

Abstract
Summary: We present BioBlend, a unified API in a high-level language (python) that wraps the functionality of Galaxy and CloudMan APIs. BioBlend makes it easy for bioinformaticians to automate end-to-end large data analysis, from scratch, in a way that is highly accessible to collaborators, by allowing them to both provide the required infrastructure and automate complex analyses over large datasets within the familiar Galaxy environment.
Availability and implementation: http://bioblend.readthedocs.org/. Automated installation of BioBlend is available via PyPI (e.g. pip install bioblend). Alternatively, the source code is available from the GitHub repository (https://github.com/afgane/bioblend) under the MIT open source license. The library has been tested and is working on Linux, Macintosh and Windows-based systems.
Contact: enis.afgan@unimelb.edu.au
1 INTRODUCTION
With the continuous influx of high-throughput genomic sequencing data, automation of complex analyses has become essential to reduce repetitive effort by researchers. Genomics increasingly involves running computationally intensive analyses, each of which may take days, over large numbers of samples. These analyses use a broad landscape of tools and usually require the skills of both biologists and bioinformaticians.
Galaxy (Nekrutenko and Taylor, 2012) is a popular application for bioinformatics analysis. It provides a web-based interface that allows interactive analysis and visualization of large complex data while automatically tracking all the analysis steps. It is thus an excellent tool for designing analyses, recording provenance and facilitating collaboration between bioinformaticians and biologists. However, the graphical point-and-click interface is less than ideal for the execution of the resulting pipelines over large numbers of samples, or for handling complex, sample-dependent workflow logic. To remedy this, Galaxy also offers a programmatic API. This makes it possible to (i) use Galaxy to design an analysis in a visual and integrated setting; (ii) automate reuse of the created pipeline in a flexible scripted manner; and (iii) retain all of the results in Galaxy’s interactive environment.
In addition to a framework for composing pipelines, there is a need for computational infrastructure capable of doing the processing and data storage in a scalable manner. Galaxy supports the notion of executing entirely within a cloud computing environment via the CloudMan platform (Afgan et al., 2011). The CloudMan platform enables a complete deployment of Galaxy, including the Galaxy application itself, the underlying bioinformatics command-line tools and the reference data, to be easily provisioned and managed on a cloud infrastructure (Afgan et al., 2010). CloudMan works with Amazon, OpenStack, OpenNebula and Eucalyptus-based clouds and can be used with applications other than Galaxy. CloudMan also exposes an API through which it can be programmatically manipulated, allowing provisioning and scaling of the compute platform for automated parallelized pipeline processing.
Currently, the Galaxy and CloudMan APIs are available as HTTP-based REST interfaces, which are arguably difficult for bioinformaticians to discover and interact with. Most bioinformaticians are comfortable writing automation scripts in a high-level scripting language. They are typically much less interested in writing direct HTTP requests or using low-level constructs. With that in mind, we have developed a Python library for Galaxy and CloudMan, called BioBlend that provides a high-level interface for interacting with the two applications. This promotes faster interaction with those applications, facilitates reuse and sharing of scripts and eases collaboration between bioinformaticians and biologists. Extensive API documentation is provided for the library while the source code repository contains specific examples.
2 METHODS
The BioBlend library is implemented in Python and provides Python bindings for Galaxy and CloudMan. The library needs to be installed on a local system and imported into a runtime environment (or a script) before use. The library functionality is then available via regular method calls on objects. When invoked, the methods return data as Python dictionaries, encapsulating the return state of the method invocation.
Structurally, at the top-most level, the library is divided into two main modules, representing the two applications, Galaxy and CloudMan. Within the module for each application, further logical structure has been created to match the concepts and functionality available within the application. Figure 1 illustrates this structure.
Fig. 1.
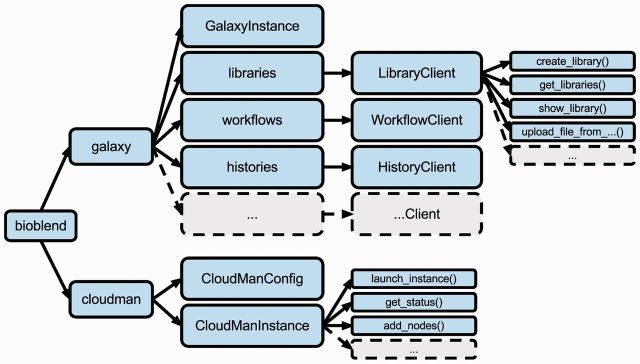
Logical structure of BioBlend library modules
The CloudMan bindings allow cloud resources to be provisioned and managed. By default, this will happen on the Amazon EC2 infrastructure but support for other clouds exists. For instance, with the following code, we launch a new CloudMan platform:

The launch_instance method is a blocking call (configurable), which will return once the CloudMan instance is provisioned and ready for use. Next, we enable CloudMan’s auto-scaling feature, which will keep the size of the compute infrastructure proportional to the given workload:

The Galaxy bindings allow users to import data, user histories or workflows to Galaxy, and to interact with existing objects. For instance, we can import a workflow that has been archived as JSON using

where my_workflow will be returned as a Python dictionary encapsulating the imported workflow’s state.
We can import data from local or remote systems. For instance, to import data into a Galaxy Data Library from a URL:
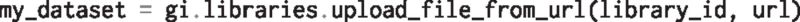
or from the local machine:
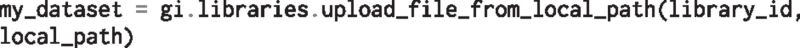
This will return a dictionary encapsulating the dataset parameters. We can then execute the workflow using:

This call mirrors the structure of the data types used by the underlying Galaxy REST API. It will call the imported workflow with the imported data as input, and the Galaxy workflow engine will execute the workflow’s tasks in a correct and, if possible, parallelized order, taking advantage of the parallel compute infrastructure. The output files can be viewed in or retrieved from the persistent user-owned History, which, in this case, we have created and named Example output. In a scripting context, we can of course scale this up to high-throughput or more complex analysis easily. The online documentation includes more complete examples. As calling run_workflow submits a series of jobs to Galaxy and returns immediately, we can leave job queuing, parallelization and dependency checking to the workflow engine and write our scripts in a straightforward procedural manner.
3 DISCUSSION
Pipeline automation is becoming a necessity to reduce repetitive effort performed by bioinformaticians [e.g. Ruffus (http://code.google.com/p/ruffus/), Nesoni (http://vicbioinformatics.com/nesoni.shtml)]. The high-level API made available by BioBlend allows researchers to combine the flexibility and automation of a scripting language with the accessibility of Galaxy’s environment and CloudMan-managed infrastructure. This makes it possible for a bioinformatician to perform a scripted analysis and then to follow it up with a biologist in the Galaxy setting, leveraging Galaxy’s capabilities for interactive analysis, visualization and direct publishing of data or workflows. This fills an increasingly important gap between the need to make analyses accessible and reproducible, and the need for an automated and flexible analysis environment that handles all the corner cases of a research project.
As a scripting library, BioBlend opens the door for the automation of not only pipeline processing but also infrastructure provisioning and management. As a result, it is uniquely positioned to streamline pipeline automation and to become a standard library for interacting with CloudMan and Galaxy: CloudMan can be used to provision and manage the infrastructure required to run a high-throughput analysis, whereas all the processing is done through Galaxy. At the end of a run, the compute infrastructure can be automatically released, whereas all the data and performed steps are retained within Galaxy, enabling easy reuse, visual interaction, sharing and further analysis. Furthermore, it is possible to envision BioBlend as a first step toward defining a Galaxy Shell, which would allow a more integrated access to Galaxy internals for advanced usage.
Since the release of BioBlend, two new complementary libraries based on BioBlend have emerged from the community: Blend4j (https://github.com/jmchilton/blend4j) and clj-blend (https://github.com/chapmanb/clj-blend). These provide comparable functionality but for different languages, Java and Clojure, respectively. BioBlend is also demonstrated in production use in BioCloudCentral (http://biocloudcentral.org/), where BioBlend is used to launch CloudMan clusters on different clouds.
ACKNOWLEDGEMENTS
Galaxy, CloudMan and BioBlend have been developed by the Galaxy (http://wiki.galaxyproject.org/GalaxyTeam) and LSCC teams (http://www.vlsci.org.au/page/lscc).
Funding: This work was, in part, supported by the Genomics Virtual Laboratory (GVL) grant from the National eResearch Collaboration Tools and Resources (NeCTAR).
Conflict of interest: None declared.
REFERENCES
- Afgan E, et al. Galaxy CloudMan: delivering cloud compute clusters. BMC Bioinformatics. 2010;11(Suppl. 12):S4. doi: 10.1186/1471-2105-11-S12-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afgan E, et al. Harnessing cloud computing with Galaxy Cloud. Nat. Biotech. 2011;29:972–974. doi: 10.1038/nbt.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrutenko A, Taylor J. Next-generation sequencing data interpretation: enhancing reproducibility and accessibility. Nat. Rev. Genet. 2012;13:667–672. doi: 10.1038/nrg3305. [DOI] [PubMed] [Google Scholar]