

Abstract
Purpose.
Progressive decline of psychophysical cone-mediated measures has been reported in type 1 (USH1) and type 2 (USH2) Usher syndrome. Conventional cone electroretinogram (ERG) responses in USH demonstrate poor signal-to-noise ratio. We evaluated cone signals in USH1 and USH2 by recording microvolt level cycle-by-cycle (CxC) ERG.
Methods.
Responses of molecularly genotyped USH1 (n = 18) and USH2 (n = 24) subjects (age range, 15–69 years) were compared with those of controls (n = 12). A subset of USH1 (n = 9) and USH2 (n = 9) subjects was examined two to four times over 2 to 8 years. Photopic CxC ERG and conventional 30-Hz flicker ERG were recorded on the same visits.
Results.
Usher syndrome subjects showed considerable cone flicker ERG amplitude losses and timing phase delays (P < 0.01) compared with controls. USH1 and USH2 had similar rates of progressive logarithmic ERG amplitude decline with disease duration (−0.012 log μV/y). Of interest, ERG phase delays did not progress over time. Two USH1C subjects retained normal response timing despite reduced amplitudes. The CxC ERG method provided reliable responses in all subjects, whereas conventional ERG was undetectable in 7 of 42 subjects.
Conclusions.
Cycle-by-cycle ERG showed progressive loss of amplitude in both USH1 and USH2 subjects, comparable to that reported with psychophysical measures. Usher subjects showed abnormal ERG response latency, but this changed less than amplitude with time. In USH syndrome, CxC ERG is more sensitive than conventional ERG and warrants consideration as an outcome measure in USH treatment trials.
Keywords: cone function, microvolt electroretinogram, Usher syndrome, Usher genes
Cone electroretinography signals in Usher (USH) syndrome demonstrate poor signal-to-noise ratio when measured by standard methodology. The cycle-by-cycle ERG provides for a more reliable measurement with potential as an indicator of disease progression and as an outcome measure in USH treatment trials.
Introduction
Human Usher syndrome (USH) is characterized by congenital or early age bilateral sensorineural deafness and later onset of visual acuity loss, impaired night vision and constricted visual field due to co-occurring retinitis pigmentosa (RP). Usher syndrome is the most common cause of deaf-blindness and the most frequent form of recessively inherited RP,1–3 comprising 18% of all RP.4 Three clinical subtypes can be recognized based on the onset age and severity of hearing loss and RP symptoms. Usher syndrome type 1 is the most severe, with deafness at birth and prepubertal RP onset, with most subjects showing vestibular dysfunction. Usher syndrome type 2 causes less severe hearing loss, and RP is diagnosed somewhat later, usually during puberty.2,3 Usher syndrome type 3 subjects show a progressive deafness and age of RP onset in the second to fourth decade, along with variable vestibular dysfunction.1,4 Usher syndrome is genetically heterogeneous, and 10 causative genes have been identified thus far. The six USH1 genes (and the proteins they encode) are MYO7A (myosin VIIA, MIM 276900), USH1C (harmonin, MIM 276904), CDH23 (cadherin 23, MIM 601067), PCDH15 (protocadherin 15, MIM 605514 [602083 is MIM for USH1F], similar issues other genes), SANS (SANS, MIM 606943), and CIB2 (CIB2, MIM 614869). There are three USH2 genes: USH2A (usherin, MIM 276901), GPR98 (G protein-coupled receptor 98, MIM 605472), and WHRN (whirlin, MIM 607696) while mutations of USH3A (clarin-1, MIM 276902) are responsible for type 3.5–8 These 10 different “USH proteins” are each required for the normal function of hair cells in the inner ear and retinal photoreceptors. In the photoreceptors, many of the Usher proteins have been immunolocalized to synaptic terminals of photoreceptors and to the pericilliary membrane zone adjacent to the connecting cilium where they may be involved in flux of molecules between the inner and outer segments. The localization of the Usher protein interactome to two common intracellular sites may explain why the retinal phenotype is similar across different genetic and clinical types of USH.5–8
The time course of USH retinal degeneration has been characterized for several of the genetic subtypes9,10 and is generally described as having rapid functional and anatomic loss of rods followed by relatively slower loss of cones. The onset and rate of clinical disease progression differ among subtypes, and the onset typically is earlier for USH type 1 (USH1) compared with types 2 (USH2) and 3 (USH3). The rate of cone functional loss in USH has been evaluated mainly by psychophysical methods (e.g., Jacobson et al.11), with annual progression in the range of 10% to 14% for both USH1 and USH2. Although cone-mediated electroretinogram (ERG) responses have been used successfully to monitor progressive functional loss in many types of RP (e.g., Berson et al.12), the ERG is used less frequently to track progression in USH. One factor limiting the use of ERG is the frequent severe reduction of amplitudes early in the disease course. For example, the cone flicker ERG can be affected so severely that it is difficult to isolate signal from noise. Seeliger et al.13 recorded 33-Hz flicker Ganzfeld ERGs and found a major amplitude loss and timing phase delays in USH2 subjects, whereas USH1 subjects showed amplitude losses but retained normal phase, leading the authors to suggest using flicker ERG phase to differentiate USH1 from USH2 subjects. Sandberg et al.14 reported the disease course of cone dysfunction in USH2 by recording cone flicker ERGs with the aid of digital filtering and extensive averaging and found that cone flicker ERG amplitude declined at 13% per year, similar to that observed for cone function in psychophysical studies of USH1 and USH2 subjects.15
The problem remains, however, that recording cone flicker ERG is difficult in USH subjects. We addressed this issue by exploring a method for recording small-amplitude flicker ERG using “CxC analysis” with discrete Fourier analysis to isolate response harmonic components.16 This (1) provides a robust statistical estimate of the measurement uncertainty, (2) is applicable to any periodic flicker response, and (3) appears particularly useful for evaluating quite small responses from advanced retinal degeneration. This study evaluated flicker ERG recordings in molecularly genotyped USH1 and USH2 subjects using CxC analysis of the fundamental harmonic component to quantify retinal cone function. We also evaluated rate of change to compare the ERG temporal properties between the two groups of subjects.
Methods
Subjects
Forty-two subjects (age 15–69 years) affected by USH1 (n = 18) or USH2 (n = 24), with identified genotypes (Table) were included in the study. All subjects were seen as part of National Eye Institute protocol 05-EI-0096/clinicaltrials.gov NCT00106743. The study protocol conformed to the tenets of the Declaration of Helsinki and was approved by the National Institutes of Health Combined Neuroscience institutional review board. Written informed consent was obtained from all adult subjects and the parents of minor subjects. Molecular genotyping was performed for all subjects, and two mutant alleles were found for 36 of 42 subjects (Table). All subjects underwent complete eye examination including Early Treatment Diabetic Retinopathy Study (ETDRS) visual acuity, Goldmann visual field, dilated fundus examination, and flash ERGs recorded according to International Society for Clinical Electrophysiology of Vision (ISCEV) standard protocols.17 Protocol participants with ISCEV 30-Hz flicker amplitudes less than 15 μV also had CxC ERG recorded. Diagnosis of RP was based on nyctalopia, visual field constriction, observable retinal pigmentary changes, and an abnormal ERG. Sensorineural hearing loss was confirmed and characterized by audiometry. Subjects were asked about the age when they first noticed vision problems of nyctalopia or visual field constriction. This was considered as the age of disease onset, and disease duration was calculated as time between onset of visual symptoms and examination date.
Table.
Demographics, Genotype, and ERG Results for the Study Population
|
PT |
Age, y |
Sex |
Presentation Age, y |
Onset of Night Blindness |
Gene* |
Mutation 1 |
Mutation 2 |
Acuity |
VF@ |
ERG Amplitude |
ERG Phase† |
| 1 | 68 | F | 23 | 23 | USH2A | c.2299delG; p.E767SfsX21 | c.2299delG; p.E767SfsX21 | HM | V | 0.95 (0.10) | 120 (10) |
| 2 | 64 | F | 27 | - | USH1C * | c.216G>A | c.216G>A | 0.6 | IVB | 4.16 (0.10) | 359 (8.4) |
| 3 | 57 | F | 26 | 21 | USH2A | c.1760_1762delACA; p.N587del | c.13316C>T; p.T4439I | 0.25 | V | 0.32 (0.02) | 255.4 (20.9) |
| 4 | 49 | M | 16 | 12 | USH2A | c.4338_4339delCT; p.C1447QfsX29 | c.4338_4339delCT; p.C1447QfsX29 | 0.625 | IVB | 0.95 (0.15) | 347.8 (2.9) |
| 5 | 61 | F | 29 | 20 | USH2A* | c.2276G>T; p.C759F | c.9815C>T; p.P3272L | 0.625 | IVB | 1.33 (0.52) | 107 (9.3) |
| 6 | 46 | F | 16 | 9 | PCDH15* | c.733C>T; p.R245X | c.2785C>T; p.R929X | 0.1 | IVB | 0.80 (0.07) | 55 (18.6) |
| 7 | 55 | F | 30 | 30 | GPR98 | c.13538_13541delCCTT; p.P4513LfsX4 | c.17668_17669delAT; p.M5890VfsX10 | 0.25 | V | 1.01 (0.27) | 350 (33.4) |
| 8 | 42 | F | 5 | 5 | PCDH15* | c.733C>T; p.R245X | c.2785C>T; p.R929X | 0.25 | V | 0.74 (0.30) | 50 (17.7) |
| 9 | 62 | F | 19 | 53 | CDH23* | c.5237G>A, p.1746Q | c.7872G>A | 0.08 | V | 1.14 (0.05) | 348 (14.5) |
| 10 | 35 | M | 10 | 6 | PCDH15* | c.733C>T; p.R245X | c.733C>T; p.R245X | HM | V | 0.91 (0.18) | 55 (32) |
| 11 | 59 | F | 37 | 10 | MYO7A | c.5392C>T, p.Q1798X | c.4951G>A; p.G1651S | 0.3 | V | 2.02 (0.06) | 350 (10.8) |
| 12 | 38 | M | 2 | 6 | SANS | c.113G>A; p.W38X | c.113G>A; p.W38X | LP | V | 0.48 (0.30) | 320 (3.7) |
| 13 | 41 | M | 24 | 16 | USH2A* | c.2299delG; p.E767SfsX21 | c.2299delG; p.E767SfsX21 | 0.5 | V | 1.38 (0.29) | 360 (2.6) |
| 14 | 54 | M | 29 | 14 | USH2A* | c.5018T>C, p.L1673P | c.8682-9A>G | 0.08 | IVB | 1.41 (0.10) | 27.1 (7.1) |
| 15 | 44 | F | - | - | USH2A | c.5018T>C, p.L1673P | c.8682-9A>G | 0.625 | IVB | 0.83 (0.33) | 342 (15.6) |
| 16 | 24 | M | 13 | 2 | CDH23 | c.6933delT; p.T2313PfsX60 | c.2135_2136delCC; p.S712FfsX23 | 0.4 | IVA | 2.4 (0.44) | 41.1 (5.8) |
| 17 | 31 | F | 13 | 11 | CDH23 | c.3016G>A; p.E1006K | c.3369+1G>A | 0.25 | V | 1.68 (1.26) | 133.3 (111) |
| 18 | 38 | F | 17 | 16 | USH2A | c.9371+1G>C | c.15063_15081del19insGC; p.T5022QfsX150 | 0.16 | V | 1.09 (0.14) | 63.6 (3.8) |
| 19 | 51 | M | 40 | - | GPR98* | c.4919delA; p.Q1640RfsX6 | c.17668_17669delAT; p.M5890VfsX10 | 0.5 | IVB | 3.42 (0.22) | 355 (12.0) |
| 20 | 36 | F | 29 | 20 | USH2A* | c.4124C>T, p.S1375L | - | 0.05 | III | 27 (2.7) | 101.7 (0.81) |
| 21 | 25 | F | 18 | 18 | CDH23* | c.5893C>T; p.Q1965X | - | 0.6 | IVA | 2.61 (0.48) | 115 (14.4) |
| 22 | 41 | F | 46 | 40 | PCDH15* | c.4733_4736delTCAG; p.V1578AfsX6 | - | 0.25 | II | 3.7 (0.22) | 44.2 (11.6) |
| 23 | 20 | M | 13 | 13 | USH2A* | c.2299delG; p.E767SfsX21 | c.1724G>A; p.C575Y | 1.25 | III | 5.61 (0.38) | 90.1 (11.5) |
| 24 | 24 | F | 17 | 14 | USH2A | c.2299delG; p.E767SfsX21 | c.1724G>A; p.C575Y | 0.8 | III | 0.84 (0.19) | 15.26 (2.6) |
| 25 | 27 | F | 26 | 21 | USH2A | c.2299delG; p.E767SfsX21 | c.1036A>C; p.N346H | 1.0 | I | 3.2 (0.23) | 106.8 (19.5) |
| 26 | 28 | M | 10 | 10 | MYO7A | c.285+2T>C | c.4489G>C; p.G1497R | 0.5 | IVB | 3.64 (0.11) | 349.4 (9.9) |
| 27 | 39 | F | 36 | 12 | USH2A | c.6601C>T; p.Q2201X | - | 0.8 | IVB | 1.6 (0.48) | 355.5 (33.5) |
| 28 | 15 | F | - | 4 | CDH23 | c.8917G>T, p.E2973X | c.8917G>T, p.E2973X | 0.4 | IVB | 2.6 (0.48) | 114.6 (14.4) |
| 29 | 32 | F | 15 | 12 | USH2A* | c.10712C>T:p.T3571M | c.4711G>C; p.A1571P | 0.4 | IVA | 2.16 (0.20) | 95.6 (2.8) |
| 30 | 30 | M | 23 | - | MYO7A | c.5428A>T; p.K1810X | c.6025G>A; p.A2009T | 0.625 | IVB | 5.5 (0.27) | 309.2 (5.5) |
| 31 | 34 | F | 25 | 21 | USH2A | c.2299delG; p.E767SfsX21 | c.10585G>A; p.G3529S | 0.5 | IVB | 1.55 (0.10) | 60.5 (14.0) |
| 32 | 36 | M | 13 | 7 | MYO7A* | c.487G>A; p.G163R | c.2904G>T; p.E968D | 0.625 | IVA | 1.47 (0.91) | 216.3 (72.6) |
| 33 | 51 | M | 13 | 13 | MYO7A* | c.2904G>T; p.E968D | c.224dupA; p.D75EfsX65 | 0.2 | V | 2.81 (1.6) | 137.9 (115) |
| 34 | 59 | M | 35 | 17 | USH2A | c.920_923dupGCCA; p.H308QfsX16 | c.11864G>A; p.W3955X | 0.2 | V | 1.04 (0.43) | 350 (32.7) |
| 35 | 15 | F | 15 | 15 | USH2A | c.2299delG; p.E767SfsX21 | c.6398G>A; p.W2133X | 1.0 | II | 1.096 (0.03) | 340 (12.4) |
| 36 | 31 | M | 23 | 14 | USH2A* | c.2299delG; p.E767SfsX21 | c.2299delG; p.E767SfsX21 | 0.4 | IVA | 1.48 (0.38) | 106.4 (34.4) |
| 37 | 61 | F | 13 | 6 | USH2A | c.13022G>T; p.C4341F | c.13022G>T; p.C4341F | LP | V | 1.51 (1.06) | 190.9 (46.2) |
| 38 | 42 | M | 16 | 16 | USH2A* | c.8682-9A>G | c.8682-9A>G | 0.625 | IVA | 1.05 (0.21) | 12.9 (2.9) |
| 39 | 39 | M | 17 | - | MYO7A | c.1556G>A; p.G519D | c.6439-2A>G | 0.5 | IVB | 7.3 (0.26) | 342 (2.8) |
| 40 | 27 | F | 19 | - | USH1C | c.216G>A | c.216G>A | 0.8 | IVA | 4.95 (0.97) | 328.6 (7.7) |
| 41 | 43 | F | 11 | 9 | USH2A | c.2299delG; p.E767SfsX21 | - | 0.4 | V | 1.6 (1.6) | 49.5 (43.9) |
| 42 | 43 | M | 21 | 18 | USH2A | c.2299delG; p.E767SfsX21 | - | 0.4 | V | 0.52 (0.07) | 355 (15.7) |
Visual field grading as per Jacobson et al.11 The cDNA GenBank accession numbers are as follows: MYO7A (NM_000260), USH1C (NM_153676), CDH23 (NM_022124), PCDH15 (NM_0033056), SANS (NM_173477), USH2A (NM_206933), and GPR98 (NM_032119). -, not determined; M, male; F, female.
Nine USH1 and nine USH2 subjects underwent two to four clinical and ERG follow-up examinations across a 2- to 8-year span, with a 2-year average interval. The Table also shows the amplitude and phase SDs derived from the three consecutive measurements obtained for each patient in each recording session (see also Results). For those subjects who underwent clinical and ERG follow-up, ERG values are reported for their final visit. The data from one eye of 12 healthy subjects (age 20–60 years) were used as control values.
ERG Recordings
Pupils were dilated with topical phenylephrine HCL 2.5% and tropicamide 1%. Ganzfeld ERGs were elicited and recorded according to a described technique,16,18 using a Burian-Allen contact lens bipolar electrode (Hansen Ophthalmic Instruments, Iowa City, IA, USA) with topical proparacain 0.5% anesthesia. The 32-Hz flash train had a time-averaged luminance of 1.92 log cd/m2 in the Ganzfeld. Subjects were light-adapted at 40 cd/m2 for 10 minutes prior to recording. The recording strategy has been described in detail elsewhere.19 Briefly, it implements a discrete Fourier transform of the response during each flash cycle, with the series coefficient limited to the first six harmonics, which contain the relevant biological information. Given the small responses of the USH cohort, the second harmonic is not reliable and was not tracked. The signal is digitized and multiplied point-for-point with the sine and cosine functions within a single 32-Hz flicker cycle (i.e., 31.25 ms). The ERG signal is digitized at 12.288 kHz across two channels (for right and left eyes, respectively), which provides 192 points/cycle. Recording periods of 15 seconds were used for each run, and three to five runs were recorded during each session.
Molecular Analysis
Genomic DNA was sheared to approximately 400 bp using a Covaris S2 sonicator (Covaris, Woburn, MA, USA) and hybridized to a custom designed SureSelect library (Agilent Technologies, Santa Clara, CA, USA) comprising 101 genes known to cause hereditary hearing loss (detailed in the Supplementary Methods). The DNA was sequenced on an AB5500 sequencer (AB SCIEX, Framingham, MA, USA),19,20 and the resulting 75 nucleotide reads mapped to the reference human genome (hg19) using LifeScope software (LifeTech, Carlsbad, CA, USA). Variants were identified using LifeScope. Suspected pathological variants were confirmed by conventional PCR and Sanger sequencing in a Clinical Laboratory Improvement Amendments (CLIA)–approved laboratory in the National Institute on Deafness and other Communication Disorders (NIDCD).21 Ninety-three percent of the targets had greater than or equal to 20-fold coverage. The average depth of target coverage was 482.
Statistical Analysis
Electroretinogram data were evaluated by elliptical statistics using the sine and cosine values of the fundamental 32-Hz harmonic derived from each ERG record. Both eyes were tested, but only right-eye data were analyzed to avoid overestimation of significance. Response amplitude and phase were compared across patient groups and with healthy controls by a multivariate analysis of variance with Hotelling's T2 statistics. Amplitude data were normalized to log10 values to better approximate a normal distribution. Phase data were converted into implicit time values using the formula (phase degrees/360) × (1000 ms/32 cycles), and values were bracketed to between 20 and 54 ms22 The ERG data were also evaluated in relation to clinical data, including age of disease onset and duration, visual field grade (assessed according to Jacobson et al.11) and ETDRS visual acuity. Pearson's correlation was employed for this analysis except for visual field grade correlation, which was assessed by nonparametric ANOVA. Longitudinal analysis was performed by plotting the log amplitude values and timing phase as a function of disease duration.11 The rate of log amplitude change was determined individually by linear regression fits to the data. In all analyses, P less than 0.05 was considered statistically significant.
Results
Typical results of CxC ERG analysis from a healthy subject and an USH1 patient (#22 in the Table) are shown in Figure 1, with three consecutive records obtained in each session that lasted 5 to 10 minutes. Recording for 5 seconds gives 160 responses for analysis. The cosine and sine values of the fundamental harmonic of each cycle of ERG response are plotted as the scatter of the 160 data points on rectangular coordinates. The average response vectors and the 95% confidence ellipses of the estimated means (based on standard error of the mean [SEM]) are also depicted in each plot. The difference in plot amplitude makes the data scatter appear larger for the USH1 subject when in reality the scatter is comparable to the controls. On a corresponding polar plot (not shown), each data point would represent a vector whose length indicates the component amplitude and the counter-clockwise orientation is the phase angle (with 360° equal to the 31.25-msec interflash interval at 32-Hz flicker). The fundamental harmonic responses of the healthy subject are clustered in the lower right quadrant, with 74-μV amplitude and 328° timing phase angle (values on three repeat measurements were 72.6, 74.5, 77.6 μV; 331.1, 327.2, 329.5° (28.7, 28.4, 28.6 msec). USH1 responses have major amplitude loss and are clustered in the upper right quadrant, corresponding to a phase angle of 360° + 30°, which represents a delay of approximately 70°, or approximately 6-ms delay, compared with the heathy subject. The confidence ellipses estimate the statistical reproducibility.16 A reliability analysis was performed for responses recorded in the same session (n = 3) for USH1 or USH2 subjects by using the intraclass correlation coefficient. The intraclass correlation coefficient was 0.903 (95% confidence interval [CI]: 0.80–0.96) for USH1 subjects and 0.98 (95% CI 0.96–0.99) for USH2 subjects. Therefore CxC analysis provided highly repeatable outcomes in the same session due to signal averaging of many responses in the face of considerable noise even for small amplitudes.
Figure 1.

Typical CxC ERG results obtained from a healthy subject and a USH1 patient (#22 in the Table). Three records obtained in the same session lasting a few minutes are shown for both the control and the patient. The patient's response are plotted either on the same scale as the control's response (top) or on a magnified scale (bottom). The cosine and sine values of the fundamental harmonic of each cycle of ERG response are plotted as scatters of data points on rectangular coordinates. The average response vectors and the 95% confidence ellipses of the estimated means are also depicted in each plot.
Figure 2 depicts amplitude and timing phase data of the cone ERG fundamental harmonic of 42 USH1 and USH2 subjects as reported in the Table. One USH2A patient (#20, age 29 years, Table) is not shown, as her 27-μV response amplitude considerably exceeded the depicted plotting range. The USH1 and USH2 groups show a substantial overlap, and MANOVA did not reveal a significant difference between them (F[1,42]: 0.04, P = 0.8). Mean normal timing phase of controls is 340° ± 20° (SD; i.e., 29.5 ± 1.7 msec). Many USH subjects of both subtypes had significant timing delay (P < 0.05). However, timing delay did not correlate with severity of amplitude loss. Many subjects with relatively larger amplitudes had timing delays, while others with relatively smaller amplitudes continued to have normal timing. Consistent with the report of Seeliger et al,13 some of our USH1 subjects (Table) retained normal timing despite substantially reduced amplitudes of less than 4 μV. However, unlike the data reported by Seeliger and coauthors,13 we also found abnormal timing in 11/18 USH1 subjects (61%) and normal timing for 9/24 USH2 subjects (37% of our pool), indicating that ERG timing provided poor diagnostic discrimination between the two subject groups.
Figure 2.
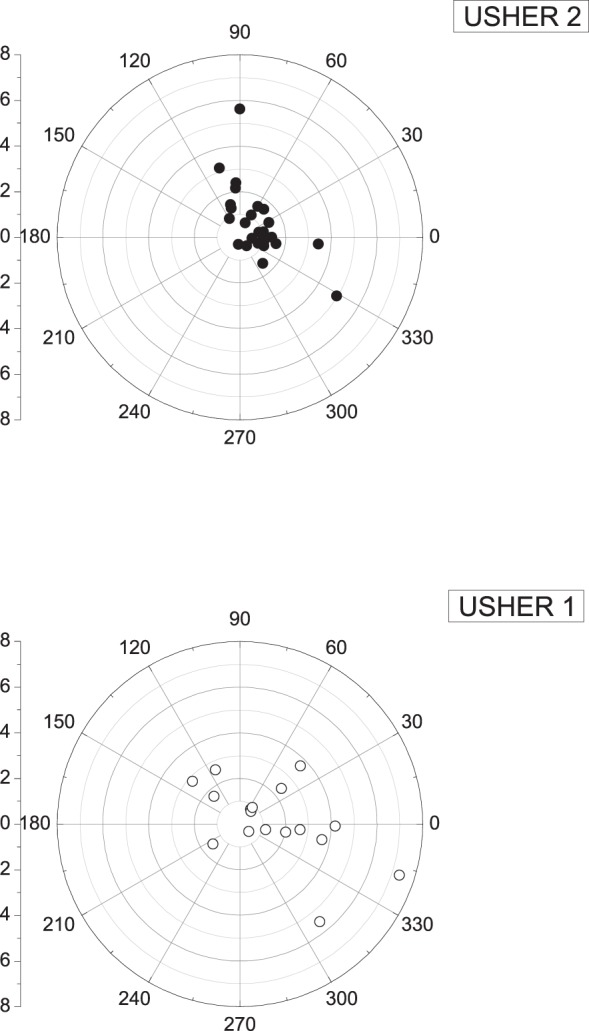
Individual amplitude and phase data of the cone ERG fundamental harmonic, recorded from all USH1 and USH2 patients. Data from one USH2A patient (#20 in the Table) are not shown since the response amplitude (27 μV) considerably exceeded the amplitudes of all other patients. Normal control timing phase was 340° ± 20° (SD), corresponding to 29.5 ± 1.7 msec (SD). Subjects from both USH subtypes showed latency timing phase delays that did not correlate with the severity of amplitude loss.
Electroretinogram amplitudes plotted on a log scale decreased progressively with disease duration for both USH1 and USH2 subjects (P < 0.05) at a rate of 0.012 log unit amplitude/y (Fig. 3). This corresponds to an annual rate of decline of 10.3% and is consistent with the range of 10% to 14% found by cone psychophysical measurements. The log-linear relation is consistent with an exponential decay model with estimated mean slopes of −0.007 (SE = 0.02) and of −0.07 (SE = 0.03) for USH1 and USH2 subjects, respectively. This difference is not significant (P < 0.07).
Figure 3.
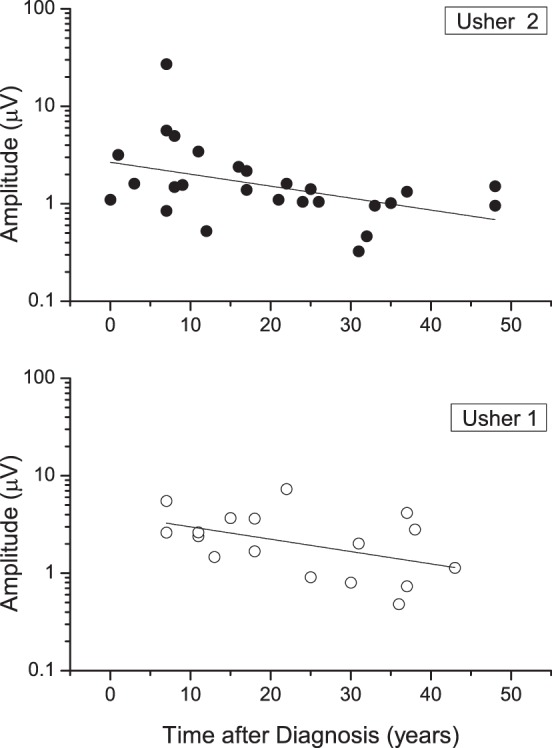
Log ERG amplitudes versus disease duration for USH1 and USH2 patients.
Implicit time measurements of the ERG fundamental harmonic of CxC recordings were highly correlated with implicit times measured for the standard ISCEV Ganzfeld flicker ERG (Fig. 4). Standard ERG flicker responses, however, measured below 1 μV in seven subjects (four USH1 and three USH2). Implicit times were uncertain in an additional six subjects (four USH1 and two USH2) because the small amplitudes and considerable noise produced ambiguous waveforms. For the remaining 26 subjects the correlation between the two techniques was significant (r = 0.72, P < 0.001). Nonetheless, the individual values showed considerable scatter, and in one case the fundamental harmonic was 40 vs. 28 msec for the standard flicker. The 40-msec value was reproducible in three subsequent recordings. As the fundamental component is thought to reflect input of cone photoreceptors to bipolar cells,23 this difference implies that proximal retinal activity varies widely across the spectrum of USH subjects.
Figure 4.
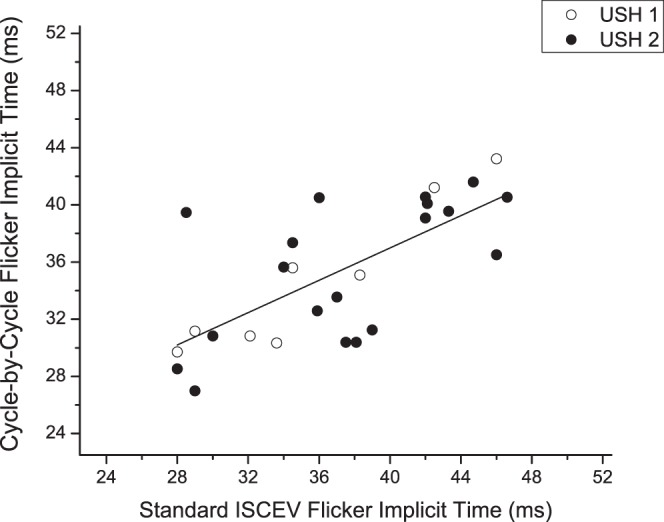
Scatterplot showing the implicit time values of the flicker response fundamental component obtained with CxC recordings in both USH1 and USH2 patients plotted as a function of the corresponding values obtained with ISCEV standard ERG. Dashed line depicts 1:1 timing.
Amplitude decline with disease duration was seen in 15 of the 18 subjects. Fewer show longer implicit time with disease duration, however, as six of nine with USH1 exhibit progressive amplitude decrease, but only four of nine have progressively longer implicit times. Figure 5 shows the longitudinal changes in amplitude and implicit time for the study population. For USH2 subjects, nine show a progressive amplitude decrease, but only four of these nine have progressively longer implicit times. A McNemar's test comparing amplitude and implicit time changes in both groups of subjects showed that amplitude was more sensitive than implicit time in detecting ERG progressive loss over time (χ2 = 5.14, P = 0.023). We conclude that amplitude is more likely than implicit time to show a change for individual subjects over shorter intervals. However, in general, the data for single subjects are too sparse to judge whether they follow linear or exponential kinetics over these short intervals.
Figure 5.

(A) Longitudinal flicker ERG response latency timing of USH1 and USH2 patients. Values were calculated from the phase measurements (phase degrees/360 × 31.25 msec) and bracketing the resulting values between 20 and 54. The horizontal dashed lines in the plots denote the normal 95% confidence limits for implicit time values. (B) Longitudinal ERG amplitude. Individual log ERG amplitude values plotted as a function of time after diagnosis for USH1 and USH2.
Electroretinogram amplitude data also correlated with visual field grade and visual acuity (Table). There was a trend (nonparametric ANOVA, P = 0.07) for amplitude to decline as visual field grade of subjects increased. Electroretinogram amplitude also tended to increase as visual acuity of subjects increased (r = 0.45, P < 0.07), suggesting that CxC flicker ERG losses are associated with clinical functional losses in USH subjects.
Discussion
This study used a CxC technique to record and analyze flicker ERG responses16 in order to assess retinal cone–mediated function in genetically characterized USH subjects. Psychophysical assessment of cone function has included visual acuity, light-adapted kinetic perimetry,24 color vision, and automated light-adapted sensitivities at different retinal loci.11 Relatively few ERG studies are available on cone function in Usher subjects,13,14,25 as they generally have small amplitude photopic responses that make differentiation of signal from noise difficult. We found that the CxC method provided a robust measurement of response amplitude and timing from the Fourier-derived fundamental harmonic compared with standard ERG recording techniques. Further, it gives a statistical estimate of measurement uncertainty. Although the ERG amplitudes were severely reduced in most of our USH1 and USH2 subjects, the measurements were reproducible and, as shown by the confidence ellipses, could be statistically differentiated from noise levels (Fig. 1). Intratest repeatability yielded a high intraclass correlation coefficient in both USH1 and USH2 subjects (0.90 and 0.98, respectively).
USH1 and USH2 both showed a decrease of log ERG amplitude with disease duration, at a rate of 0.012 log unit/y. Similarly, for most individual subjects, longitudinal measures showed a progressive loss of log amplitude that was independent of genotype. This progressive loss of cone ERG function is comparable to that observed with psychophysical measures of cone function, including kinetic perimetry for USH2 subjects26,27 and light-adapted cone sensitivity for USH 1C subjects.11 Our rate measurement also corresponds well with that from the only other study using the cone ERG14 (USH2 subjects). Electroretinogram latency timing for the fundamental showed less change over time. Note also that this rate of decrease is comparable to that found for a generalized RP population.15
Most USH1 and USH2 subjects showed delayed implicit time compared with those of controls. A notable exception was seen for two unrelated subjects with mutations in USH1C encoding harmonin. These two USH1C subjects had an amplitude loss without a delay. A previous study28 reported that two siblings with homozygous 238_239insC USH1C mutations had reduced cone ERG amplitude without delay when tested at age 25 and 32 years. Further study is needed to learn whether USH1C cone dysfunction presents with specific pathophysiological aspects that could be used to differentiate mutations of USH1C from other causes of USH and thus may serve as a clinical marker.
The mean implicit time for the USH1 population (n = 18) was not different from that of the USH2 cohort (n = 24). Our data disagree with those of Seeliger et al.13 who reported that USH1 subjects have normal timing of the 33-Hz cone flicker, unlike USH2 and typical RP subjects. Our results indicate a more complex picture, as only seven (38.9%) of 18 USH1 subjects with specific mutant alleles involving USH1C and USH1B may show normal response timing of their residual cone signals.
The ERG implicit time estimated using the CxC technique was significantly correlated with implicit times measured by the standard ISCEV flicker ERG. However, several subjects in both the USH1 and USH2 groups, for whom the ISCEV standard ERG was submicrovolt or the waveform was ambiguous leading to an uncertain measurement of implicit time, had reproducible values of CxC ERG. The CxC approach yielded far fewer of these results and was more sensitive in following cone function over time.
In considering the cellular locus of the prolonged response timing, this might originate with the cone photoreceptors, in which case one would expect to see change over time in USH subjects. Some models of flicker ERG analysis posit that interaction of signals from depolarizing and hyperpolarizing bipolar cells, located postsynaptically to the cones themselves, contributes to implicit time of the primate 30-Hz flicker response.23 If this is the case, the stability of latency with disease progression in USH may reflect a stable synapse-level abnormality, consistent with some studies that indicate that expression of USH proteins occurs in the synaptic terminal.7
A limitation of our study is that we identified only a single allele mutation in 6 of 42 subjects (14%). This is not surprising, as the large size and complexity of the USH genes limits identifying biallelic mutations to 80% to 90% of cases.29–31 Screening for duplications, deletions and intronic mutations finds a second allele in approximately 35% of USH2A patients only.32 The yield with next generation sequencing is comparable to Sanger sequencing.30 For this study, we made the assumption that subjects with a characteristic clinical Usher phenotype, plus at least one mutation allele in a known Usher gene, were suitable for inclusion.
The present study presents several novel outcomes. To our knowledge, this is the first longitudinal study to use the cone flicker ERG to follow USH1 subjects with known mutations of genes reported to be associated with USH. This complements a previous study of USH2A.14 It also provides new information on cone ERG timing versus genotype, as subjects were not genotyped in the study by Seeliger et al.13 No previous Usher syndrome study reports ERG latency phase changes over time. The present results support the use of CxC ERG to evaluate retinal cone dysfunction and monitor its progression in USH subjects. This could be a useful adjunct to standard psychophysical techniques, and offers the opportunity to evaluate some aspects of retinal cone function, such as processing phase delay, which cannot be captured with other methods. The significant delays found in both USH1 and USH2 subjects, together with relative stability over time, suggest a temporal abnormality at the synaptic level that merits further investigation. Cycle-by-cycle ERG is a reliable method to measure and follow small amplitude cone responses and is therefore valuable as an outcome measure in USH clinical trials.
Acknowledgments
The authors thank the subjects who participated in this study. They also thank J. Sarchet, A. Bamji, P. Lopez, L. Reuter, and J. Rowan for their excellent assistance and technical expertise, and Brian P. Brooks and Lisa Wei for their critiques of the manuscript.
Supported by grants from the National Institutes of Health Intramural Research Programs of the National Eye Institute (Protocol 05-EI-0096; Bethesda, MD, USA) and National Institute on Deafness and Other Communication Disorders (DC000060-12 [AJG] and DC000039-16 [TBF]; Bethesda, MD, USA).
Disclosure: W.M. Zein, None; B. Falsini, None; E.T. Tsilou, None; A.E. Turriff, None; J.M. Schultz, None; T.B. Friedman, None; C.C. Brewer, None; C.K. Zalewski, None; K.A. King, None; J.A. Muskett, None; A.U. Rehman, None; R.J. Morell, None; A.J. Griffith, None; P.A. Sieving, None
References
- 1. Pakarinen L, Tuppurainen K, Laippala P, Mantyjarvi M, Puhakka H. The ophthalmological course of Usher syndrome type III. Int Ophthalmol. 1995; 19: 307–311. [DOI] [PubMed] [Google Scholar]
- 2. Reisser CF, Kimberling WJ, Otterstedde CR. Hearing loss in Usher syndrome type II is nonprogressive. Ann Otol Rhinol Laryngol. 2002; 111: 1108–1111. [DOI] [PubMed] [Google Scholar]
- 3. Tsilou ET, Rubin BI, Caruso RC, et al. Usher syndrome clinical types I and II: could ocular symptoms and signs differentiate between the two types? Acta Ophthalmol Scand. 2002; 80: 196–201. [DOI] [PubMed] [Google Scholar]
- 4. Petit C. Usher syndrome: from genetics to pathogenesis. Annu Rev Genomics Human Genet. 2001; 2: 271–297. [DOI] [PubMed] [Google Scholar]
- 5. Cosgrove D, Zallocchi M. Usher protein functions in hair cells and photoreceptors. Int J Biochem Cell Biol. 2014; 46: 80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cremers FP, Kimberling WJ, Kulm M, et al. Development of a genotyping microarray for Usher syndrome. J Med Genet. 2007; 44: 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Reiners J, Nagel-Wolfrum K, Jurgens K, Marker T, Wolfrum U. Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp Eye Res. 2006; 83: 97–119. [DOI] [PubMed] [Google Scholar]
- 8. Williams DS. Usher syndrome: animal models, retinal function of Usher proteins, and prospects for gene therapy. Vision Res. 2008; 48: 433–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schwartz SB, Aleman TS, Cideciyan AV, et al. Disease expression in Usher syndrome caused by VLGR1 gene mutation (USH2C) and comparison with USH2A phenotype. Invest Ophthalmol Vis Sci. 2005; 46: 734–743. [DOI] [PubMed] [Google Scholar]
- 10. Fakin A, Jarc-Vidmar M, Glavac D, Bonnet C, Petit C, Hawlina M. Fundus autofluorescence and optical coherence tomography in relation to visual function in Usher syndrome type 1 and 2. Vision Res. 2012; 75: 60–70. [DOI] [PubMed] [Google Scholar]
- 11. Jacobson SG, Cideciyan AV, Gibbs D, et al. Retinal disease course in Usher syndrome 1B due to MYO7A mutations. Invest Ophthalmol Vis Sci. 2011; 52: 7924–7936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Berson EL. Retinitis pigmentosa. The Friedenwald Lecture. Invest Ophthalmol Vis Sci. 1993; 34: 1659–1676. [PubMed] [Google Scholar]
- 13. Seeliger MW, Zrenner E, Apfelstedt-Sylla E, Jaissle GB. Identification of Usher syndrome subtypes by ERG implicit time. Invest Ophthalmol Vis Sci. 2001; 42: 3066–3071. [PubMed] [Google Scholar]
- 14. Sandberg MA, Rosner B, Weigel-DiFranco C, McGee TL, Dryja TP, Berson EL. Disease course in patients with autosomal recessive retinitis pigmentosa due to the USH2A gene. Invest Ophthalmol Vis Sci. 2008; 49: 5532–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jacobson SG, Cideciyan AV, Aleman TS, et al. Usher syndromes due to MYO7A, PCDH15, USH2A or GPR98 mutations share retinal disease mechanism. Hum Mol Genet. 2008; 17: 2405–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sieving PA, Arnold EB, Jamison J, Liepa A, Coats C. Submicrovolt flicker electroretinogram: cycle-by-cycle recording of multiple harmonics with statistical estimation of measurement uncertainty. Invest Ophthalmol Vis Sci. 1998; 39: 1462–1469. [PubMed] [Google Scholar]
- 17. Marmor MF, Fulton AB, Holder GE, et al. ISCEV Standard for full-field clinical electroretinography (2008 update). Doc Ophthalmol. 2009; 118: 69–77. [DOI] [PubMed] [Google Scholar]
- 18. Sieving PA, Bingham EL, Kemp J, Richards J, Hiriyanna K. Juvenile X-linked retinoschisis from XLRS1 Arg213Trp mutation with preservation of the electroretinogram scotopic b-wave. Am J Ophthalmol. 1999; 128: 179–184. [DOI] [PubMed] [Google Scholar]
- 19. Jenkinson EM, Rehman AU, Walsh T, et al. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am J Hum Genet. 2013; 92: 605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rehman AU, Santos-Cortez RLP, Morell RJ, et al. Mutations in TBC1D24, a gene associated with epilepsy, also cause nonsyndromic deafness DFNB86. Am J Hum Genet. 2014; 94: 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bork JM, Peters LM, Riazuddin S, et al. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet. 2001; 68: 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Iarossi G, Falsini B, Piccardi M. Regional cone dysfunction in retinitis pigmentosa evaluated by flicker ERGs: relationship with perimetric sensitivity losses. Invest Ophthalmol Vis Sci. 2003; 44: 866–874. [DOI] [PubMed] [Google Scholar]
- 23. Kondo M, Sieving PA. Post-photoreceptoral activity dominates primate photopic 32-Hz ERG for sine-, square-, and pulsed stimuli. Invest Ophthalmol Vis Sci. 2002; 43: 2500–2507. [PubMed] [Google Scholar]
- 24. Sadeghi AM, Eriksson K, Kimberling WJ, Sjostrom A, Moller C. Longterm visual prognosis in Usher syndrome types 1 and 2. Acta Ophthalmol Scand. 2006; 84: 537–544. [DOI] [PubMed] [Google Scholar]
- 25. Malm E, Ponjavic V, Moller C, Kimberling WJ, Andreasson S. Phenotypes in defined genotypes including siblings with Usher syndrome. Ophthalmic Genet. 2011; 32: 65–74. [DOI] [PubMed] [Google Scholar]
- 26. Iannaccone A, Kritchevsky SB, Ciccarelli ML, et al. Kinetics of visual field loss in Usher syndrome Type II. Invest Ophthalmol Vis Sci. 2004; 45: 784–792. [DOI] [PubMed] [Google Scholar]
- 27. Fishman GA, Bozbeyoglu S, Massof RW, Kimberling W. Natural course of visual field loss in patients with Type 2 Usher syndrome. Retina. 2007; 27: 601–608. [DOI] [PubMed] [Google Scholar]
- 28. Williams DS, Aleman TS, Lillo C, et al. Harmonin in the murine retina and the retinal phenotypes of Ush1c-mutant mice and human USH1C. Invest Ophthalmol Vis Sci. 2009; 50: 3881–3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bonnet C, Grati M, Marlin S, et al. Complete exon sequencing of all known Usher syndrome genes greatly improves molecular diagnosis. Orphanet J Rare Dis. 2011; 6: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krawitz PM, Schiska D, Kruger U, et al. Screening for single nucleotide variants, small indels and exon deletions with a next-generation sequencing based gene panel approach for Usher syndrome. Mol Genet Genomic Med. 2014; 2: 393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Le Quesne Stabej P, Saihan Z, Rangesh N, et al. Comprehensive sequence analysis of nine Usher syndrome genes in the UK National Collaborative Usher Study. J Med Genet. 2012; 49: 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Steele-Stallard HB, Le Quesne Stabej P, Lenassi E, et al. Screening for duplications, deletions and a common intronic mutation detects 35% of second mutations in patients with USH2A monoallelic mutations on Sanger sequencing. Orphanet J Rare Dis. 2013; 8: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]