

Abstract
Lycogarubin C, permethyl storniamide A and lamellarin G trimethyl ether are pyrrole containing, natural products, which exhibit interesting biological properties. Such properties include anti-tumor activity on a variety of cancer cell lines including those that confer drug resistance, inhibition of HIV integrase and vascular disrupting activity. We now describe the use of methyl and ethyl 3-bromo-2-formylpyrrole-5-carboxylate as building blocks for the formal synthesis of these three highly functionalized, bioactive pyrroles. These new building blocks will now provide ready access to the natural products and many novel analogs due to the ability to easily modify positions 2,3,4 and 5 of the pyrrole core.
Keywords: Suzuki Cross-Coupling, Pyrrole, Marine Natural Products
1. Introduction
We have recently reported1 the use of ethyl 3-bromo-2-formylpyrrole-5-carboxylate (1) as a very flexible and efficient pyrrole building block, which allows for Suzuki cross-coupling reactions. This becomes important, since various non-activated bromopyrroles have been reported2–8 to be problematic in such cross-coupling reactions. In addition to the formyl group activating these transformations, a versatile carbonyl function is also introduced at the 2-position of the pyrrole and this is common to many pyrrole containing natural products. In our recent report, we described the utilization of this strategy for the formal synthesis of the natural products polycitone A and B and polycitrin A (Scheme 1).
Scheme 1.

Formyl Group Activation in the Formal Synthesis of Polycitone A and B and Polycitrin A
Lycogarubin C, permethyl storniamide A and lamellarin G trimethyl ether (Fig. 1) also represent novel and biologically significant, pyrrole containing natural products and we now describe our use of the formyl group activation strategy for the completion of formal syntheses of these substances.
Figure 1.
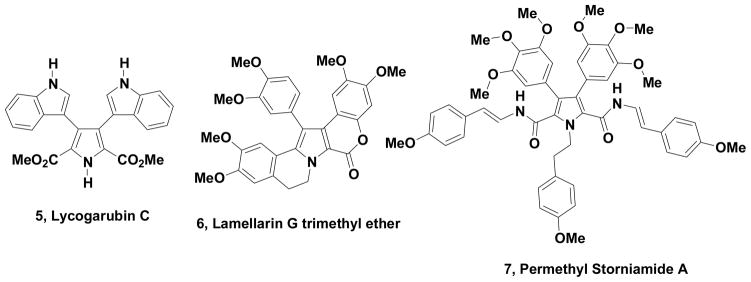
Pyrrole Containing Natural Products
2. Results and Discussions
Steglich9 and Asakawa10 simultaneously reported the isolation of various members of the lycogarubin/lycogallic acid family of natural products from slime molds in 1994. It is interesting to note that Sherman11 (2005) and Walsh12 (2007) have suggested that lycogallic acid is a biosynthetic precursor to the important naturally occurring, antitumor agents rebeccamycin and staurosporin. Previous syntheses of members of the lycogarubin/lycogallic acid family of natural products have been accomplished by Steglich9 (1994), Furstner2 (2002), Onaka13 (2006), Boger14 (2010), Gribble15 (2010) and more recently by Xie16 (2014). The Boger and Gribble syntheses both rely on the Kornfeld-Boger ring contraction methodology to generate the pyrrole core. In the Gribble synthesis of lycogarubin C, the final precursor to the natural product was a bis-N, N-sulfonylated derivative of lycogarubin C and this material subsequently became our synthetic target. We initiated our synthetic strategy by preparing methyl 3-bromo-2-formylpyrrole-5-carboxylate (8) in a three step process analogous to ethyl 3-bromo-2-formylpyrrole-5-carboxylate, which we previously reported1. Such transformations proceed in very high yield with little if any purification required of the intermediate products and very significant amounts of the pyrrole building block (8) can be obtained very rapidly. It is fortunate that 1-(phenylsulfonyl)-3-indolylboronic acid pinacol ester (9) is commercially available and it was employed in our Suzuki cross-coupling reaction (Scheme 2) with our pyrrole building block (8) using reaction conditions described in our previous report1. The resulting cross-coupled product (10) was then oxidized with sodium chlorite in water/DMSO to produce the corresponding acid (11) in good yield. Subsequent treatment of this acid (11) with dicabonylimidazole and methanol generated the corresponding bis-methyl ester (12) also in good yield. This diester (12) was then brominated with KOH/NBS in DMF producing the corresponding 4-bromo compound (13) and this material was cross-coupled with 1-(phenylsulfonyl)-3-indolylboronic acid pinacol ester (9) under previously detailed conditions. The resulting product (14) was the Gribble lycogarubin C precursor, which was previously bis de-sulfonylated with magnesium/ammonium chloride in methanol by the Gribble group to yield the natural product (5). The overall yield of the Gribble precursor (14) in five steps from our pyrrole building block (8) was 37%. The proton and carbon NMR spectra for compound 14 were identical to the values reported by Gribble and coworkers.
Scheme 2.

Gupton Group Synthesis of Gribble Precursor to Lycogarubin C
In order to further demonstrate the utility of this synthetic strategy as it relates to rapidly generating analogs, which might be required in a biologically directed SAR study, we decided to utilize the mono cross-coupled aldehyde (10) as a precursor to such compounds. We selected boronic acids (16a, 16b and 16c), which contain highly oxygenated phenyl groups given that many of the related pyrrole containing natural products possess such functionality. The resulting synthetic process is presented in Scheme 3. The presence of the aldehyde group in the final products (17) allows great flexibility for further synthetic manipulations.
Scheme 3.
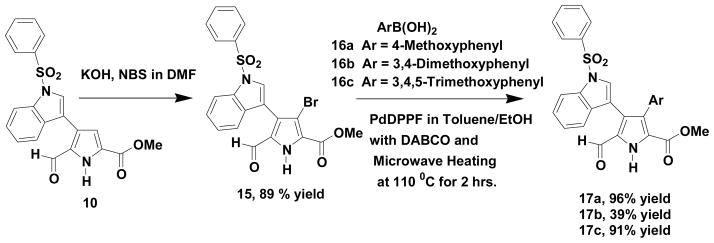
Synthesis of Lycogarubin C analogs
Another very significant natural product derived compound is permethyl storniamide A (7, Fig. 1). The storniamide marine alkaloids were first isolated in 1996 by Seldes and coworkers17 and subsequently Takamura18 (2009), Iwao19 (2003), Furstner2 (2002), Boger20 (1999), Steglich21 (1998) and our group22 (2008) reported either total or formal syntheses of permethyl storniamide A (7, Fig. 1).
Scheme 4 represents our bromoformylpyrrole methyl ester building block approach to the synthesis of the Boger storniamide intermediate20 (23). We have previously prepared22 this same compound via a vinamidinium salt based approach and compounds 21, 22 and 23 in Scheme 4 have been previously synthesized and fully characterized. The synthetic steps used in our approach are analogous to those used in our formal synthesis of lycogarubin C (Scheme 2). After an initial cross-coupling reaction of the bromoformylpyrrole ester (8) with 3,4,5-trimethoxyphenylboronic acid (18), the resulting pyrrole (19) was oxidized to the corresponding acid (20) and subsequently esterified (21) with iodomethane and base. Iodination of the pyrrole diester (21) generated the 3-iodopyrrole (22), which was then cross-coupled with 3,4,5-trimethoxyphenylboronic acid (18) using our standard conditions for this transformation. The resulting product (23) from the cross-coupling reaction exhibited spectral properties identical to those reported for the Boger storniamide intermediate20 and also to a sample of the compound, which we previously prepared by the alternate route22.
Scheme 4.

Gupton Group Synthesis of Boger Intermediate to Permethyl Storniamide A
The lamellarin class of pyrrole-containing marine alkaloids has been studied in some detail23 as a result of potent antitumor activity. Lamellarin G trimethyl ether (6) is the substance often used as a synthetic target for proof of concept for new synthetic methods and approaches. We have previously reported24 a vinamidinium salt based approach to this important target and we decided to use our new synthetic methodology (Scheme 5) for the preparation of a key pyrrole intermediate (28), which we previously described24 in our formal synthesis of lamellarin G trimethyl ether (6).
Scheme 5.

Synthesis of Gupton Group Lamellarin G Trimethyl Ether Intermediate
The synthetic process (Scheme 5) begins with an ethyl ester analog (24) of our pyrrole methyl ester building block (8), which is cross-coupled with potassium 3,4-dimethoxyphenyltrifluoroborate (25) under our optimized conditions. The resulting product is iodinated at C-3 (27) and this material undergoes cross-coupling to produce a tetrasubstituted pyrrole (28), which we previously prepared24 via the vinamidinium salt route and was fully characterized. The spectral properties for the product (28) of our new methodology were identical to those we reported earlier.
3. Conclusions
Herein we have reported formal syntheses of lycogarubin C and permethyl storniamde A via our bromoformylpyrrole methyl ester building block (8). The products (14 and 23) from both reaction schemes (Scheme 2 and 4) were obtained in five steps from the pyrrole building block (8) in a very efficient manner. In addition, a key lamellarin G trimethyl ether intermediate (28) was prepared from a similar pyrrole building block (24) in 3 synthetic steps.
Regarding the lycogarubin C strategy, three analogs were easily prepared (Scheme 3) demonstrating that this methodology should prove very useful for the synthesis of libraries of highly functionalized pyrroles. The examples depicted in Scheme 3 also demonstrate the regiocontolled nature of introducing different groups at positions 3 and 4 of the pyrrole core in a sequential fashion. In addition, it should be noted that the aldehyde group at C-2 of the various pyrroles (compounds 10, 19 and 28) will allow for a variety of functional group manipulations necessary for SAR guided synthesis of analogs, which may possess enhanced biological properties. The vast number of boronic acid derivatives, which are now commercially available, suggests that the formyl group activation strategy may have wide ranging utility for the preparation of highly functionalized pyrroles with complete control of regiochemistry.
4. Experimental
4.1 General
All chemicals were used as received from the manufacturer (Aldrich Chemicals and Fisher Scientific). All solvents were dried over 4 angstrom molecular sieves prior to their use. NMR spectra were obtained on either a Bruker 300 MHz spectrometer, or a Bruker 500 MHz spectrometer in either CDCl3, d6-DMSO or d6-acetone solutions. IR spectra were recorded on a Nicolet Avatar 320 FT-IR spectrometer with an HATR attachment. High resolution mass spectra were obtained on a Shimadzu IT-TOF mass spectrometer at the University of Richmond. Low resolution GC-MS spectra were obtained on a Shimadzu QP 5050 instrument. Melting points and boiling points are uncorrected. Chromatographic purifications were carried out on a Biotage SP-1 instrument or a Biotage Isolera instrument (both equipped with a silica cartridge). Gradient elution with ethyl acetate/hexane was accomplished in both instances. The reaction products were normally eluted within the range of 4–8 column volumes of eluant with a gradient mixture of 60–80% ethyl acetate in hexane. TLC analyses were conducted on silica plates with hexane/ethyl acetate as the eluant. All purified reaction products gave TLC results, flash chromatograms, and 13C NMR spectra consistent with a sample purity of >95%.
4.1.1 Methyl 3-Bromo-2-formylpyrrole-5-carboxylate (8)
Into a 100 mL round bottom flask equipped with magnetic stirring and a rubber septum cap was placed 10 mL of anhydrous dichloromethane, 1.61 g (0.022 mol) of dry DMF, 2.90 g (0.019 mol) of phosphorus oxychloride and the resulting mixture was stirred in an ice bath for 10 mins. To this flask was then added 1.28 g (0.0063 mol) of methyl 4-bromopyrrole-2-carboxylate in 10 mL of anhydrous dichloromethane and the resulting mixture was stirred overnight at room temperature. The reaction was worked up by the addition of 50 mL of water and separation of the two phases. The aqueous phase was extracted with additional dichloromethane (3 × 15 mL) and the combined dichloromethane phases were washed with brine (1 × 15 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to yield 1.20 g (82% yield) of a light brown solid. This material was of sufficient purity to be used in subsequent experiments but an analytical sample was prepared by purification via flash chromatography on a Biotage Isolera system in which case a light colored solid was obtained, which exhibited the following physical properties: mp 169–172 °C; 1H NMR (CDCl3) δ 9.75 (s, 1H), 6.97 (d, J = 3.0 Hz, 1H) and 3.94 (s, 3H); 13C NMR (CDCl3) δ 179.3, 159.9, 130.7, 127.5, 117.9, 107.8 and 52.5; IR (neat) 1704 and 1663 cm−1; HRMS (ES, M+H) m/z calcd for C7H7BrNO3 231.9609, found 231.9609.
4.1.2 4-(1-Benzenesulfonyl-1H-indol-3-yl)-5-formyl-1H-pyrrole-2-carboxylic acid methyl ester (10)
Into a 20 mL microwave reaction tube containing a stir bar was placed methyl 3-bromo-2-formylpyrrole-5-carboxylate (0.250 g, 1.22 mmol), 1-(phenylsulfonyl)-3-indolylboronic acid pinacol ester (0.468 g, 1.22 mmol), DABCO (0.160 g, 1.43 mmol) along with 9 mL of toluene and 3 mL of ethanol. After stirring the resulting mixture for several minutes, dichloro[1,1′-bis-(diphenyl-phosphino)ferrocene]palladium(II) dichloromethane adduct (0.037 g, 0.031 mmol) was added to the microwave reaction tube followed by the addition of 20 drops of water and the tube was capped and sealed with a crimping tool. The reaction mixture was heated in a Biotage Initiator microwave system for 2 hrs at 110 °C. After cooling to room temperature, the reaction mixture was filtered through a short plug of silica gel and the silica was subsequently washed with 3 × 20 mL of ethyl acetate and the combined organic materials were concentrated in vacuo to give a dark solid (0.598 g). The solid was subjected to flash chromatographic purification on a Biotage Isolera system with a SNAP 25 g silica column in which case an orange-red solid was obtained (0.374 g, 90% yield). This material exhibited the following physical properties: mp 160–162 °C; 1H NMR (acetone-d6) δ 9.85 (s, 1H), 8.10–8.14 (m, 4H), 7.68–7.73 (m, 2H), 7.62 (t, J = 6.3 Hz, 2H), 7.45 (t, J = 6.9 Hz, 1H), 7.36 (t, J = 6.9 Hz, 1H), 7.20 (s, 1H) and 3.91 (s, 3H); 13C NMR (CDCl3) δ 185.0, 165.4, 143.1, 140.3, 139.6, 136.9, 135.2, 134.9, 133.1, 132.2, 130.8, 130.4, 129.3, 129.2, 125.5, 120.9, 120.2, 118.9 and 56.6; IR (neat) 1720 and 1658 cm−1; HRMS (ES, M+Na) m/z calcd for C21H16N2NaO5S 431.0678 found 431.0641.
4.1.3 3-(1-Benzenesulfonyl-1H-indol-3-yl)-1H-pyrrole-2,5-dicarboxylic acid 5-methyl ester (11)
Into a 100 mL round bottomed flask equipped with a magnetic stir bar was placed 4-(1-benzenesulfonyl-1H-indol-3-yl)-5-formyl-1H-pyrrole-2-carboxylic acid methyl ester (0.120 g, 0.376 mmol) along with 35 mL of DMSO. The flask was placed in an ice bath and sodium dihydrogen phosphate (0.204 g, 2.25 mmol), which had been dissolved in 10 mL of water, was added followed by the dropwise addition of sodium chlorite (0.080 g, 0.881 mmol) in 10 mL of water. The mixture was allowed to stir overnight. The same reaction conditions were repeated exactly for a second time and both reaction mixtures were combined and worked up together. The combined reaction mixtures were adjusted to pH 2 with 6M hydrochloric acid and then diluted with 200 mL of water and then extracted with ethyl acetate (3 × 25 mL). The combined organic phases were extracted with brine (2 × 50 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to yield 0.270 g (93 % yield) of a yellow solid. This material was of sufficient purity to be used in subsequent experiments but an analytical sample was prepared by purification via flash chromatography on a Biotage Isolera system in which case the resulting solid exhibited the following physical properties: mp 205–207 °C; 1H NMR (acetone-d6) δ 8.19 (s, 1H), 8.03–8.06 (m, 3H), 7.67 (d, J = 7.8 Hz, 1H), 7.52–7.59 (m, 3H), 7.34 (t, J = 7.8 Hz, 1H), 7.30 (t, J = 7.8 Hz, 1H), 7.14 (s, 1H) and 3.88 (s, 3H); 13C NMR (acetone-d6) δ 160.2, 138.0, 134.8, 134.1, 130.7, 129.5, 127.0, 126.4, 124.5, 124.4, 123.5, 120.6, 120.4, 116.2, 116.1, 113.5 and 51.1; IR (neat) 1717 and 1677 cm−1; HRMS (ES, M+Na) m/z calcd for C21H16N2NaO6S 447.0627 found 447.0605.
4.1.4 3-(1-Benzenesulfonyl-1H-indol-3-yl)-1H-pyrrole-2,5-dicarboxylic acid dimethyl ester (12)
Into a 100 mL round bottomed flask equipped with a magnetic stir bar, reflux condenser, heating mantel and under a nitrogen atmosphere was placed carbonyl diimidazole (0.077 g, 0.470 mmol) and 3-(1-benzenesulfonyl-1H-indol-3-yl)-1H-pyrrole-2,5-dicarboxylic acid 5-methyl ester (0.200 g, 0.470 mmol) in 2 mL of DMF. The reaction mixture was stirred at 40 °C for 1 hour. Methanol (0.003 mL, 0.94 mmol) was then added to the reaction mixture and the subsequent mixture was maintained at 40 °C for 24 hrs. After the reaction mixture had cooled to room temperature it was diluted with ethyl acetate (20 mL) and the resulting solution was extracted with 10% hydrochloric acid (1 × 20 mL), saturated aqueous bicarbonate (1 × 20 mL) and water (1 × 20 mL). The resuitng organic phase was dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to yield a light yellow solid (0.180 g, 87% yield). This material was of sufficient purity to be used in subsequent experiments but an analytical sample was prepared by purification via flash chromatography on a Biotage Isolera system in which case the resulting solid exhibited the following physical properties: mp 130–132 °C; 1H NMR (acetone-d6) δ 8.06–8.09 (m, 4H), 7.68 (t, J = 7.2 Hz, 1H), 7.61–7.64 (m, 3H), 7.40 (t, J = 7.8 Hz, 1H), 7.31 (t, J = 7.8 Hz, 1H), 7.14 (s, 1H), 3.88 (s, 3H) and 3.77 (s, 3H); 13C NMR (acetone-d6) δ 160.2, 160.8, 138.1, 134.8, 134.2, 130.5, 129.5, 126.9, 126.1, 125.6, 124.7, 123.6, 122.8, 121.5, 120.5, 116.3, 115.7, 113.5, 51.2 and 51.0; IR (neat) 1695 cm−1; HRMS (ES, M+Na) m/z calcd for C22H18N2NaO6S 461.0783 found 461.0777.
4.1.5 3-(1-Benzenesulfonyl-1H-indol-3-yl)-4-bromo-1H-pyrrole-2,5-dicarboxylic acid dimethyl ester (13)
Into a 100 mL round bottomed flask equipped with a magnetic stir bar was placed 3-(1-benzenesulfonyl-1H-indol-3-yl)-1H-pyrrole-2,5-dicarboxylic acid dimethyl ester (0.090 g, 0.205 mmol), KOH (0.023 g, 0.410 mmol) and 10 mL of DMF. The resulting mixture was stirred for 15 minutes and N-bromosuccinimide (0.037 g, 0.205 mmol) was added in one portion. The flask was covered with aluminum foil and the reaction mixture was stirred overnight at room temperature. The reaction was subsequently worked up by dilution with water (20 mL) and a 10% aqueous solution of sodium thiosulfate (20 mL) followed by extraction with ethyl acetate (3 × 15 mL). The combined organic phases were washed with water (1 × 20 mL), brine (1 × 15 mL) and dried over anhydrous sodium sulfate. After removal of the drying agent by filtration, the organic phase was concentrated in vacuo to yield a light yellow solid (0.086 mg, 81 % yield). This material exhibited the following physical properties: mp 182–184 °C; 1H NMR (acetone-d6) δ 8.04–8.08 (m, 3H), 7.79 (s, 1H), 7.69 (t, J = 7.8 Hz, 1H), 7.62 (t, J = 7.8 Hz, 2H), 7.37 (t, J = 6.9 Hz, 1H), 7.32 (d, J = 6.9 Hz, 1H), 7.28 (t, J = 6.9 Hz, 1H), 3.91 (s, 3H) and 3.56 (s, 3H); 13C NMR (acetone-d6) δ 159.3, 159.1, 138.1, 134.8, 134.2, 130.7, 129.5, 126.9, 126.6, 124.6, 123.9, 123.3, 123.2, 122.0, 121.0, 115.0, 113.5, 105.7, 51.3 and 51.1; IR (neat) 1699 cm−1; HRMS (ES, M+Na) m/z calcd for C22H17BrN2NaO6S 538.9888 found 538.9895.
4.1.6 3,4-Bis-(1-benzenesulfonyl-1H-indol-3-yl)-1H-pyrrole-2,5-dicarboxylic acid dimethyl ester (14)
Into a 20 mL microwave reaction tube containing a stir bar was placed 3-(1-benzenesulfonyl-1H-indol-3-yl)-4-bromo-1H-pyrrole-2,5-dicarboxylic acid dimethyl ester (0.100 g, 0.190 mmol), 1-(phenylsulfonyl)-3-indolylboronic acid pinacol ester (0.145 g, 0.380 mmol), and DABCO (0.031 g, 0.270 mmol) along with 9 mL of toluene and 3 mL of ethanol. After stirring the resulting mixture for several minutes, dichloro[1,1′-bis-(diphenylphosphino)ferrocene]palladium(II) dichloromethane adduct (0.007 g, 0.009 mmol) was added to the microwave reaction tube followed by the addition of 20 drops of water and the tube was capped and sealed with a crimping tool. The reaction mixture was then heated in a Biotage Initiator microwave system for 2 hrs at 110 °C. After cooling to room temperature, the reaction mixture was filtered through a short plug of silica gel and the silica was subsequently washed with ethyl acetate (2 × 20 mL) and the combined organic phases were concentrated in vacuo to yield a yellow-orange solid (0.210 g). This material was purified via flash chromatography on a Biotage Isolera system in which case a yellow-orange solid (0.082 g, 63% yield) resulted. This material exhibited spectral properties identical with those reported by Gribble and coworkers for bis-N-Benzenesulfonyllycogarubin C: mp 204–206°C (lit. 205–207°C); 1H NMR (acetone-d6) δ 7.85 (d, J = 8.1 Hz, 2H), 7.66–7.71 (m, 6H), 7.58 (t, J = 7.5 Hz, 2H), 7.46 (t, J = 7.5 Hz, 4H), 7.16–7.24 (m, 4H), 7.01 (t, J = 7.5 Hz, 2H) and 3.56 (s, 6H); 13C NMR (acetone-d6) δ 160.0, 138.0, 134.3, 134.0, 131.0, 129.5, 126.4, 126.2, 124.4, 123.8, 123.1, 121.7, 120.5, 115.5, 113,1 and 50.9; IR (neat) 1701 cm−1; HRMS (ES, M+Na) m/z calcd for C36H27N3NaO8S2 716.1137 found 716.1129.
4.1.7 4-(1-Benzenesulfonyl-1H-indol-3-yl)-3-bromo-5-formyl-1H-pyrrole-2-carboxylic acid methyl ester (15)
Into a 100 mL round bottomed flask equipped with a magnetic stir bar was placed 4-(1-benzenesulfonyl-1H-indol-3-yl)-5-formyl-1H-pyrrole-2-carboxylic acid methyl ester (.100 g, 0.245 mmol), KOH (0.014 g, 0.245 mmol) and 10 mL of DMF. The resulting mixture was stirred for 45 minutes and N-bromosuccinimide (0.087 g, 0.45 mmol), which had been dissolved in 5 mL of DMF, was added in one portion. The flask was covered with aluminum foil and the reaction mixture was stirred overnight at room temperature. The reaction was subsequently worked up by dilution with water (40 mL) and a 10% aqueous solution of sodium thiosulfate (20 mL) followed by extraction with ethyl acetate (3 × 15 mL). The combined organic phases were washed with brine (1 × 30 mL) and dried over anhydrous magnesium sulfate. After removal of the drying agent by filtration, the organic phase was concentrated in vacuo to yield a solid (0.340 g). This material was purified via flash chromatography on a Biotage Isolera system in which case a white solid (0.105 g, 89% yield) resulted. This material exhibited the following physical properties: mp 211–213 °C; 1H NMR (acetone-d6) δ 9.57 (1H), 8.10–8.12 (m, 3H), 8.02 (s, 1H), 7.71 (t, J = 7.2 Hz, 1H), 7.63 (t, J = 7.2 Hz, 2H), 7.40–7.48 (m, 2H), 7.31 (t, J = 8.7 Hz, 1H) and 3.92 (s, 3H); 13C NMR (acetoned6) δ 179.7, 158.3, 137.7, 134.9, 134.5, 131.5, 130.4, 129.7, 129.6, 127.1, 127.0, 125.2, 125.1, 125.0, 123.8, 121.0, 113.6, 113.0 and 51.5; IR (neat) 1713 1672 cm−1; HRMS (ES, M+Na) m/z calcd for C21H15N2NaO5SBr 508.9777 found 508.9729.
4.1.8 4-(1-Benzenesulfonyl-1H-indol-3-yl)-5-formyl-3-(4-methoxyphenyl)-1H-pyrrole-2-carboxylic acid methyl ester (17a)
Into a 20 mL microwave reaction tube containing a stir bar was placed 4-(1-benzenesulfonyl-1H-indol-3-yl)-3-bromo-5-formyl-1H-pyrrole-2-carboxylic acid ethyl ester (0.080 g, 0.164 mmol), 4-methoxyphenylboronic acid (0.030 g, 0.197 mmol), DABCO (0.026 g, 0.230 mmol), dichloro[1,1′-bis-(diphenyl-phosphino)ferrocene]palladium(II) dichloromethane adduct (.006 g, 0.008 mmol), toluene (9 mL), methanol (3 mL) and water (5 drops). The resulting mixture was stirred and heated in a Biotage Initiator microwave system for 2 hrs at 110 °C. After cooling to room temperature, the reaction mixture was filtered through a short plug of silica gel and the silica was subsequently washed with ethyl acetate (25 mL) and the combined organic phases were concentrated in vacuo to yield an orange solid. The crude product was purified via flash chromatography on a Biotage Isolera system in which case a yellow-orange solid (0.081 g, 96% yield) was obtained and exhibited the following physical properties: mp 75–78 °C; 1H NMR (acetone-d6) δ 9.61 (s, 1H), 7.98 (d, J = 6.0 Hz, 1H), 7.90 (d, J = 8.4 Hz, 2H), 7.75 (s, 1H), 7.72 (t, J = 7.5 Hz, 1H), 7.59 (t, J = 6.9 Hz, 2H), 7.28 (t, J = 8.1 Hz 1H), 7.07–7.16 (m, 4H), 6.68 (d, J = 8.7 Hz, 2H), 3.77 (s, 3H) and 3.74 (s, 3H); 13C NMR (acetone-d6) δ 180.5, 160.4, 158.8, 137.7, 134.8, 134.3, 131.4, 131.3, 131.0, 129.7, 129.6, 127.0, 126.8, 126.7, 125.0, 124.5, 123.9, 123.6, 120.5, 114.6, 113.4, 112.8, 54.5 and 51.1; IR (neat) 1714 and 1666 cm−1; HRMS (ES, M+Na) m/z calcd for C28H22N2NaO6S 537.1091 found 537.1054.
4.1.9 4-(1-Benzenesulfonyl-1H-indol-3-yl)-5-formyl-3-(3,4,5-trimethoxyphenyl)-1H-pyrrole-2-carboxylic acid methyl ester (17b)
Into a 20 mL microwave reaction tube containing a stir bar was placed 4-(1-benzenesulfonyl-1H-indol-3-yl)-3-bromo-5-formyl-1H-pyrrole-2-carboxylic acid ethyl ester (0.150 g, 0.307 mmol), 3,4,5-trimethoxyphenylboronic acid (0.080 g, 0.369 mmol), DABCO (0.048 g, 0.430 mmol), dichloro[1,1′-bis-(diphenyl-phosphino)ferrocene]palladium(II) dichloro-methane adduct (.011 g, 0.015 mmol), toluene (9 mL), methanol (3 mL) and water (5 drops). The resulting mixture was stirred and heated in a Biotage Initiator microwave system for 2 hrs at 110 °C. After cooling to room temperature, the reaction mixture was filtered through a short plug of silica gel and the silica was subsequently washed with ethyl acetate (25 mL) and the combined organic phases were concentrated in vacuo to yield a solid. The crude product was purified via flash chromatography on a Biotage Isolera system in which case 0.161 g (91% yield) of an orange solid was obtained and exhibited the following properties: mp 71–74 °C; 1H NMR (acetone-d6) δ 9.66 (s, 1H), 7.98 (d, J = 8.4 Hz, 1H), 7.93 (d, J = 7.2 Hz, 2H), 7.84 (s, 1H), 7.71 (t, J = 7.5 Hz, 1H), 7.60 (t, J = 7.2 Hz, 2H), 7.30 (t, J = 8.4 Hz, 1H), 7.07–7.14 (m, 2H), 6.55 (s, 2H), 3.81 (s, 3H), 3.67 (s, 3H) and 3.39 (s, 6H); 13C NMR (acetone-d6) δ 180.2, 160.4, 152,5, 137.6, 134.5, 131.3, 131.1, 130.9, 129.7, 128.1, 126.8, 126.6, 124.9, 124.1, 123.9, 123.5, 120.6, 114.2, 113.2, 108.3, 93.0, 59.7, 55.3 and 51.3; IR (neat) 1714 and 1666 cm−1; HRMS (ES, M+Na) m/z calcd for C30H26N2NaO8S 597.1302 found 597.1237.
4.1.10 4-(1-Benzenesulfonyl-1H-indol-3-yl)-3-(3,4-dimethoxyphenyl)-5-formyl-1H-pyrrole-2-carboxylic acid methyl ester (17c)
Into a 20 mL microwave reaction tube containing a stir bar was placed 4-(1-benzenesulfonyl-1H-indol-3-yl)-3-bromo-5-formyl-1H-pyrrole-2-carboxylic acid ethyl ester (0.100 g, 0.205 mmol), 3,4-dimethoxyphenylboronic acid (0.045 g, 0.246 mmol), DABCO (0.032 g, 0.287 mmol), dichloro[1,1′-bis-(diphenyl-phosphino)ferrocene]palladium(II) dichloro-methane adduct (.008 g, 0.010 mmol), toluene (9 mL), methanol (3 mL) and water (5 drops). The resulting mixture was stirred and heated in a Biotage Initiator microwave system for 2 hrs at 110 °C. After cooling to room temperature, the reaction mixture was filtered through a short plug of silica gel and the silica was subsequently washed with ethyl acetate (25 mL) and the combined organic phases were concentrated in vacuo to yield a solid. The crude product was purified via flash chromatography on a Biotage Isolera system in which case 0.043 g (39% yield) of a red solid was obtained and exhibited the following properties: mp 68–71 °C; 1H NMR (acetone-d6) δ 9.60 (s, 1H), 7.98 (d, J = 8.4 Hz, 1H), 7.90 (d, J = 8.4 Hz, 2H), 7.78 (s, 1H), 7.72 (t, J = 7.8 Hz, 1H), 7.59 (t, J = 7.2 Hz, 2 H), 7.30 (t, J = 8.4 Hz, 1H), 7.17 (d, J = 8.7 Hz, 1H), 7.13 (t, J = 8.1 Hz, 1H), 6.70–6.80 (m, 3H), 3.78 (s, 3H), 3.76 (s, 3H) and 3.35 (s, 3H); 13C NMR (acetone-d6) δ 180.1, 160.4, 148.6, 148.3, 137.7, 134.6, 134.3, 131.3, 131.0, 129.6, 126.7, 126.6, 125.3, 124.9, 124.2, 123.9, 123.6, 122.9, 120.6, 114.5, 114.4, 113.3, 110.8, 55.0, 54.5 and 51.1; IR (neat) 1708 and 1663 cm−1; HRMS (ES, M+Na) m/z calcd for C29H24N2O7S 567.1196 found 567.1135.
4.1.11 5-Formyl-4-(3,4,5-trimethoxyphenyl)-1H-pyrrole-2-carboxylic acid methyl ester (19)
Into a 20 mL microwave reaction tube containing a stir bar was placed 4-bromo-5-formyl-1H-pyrrole-2-carboxylic acid methyl ester (0.400 g, 1.72 mmol), potassium 3,4,5-trimethoxyphenyltrifluoroborate (0.570 g, 2.07 mmol), DABCO (0.270 g, 2.41 mmol) and dichloro[1,1′-bis-(diphenylphosphino)ferrocene]palladium(II) dichloromethane adduct (0.063 g, 0.086 mmol). To this mixture was added 9 mL of toluene, 3 mL of ethanol and 20 drops of water and the resulting mixture was stirred and heated in a Biotage Initiator microwave system for 2 hrs at 110 °C. After cooling to room temperature, the reaction mixture was filtered through a short plug of silica gel and the silica was subsequently washed with ethyl acetate (25 mL) and the combined organic phases were concentrated in vacuo to yield a dark solid (0.500 g). The crude product was purified via flash chromatography on a Biotage Isolera system in which case a yellow-orange solid (0.31 g, 56% yield) was obtained. This material exhibited the following physical properties: mp 150–152 °C; 1H NMR (CDCl3) δ 9.83 (s, 1H), 7.02 (d, J = 2.4 Hz, 1H), 6.89 (s, 2H), 3.96 (s, 3H) and 3.92 (s, 9H); 13C NMR (CDCl3) δ 180.6, 160.6, 153.6, 138.5, 138.0, 130.3, 128.1, 127.2, 115.3, 106.5, 61.0, 56.3 and 52.3; IR (neat) 1695 and 1648 cm−1; HRMS (ES, M+H) m/z calcd for C16H18NO6 320.1134 found 320.1144.
4.1.12 3-(3,4,5-Trimethoxyphenyl)-1H-pyrrole-2,5-dicarboxylic acid 5-methyl ester (20)
Into a 100 mL round bottomed flask equipped with a magnetic stir bar was placed 5-formyl-4-(3,4,5-trimethoxyphenyl)-1H-pyrrole-2-carboxylic acid methyl ester (0.280 g, 0.870 mmol), 35 mL of DMSO and sodium dihydrogen phosphate (0.120 g, 0.870 mmol), which had been dissolved in 10 mL of water. The resulting solution was cooled in an ice bath for 5 minutes and a solution of sodium chlorite (0.236 g, 2.61 mmol) in 10 mL of water was added dropwise and the reaction mixture was allowed to stir overnight at room temperature. The cooled reaction mixture was worked up by acidification with 6 M hydrochloric acid and was then diluted with 150 mL of water. Extraction of the aqueous mixture with ethyl acetate (3 × 25 mL) followed by drying over anhydrous sodium sulfate, filtration and concentration in vacuo produced a light yellow solid (0.290 g, 99% yield). This material was of sufficient purity to be used in subsequent experiments but an analytical sample was prepared by purification via flash chromatography on a Biotage Isolera system in which case the resulting solid exhibited the following physical properties: mp 201–203 °C; 1H NMR (CDCl3) δ 7.00 (d, J = 3.0 Hz, 1H), 6.84 (s, 2H), 3.95 (s, 3H), 3.92 (s, 3H) and 3.90 (s, 6H); 13C NMR (CDCl3) δ 164.1, 160.6, 152.8, 137.9, 133.8, 128.9, 125.5, 120.4, 116.9, 107.0, 60.9, 56.2 and 52.2; IR (neat) 1717 and 1677 cm−1; HRMS (ES, M+Na) m/z calcd for C16H17NNaO7 358.0903 found 358.0913.
4.1.13 3-(3,4,5-Trimethoxyphenyl)-1H-pyrrole-2,5-dicarboxylic acid dimethyl ester (21)
Into a 100 mL round bottomed flask equipped with a magnetic stir bar was placed 3-(3,4,5-trimethoxyphenyl)-1H-pyrrole-2,5-dicarboxylic acid 5-methyl ester (0.150g, 0.447 mmol), ethyldiisopropylamine (0.170 g, 1.34 mmol), and 35 mL of anhydrous dichloromethane. The mixture was stirred for 30 minutes and iodomethane (0.25 g, 1.79 mmol) was added in one portion. The resulting mixture was stirred overnight at room temperature and concentrated in vacuo to yield a viscous orange product. The material was subjected to flash chromatography on a Biotage Isolera system in which case the resulting solid (0.110 g, 71% yield) exhibited spectral properties identical to material, which we have previously prepared22 and characterized via a different route: 1H NMR (CDCl3) δ 6.98 (d, J = 3.0 Hz, 1H), 6.83 (s, 2H), 3.95 (s, 3H),3.91 (s, 3H), 3.90 (s, 6H) and 3.87 (s, 3H).
4.1.14 3-Iodo-4-(3,4,5-trimethoxyphenyl)-1H-pyrrole-2,5-dicarboxylic acid dimethyl ester (22)
Into a 100 mL round bottomed flask equipped with a magnetic stir bar was placed 3-(3,4,5-trimethoxyphenyl)-1H-pyrrole-2,5-dicarboxylic acid dimethyl ester (0.190 g, 0.543 mmol), potassium hydroxide (0.087 g, 1.63 mmol) and 20 mL of DMF. The reaction mixture was stirred at room temperature for 30 minutes and iodine (0.207 g, 0.816 mmol) was added, the reaction flask was covered with aluminum foil and the resulting mixture was stirred overnight at room temperature. The reaction mixture was quenched with 25 mL of sodium thiosulfate and extracted with ethyl acetate (2 × 25 mL). The combined organic phases were washed with water (2 × 25 mL) and brine (1 × 15 mL) and dried over anhydrous sodium sulfate. After removing the drying agent by filtration and concentration in vacuo a solid product (0.160 g, 62% yield) was obtained, which we have previously prepared22 and characterized via a different route: 1H NMR (CDCl3) δ 6.55 (s, 2H), 3.98 (s, 3H), 3.94 (s, 3H), 3.89 (s, 6H) and 3.10 (s, 3H).
4.1.15 3,4-Bis-(3,4,5-trimethoxyphenyl)-1H-pyrrole-2,5-dicarboxylic acid dimethyl ester (23)
Into a 20 mL microwave reaction tube containing a stir bar was placed 3-iodo-4-(3,4,5-trimethoxyphenyl)-1H-pyrrole-2,5-dicarboxylic acid dimethyl ester (0.100 g, 0.210 mmol), potassium 3,4,5-trimethoxyphenyltrifluoroborate (0.069 g, 0.25 mmol), DABCO (0.033 g, 0.29 mmol), dichloro[1,1′-bis-(diphenylphosphino)ferrocene]palladium(II) dichloromethane adduct (0.00 7 g, 0.009 mmol) along with 9 mL of toluene, 3 mL of ethanol and 20 drops of water. The resulting mixture was stirred and heated in a Biotage Initiator microwave system for 2 hrs at 110 °C. After cooling to room temperature, the reaction mixture was filtered through a short plug of silica gel and subsequently washed with ethyl acetate (25 mL) and the combined organic phases were concentrated in vacuo to yield a a dark solid. The crude material was purified via flash chromatography on a Biotage Isolera system in which case a yellow solid (0.070 g, 65% yield) was obtained, which we have previously prepared22 and characterized via a different route: 1H NMR (CDCl3) δ 6.40 (s, 4 H), 3.85 (s, 12H) and 3.67 (s, 12H). We have previously reported22 that this material was identical to a key synthon used by Boger and coworkers to prepare permethylstorniamide A.
4.1.16 4-(3,4-Dimethoxyphenyl)-5-formyl-1H-pyrrole-2-carboxylic acid ethyl ester (26)
Into a 20 mL microwave reaction tube containing a stir bar was placed 4-bromo-5-formyl-1H-pyrrole-2-carboxylic acid ethyl ester (0.250 g, 1.02 mmol), potassium 3,4-dimethoxyphenyltrifluoroborate (0.316 g, 1.32 mmol), DABCO (0.229 g, 2.04 mol) and a mixture of 3:1 toluene:ethanol (12 mL) along with 20 drops of water. After stirring for several minutes, dichloro[1,1′bis(diphenyl-phosphino)ferrocene]palladium(II) dichloromethane adduct (0.037 g, 0.051 mmol) was added and the resulting mixture was stirred and heated in a Biotage Initiator microwave system for 2 hrs at 110 °C. After cooling to room temperature, the reaction mixture was filtered through a short plug of silica gel and was subsequently washed with ethyl acetate (2×30 mL) and the combined organic phases were concentrated in vacuo to yield a dark solid. The crude material was purified via flash chromatography on a Biotage Isolera system in which case a light colored solid (0.300 g, 70% yield) was obtained, which have previously prepared24 and characterized via a different route: 1H NMR (CDCl3) δ 9.79 (s, 1H), 7.05 (dd, J = 3.0 Hz, J = 9.0 Hz, 1H), 7.02 (d, J = 3.0 Hz, 1H), 7.00 (d, J = 1.5 Hz, 1H), 6.97(d, J = 9.0 Hz, 1H), 4.42 (q, J = 6.0 Hz, 2H), 3.95 (broad s, 6H) and 1.42(t, J = 6.0 Hz, 3H).
4.1.17 4-(3,4-Dimethoxyphenyl)-5-formyl-3-iodo-1H-pyrrole-2-carboxylic acid ethyl ester (27)
Into a 100 mL round bottomed flask equipped with a magnetic stir bar was placed 4-(3,4-dimethoxy-phenyl)-5-formyl-1H-pyrrole-2-carboxylic acid ethyl ester (0.340 g, 1.11 mmol), potassium hydroxide (0.250 g, 4.46 mmol), and 15 mL of DMF. The mixture was stirred for 30 minutes and iodine (0.566 g, 2.23 mmol) was added in one portion. The reaction flask was covered with aluminum foil and the resulting mixture was stirred overnight at room temperature. The reaction mixture was quenched with 45 mL of sodium thiosulfate and extracted with ethyl acetate (3 × 30 mL). The combined organic phases were washed with aqueous lithium chloride solution (3 × 15 mL) and dried over anhydrous sodium sulfate. After removing the drying agent by filtration and concentration in vacuo a dark solid product was obtained. The crude material was purified via flash chromatography on a Biotage Isolera system in which case an orange solid (0.162 g, 70% yield) was obtained, which we have previously prepared24 and characterized via a different route: 1H NMR (DMSO-d6) δ 9.45 (s, 1H), 7.04 (d, J = 8.4 Hz, 1H), 6.99 (d, J = 2.1 Hz, 1H), 6.92 (dd, J = 2.1 Hz, J = 8.4 Hz, 1H), 4.32 (q, J = 7.2 Hz, 2H), 3.81 (s, 3H), 3.77 (s, 3H) and 1.33 (t, J = 7.2 Hz, 3H).
4.1.18 3,4-Bis-(3,4-dimethoxyphenyl)-5-formyl-1H-pyrrole-2-carboxylic acid ethyl ester (28)
Into a 20 mL microwave reaction tube containing a stir bar was placed potassium 4-(3,4-dimethoxyphenyl)trifluoroborate (0.198 g, 0.811 mmol), 5-formyl-3-iodo-1H-pyrrole-2-carboxylic acid ethyl ester (0.290 g, 0.676 mmol), DABCO (0.106 g, 0.946 mmol), dichloro[1,1′-bis-(diphenylphosphino)ferrocene]palladium(II) dichloromethane adduct (0.025 g, 0.033 mmol) along with 9 mL of toluene, 3 mL of ethanol and 20 drops of water. The resulting mixture was stirred and heated in a Biotage Initiator microwave system for 2 hrs at 110 °C. This reaction was duplicated and both reaction mixtures were combined after the microwave heating was completed. The combined organic phases were filtered through a short plug of silica gel and subsequently washed with ethyl acetate (3 × 20 mL) and concentrated in vacuo to yield a dark solid. The crude material was purified via flash chromatography on a Biotage Isolera system in which case a yellow-orange solid (0.33 g, 56% yield) was obtained, which we have previously prepared24 and characterized via a different route: 1H NMR (DMSO-d6) δ 9.61 (s, 1H), 6.87 (d, J = 8.1 Hz, 1H), 6.83 (d, J = 8.1 Hz, 1H), 6.76 (dd, J = 2.1 Hz, J = 8.1 Hz, 1H), 6.73 (d, J = 2.1 Hz, 1H), 6.70 (d, J = 2.1 Hz, 1H), 6.64 (dd, J = 2.1 Hz, J = 7.2 Hz, 1H), 4.14 (q, J = 6.9 Hz, 2H), 3.73 (s, 3H), 3.72 (s, 3H), 3.58 (s, 3H), 3.53 (s, 3H) and 1.13 (t, J = 7.2 Hz, 3H). We have previously reported that this material was identical to a key synthon used by our research group in the formal synthesis of lamellarin G trimethyl ether.
Acknowledgments
We gratefully thank the National Institutes of Health (grant no. R15-CA67236) for support of this research project. Thanks also to the Floyd D. and Elisabeth S. Gottwald endowment for support to JTG. We are also very appreciative of Advion Inc. for the generous donation of a CMS electrospray mass spectrometer. Previous grants from the MRI program of the National Science Foundation for the purchase of a 500 MHz NMR spectrometer (CHE-0116492) and a high resolution electrospray mass spectrometer (CHE-0320669) are also gratefully acknowledged. We are also very appreciative of Professor Gordon Gribble for helpful discussions regarding the spectral properties of the Gribble precursor to lycogarubin C (14).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Gupton J, Telang N, Wormald M, Lescalleet K, Patteson J, Curry W, Harrison A, Hoerrner M, Sobieski J, Kimmel M, Kluball E, Perry T. Tetrahedron. 2014;70:2738. doi: 10.1016/j.tet.2014.02.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Furstner A, Krause H, Thiel O. Tetrahedron. 2002;58:6373. [Google Scholar]
- 3.Pla D, Marchal A, Olsen C, Albericio F, Alavarez M. J Org Chem. 2005;70:8231. doi: 10.1021/jo051083a. [DOI] [PubMed] [Google Scholar]
- 4.(a) Banwell M, Bray A, Edwards A, Wong D. J Chem Soc, Perkin Trans. 2002;1:1340. [Google Scholar]; (b) Banwell M, Goodwin T, Ng S, Smith J, Wong D. Eur J Org Chem. 2006;14:3043. [Google Scholar]
- 5.Handy S, Zhang Y, Bregman H. J Org Chem. 2004;69:2362. doi: 10.1021/jo0352833. [DOI] [PubMed] [Google Scholar]
- 6.Serge-Mitherand T, Fatunsin O, Villinger A, Langer P. Tetrahedron Lett. 2011;52:3732. [Google Scholar]
- 7.Fukuda T, Sudo E, Shimokawa K, Iwao M. Tetrahedron. 2008;64:328. [Google Scholar]
- 8.Handy S, Bregman H, Lewis J, Zhang X, Zhang Y. Tetrahedron Lett. 2003;44:427. [Google Scholar]
- 9.Frode R, Hinze C, Josten I, Schmidt B, Steffan B, Steglich W. Tetrahedron Lett. 1994;11:1689. [Google Scholar]
- 10.Asakawa Y, Hashimoto T, Yasuda A, Akasawa K, Takaoka S, Tori M. Tetrahedron Lett. 1994;35:2559. [Google Scholar]
- 11.Sherman D, Nishizawa T, Gruschow S, Jayamaha D, Nishizawa-Harada C. J Am Chem Soc. 2006;128:724. doi: 10.1021/ja056749x. [DOI] [PubMed] [Google Scholar]
- 12.Walsh C, Howard-Jones A. J Am Chem Soc. 2007;129:11016. doi: 10.1021/ja0743801. [DOI] [PubMed] [Google Scholar]
- 13.Asamizu S, Kato Y, Igarashi Y, Furumai T, Onaka H. Tetrahedron Lett. 2006;47:473. [Google Scholar]
- 14.Oakdale J, Boger D. Org Lett. 2010;12:1132. doi: 10.1021/ol100146b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu L, Gribble G. Tetrahedron Lett. 2010;51:537. [Google Scholar]
- 16.Zhou N, Xie T, Liu L, Xie Z. J Org Chem. 2014;79:6061. doi: 10.1021/jo500740w. [DOI] [PubMed] [Google Scholar]
- 17.Palermo J, Brasco M, Seldes A. Tetrahedron Lett. 1996;52:2727. [Google Scholar]
- 18.Yasui E, Wada M, Takamura N. Tetrahedron Lett. 2009;50:4762. [Google Scholar]
- 19.Iwao M, Takeuchi T, Fujikawa N, Fukuda T, Ishibashi F. Tetrahedron Lett. 2003;44:4443. [Google Scholar]
- 20.Boger D, Boyce C, Labroli M, Sehon C, Jin Q. J Am Chem Soc. 1999;121:54. [Google Scholar]
- 21.Ebel H, Terpin A, Steglich W. Tetrahedron Lett. 1998;39:9165. [Google Scholar]
- 22.Gupton J, Banner E, Sartin M, Fisher D, Giglio B, Smith K, Keough M, Smith T, Kanters R, Dominey R, Sikorski J. Tetrahedron. 2008;64:5246. doi: 10.1016/j.tet.2008.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hasse K, Willis A, Banwell M. Eur J Org Chem. 2011:88. references therein. [Google Scholar]
- 24.Gupton J, Giglio B, Eaton J, Rieck E, Smith K, Keough M, Barelli P, Firich L, Hempel J, Smith T, Kanters Tetrahedron. 2009;65:4283. doi: 10.1016/j.tet.2009.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]