

Abstract
Amyotrophic lateral sclerosis (ALS) is a late-onset neurodegenerative disorder characterized by the loss of motor neurons. Fused in sarcoma/translated in liposarcoma (FUS/TLS) and TAR DNA-binding protein (TDP)-43 are DNA/RNA-binding proteins found to be mutated in sporadic and familial forms of ALS. Ectopic expression of human ALS-causing FUS/TLS mutations in Drosophila caused an accumulation of ubiquitinated proteins, neurodegeneration, larval-crawling defect and early lethality. Mutant FUS/TLS localized to both the cytoplasm and nucleus, whereas wild-type FUS/TLS localized only to the nucleus, suggesting that the cytoplasmic localization of FUS/TLS is required for toxicity. Furthermore, we found that deletion of the nuclear export signal strongly suppressed toxicity, suggesting that cytoplasmic localization is necessary for neurodegeneration. Interestingly, we observed that FUS/TLS genetically interacts with TDP-43 in a mutation-dependent fashion to cause neurodegeneration in vivo. In summary, we demonstrate that ALS-associated mutations in FUS/TLS cause adult-onset neurodegeneration via a gain-of-toxicity mechanism that involves redistribution of the protein from the nucleus to the cytoplasm and is likely to involve an interaction with TDP-43.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disease of motor neurons characterized by cytoplasmic ubiquitin-positive inclusions. Recently, TAR DNA-binding protein (TDP)-43 and fused in sarcoma/translocated-in-liposarcoma (FUS/TLS) have been identified as major genes mutated in both familial and sporadic ALS. Familial ALS (FALS) accounts for ∼10% of all cases with mutations in TDP-43 accounting for ∼30–40% of FALS cases and mutations in FUS/TLS accounting for 40–50% of FALS cases (1–4). TDP-43 and FUS/TLS are RNA-binding proteins implicated in multiple aspects of RNA metabolism, including microRNA processing, RNA splicing, trafficking and translation (5). FUS/TLS is a ubiquitously expressed, 526 amino acid nuclear protein with several distinct domains, including an N-terminal domain enriched in glutamine, glycine, serine and tyrosine residues (QGSY region); a glycine-rich region (GRR domain); a RNA recognition motif (RRM domain); multiple arginine/glycine/glycine (RGG) repeats in an arginine- and glycine-rich region; and a C-terminal zinc finger motif. Most of the pathogenic mutations in FUS/TLS are missense mutations in the C-terminal 13 amino acids, where the nuclear localization signal (NLS) is located (3–5). R521H and R521C mutations in FUS/TLS are the most common mutations in human ALS patients (3,4). FUS/TLS shows nuclear and cytoplasmic expression and shuttles between the cytoplasm and the nucleus (6,7). ALS-causing mutations in either TDP-43 or FUS/TLS lead to redistribution of the protein to the cytoplasm from the nucleus (3,4,8). It is still not clear how ALS-causing mutations disrupt FUS/TLS function.
Postmortem analysis of brain and spinal cord tissues from patients with FUS/TLS mutations demonstrated abnormal FUS/TLS cytoplasmic inclusions in neurons and glial cells (3,4). These inclusions were reported to be immunoreactive for FUS/TLS, GRP78/BiP, p62 and ubiquitin, but not for TDP-43, suggesting that neurodegeneration caused by FUS/TLS mutations are independent of TDP-43 pathology (4,9). However, a recent study demonstrated that FUS-immunoreactive ‘skein-like' inclusions are present in a patient with a TDP43 G298S mutation that also stained positive for TDP-43 (10). The structure and distribution pattern of the FUS/TLS inclusions in TDP-43 related ALS patients had significant resemblance to those of TDP43 inclusions observed in ALS patients, suggesting a common pathway involved in causing FUS/TLS and TDP-43-related ALS (10).
Furthermore, proteomic and biochemical approaches combined with deep sequencing demonstrated that FUS/TLS and TDP-43 interact in vitro (11–13). Interestingly, the FUS/TLS interaction with TDP-43 is enhanced by ALS-associated mutations in TDP-43, suggesting that mutations in TDP-43 may perturb FUS/TLS function (13). Although these cell culture-based studies suggest that TDP-43 and FUS/TLS physically and biochemically interact, the physiological relevance of these studies is unclear.
Here, we describe a Drosophila model of FUS/TLS-mediated ALS that recapitulates several key features of human ALS pathology. Targeted expression of mutant human FUS/TLS in Drosophila eyes and motor neurons led to eye degeneration and locomotor dysfunction, respectively. Wild-type (WT) FUS predominantly localized in the nucleus, whereas mutant FUS/TLS was distributed in both the cytoplasm and nucleus, suggesting that cytoplasmic localization is required for causing toxicity. Interestingly, deletion of the nuclear export signal (ΔNES) rescued toxicity associated with mutant FUS/TLS, suggesting that cytoplasmic localization of mutant FUS/TLS is required for causing ALS pathogenesis. Additionally, we found that interaction of WT FUS/TLS with mutant TDP-43 strongly enhances neurodegeneration associated with mutant TDP-43 and occurs in a synergistic manner. Similarly, co-expression of WT TDP-43 with mutant FUS/TLS strongly enhances neurodegeneration associated with mutant FUS/TLS.
RESULTS
Ectopic expression of mutant FUS/TLS leads to neurodegeneration in Drosophila
To investigate the molecular mechanisms of FUS/TLS-related neurodegeneration, we generated transgenic flies over expressing HA-tagged WT or mutant human FUS using the highly versatile UAS/GAL4 system (14). We found that targeted expression of mutant human FUS/TLS (R518K, R521C and R521H) caused severe neurodegeneration in Drosophila eyes characterized by disorganized ommatidia and loss of mechanosensory bristles (Fig. 1A–C), whereas expression of WT human FUS/TLS resulted in very mild eye degeneration (Fig. 1). We did quantitation of the eye phenotype in the flies expressing WT and mutant FUS/TLS (Fig. 1C) using our previously published criteria (15). The eyes appear to be degenerated externally in flies ectopically expressing FUS/TLS containing ALS-associated R518K, R521C and R521H mutations, and this correlates with vacuolar neurodegeneration with severe disruption of the retinal organization and thinning of the retina (Fig. 1B). We used two independent insertion lines for validation of the mutation-dependent phenotype (Supplementary Material, Fig. S1A). To understand the consequences of mutant FUS/TLS in the nervous system, we targeted FUS/TLS expression to neurons using a pan-neuronal driver (appl-GAL4) and found that mutant FUS/TLS (R518K, R521H and R521C) caused pupal lethality, whereas the WT FUS/TLS expressing flies eclosed normally (Fig. 1D). These findings further confirm that the neurons are highly vulnerable to expression of mutant FUS/TLS. Interestingly, ectopic expression of mutant FUS using the Appl-GAL4 (pan-neuronal expression) and OK371-GAL4 (motor neurons expression) caused pupal lethality. We observed a mutation-dependent degenerative phenotype despite almost equal FUS/TLS protein expression levels (Fig. 1E and Supplementary Material, Fig. S1B). Interestingly, we observed a mutation-dependent accumulation of ubiquitin-positive proteins in FUS-expressing animals (Supplementary Material, Fig. S2). Taken together, these results demonstrate that expression of human ALS-related FUS/TLS mutations cause mutation-dependent neurodegenerative phenotypes, consistent with a gain-of-toxicity mechanism that has been proposed for this disease.
Figure 1.

Human ALS-causing mutations in FUS lead to neurodegeneration in Drosophila. (A) Scanning electron micrographs of Day 1 adult fly eyes in which expression of WT or mutant FUS is targeted by the eye-specific promoter GMR-GAL4. The eyes of control flies (GMR-GAL4/+) and flies expressing WT FUS are almost normal and have an organized ommatidial architecture [(A) and (B)]. The eyes of flies expressing mutant FUS R518K or R521H show ommatidial degeneration, partial collapse and loss of eye pigmentation. The eyes of flies expressing mutant FUS R521C are severely degenerated with complete loss of eye pigmentation. (B) Light micrographs of Day 1 adult fly eyes expressing WT or mutant FUS under the control of the GMR-GAL4 driver. Bottom row: corresponding Richardson's stained frontal sections showing a mutation-dependent severe internal degeneration. (C) Quantitative analysis of eye phenotypes. Flies from each genotype were randomly selected for objective scoring according to the criteria described previously (15). Flies carrying a mutated FUS R518K, R521H or R521C all display a more severe phenotype. Values are the means ± SEM. The severity of the phenotype between FUS-WT (*) and each of the individual mutants (**) was significant (P < 0.0001). (D) Ectopic expression of FUS in the fly neurons results in mutation-dependent loss of viability. WT or mutant (R518K, R521C and R521H) FUS was expressed by using the pan-neuronal driver Appl-GAL4. Mean eclosion rates were drastically reduced in pupae with pan-neuronal expression of mutant FUS R521H and no R518K or R521H flies eclosed. However, pupae expressing WT FUS eclosed normally when compared with the control (Appl-GAL4 driver alone). Values are the means ± SEM. Although control (*) and FUS-WT (*) are not significantly different from each other, they are each significantly different from each of the FUS mutants (**) (P < 0.0001). (E) Western blot showing expression levels of WT and mutant forms of FUS in the eyes of Day 1 adult flies (top panel). Tubulin served as a loading control (lower panel).
Conditional expression of mutant FUS/TLS in adult flies causes a severe locomotor defect and increases mortality
Because targeted expression of mutant FUS/TLS in the fly nervous system during development caused pupal lethality, we decided to use the conditional expression system elav-GeneSwitch (elav-GS) to target expression of the WT and mutant FUS/TLS to the neurons of the adult fly (15–17). We determined the effects of FUS/TLS expression on life span of the flies by treating the elavGS/+ (control), elavGS; UAS-FUS WT and elavGS; UAS-FUS R521C with RU486 (20 mm) from Day 1 adult stage on and monitoring their survival rate. There was very low mortality in untreated FUS WT, FUS R521C and driver alone (Fig. 2A and B). However, induction of FUS R521C in adult neurons caused a severe decline in the survival rate after ∼10 days of RU486 treatment and drastically shortened life span when compared with untreated FUS R521C and driver alone. Approximately 50% of the FUS R521C flies were dead within 18 days of RU486 treatment with all of the flies dead in 30 days (average life span 17.4 days). Flies expressing FUS WT started dying after 10 days of RU486 treatment, but their rate of death was significantly slower when compared with the FUS R521C expressing animals (50% of FUS WT flies were dead in 28 days versus 50% of FUS R521C flies were dead after 18 days) (Fig. 2A and B). We found that the overall mutation-dependent reduction in life span was similar in males and females with all of the male flies dead in 29 days. Flies expressing the driver alone did not have a significant difference in life span after exposure to RU-486, as >90% flies were viable after 30 days of RU486 exposure (Fig. 2A and B). These findings further demonstrate a mutation-dependent decline in the life span of the FUS R521C expressing animals. However, WT FUS expressing animals also showed a decline in life span, but the degree of severity was less than mutant FUS animals.
Figure 2.

Conditional expression of mutant FUS in the adult nervous system leads to a severe reduction in life span and climbing ability. Driver lines expressing the transcriptional activator Gene Switch under control of the neuron-specific promoter elav-GS were crossed with either UAS-FUS WT or UAS-FUS R521C flies. Following treatment with RU486, the Gene Switch protein is transcriptionally activated and binds to UAS and thus induces expression of the respective FUS protein specifically in the fly nervous system. (A and B) Lifespan of female and male flies expressing FUS WT and FUS R521C transgenes under the control of elav-GS. The survival curves show the percentage of living flies as a function of age. Neuronal expression of FUS R521C strongly reduces lifespan, whereas FUS WT expression has a significantly weaker effect on the reduction of life span. Control animals (Elav-GS/+) raised and maintained with RU486 (+) or without RU486 (−) showed normal life span with many flies surviving beyond the end of the study. (C) Expression of FUS R521C in the adult fly nervous system caused age-dependent and mutation-dependent locomotor dysfunction. Climbing ability of Elav-GS/+, UAS-FUS WT/+;elavGS/+ and UAS-R521C/+;elavGS/+ flies raised with RU486 (+) or without RU486 (−) were assessed at the indicated time-points. Neuronal expression of FUS R521C caused a severe locomotor defect when compared with age-matched FUS WT animals (<3% of the FUS R521C flies were able to climb 10 cm when compared with 41% of FUS WT expressing flies on Day 15). Control animals (Elav-GS/+) raised and maintained with RU486 (+) and without RU486 (−) showed normal climbing behavior. (D) Western blot analyses of total protein extracts prepared from heads of new born flies in which expression of UAS-FUS was induced (+) or not (−) with RU486 for 1 day and 15 days. As reported previously, a thin band of FUS is detected in the absence of RU486, indicating a leak in the control of expression (21). Expression of both WT and mutant FUS was significantly increased on Day 15. Tubulin was used as a loading control.
Because ALS is an age-related motor neuron disease, we sought to determine the effect of FUS/TLS expression on adult locomotor function using a well-established adult climbing assay that has extensively been used to characterize Drosophila models of neurodegenerative diseases (18,19). We found that induction of FUS/TLS by RU486 from Day 1 adult stage caused an age-dependent decline in climbing ability (Fig. 2C), and this phenotype is more severe in flies expressing mutant FUS/TLS when compared with WT. The FUS R521C flies induced with RU486 started showing impairment in the climbing behavior by Day 10 and at Day 20 <3% of the FUS R521C flies treated with RU486 were able to climb when compared with 41% of the FUS WT flies treated with RU486 (Fig. 2C). As expected, there was no FUS protein expression in Day 1 elav-GS; UAS-FUS WT and elav-GS; UAS-FUS R521C flies before exposure to the inducing agent RU486. However, we observed robust FUS/TLS protein expression in Day 15 elav-GS; UAS-FUS WT and elav-GS; UAS-FUS R521C flies exposed to RU486 (Fig. 2D). We observed a very mild leaky expression of FUS/TLS in Day 15 flies even when not exposed to RU486 as reported previously (20). Collectively, our data demonstrate that expression of mutant FUS/TLS specifically in the neurons of the adult fly leads to a severe decline in life span and a progressive adult locomotor defect. Hence, we developed a conditional Drosophila model of ALS that can potentially serve as a useful system for further investigating the potential mechanisms underlying pathogenesis in ALS, without any confounding effects on neuronal development.
Motor neuron expression of mutant FUS/TLS causes a larval-crawling defect without apparent morphological defects at the neuromuscular junction
To investigate the mechanisms underlying locomotor dysfunction with mutant FUS expression, we targeted expression of FUS/TLS to motor neurons using the OK371-GAL4 driver. We performed a larval-crawling assay to determine the locomotor function of third instar larvae expressing WT and mutant forms of FUS/TLS in motor neurons (22). We found that expression of FUS R518K, FUS R521C and FUS R521H in motor neurons caused a larval-crawling defect when compared with FUS WT and driver alone (Fig. 3A). These observations prompted us to investigate whether the larval-crawling defect could be explained by morphological defects at the neuromuscular junction (NMJ). To determine the morphology of synaptic boutons at the NMJ, we used the cysteine-string protein (CSP) and the discs large (DLG) protein as markers for pre- and post-synaptic compartments, respectively, and counted the number of synaptic boutons at the NMJ. The animals expressing FUS R521C and FUS R521H displayed normal numbers of synaptic boutons with no apparent differences with FUS WT and driver alone (Fig. 3B). We also found that animals expressing FUS R521C and FUS R521H have a similar number of synaptic boutons when compared with FUS WT and driver alone (Fig. 3C). This suggests that the ALS-linked FUS mutations do not disrupt formation of the NMJ.
Figure 3.
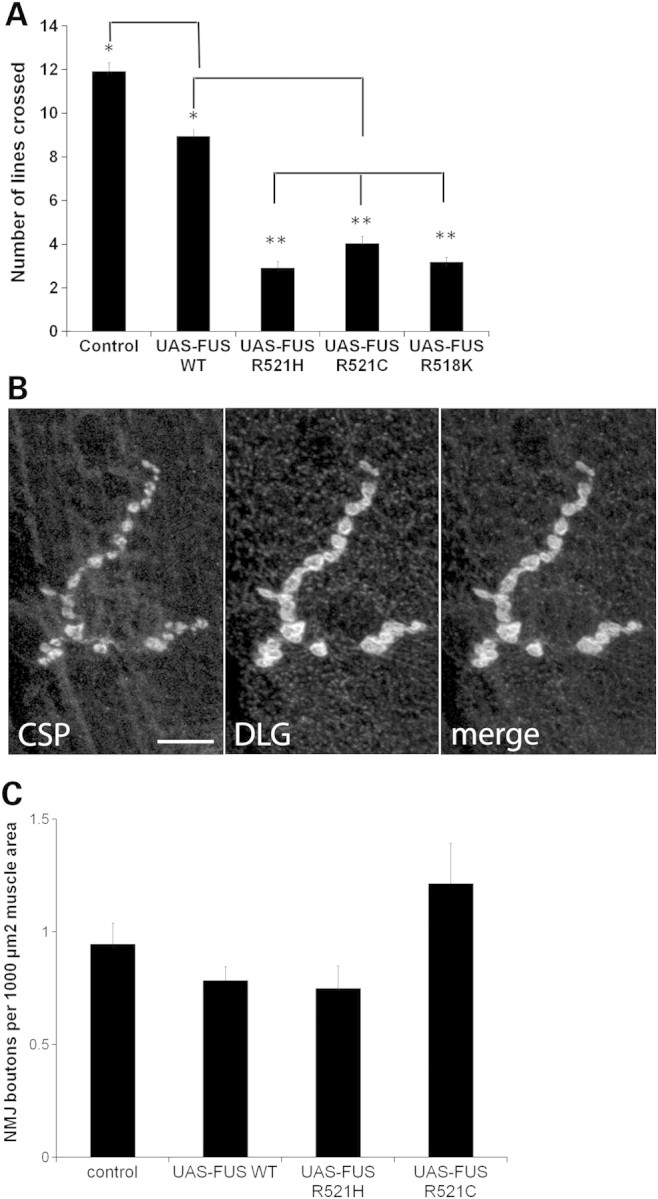
Expression of mutant FUS in motor neurons leads to a larval crawling defect without affecting neuromuscular junction structure. (A) Ectopic expression of FUS R518K, FUS R521C and FUS R521H in motor neurons results in a larval crawling defect when compared with FUS WT expressing animals or driver alone. Values are the means ± SEM. FUS-WT (*) is significantly different from the control (*) (P < 0.0001). Both the control and FUS-WT are significantly different from each of the FUS mutants (**) (P < 0.0001). (B) Muscle 4, segment A3 larval neuromuscular junction of larva expressing FUS R521C under the control of OK371-GAL4 display normal morphology of synaptic boutons as determined by immunohistochemistry with antibodies against pre-synaptic (CSP) and post-synaptic (discs large, DLG) proteins. Scale bar is 10 µm. (C) FUS WT, FUS R521C and FUS R521H expressing animals have a normal number of synaptic boutons when compared with the non-transgenic animals. There is no significant difference between any of the groups. N = 15 synapses from four animals of each genotype.
Deletion of the NES strongly suppresses toxicity associated with mutant FUS/TLS
FUS/TLS is predominantly a nuclear protein and it has been demonstrated that FUS/TLS protein shuttles between the nucleus and cytoplasm (3,4). Abnormal cytoplasmic mislocalization of mutant FUS has been shown in human ALS patients’ neuronal and glial tissues as well as in cell culture-based assays (3,4,23). We reasoned that if cytoplasmic localization of mutant FUS/TLS is an essential step in causing ALS pathogenesis, we should be able to suppress the disease by trapping the mutant FUS/TLS in the nucleus. We deleted the NES to generate an FUS mutant that contained only the NES deletion (FUS ΔNES) as well as double mutants (FUS ΔNES R518K and FUS ΔNES R521C). We observed that targeted expression of FUS ΔNES in Drosophila eyes did not cause any external eye degeneration (Fig. 4A and B). Interestingly, targeted expression of double mutants (FUS ΔNES R518K and FUS ΔNES R521C) also showed almost normal ommatidial architecture externally and internally as evident from light microscopy and histology (Fig. 4A and B). These data demonstrate that the deletion of the conserved NES strongly mitigates toxicity. We validated these findings using three independent transgenic lines as shown in Supplementary Material, Figure S3.
Figure 4.
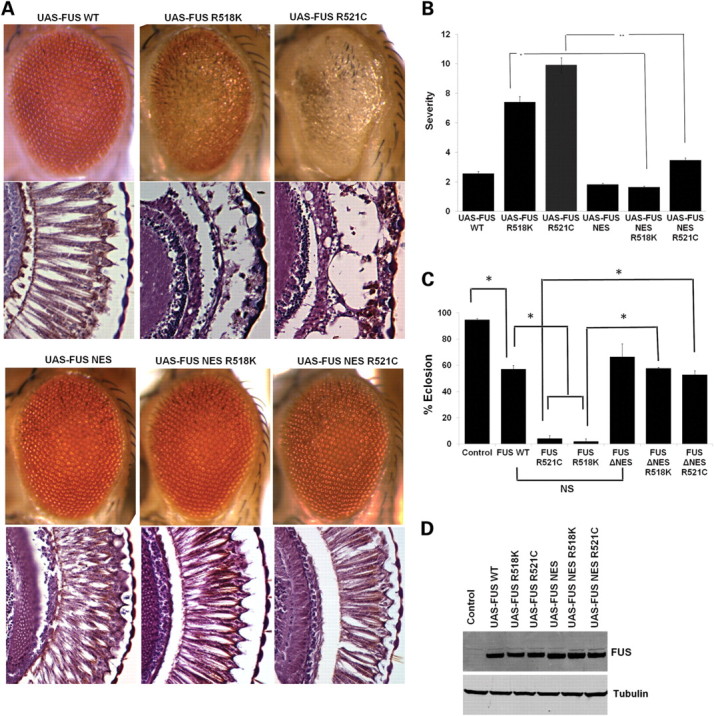
FUS NES deletion (FUS ΔNES) rescues the degenerative phenotype associated with ALS-causing FUS mutations. (A) Light micrographs of eyes from 1-day-old flies in which FUS WT, FUS R518K and FUS R521C were expressed by using the GMR-GAL4 driver. Eyes from the line expressing FUS WT showed almost normal morphology by light microscopy and histology. However, the FUS R518K and FUS R521C lines showed moderate and severe eye degeneration, respectively. Ectopic expression of FUS ΔNES does not cause any external eye degeneration, similar to the FUS-WT line. Strikingly, deletion of the NES strongly suppressed neurodegeneration associated with R518K and R521C mutations. (B) Quantitative analysis of eye phenotypes. Flies from each genotype were randomly selected for objective scoring according to criteria described previously (15). While R518K and R521C induced severe degeneration of the eyes, the combination of these mutations with deletion of the NES resulted in an almost total reduction in severity. Values are the means ± SEM. The single asterisk denotes the significant difference between the indicated pairs (P < 0.001). The double asterisks denote the significant difference between the indicated pairs (P < 0.001). The difference between FUS WT and FUS ΔNES is less significant (P < 0.05). (C) Ectopic expression of FUS in fly neurons results in mutation-dependent loss of viability. WT or mutant (R518K, R521C or R521H) FUS was expressed by using the pan-neuronal driver Appl-GAL4. Deletion of the NES strongly suppressed the mutation-dependent viability defect and flies expressing FUS ΔNES R521C and FUS ΔNES R518K eclosed normally when compared with FUS WT and the driver alone. Mean eclosion rates were drastically reduced in pupae with pan-neuronal expression of either FUS R521C or R518K. Values are the means ± SEM. The single asterisk denotes the significant difference between the indicated pairs (P < 0.01 for each matching). There is no significant difference between FUS WT and FUS ΔNES (NS). (D) Western blot analysis of FUS expression in fly heads from FUS WT, FUS-R518K, FUS-R521C, FUS ΔNES, FUS ΔNES R518K and FUS ΔNES R521C expressing transgenic flies. Tubulin served as a loading control.
Next, we determined whether deletion of the NES could abrogate the pupal lethality associated with neuronal expression of mutant FUS/TLS (Fig. 1D). We found that flies expressing the single mutation (FUS ΔNES) and the double mutations (FUS ΔNES R518K and FUS ΔNES R521C) in neurons eclosed normally, just as the FUS WT and driver alone do (Fig. 4C). These findings suggest that deletion of NES can suppress mutation-dependent FUS/TLS toxicity in Drosophila eyes and neurons. We further determined the expression level of FUS/TLS protein in FUS ΔNES, FUS ΔNES R518K and FUS ΔNES R521C mutants and found that these animals express comparable FUS protein levels with the FUS WT, FUS R518K and FUS R521C (Fig. 4D). These observations suggest that deletion of NES does not destabilize the expression level of FUS/TLS protein. Together, these findings suggest that the NES domain is required for mutation-dependent FUS/TLS toxicity in Drosophila eyes and neurons.
WT FUS/TLS is predominantly nuclear whereas mutant FUS/TLS is also distributed into the cytoplasm. Deletion of the NES restricts FUS/TLS expression to the nucleus
To determine the sub-cellular localization of FUS/TLS, we targeted the expression of FUS WT and FUS R518K transgenes to motor neurons using the driver OK371-GAL4. We used anti-lamin antibody to highlight the nuclear envelope, and thus staining inside the lamin ring is nuclear and outside the ring is cytoplasmic. We found that WT and mutant FUS/TLS display a different sub-cellular localization in Drosophila third instar larvae motor neurons. WT FUS/TLS localized exclusively to the nucleus while FUS R518K was present in the cytoplasm as well as in the nucleus (Fig. 5). WT FUS/TLS (Fig. 5B) can clearly be seen localized to the nucleus while mutant FUS R518K staining is also seen in the cytoplasm, outside of the lamin ring (Fig. 5C, magnified image).
Figure 5.
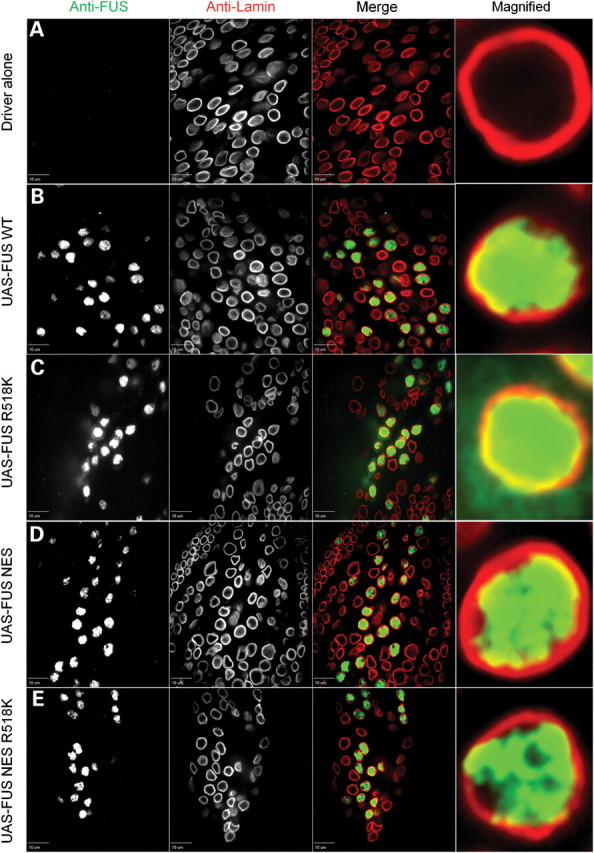
WT human FUS is predominantly nuclear, but mutant FUS is also redistributed to the cytoplasm. FUS mutants with deletion of the NES are retained exclusively in the nucleus. Lamin was used as a nuclear marker to highlight the nuclear envelope membrane, and thus immunostaining inside the lamin ring is nuclear and staining outside is cytoplasmic. (A) OK371-GAL4 (control) third instar larvae stained with both anti-FUS and anti-lamin antibody clearly showed a nuclear envelope membrane. The anti-FUS antibody resulted in a non-specific tiny punctuate staining pattern in control animals mostly outside the nucleus. (B) WT FUS protein was predominantly in the nucleus. (C) FUS R518K, besides the nucleus, was also redistributed to the cytoplasm. FUS R518K staining (C, arrows) was also in the cytoplasm, outside of the lamin ring. (D) When FUS had the NES deleted (FUS ΔNES), FUS protein predominantly localized inside the nucleus similar to the FUS WT alone [compare (B) with (D)]. (E) The NES double-mutant (FUS- ΔNES R518K) was also retained in the nucleus similar to the FUS WT and FUS ΔNES. Genotypes used here are OK371-GAL4/+, OK371-GAL4;UAS-FUS WT, OK371-GAL4;UAS-FUS R18K, OK371-GAL4;UAS-FUS ΔNES and OK371-GAL4;UAS-FUS ΔNES R518K.
Because we observed a strong suppression of mutant FUS/TLS toxicity by deleting the NES, we sought to determine the sub-cellular distribution of FUS/TLS in the FUS ΔNES single and double mutant lines. We observed that when the NES was deleted from FUS WT or FUS R518K, then both FUS ΔNES (Fig. 5D) and the mutant FUS ΔNES R518K (Fig. 5E) were predominantly retained inside the nucleus, suggesting that cytoplasmic localization of mutant FUS/TLS is important for causing ALS pathogenesis. We performed a quantification of cytoplasmic and nuclear localization in motor neurons that showed a significant localization of mutant FUS in the cytoplasm when compared with WT and ΔNES [single and double mutants (Supplementary Material, Fig. S4)].
Genetic interaction between human FUS/TLS and TDP-43
TDP-43 and FUS/TLS are structurally and functionally similar RNA-binding proteins and both proteins have been implicated in ALS pathogenesis. Recent studies demonstrated that ALS-causing mutations in TDP-43 strongly enhance its complex formation with FUS/TLS (24). However, the functional consequences of increased complex formation between mutant TDP-43 and FUS/TLS are still not clear. To investigate these interactions, we co-expressed both of these ALS-causing proteins in Drosophila eyes. Ectopic expression of FUS WT (Fig. 1A) or TDP-43 WT (25) alone does not cause significant external eye degeneration. However, co-expression of both FUS WT and TDP-43 WT leads to moderate eye degeneration in Drosophila (Fig. 6A). Interestingly, ectopic co-expression of FUS WT and TDP-43 M337V led to severe eye degeneration when compared with the FUS WT and TDP-43 WT co-expression. Similarly, co-expression of FUS R521H and TDP-43 WT synergistically enhanced the degenerative phenotype suggesting a possibility that mutation in either of these proteins can exaggerate the neurodegenerative phenotype (Fig. 6). We did a quantitation of eye degeneration using previously published criteria and found that either FUS WT and TDP-43 M337V or FUS R521H and TDP-43 WT interaction enhanced neurodegeneration in a synergistic manner and it was not merely an additive effect (Fig. 6B).
Figure 6.
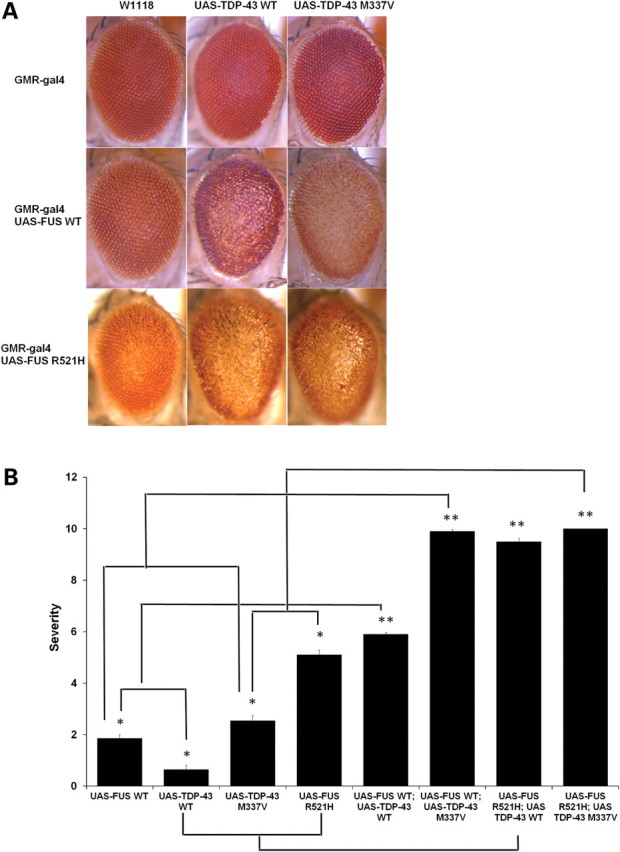
Ectopic expression of WT FUS/TLS enhances neurodegeneration associated with the disease-causing TDP-43 mutation M337V. (A) Light micrographs of Day 1 adult eyes expressing GMR-GAL4/+, TDP-43 or TDP-43 M337V (top panel). The eyes of control flies (GMR-GAL4/+) and flies expressing TDP-43 WT had almost normal and organized ommatidial architecture. The eyes of flies expressing mutant TDP-43 M337V showed mild eye degeneration as reported previously (25). Co-expression of FUS WT and TDP-43 WT caused a rough eye phenotype with ommatidial degeneration, and ommatidial fusion suggesting a synergistic enhancement in the eye degeneration. In addition, co-expression of FUS WT and TDP-43 M337V caused a severe rough eye phenotype (much rougher when compared with the flies co-expressing FUS WT and TDP-43 WT). (B) Quantitative analysis of eye phenotypes. Flies from each genotype were randomly selected for objective scoring according to the criteria described in Materials and Methods. It was clear that coexpression of WT FUS with mutated TDP-43 M337V or co-expression of mutated FUS R521H with WT TDP-43 resulted in a much more severe phenotype than co-expression of WT FUS and WT TDP-43 (based on a scale of 1–10 where 10 is the maximum score). Values are the means ± SEM. The dendrograms illustrate the significant differences between the two genotypes used for the respective crosses (*) with each genotype being significantly different from the other genotype in eye degeneration severity (P < 0.01 in all cases). It also illustrates the significant difference between the resulting cross (**) with each of the genotypes (*) used for the respective cross (P < 0.01 in all cases).
DISCUSSION
Ectopic expression of FUS/TLS carrying ALS-causing mutations leads to neurodegeneration in Drosophila
FUS/TLS and TDP-43, two major genes implicated in ALS pathogenesis, share significant structural and functional similarity. In FUS/TLS, the highly conserved C-terminal region, encoded by exons 14 and 15, appears to be the mutational hotspot associated with ALS (3,4,13,26,27). Although these mutations in FUS/TLS clearly demonstrate that mutations in FUS/TLS lead to ALS pathogenesis, the molecular mechanisms of ALS-causing mutations are still not clear. In particular, because animal models of FUS-related disease have not yet been generated, it is unclear whether mutations in FUS/TLS cause a gain- or loss-of-function. Here, we report the generation and characterization of transgenic flies expressing both WT and ALS-causing mutant forms of human FUS/TLS. Our FUS/TLS transgenic flies have several features strikingly similar to neuropathological characteristics of FUS/TLS proteinopathy and functional defects in these flies have similarities with those in ALS patients. We found several intriguing phenotypes that make our Drosophila model a highly tractable system for dissecting molecular mechanisms of FUS-related ALS (Figs 1 and 2). We showed that ectopic expression of ALS-causing FUS mutations in Drosophila lead to a mutation-dependent neurodegenerative phenotype such as retinal degeneration, locomotor dysfunction and lethality. In addition, overexpression of mutant FUS/TLS caused an accumulation of ubiquitinated proteins, one of the pathological hallmarks of ALS. Therefore, our results recapitulate unique features of pathological FUS/TLS that are hallmarks of ALS. These findings are consistent with a gain-of-toxicity mechanism for FUS-related neurodegenerative disease, similar to that observed in the Drosophila model of TDP-43 proteinopathy, where ectopic expression of a disease protein causes a mutation-dependent toxicity (25).
A conditional model of FUS/TLS-related ALS showed reduced longevity and locomotor defect
We have developed a conditional Drosophila model of FUS/TLS-related ALS using an inducible neuronal driver (Elav-GS) (15,16,22). Over expression of FUS R521C in adult fly neurons drastically reduced lifespan and also led to locomotor dysfunction when compared with FUS WT and driver alone. These findings are consistent with previous reports, showing that conditional expression of neurodegeneration-causing proteins can cause a reduced life span and behavioral abnormalities in Drosophila models of neurodegenerative diseases, such as Alzheimer's disease and polyglutamine diseases (15,28–30). Interestingly, FUS WT expressing animals also showed a decreased life span and an aging-associated climbing defect, although the effect is less severe than that with mutant FUS/TLS, suggesting that WT FUS/TLS is also toxic when overexpressed in the nervous system. These findings are similar to those previously shown, i.e. overexpression of WT Tau or TDP-43 causes neurodegeneration in Drosophila, suggesting that even overexpression of WT protein is sufficient to cause toxicity (31–33). Our observations are consistent with the previously published animal models (flies and mice) of other neurodegenerative disease-related genes such as TDP-43, tau and α-synuclein (18,31,34,35). Thus, our data suggest that overexpression of WT FUS/TLS is also toxic, although significantly less toxic than mutant FUS/TLS and that neuronal tissues are particularly vulnerable to the FUS/TLS levels.
Mutant FUS/TLS-expressing animals develop larval-crawling defect without apparent morphological defects at the NMJ
The Drosophila NMJ is a versatile model for investigating fundamental questions about synapses (36). The development of both Drosophila and vertebrate synapses is similar at the cellular and molecular levels. Several proteins with structural or functional similarity to vertebrate proteins are expressed at developing Drosophila synapses. Abnormal NMJ morphology and behavioral defects have been implicated in many Drosophila models of neurodegenerative diseases such as Alzheimer's disease, Fragile X syndrome, spinal muscular atrophy and tauopathies (37–44).
We found that targeted expression of FUS/TLS in motor neurons leads to a mutation-dependent larval crawling defect in third instar larvae. However, our examination of NMJ structure in a Drosophila model of FUS/TLS-related ALS using the respective pre- and post-synaptic markers, CSP and DLG, revealed normal number of synaptic boutons in both WT and mutant FUS/TLS expressing animals. This suggests that the decline in motor neuron function starts before synaptic retraction at the NMJ (Fig. 3). Future electrophysiological studies will be necessary to understand more thoroughly the functional consequences of mutant FUS/TLS expression in our Drosophila model of ALS.
Deletion of the NES blocks toxicity associated with mutant FUS/TLS
FUS/TLS localizes in the nucleus, but due to ALS-causing mutations, FUS/TLS is found in the nucleus and the cytoplasm in affected neurons. We found that WT FUS/TLS predominantly localizes in the nucleus, whereas FUS/TLS with ALS-causing mutations is found to be in both the cytoplasm as well as remaining in the nucleus (Fig. 6). These findings are consistent with previous reports showing cytoplasmic mislocalization of mutant FUS/TLS in human ALS patients as well as in cell culture models (3,4,45). These findings also extend and confirm observations in TDP-43 proteinopathy models, showing that WT TDP-43 predominantly localized in the nucleus and that mutant TDP-43 is redistributed to the cytoplasm. The cytoplasmic localization of mutant forms of both FUS/TLS and TDP-43 proteins involved in ALS pathogenesis further suggests a common mechanism of disease pathogenesis.
We found that deletion of the NES from mutant FUS/TLS strongly suppressed the degenerative phenotype associated with mutant FUS/TLS. Thus, our data strongly support the hypothesis that ALS-associated mutations in the NLS of FUS cause toxicity by causing the protein to mislocalize in the cytoplasm. Interestingly, deletion of the NES domain of WT FUS/TLS did not cause any protein destabilization as the FUS lines with the NES deletion showed high FUS protein expression levels (Fig. 4C). The NES deletion can also suppress the eclosion rate defect associated with targeted expression of mutant FUS in neurons (Fig. 4D). We observed that WT FUS/TLS overexpression also caused very mild toxicity when expressed in Drosophila eyes, suggesting that ectopic expression of even WT FUS/TLS may cause mild toxicity (Figs 1, 5 and Supplementary Material, Fig. S1). Furthermore, conditional expression of WT FUS/TLS also caused a significant reduction in life span and locomotor function when compared with the driver alone, further suggesting that WT FUS/TLS is also toxic (but less toxic than mutant FUS/TLS). In contrast, ectopic expression of FUS/TLS with only the NES deletion in Drosophila eyes did not cause any external eye degeneration. We tested the toxicities of WT and mutant FUS with multiple tissue-specific drivers and found that ectopic expression of mutant FUS is more toxic when compared with the WT FUS. NES deletion in mutant FUS/TLS lines strongly suppressed the neurodegeneration phenotype, suggesting that the NES deletion can suppress cytoplasmic as well as nuclear toxicity associated with FUS/TLS expression by preventing the nucleocytoplasmic shuttling of FUS. Interestingly, deletion of the NES strongly suppresses the mutant FUS toxicity but has a less pronounced affect on the milder toxicity of WT-FUS. Of note, toxicity of FUS R518K-ΔNES and FUS R521C-ΔNES remains the same as FUS- ΔNES. This observation suggests that all versions of FUS cause a modest nuclear toxicity (perhaps by impairing a nuclear function of FUS such as splicing and repair), but a more severe toxicity is caused by excess cytoplasmic FUS associated with disease-causing mutations. It is difficult to determine whether deletion of the NES domain in FUS/TLS suppresses nuclear toxicity associated with WT FUS/TLS more strongly when compared with suppression of the cytoplasmic toxicity caused by mutant FUS/TLS. These findings further support the hypothesis that cytoplasmic localization of mutant FUS/TLS is an important step in causing ALS pathogenesis and that trapping mutated protein in the nucleus may help in preventing neurodegeneration associated with mutated FUS/TLS. These observations are consistent with previous reports showing that deletion of the NES domain of other proteins can suppress their toxicity involved in causing neurodegenerative diseases (21,41).
ALS-causing mutations in FUS/TLS redistribute it to the cytoplasm and deletion of the NES domain locks mutant FUS/TLS in the nucleus
Previous reports have shown the cytoplasmic mislocalization of mutant FUS/TLS in human ALS patients as well as in cell culture models (3,4,45). But the significance of this mutation-dependent redistribution is still not clear. Our findings are consistent with these previous reports in that in Drosophila WT FUS/TLS predominantly localizes in the nucleus whereas FUS/TLS with ALS-causing mutations is redistributed to both the cytoplasm as well as remaining in the nucleus (Fig. 5). These findings also extend and confirm observations in TDP-43 proteinopathy models, showing that WT TDP-43 predominantly localized in the nucleus and that mutant TDP-43 is redistributed to the cytoplasm (25,31). The cytoplasmic localization of mutant forms of both FUS/TLS and TDP-43 proteins involved in ALS pathogenesis further indicates a common mechanism of disease pathogenesis. However, we cannot determine whether mislocalization of mutant FUS/TLS to the cytoplasm leads to a loss of nuclear function or a gain of cytoplasmic function or both. We further determined whether suppression of neurodegeneration associated with NES deletion in mutant FUS/TLS expressing animals could be explained by sub-cellular localization of the protein. We found that the NES deletion in both WT and mutant FUS/TLS expressing animals leads to predominantly nuclear localization of the protein. Therefore, these findings indicate that, as expected for an NES, deletion of the NES domain of mutant FUS/TLS traps the protein in the nucleus. Nuclear localization of mutant FUS/TLS is sufficient to suppress degenerative phenotypes such as external eye degeneration as well as eclosion rate defect (Fig. 4).
FUS/TLS genetically interacts with TDP-43 and synergistically enhances neurodegeneration in a mutation-dependent manner
It has previously been demonstrated that FUS/TLS and TDP-43 form a complex suggesting the possibility of a common link underlying disease pathogenesis (24). It has also been shown that ALS-causing mutations in TDP-43 strongly enhance the association with WT FUS/TLS, whereas only a small fraction of WT FUS/TLS (<1%) associates with WT TDP-43 (24). However, functional consequences of FUS/TLS and TDP-43 interaction are still not clear. We found that co-expression of WT FUS/TLS and WT TDP-43 causes mild eye degeneration (Fig. 6). More importantly, we observed that co-expression of WT TDP-43 and FUS/TLS containing ALS-causing mutations (R521H or R518K) in Drosophila eyes strongly enhance the neurodegenerative phenotype. Similarly, co-expression of mutant TDP-43 (M337V), which has a mild external eye degeneration phenotype, and WT FUS/TLS, which also has a very mild external eye degeneration phenotype, caused severe external eye degeneration, thus, suggesting that co-expression of WT FUS/TLS or WT TDP-43 with ALS-causing mutations in either of these two proteins strongly exaggerate the neurodegeneration phenotype. Interestingly, we found that the enhancement in the neurodegenerative phenotype with either WT FUS/TLS and mutant TDP-43 or mutant FUS/TLS and WT TDP-43 are not just an additive effect but is a synergistic effect, as evident from our quantitative analysis of eye degenerative phenotype using our previously established criteria (15,25). Because the co-expression of WT TDP-43 with WT FUS caused only a mild external eye degeneration, it is reasonable that enhancement in neurodegeneration seen by co-expression of both proteins in which one is mutated is caused by the mutated protein. Our findings indicate that several potential molecular mechanisms of FUS/TLS and TDP-43 mediated ALS pathogenesis. It is possible that the enhancement in neurodegenerative phenotype caused by co-expression of mutant TDP-43 and WT FUS/TLS (and vice versa) might be mediated by the increased stability of mutant TDP-43 as shown previously (24). Alternatively, genetic interaction between WT FUS/TLS and the dominant mutations in TDP-43 could cause perturbations of the physiological functions of both TDP-43 and FUS/TLS. It is reasonable to hypothesize that ALS-causing mutations in TDP-43 may perturb normal FUS/TLS function and similarly pathological mutations in FUS/TLS may disrupt the normal function of TDP-43, thus providing possible convergence of pathogenic pathways in ALS by mutant TDP-43 and FUS/TLS. Alternatively, it is possible that TDP-43 and FUS/TLS proteins can influence each other's function in a dose-dependent manner. We cannot exclude the possibility that FUS/TLS and TDP-43 are in the same pathway or that they share a parallel pathway with potential intersection that allows them to influence each other's function.
Recently, FUS-positive inclusions have been demonstrated in anterior horn neurons and their neurites in the spinal cord of several sporadic ALS patients, in patients with TDP43 or FUS/TLS mutations, and in several other FALS cases with unknown genetic defects (10). Interestingly, FUS-positive inclusions were not present in ALS patients with superoxide dismutase 1 (SOD1) mutations or in a transgenic mouse mode of SOD1-related ALS (10). It has been suggested that FUS inclusions are involved not only in mutant FUS-linked FALS, but also in sporadic ALS and non-SOD1 FALS. These data also suggest that the pathogenic pathway of mutant SOD1-mediated ALS is largely independent from other forms of ALS, including mutant FUS-linked ALS. Interestingly, Deng et al. were the first to demonstrate co-localization of FUS and TDP43 in neuronal inclusions, raising the possibility that interactions of FUS and TDP43 with other cellular components may be a common mechanism in motor neuron degeneration in SOD1-negative forms of ALS. However, more studies are needed to establish whether TDP-43 and FUS/TLS are the components of protein inclusions in sporadic ALS patients.
Recently, it has been demonstrated that TDP-43 and FUS/TLS form a complex with HDAC6 (46). HDAC6 has recently been identified as an mRNA substrate of TDP-43 and its function is regulated by TDP-43 and FUS/TLS (47). We and others previously demonstrated that ectopic expression of HDAC6 suppresses neurodegeneration associated with spinal and bulbar muscular atrophy, Alzheimer's disease and Parkinson's disease (15,46,48–50). We also demonstrated that HDAC6 functions as a linker between two important protein degradation pathways, i.e. the ubiquitin proteasome system and autophagy, and promotes degradation of misfolded proteins in an autophagy-dependent manner (15,49). Regulation of HDAC6 activity by TDP-43 and FUS/TLS suggests a possibility that mutations in either of these two ALS-causing proteins might disrupt or misregulate HDAC6 function that in turn may lead to ALS pathogenesis. C-terminal fragments of TDP-43 protein have been reported to accumulate in human TDP-43-related ALS patients and animals models of TDP-43 proteinopathy (25,51). It has been shown that the C-terminal fragments of TDP-43 can interact with FUS/TLS and it is possible that the accumulation of C-terminal TDP-43 fragments may compromise TDP-43-FUS/TLS complex function, supporting the hypothesis that TDP-43 and FUS/TLS operate together in a common biochemical pathway. Further studies will be needed to identify molecular mediators of TDP-43 and FUS/TLS interaction and if mutation in either TDP-43 or FUS/TLS can affect their interaction with other interacting partners such as hnRNAPs, Drosha and splicing factors (52,53). Delineation of molecular mechanisms of TDP-43 and FUS/TLS interaction would be an important step towards identifying novel genetic or small molecule targets of TDP-43 and FUS/TLS-mediated ALS, a disorder for which there is currently no available cure.
MATERIALS AND METHODS
Plasmid construction
To generate the pUAST–FUS constructs, human FUS cDNA was excised from pCI-Neo FUS WT, pCI-Neo-FUS R521C and pCI-Neo-FUS R521H vectors by XhoI–XbaI restriction enzyme digestion and sub-cloned into the XhoI–XbaI sites of the pUAST vector. All FUS constructs had an HA tag (YPYDVPDYA) coding sequence previously inserted at their 5′ ends. FUS mutant R518K was generated with the QuikChange II Site-Directed Mutagenesis Kit (Stratagene). The template DNA was pUAST-FUS WT with the primers R518K-F and R518K-R shown in Supplementary Material, Table S1. The manufacturer's suggested protocol was followed. All Table S1. mutations and deletions were verified by sequencing.
For the deletion of the NES domain of human FUS (amino acids VQGLGENVTI) the Phusion Site-Directed Mutagenesis Kit was used with the primers ΔNES-F and ΔNES-R following the suggested protocol. The FUS ΔNES construct was excised from the pCIneo vector and inserted into the same location in pUAST.
The double mutants pUAST-FUS ΔNES R518K and pUAST-FUS ΔNES R521C were derived from pCIneo ΔNES in the following manner. The enzyme EcoRI cuts out a fragment that spans the entire RRM domain, which includes the NES domain. The NES-deleted regions from pCIneo ΔNES were cut in this manner and then inserted into both pUAST-FUS R518K and pUAST R521C vectors from which the EcoRI fragment had been removed. The correct insertion and orientation of the subcloned region containing the ΔNES mutation was verified by sequencing.
Fly culture
All Drosophila stocks were maintained on standard Bloomington medium at 25°C in light/dark-controlled incubators. Flies transgenic for UAS-FUS WT, UAS-FUS R518K, UAS-FUS R521H, UAS-FUS R521C, UAS-FUS ΔNES, UAS-FUS ΔNES R518K and FUS ΔNES R521C were generated by injecting the constructs described above into w1118 embryos using standard techniques at the Best Gene Inc. The UAS-TDP-43 WT and UAS-TDP-43 M337V lines were described previously (25).
Light microscopy of fly eyes
Eye phenotypes of 1-day-old CO2-anesthetized flies were evaluated with an Leica M 205C stereomicroscope and photographed with an Leica DFC420 digital camera. For each genotype and condition 100 to >1000 flies were evaluated and quantitation of eye phenotypes was done as described previously (15).
Scanning electron microscopy
Samples for scanning electron microscopy (SEM) were collected and fixed in 2.5% gluteraldehyde (Ted Pella, Inc.) in PBS and post-fixed for 15–30 min in 1.5% osmium tetroxide (Ted Pella, Inc.) in PBS. Samples were dehydrated in ethanol, washed in hexamethyldisilazane (Ted Pella, Inc.) and dried in a dessicator for 3 days. Specimens were coated with gold:palladium using a Denton DV-503 vacuum evaporator, and analyzed using an AMRAY 1820D scanning electron microscope.
Histology
For histology of fly eyes, heads of the appropriate genotype were collected and fixed in 4% buffered paraformaldehyde in PBS for 2 h at room temperature. Samples were serially dehydrated in ethanol and processed as described previously (15).
Larval crawling assay and eclosion rate assay
Third instar wandering larvae of the indicated genotypes (>30 larvae/genotype) were washed in PBS and placed on a 1% agarose gel in a Petri dish with gridlines spaced at 5 mm. The larvae were allowed to acclimate for 1 min, and the number of grid lines that the posterior end of the larvae passed in 1 min was determined as described previously (22). The eclosion rate of WT and mutant FUS expressing flies were determined as described previously (25).
Life span and adult climbing assays
Flies were crossed in the absence of RU486 on standard fly food. Day 1 adult males and females were separated and transferred on to experimental vials containing fly food mixed with or without RU486 (20 mm) at a density of 25 flies per vial (n > 200). Deaths were scored every other day and flies were transferred to fresh food three times a week. Adult locomotor function was assessed by a previously described method (18,19).
Western blotting
Western blotting was performed as described previously (15). Briefly, three fly heads were used for protein extraction in RIPA buffer followed by sonication and separation on 4–12% NuPage gels (Invitrogen) following the manufacturer's suggested protocol (1× LDS sample buffer with reducing agent and heated at 70°C for 10 min; running buffer of 1× MOPS with antioxidant in the upper chamber). Transfer was done onto PVDF membranes using the iBlotter (Invitrogen). The mouse monoclonal anti-HA antibody (Sigma, cat. # H3663) was used at 1:1500; rabbit polyclonal anti-FUS (Bethyl Labs, cat. # A300-302A) at 1:1000; rabbit polyclonal anti-N-terminal TDP-43 antibody (Proteintech Group, cat. # 10782-2-AP) at 1:1500; mouse monoclonal anti-tubulin (Sigma, cat. # T5168) at 1:10 000; and mouse monoclonal anti-FLAG (Sigma, cat. # F1804) at 1:1000. Secondary antibodies were from Li-Cor Biosciences and were used at 1:15 000. Scanning of western blots was performed using the Odyssey Infrared Imaging System (LI-COR Biosciences).
Immunohistochemistry and NMJ analysis
Wandering third instar larvae were dissected and fixed with 4% formaldehyde for 15–25 min. Antibodies used included anti-FUS (Bethyl Labs, cat. # A300-302A, 1:1000), anti-GFP (Invitrogen, cat. # 3E6,1:250) and anti-HA (Roche, cat # 3F10, 1:125). Mouse monoclonal antibodies anti-DCSP-2 (6D6, 1:250), anti-DLG (4F3, 1:250) and anti-lamin DmO (ADL67.10, at 1:100) were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa, Department of Biology, Iowa City, IA. Double labeling of anti-CSP and anti-DLG was accomplished with isotype-specific Alexa-conjugated secondary antibodies (Jackson Immunohistochemistry). After staining, the larval pelts were mounted in Fluoromount-G (Southern Biotech, cat# 0100-01). Images were captured on a Zeiss LSM510 confocal or Marianas system spinning disc confocal (Intelligent Imaging Innovations, Denver, CO, USA) with a ×63, NA 1.4 oil-immersion objective. Spinning disc confocal images were analyzed with SlideBook 5.0 software (Intelligent Imaging Innovations). All motor neurons imaged were located in the dorsal midline of the ventral ganglion. For NMJ bouton quantitation, the number of synaptic boutons (type 1b) was counted in muscle 4, segments A2 and A3, and the total number of boutons at each synapse was divided by the muscle area.
For sub-cellular localization analysis, all motor neurons imaged and counted were located in the dorsal midline of the ventral ganglion (54). For the quantification, cytoplasmic staining was counted as present when it was as at least two times the background fluorescence. Quantifications were completed in Slidebook 5.0 software.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by The Robert Packard Center for ALS at Johns Hopkins Medical Center (U.B.P. and T.E.L.).
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Dr Christopher Shaw for generously providing us the pCI-Neo-FUS WT, pCI-Neo-FUS R521C and pCI-Neo-FUS R521H plasmids. We would also like to thank Dr Charles Nichols for use of his confocal microscope, the Morphology and Imaging Core at LSU Health Sciences Center in New Orleans for their expert technical assistance and Dr Jibao He at Tulane University for the SEM analysis.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Kabashi E., Valdmanis P.N., Dion P., Spielgelman D., McConkey B.J., Vande Velde C., Bouchard J.P., Lacomblez L., Pochigaeva K., Salachas F., et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- 2.Sreedharan J., Blair I.P., Tripathi V.B., Hu X., Vance C., Rogelj B., Ackerley S., Durnall J.C., Williams K.L., Buratti E. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kwiatkowski T.J., Jr., Bosco D.A., Leclerc A.L., Tamrazian E., Vanderburg C.R., Russ C., Davis A., Gilchrist J., Kasarskis E.J., Munsat T., et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 4.Vance C., Rogelj B., Hortobágyi T., De Vos K.J., Nishimura A.L., Sreedharan J., Hu X., Smith B., Ruddy D., Wright P., et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lagier-Tourenne C., Cleveland D.W. Rethinking ALS: the FUS about TDP-43. Cell. 2009;136:1001–1004. doi: 10.1016/j.cell.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andersson M.K., Stahlberg A., Arvidsson Y., Olofsson A., Semb H., Stenman G., Nilsson O., Aman P. The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol. 2008;9:37–54. doi: 10.1186/1471-2121-9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zinszner H., Sok J., Immanuel D., Yin Y., Ron D. TLS (FUS) binds RNA in vivo and engages in nucleo-cytoplasmic shuttling. J. Cell. Sci. 1997;110:1741–1750. doi: 10.1242/jcs.110.15.1741. [DOI] [PubMed] [Google Scholar]
- 8.Dormann D., Rodde R., Edbauer D., Bentmann E., Fischer I., Hruscha A., Than M.E., Mackenzie I.R., Capell A., Schmid B., Neumann M., Haass C. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010;29:2841–2857. doi: 10.1038/emboj.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Langenhove T., van der Zee J., Sleegers K., Engelborghs S., Vandenberghe R., Gijselinck I., Van den Broeck M., Mattheijssens M., Peeters K., De Deyn P.P., et al. Genetic contribution of FUS to frontotemporal lobar degeneration. Neurology. 2010;74:366–371. doi: 10.1212/WNL.0b013e3181ccc732. [DOI] [PubMed] [Google Scholar]
- 10.Deng H.X., Zhai H., Bigio E.H., Yan J., Fecto F., Ajroud K., Mishra M., Ajroud-Driss S., Heller S., Sufit R., et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann. Neurol. 2010;67:739–748. doi: 10.1001/archneurol.2010.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Freibaum B.D., Chitta R.K., High A.A., Taylor J.P. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J. Proteome Res. 2010;9:1104–1120. doi: 10.1021/pr901076y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sephton C.F., Cenik C., Kucukural A., Dammer E.B., Cenik B., Han Y.H., Dewey C.M., Roth F.P., Herz J., Peng J., et al. Identification of neuronal RNA targets of TDP-43-containing Ribonucleoprotein complexes. J. Biol. Chem. 2011;286:1204–1215. doi: 10.1074/jbc.M110.190884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lagier-Tourenne C., Polymenidou M., Cleveland D.W. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010;19:R46–R64. doi: 10.1093/hmg/ddq137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brand A.H., Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 15.Pandey U.B., Nie Z., Batlevi Y., McCray B.A., Ritson G.P., Nedelsky N.B., Schwartz S.L., DiProspero N.A., Knight M.A., Schuldiner O., et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 16.Osterwalder T., Yoon K.S., White B.H., Keshishian H. A conditional tissue-specific transgene expression system using inducible GAL4. Proc. Natl Acad. Sci. USA. 2001;98:12596–12601. doi: 10.1073/pnas.221303298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Todd P.K., Oh S.Y., Krans A., Pandey U.B., Di Prospero N.A., Min K.T., Taylor J.P., Paulson H.L. Histone deacetylases suppress CGG repeat-induced neurodegeneration via transcriptional silencing in models of fragile X tremor ataxia syndrome. PLoS Genet. 2010;6:1–17. doi: 10.1371/journal.pgen.1001240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feany M.B., Bender W.W. A Drosophila model of Parkinson's disease. Nature. 2000;404:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- 19.Crowther D.C., Kinghorn K.J., Miranda E., Page R., Curry J.A., Duthie F.A., Gubb D.C., Lomas D.A. Intraneuronal Abeta, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer's disease. Neuroscience. 2005;132:123–135. doi: 10.1016/j.neuroscience.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 20.Wu C., Wairkar Y.P., Collins C.A., DiAntonio A. Highwire function at the Drosophila neuromuscular junction: spatial, structural, and temporal requirements. J. Neurosci. 2005;25:9557–9566. doi: 10.1523/JNEUROSCI.2532-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miguel L., Frébourg T., Campion D., Lecourtois M. Both cytoplasmic and nuclear accumulations of the protein are neurotoxic in Drosophila models of TDP-43 proteinopathies. Neurobiol. Dis. 2011;41:398–406. doi: 10.1016/j.nbd.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 22.Batlevi Y., Martin D.N., Pandey U.B., Simon C.R., Powers C.M., Taylor J.P., Baehrecke E.H. Dynein light chain 1 is required for autophagy, protein clearance, and cell death in Drosophila. Proc. Natl Acad. Sci. USA. 2010;107:742–747. doi: 10.1073/pnas.0907967107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tateishi T., Hokonohara T., Yamasaki R., Miura S., Kikuchi H., Iwaki A., Tashiro H., Furuya H., Nagara Y., Ohyagi Y., et al. Multiple system degeneration with basophilic inclusions in Japanese ALS patients with FUS mutation. Acta Neuropathol. 2010;119:355–364. doi: 10.1007/s00401-009-0621-1. [DOI] [PubMed] [Google Scholar]
- 24.Ling S.C., Albuquerque C.P., Han J.S., Lagier-Tourenne C., Tokunaga S., Zhou H., Cleveland D.W. ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc. Natl Acad. Sci., USA. 2010;107:13318–13323. doi: 10.1073/pnas.1008227107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ritson G.P., Custer S.K., Freibaum B.D., Guinto J.B., Geffel D., Moore J., Tang W., Winton M.J., Neumann M., Trojanowski J.Q., et al. TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J. Neurosci. 2010;30:7729–7739. doi: 10.1523/JNEUROSCI.5894-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belzil V.V., Valdmanis P.N., Dion P.A., Daoud H., Kabashi E., Noreau A., Gauthier J. Hince P., Desjarlais A., et al. S2D team. Mutations in FUS cause FALS and SALS in French and French Canadian populations. Neurology. 2009;73:1176–1179. doi: 10.1212/WNL.0b013e3181bbfeef. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sleegers K., Van Broeckhoven C. Motor-neuron disease: Rogue gene in the family. Nature. 2009;458:415–417. doi: 10.1038/458415a. [DOI] [PubMed] [Google Scholar]
- 28.Sofola O., Kerr F., Rogers I., Killick R., Augustin H., Gandy C., Allen M.J., Hardy J., Lovestone S., Partridge L. Inhibition of GSK-3 ameliorates Abeta pathology in an adult-onset Drosophila model of Alzheimer's disease. PLoS Genet. 2010;6:1–18. doi: 10.1371/journal.pgen.1001087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Latouche M., Lasbleiz C., Martin E., Monnier V., Debeir T., Mouatt-Prigent A., Muriel M.P., Morel L., Ruberg M., Brice A., et al. A conditional pan-neuronal Drosophila model of spinocerebellar ataxia 7 with a reversible adult phenotype suitable for identifying modifier genes. J. Neurosci. 2007;27:2483–2492. doi: 10.1523/JNEUROSCI.5453-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marsh J.L., Pallos J., Thompson L.M. Fly models of Huntington's disease. Hum. Mol. Genet. 2003;12:R187–R193. doi: 10.1093/hmg/ddg271. [DOI] [PubMed] [Google Scholar]
- 31.Li Y., Ray P., Rao E.J., Shi C., Guo W., Chen X., Woodruff E.A., 3rd, Fushimi K., Wu J.Y. A Drosophila model for TDP-43 proteinopathy. Proc. Natl Acad. Sci. USA. 2010;107:3169–3174. doi: 10.1073/pnas.0913602107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mudher A., Shepherd D., Newman T.A., Mildren P., Jukes J.P., Squire A., Mears A., Drummond J.A., Berg S., MacKay D., et al. GSK-3 beta inhibition reverses axonal transport defects and behavioural phenotypes in Drosophila. Mol. Psychiatry. 2004;9:522–530. doi: 10.1038/sj.mp.4001483. [DOI] [PubMed] [Google Scholar]
- 33.Chee F.C., Mudher A., Cuttle M.F., Newman T.A., MacKay D., Lovestone S., Shepherd D. Over-expression of tau results in defective synaptic transmission in Drosophila neuromuscular junctions. Neurobiol. Dis. 2005;20:918–928. doi: 10.1016/j.nbd.2005.05.029. [DOI] [PubMed] [Google Scholar]
- 34.Hanson K.A., Kim S.H., Wassarman D.A., Tibbetts R.S. Ubiquilin modifies TDP-43 toxicity in a Drosophila model of amyotrophic lateral sclerosis (ALS) J. Biol. Chem. 2010;285:11068–11072. doi: 10.1074/jbc.C109.078527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams S.J., Crook R.J., Deture M., Randle S.J., Innes A.E., Yu X.Z., Lin W.L., Dugger B.N., McBride M., Hutton M., et al. Overexpression of wild-type murine tau results in progressive tauopathy and neurodegeneration. Am. J. Pathol. 2009;175:1598–1609. doi: 10.2353/ajpath.2009.090462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lloyd T.E., Taylor J.P. Flightless flies: Drosophila models of neuromuscular disease. Ann. N. Y. Acad. Sci. 2010;1184:E1–E20. doi: 10.1111/j.1749-6632.2010.05432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pepper A.S., Beerman R.W., Bhogal B., Jongens T.A. Argonaute2 suppresses Drosophila fragile X expression preventing neurogenesis and oogenesis defects. PLoS One. 2009;4:1–9. doi: 10.1371/journal.pone.0005361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang H.C., Dimlich D.N., Yokokura T., Mukherjee A., Kankel M.W., Sen A., Sridhar V., Fulga T.A., Hart A.C., Van Vactor D., Artavanis-Tsakonas S. Modeling spinal muscular atrophy in Drosophila. PLoS One. 2008;3:1–18. doi: 10.1371/journal.pone.0003209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gunawardena S., Goldstein L.S. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron. 2001;32:389–401. doi: 10.1016/S0896-6273(01)00496-2. [DOI] [PubMed] [Google Scholar]
- 40.Chee F., Mudher A., Newman T.A., Cuttle M., Lovestone S., Shepherd D. Overexpression of tau results in defective synaptic transmission in Drosophila neuromuscular junctions. Biochem. Soc. Trans. 2006;34:88–90. doi: 10.1042/BST0340088. [DOI] [PubMed] [Google Scholar]
- 41.Al-Ramahi I., Pérez A.M., Lim J., Zhang M., Sorensen R., de Haro M., Branco J., Pulst S.M., Zoghbi H.Y., Botas J. dAtaxin-2 mediates expanded Ataxin-1-induced neurodegeneration in a Drosophila model of SCA1. PLoS Genet. 2007;3:2551–2564. doi: 10.1371/journal.pgen.0030234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Vos K.J., Grierson A.J., Ackerley S., Miller C.C. Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci. 2008;31:151–173. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- 43.Duncan J.E., Goldstein L.S. The genetics of axonal transport and axonal transport disorders. PLoS Genet. 2006;2:1275–1284. doi: 10.1371/journal.pgen.0020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Falzone T.L., Gunawardena S., McCleary D., Reis G.F., Goldstein L.S. Kinesin-1 transport reductions enhance human tau hyperphosphorylation, aggregation and neurodegeneration in animal models of tauopathies. Hum. Mol. Genet. 2010;19:4399–4408. doi: 10.1093/hmg/ddq363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ito D., Seki M., Tsunoda Y., Uchiyama H., Suzuki N. Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann. Neurol. 2011;69:152–162. doi: 10.1002/ana.22246. [DOI] [PubMed] [Google Scholar]
- 46.Kim S.H., Shanware N.P., Bowler M.J., Tibbetts R.S. Amyotrophic lateral sclerosis-associated proteins TDP-43 and FUS/TLS function in a common biochemical complex to co-regulate HDAC6 mRNA. J. Biol. Chem. 2010;28:34097–34105. doi: 10.1074/jbc.M110.154831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fiesel F.C., Voigt A., Weber S.S., Van den Haute C., Waldenmaier A., Görner K., Walter M., Anderson M.L., Kern J.V., Rasse T.M., et al. Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6. EMBO J. 2010;29:209–221. doi: 10.1038/emboj.2009.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Du G., Liu X., Chen X., Song M., Yan Y., Jiao R., Wang C.C. Drosophila histone deacetylase 6 protects dopaminergic neurons against {alpha}-synuclein toxicity by promoting inclusion formation. Mol. Biol. Cell. 2010;21:2128–2137. doi: 10.1091/mbc.E10-03-0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee J.Y., Koga H., Kawaguchi Y., Tang W., Wong E., Gao Y.S., Pandey U.B., Kaushik S., Tresse E., Lu J., et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010;29:969–980. doi: 10.1038/emboj.2009.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pandey U.B., Batlevi Y., Baehrecke E.H., Taylor J.P. HDAC6 at the intersection of autophagy, the ubiquitin-proteasome system and neurodegeneration. Autophagy. 2007;3:643–645. doi: 10.4161/auto.5050. [DOI] [PubMed] [Google Scholar]
- 51.Neumann M., Sampathu D.M., Kwong L.K., Truax A.C., Micsenyi M.C., Chou T.T., Bruce J., Schuck T., Grossman M., Clark C.M., McCluskey L.F., et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 52.Allemand E., Gattoni R., Bourbon H.M., Stevenin J., Cáceres J.F., Soret J., Tazi J. Distinctive features of Drosophila alternative splicing factor RS domain: implication for specific phosphorylation, shuttling, and splicing activation. Mol. Cell. Biol. 2001;4:1345–1359. doi: 10.1128/MCB.21.4.1345-1359.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gregory R.I., Yan K.P., Amuthan G., Chendrimada T., Doratotaj B., Cooch N., Shiekhattar R. The microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–240. doi: 10.1038/nature03120. [DOI] [PubMed] [Google Scholar]
- 54.Smith R., Taylor J.P. Dissection and Imaging of Active Zones in the Drosophila Neuromuscular Junction. J. Vis. Exp. 2011 doi: 10.3791/2676. . Aailable at: http://www.jove.com/details.stp?id=2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.