Biol. 2008;20:533–40. doi: 10.1016/j.ceb.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Feng D, Nagy JA, Pyne K, Dvorak HF, Dvorak AM. Neutrophils emigrate from venules by a transendothelial cell pathway in response to FMLP. J Exp Med. 1998;187:903–15. doi: 10.1084/jem.187.6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–5. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 64.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 65.Nagy JA, Chang SH, Shih SC, Dvorak AM, Dvorak HF. Heterogeneity of the tumor vasculature. Semin Thromb Hemost. 2010;36:321–31. doi: 10.1055/s-0030-1253454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nagy JA, Dvorak HF, Dvorak AM. VEGF-A and the induction of pathological angiogenesis. Annu Rev Pathol Mech Dis. 2007;2:251–75. doi: 10.1146/annurev.pathol.2.010506.134925. [DOI] [PubMed] [Google Scholar]
- 67.Nagy JA, Feng D, Vasile E, Wong WH, Shih SC, Dvorak AM, Dvorak HF. Permeability properties of tumor surrogate blood vessels induced by VEGF-A. Lab Invest. 2006;86:767–80. doi: 10.1038/labinvest.3700436. [DOI] [PubMed] [Google Scholar]
- 68.Pettersson A, Nagy JA, Brown LF, Sundberg C, Morgan E, Jungles S, et al. Heterogeneity of the angiogenic response induced in different normal adult tissues by vascular permeability factor/vascular endothelial growth factor. Lab Invest. 2000;80:99–115. doi: 10.1038/labinvest.3780013. [DOI] [PubMed] [Google Scholar]
- 69.Chang SH, Kanasaki K, Gocheva V, Blum G, Harper J, Moses MA, et al. VEGF-A induces angiogenesis by perturbing the cathepsin-cysteine protease inhibitor balance in venules, causing basement membrane degradation and mother vessel formation. Cancer Res. 2009;69:4537–44. doi: 10.1158/0008-5472.CAN-08-4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Heer K, Kumar H, Read JR, Fox JN, Monson JR, Kerin MJ. Serum vascular endothelial growth factor in breast cancer: its relation with cancer type and estrogen receptor status. Clin Cancer Res. 2001;7:3491–4. [PubMed] [Google Scholar]
- 71.Straume O, Chappuis PO, Salvesen HB, Halvorsen OJ, Haukaas SA, Goffin JR, et al. Prognostic importance of glomeruloid microvascular proliferation indicates an aggressive angiogenic phenotype in human cancers. Cancer Res. 2002;62:6808–11. [PubMed] [Google Scholar]
- 72.Ren G, Michael LH, Entman ML, Frangogiannis NG. Morphological characteristics of the microvasculature in healing myocardial infarcts. J Histochem Cytochem.y. 2002;50:71–9. doi: 10.1177/002215540205000108. [DOI] [PubMed] [Google Scholar]
- 73.Saint-Geniez M, Ghelfi E, Liang X, Yu C, Spencer C, Abend S, et al. Fatty acid binding protein 4 deficiency protects against oxygen-induced retinopathy in mice. PloS One. 2014;9:e96253. doi: 10.1371/journal.pone.0096253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhu J, Clark RA. Fibronectin at select sites binds multiple growth factors and enhances their activity: expansion of the collaborative ECM-GF paradigm. J Invest Dermatol. 2014;134:895–901. doi: 10.1038/jid.2013.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dvorak HF, Dickersin GR, Dvorak AM, Manseau EJ, Pyne K. Human breast carcinoma: fibrin deposits and desmoplasia. Inflammatory cell type and distribution. Microvasculature and infarction. J Natl Cancer Inst. 1981;67:335–45. [PubMed] [Google Scholar]
- 76.Brown LF, Lanir N, McDonagh J, Tognazzi K, Dvorak AM, Dvorak HF. Fibroblast migration in fibrin gel matrices. Am J Pathol. 1993;142:273–83. [PMC free article] [PubMed] [Google Scholar]
- 77.Lanir N, Ciano PS, Van de Water L, McDonagh J, Dvorak AM, Dvorak HF. Macrophage migration in fibrin gel matrices. II. Effects of clotting factor XIII, fibronectin, and glycosaminoglycan content on cell migration. J Immunol. 1988;140:2340–9. [PubMed] [Google Scholar]
- 78.Nakatsu MN, Sainson RC, Aoto JN, Taylor KL, Aitkenhead M, Perez-del-Pulgar S, et al. Angiogenic sprouting and capillary lumen formation modeled by human umbilical vein endothelial cells (HUVEC) in fibrin gels: the role of fibroblasts and Angiopoietin-1. Microvasc Res. 2003;66:102–12. doi: 10.1016/s0026-2862(03)00045-1. [DOI] [PubMed] [Google Scholar]
- 79.Falanga V, Moosa HH, Nemeth AJ, Alstadt SP, Eaglstein WH. Dermal pericapillary fibrin in venous disease and venous ulceration. Arch Dermatol. 1987;123:620–3. [PubMed] [Google Scholar]
- 80.Bobba RK, Holly JS, Loy T, Perry MC. Scar carcinoma of the lung: a historical perspective. Clin Lung Cancer. 2011;12:148–54. doi: 10.1016/j.cllc.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 81.Sitohy B, Nagy JA, Dvorak HF. Anti-VEGF/VEGFR therapy for cancer: reassessing the target. Cancer Res. 2012;72:1909–14. doi: 10.1158/0008-5472.CAN-11-3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Van Cutsem E, Lambrechts D, Prenen H, Jain RK, Carmeliet P. Lessons from the adjuvant bevacizumab trial on colon cancer: what next? J Clin Oncol. 2011;29:1–4. doi: 10.1200/JCO.2010.32.2701. [DOI] [PubMed] [Google Scholar]