


Abstract
Many long noncoding RNAs (lncRNAs) are constrained to the nucleus to exert their functions. However, commonly used vectors that were designed to express mRNAs have not been optimized for the study of nuclear RNAs. We reported recently that sno-lncRNAs are not capped or polyadenylated but rather are terminated on each end by snoRNAs and their associated proteins. These RNAs are processed from introns and are strictly confined to the nucleus. Here we have used these features to design expression vectors that can stably express virtually any sequence of interest and constrain its accumulation to the nucleus. Further, these RNAs appear to retain normal nuclear associations and function. SnoVectors should be useful in conditions where nuclear RNA function is studied or where export to the cytoplasm needs to be avoided.
INTRODUCTION
Long noncoding RNAs (lncRNAs) are transcribed from thousands of loci of mammalian genomes and have been implicated in many important biological processes and act in a variety of ways to regulate gene expression (for reviews, see (1,2)). Although a number of mechanisms have been demonstrated for the mode of action of lncRNAs, these molecules often function by recruiting, assembling, modifying or scaffolding proteins (for example NEAT1 (3), HOTAIR (4,5), ANRIL (6), sno-lncRNAs (7)). Interestingly, many such lncRNAs are constrained to the nucleus to exert their functions. This means that some approaches to study their function require that expressed mutant or wild-type lncRNA sequences exist in the correct cellular compartment.
Like coding genes, the study of noncoding RNA function often requires the introduction of these molecules into cells by transfection of expression plasmids. However, most commercial and available eukaryotic expression vectors are designed primarily for the expression of coding sequences and thus harbor transcription elements, translation elements and often elements that promote nucleocytoplasmic export. The result is that many transcripts produced from these vectors are processed as mRNAs and exported to the cytoplasm. Even RNAs that are normally found only in the nucleus are frequently cytoplasmic when expressed from such vectors.
We have been interested in the development of a distinct class of vectors that do not allow nucleocytoplasmic export of expressed transcripts of interest. Here we describe the development of ‘snoVectors’ by taking advantage of our recent discovery of nuclear retained lncRNAs that are processed from introns by the snoRNA machinery (sno-lncRNAs (7)). We demonstrate that snoVectors can stably express many sequences of interest and constrain their accumulation to the nucleus. Furthermore, these nuclear expressed RNAs appear to retain normal nuclear associations and function. Thus, snoVectors should be useful in conditions where nuclear RNA function is studied or where export to the cytoplasm needs to be avoided.
MATERIALS AND METHODS
Cell culture, transfection and stable cell line selection
Human cell lines including HeLa cells, HEK293 cells and embryonal carcinoma (EC) line PA1cells were cultured using standard protocols provided by ATCC. Transfection of pZW1-snoVectors and pEGFP-C1 series of constructs was carried out with X-tremeGENE 9 (Roche) for HeLa cells, or by nucleofection (Nucleofector™ 2b Device, Lonza) with an optimized nucleofector program B-016 for PA1 cells. Normally about 70–80% transfection efficiency was achieved. For the stable cell line selection, PA1 cells were transfected with pZW1-snoVectors, followed by the addition of 1 mg/ml G418 to select single clones that are Kan/Neo resistant.
Plasmids construction
The expression vector of pcDNA3.0-sno-lncRNA2 was described previously (7). All deletions of pcDNA3-sno-lncRNA2 described in Figure 1 including LA20, LD20, LD25, LD30, LD35, RD200, RD400, RD600, RD700, RD750, ID200, ID400, ID600, ID800, ID990 were generated using the QuickChange Site-Directed Mutagenesis Kit (Stratagene). Insertions of pcDNA3.0-sno-lncRNA2 described in Figure 1D, including IN1K and IN2K (sequences of hNEAT1 RNA), were generated by using ClonExpressTM One Step Cloning Kit (Vazyme) with the Blp I site of pcDNA3-sno-lncRNA2.
Figure 1.
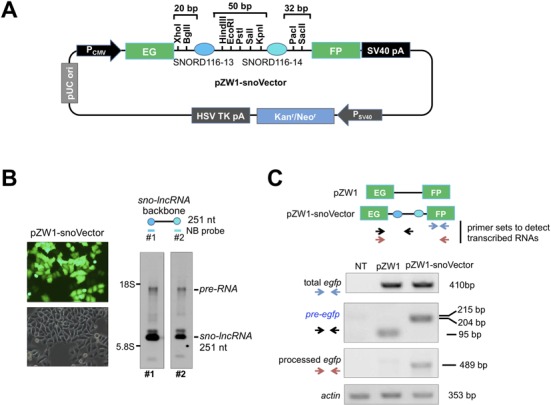
Design of the snoVector. (A) A schematic view of the snoVector (pZW1-snoVector). Two snoRNA genes SNORD116-13 (94 bp, darker blue circle) and SNORD116-14 (107 bp, lighter blue circle) from the Prader–Willi syndrome deletion region flanked by multiple cloning sites were inserted into the weak intron of Enhanced Green Fluoresence Protein (EGFP) such that only proper splicing leads to EGFP fluorescence (8) and the sno-lncRNA expression. The pZW1-snoVector carries minimal spacer sequences; and the pZW1 backbone carries the Kanamycin/Neomycin resistance gene to allow the selection of stable cell lines. (B) NB revealed the production of sno-lncRNA from pZW1-snoVector. Transfection of the pZW1-snoVector into HeLa cells resulted in the generation of sno-lncRNA 251 nt in length. Left, transfection of the empty pZW1-snoVector and the efficiency of the processing of transcribed RNAs from transfected plasmids were indicated by EGFP fluorescence intensity. Right, NB revealed that sno-lncRNA in pZW1-snoVector is efficiently processed with two snoRNA probes. (C) The processing efficiency of transcribed RNAs from pZW1 and pZW1-snoVector. Top, a schematic view of PCR primer sets used to identify different isoforms of egfp produced from pZW1 and pZW1-snoVector. Bottom, the processing of the spliced egfp is efficient in pZW1-snoVector. Total RNAs were collected from HeLa cells transfected with each individual indicated plasmid and semi-quantitative RT-PCR was used to measure the relative abundance of different isoforms of egfp RNAs. Also see supplemental information Figure S1.
For pZW1-snoVector, the sequences of SNORD116-13 and SNORD116-14 were inserted sequentially into the Xho I/EcoR I sites and EcoR I/Sac II sites of the intron region (between the two EGFP exons) of pZW1 (a gift from Dr Zefeng Wang, UNC School of Medicine) (8). Then multiple cloning sites including Bgl II, Hind III, Pst I/Sal I/Kpn I and Pac I were introduced sequentially by polymerase chain reaction (PCR) primer SNORD116-13-F(Xho I), SNORD116-13-R(EcoR I), SNORD116-14-F(EcoR I) and SNORD116-14-R(Sac II).
A stop codon was inserted into pEGFP-C1 to generate a new pEGFP-C1 with a stop codon at egfp 3’-UTR. Then, the full length or four fragments of mNEAT1 (9) were individually inserted into pZW1-snoVector or this engineered pEGFP-C1 between the Sal I and Kpn I restriction sites to generate pZW1-snoVecto-mNEAT1-FL (full length)/v1/v2/v3/v4 or pEGFP-C1-mNEAT1-FL/v1/v2/v3/v4. Flag and p54nrb were separately inserted in pcDNA3.1(+) backbone between HindIII/BamHI and BamHI/XhoI. Inverted repeated Alu elements (IRAlus) were amplified from NICN1 3’UTR (11) and inserted into pmCherry-C1 empty vector between EcoRI and XhoI restriction site.
For other constructs used in Figure 4, the second intron and the fourth intron sequences of ANKRD52 were individually inserted into pZW1-snoVector, pEGFP-C1 or pcDNA3.0 by Hind III/EcoR I. The open reading frame regions of Blasticidin, Cebpa and Snrpn were inserted into these vectors with Hind III/EcoR I sites as well. The full length of HOTAIR was inserted into these two vectors with Sal I/Kpn I restriction sites. In addition, the full length of HOTAIR and mNEAT1 sequences were inserted into pcDNA3.0 by Kpn I/Not I. Finally, we also ligated the antisense sequence of pre-let-7g into pZW1-snoVector to generate pZW1-snoVector-pre-let-7g by Hind III/EcoR I.
Figure 4.
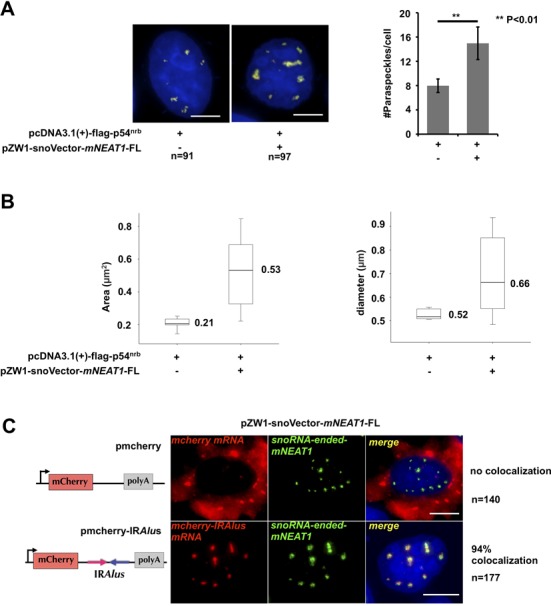
Characterization of mNEAT1 expressed from snoVector. (A) Staining of paraspeckles in cells transfected with or without snoRNA-ended-mNEAT1. Left, HeLa cells were co-transfected with Flag-p54nrb and pZW1-snoVector-mNEAT1-FL or Flag-p54nrb alone, followed by co-staining of Flag-p54nrb (red) and mNEAT1 (green) or endogenous hNEAT1 RNA (green). Both hNEAT1 and mNEAT1 probes were added (green) at the same time to indicate paraspeckles. All nuclei were counterstained with DAPI. Scale bars indicate 5 μm in all panels. Right, quantitative analysis of the average number of paraspeckles per cell by Image pro plus. Ninety-one cells (without sno-ended-mNEAT1) and 97 cells (with sno-ended-mNEAT1) were randomly selected. P values from one-tailed t-test in the pairwise comparison are shown (**P < 0.01). (B) Quantitative analysis of the average area (left) and diameter (right) of individual paraspeckles in cells transfected with or without snoRNA-ended-mNEAT1. Pictures acquired from (A) were analyzed by Image pro plus using the same parameters. (C) SnoRNA-ended-mNEAT1 co-localized with mcherry-IRAlus. HeLa cells were co-transfected with pZW1-snoVector-mNEAT1-FL and pmCherry-C1 or pmCherry-C1-IRAlus containing a pair of inverted repeated Alu elements (IRAlus) in the 3’UTR of mcherry mRNA, followed by co-staining of mcherry mRNA (red) and snoRNA-ended-mNEAT1 (green). One hundred forty cells (transfected with pmCherry-C1) and 177 cells (transfected with pmCherry-C1-IRAlus) were randomly selected.
PCR was done with Phanta Super-Fidelity DNA Polymerase (Vazyme biotech). All primers for plasmid construction are listed in Supplementary Table S1.
Total RNA isolation and semi-quantitative reverse transcriptase-polymerase chain reaction
Total RNAs from each cultured cell line or cultured cells with different transfection were extracted with Trizol Reagent (Invitrogen) according to the manufacturer's protocol. The cDNA was transcribed with SuperScript II (Invitrogen) with random hexamers. Primers for semi-quantitative PCR used were listed in Supplementary Table S1.
Nuclear/cytoplasmic RNA fractionation and Northern blotting
For Northern blotting (NB), equal amounts of total RNAs collected from cultured cells or transfected cells were resolved on 1.5% agarose gels and NB was carried out as described previously (10). Digoxigenin-labeled antisense individual probes were made using either SP6 or T7 RNA polymerases by in vitro transcription with the DIG Northern Starter Kit (Roche).
Nuclear and cytoplasmic RNA fractionations in pZW1-snoVectors and pEGFP-C1 series of constructs transfected cells were performed as described in HeLa cells with slight modifications (11). Following fractionations, NB was then used to evaluate the relative abundance in each fractionation of transfected RNAs. Equal amounts of total RNA, nuclear and cytoplasmic RNA from each transfection were resolved on 1.5% agarose gels and NB was done according to the manufacturer's protocol. All primers for NB were listed in Supplementary Table S1. tRNAlys was used as a marker for nuclear and cytoplasmic RNA fractionations. All experiments were repeated at least three times.
Half-life analysis of sno-lnRNAs by NB
For half-life analysis of the overexpressed sno-lncRNA from pZW1-snoVectors, HeLa cells were transfected with plasmids pZW1-snoVector-mNEAT1-v2 and v3 for 24 h and were then treated with actinomycin D (10 μg/ml) for 6, 12 and 18 h. Total RNAs collected from equal number of cells at the different actinomycin D treatment time were then resolved on an agarose gel and hybridized with SNORD116-13 antisense probes in Northern blots. The endogenous actin mRNA was also probed for half-life after each transfection.
RNA in situ hybridization and immunofluorescence microscopy
To detect subcellular distributions of transfected RNAs from either pZW1-snoVectors or pEGFP-C1 vectors, RNA in situ hybridization was carried out as previously described (11) with slight modifications. Briefly, untreated cells, sno-lncRNAs stably expressed cells or cells 24 h after transfection, were rinsed briefly in phosphate buffered saline (PBS) and fixed in 3.6% formaldehyde and 10% acetic acid in PBS for 15 min on ice and then permeabilized with 0.5% Triton X-100 and 2 mM Ribonucleoside Vanadyl Complex (VRC) for 5 min at room temperature. After washes with PBS, cells were precipitated with 75% ethanol at 4°C overnight. RNA hybridization was carried out using in vitro transcribed digoxigenin-labeled antisense probes for different mNEAT1 fragments. Primers were listed in Supplementary Table S1. The following detections were carried out with primary sheep anti-Dig antibody (1:500; Roche) and secondary Alexa555-conjugated donkey anti-sheep IgG (1:1000; Invitrogen). For statistical analyses of the nuclear localization pattern of each transfection, transfected cells were counted randomly under the microscope after each indicated transfection. For co-localization studies, after RNA ISH, cells were again fixed for 5 min in 2% formaldehyde, and immunofluorescence and imaging were performed as described (11). Mouse anti-flag (1:400; Sigma) was used to detect co-transfected flag-p54nrb in HeLa cells. The nuclei were counterstained with DAPI. Images were taken with an Olympus IX70 DeltaVision RT Deconvolution System microscope. All experiments were repeated at least three times.
RNA/DNA FISH
Sequential RNA/DNA FISH experiments were carried out as described previously (7). After RNA ISH of snoRNA-ended-mNEAT1 RNA, HeLa cells stably expressing snoRNA-ended mNEAT1 were denatured and then hybridized with denatured fluorescence (Cy3) labeled probes produced by nick translation of 1 μg pZW1-snoVector-mNEAT1-FL plasmid according to the standard protocol (Abbott Molecular Inc.) overnight. After hybridization and washes, image analyses were performed on single Z stacks acquired with an Olympus IX70 DeltaVision RT Deconvolution System microscope.
Double RNA FISH
HeLa cells co-transfected with pZW1-snoVector-mNEAT1-FL and pmCherry-C1-IRAlus or pmCherry-C1 were fixed as described (15). Cells were then hybridized with denatured fluorescence-labeled mNEAT1 and mcherry probes at the same time in a moist chamber at 37°C for 12 h. The mNEAT1 and mcherry probes were made by nick translation of PCR products of full-length mNEAT1 and mcherry (Abbott Molecular lnc.). After hybridization and washes, the nuclei were counterstained with DAPI. Images were acquired with an Olympus IX70 DeltaVision RT Deconvolution System microscope.
Statistical analyses of paraspeckle number, diameter and area
Image analyses of paraspeckle number, diameter and area were carried out by Image-Pro Plus according to standard protocol. Fifteen pictures randomly taken from cells transfected with or without snoRNA-ended-mNEAT1 were analyzed using the same parameters. In total, 91 cells (without transfection of sno-ended-mNEAT1) and 97 cells (with transfection of sno-ended-mNEAT1) were calculated and were used for statistical analysis. Paired t-test was applied for the statistical analysis of paraspeckle number, and Wilcoxon rank-sum test was used for the statistical analysis of paraspeckle area and diameter.
NB for miRNAs
Ten microgram of each total RNA sample collected from cultured cell or cultured transfected cells was separated on 8 M Urea-12% PAGE gel, stained with Ethidium Bromide (EB), transferred onto a nylon membrane and UV-cross linked. DNA probes complementary to the mature miRNAs were synthesized and labeled with [γ-32P] ATP. DNA oligos with the sequence same to the miRNAs were used as positive controls. The membrane was pre-hybridized in PerfectHybTM Hybridization Solution (TOYOBO) at 48°C for 20 min, and then the labeled probes were added and the hybridization was carried out at 48°C for 2 h. Then the membrane was washed with 2xSSC, 0.1% sodium dodecyl sulphate and exposed for several days to phosphor screen and read out by Isotope scanning analyzer FLA-9000 for visualization and analyzed by densitometry.
RESULTS
Characterization of sno-lncRNA processing
We previously demonstrated that sno-lncRNAs are derived from introns and are processed on both ends by the snoRNA machinery (7). The endogenous sno-lncRNAs are retained in the nucleus, but do not localize to Cajal Bodies or nucleoli (7). In order to construct a snoVector that carries the minimum essential sequences but also allows the efficient expression of snoRNA-ended RNAs, we first investigated a series of organizational features in the pcDNA3.0 backbone, starting with a wild-type sno-lncRNA (Supplementary Figure S1) that is derived from the Prader–Willi syndrome (PWS) minimal deletion region in human 15q11.3 (7). Deletion analysis revealed that the processing of this sno-lncRNA is relatively insensitive to the distance from either splice site to the nearest intronic snoRNA (Supplementary Figures S1A and S1B). In addition, snoVectors appear not to be severely restricted in the size or sequence of inserts allowed (Supplementary Figures S1C and S1D). Reducing insert size from 990 to 0 bp had little effect on sno-lncRNA expression. However, when we inserted a sequence of 3200 bp into the vector, we noticed somewhat reduced expression of sno-lncRNA (Supplementary Figure S1D). This could be due either to a modest contribution of insert length or to sequences within the insert that impede processing or promote decay.
Design of a snoVector that allows efficient expression of RNAs
To develop a snoVector that allows not only the efficient expression of processed RNA from an intron, but also an efficient readout to measure transfection efficiency and to select stable cell lines, we applied the above characterized features within the intron of a split EGFP mRNA to generate a snoVector backbone from plasmid pZW1 (8) (pZW1-snoVector, Figure 1A). In the empty pZW1 plasmid, the EGFP coding region is interrupted by a weak and very short intron, which splices inefficiently and produces only low levels of EGFP protein (8) (Figure 1B and C). However, EGFP can be spliced and produced by insertion of sequences flanking sno-lncRNAs within the weak intron (Figure 1A and B). In this way, the production of EGFP from the transfected cells can be used as an indicator of efficient transfection and the corresponding processing of the egfp mRNA and sno-lncRNA. To optimize the application of snoVectors, we have also engineered a number of unique restriction enzyme sites within the intron (Figure 1A). Two snoRNA sequences used in the snoVector are the 13th and the 14th snoRNAs from the SNORD116 box C/D snoRNA cluster in human chromosome 15q11.3 (7).
Since the sno-lncRNA processing is relatively insensitive to the distance from either splice site to the nearest intronic snoRNA (Supplementary Figures S1A and S1B), the pZW1-snoVector was designed to contain the minimal sequences that would allow the processing of the egfp mRNA and sno-lncRNA. When transfected into cells (Figure 1B), this vector expresses EGFP (Figure 1B and C) and also a sno-lncRNA of 251 nt (Figure 1B). We observed that the pZW1-snoVector could efficiently produce more mature sno-lncRNA than the unprocessed pre-RNA as revealed by NB with two different probes recognizing snoRNAs at the ends (Figure 1B) and semi-quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) of the efficient processing of the spliced egfp mRNA (Figure 1C). We have designed a number of pZW1-snoVectors with different length of the intronic sequences from splice sites to proximal and distal snoRNAs. Although the mature sno-lncRNA could be formed from all cases, the pZW1-snoVector shown in Figure 1A represents the most efficient snoVector that allows the process of mature sno-lncRNA. We thus used this pZW1-snoVector containing the minimal sequences that would allow the processing of the egfp mRNA and sno-lncRNA in our subsequent studies.
snoVectors can express nuclear retained mNEAT1 and its truncated isoforms
As a first test using a biologically relevant RNA, we introduced into the snoVector the full-length sequence of mouse NEAT1 lncRNA (mNEAT1 or Menϵ, 3.2 kb) (Figure 2A), along with four fragments of mNEAT1 RNA (9). As control, we inserted the same sequences into the 3’-UTR of a commonly used expression vector, pEGFP-C1 (Clontech). NEAT1 is a well characterized nuclear lncRNA that is involved in paraspeckle assembly and function (3,12–15). Although both human and mouse NEAT1 localize to paraspeckles (3,12–15), the primary sequence of NEAT1 is not well conserved (9,16). These unique characteristics of mNEAT1 allowed us to (i) design probes to discriminate the vector-expressed mNEAT1 from the endogenous human NEAT1 (hNEAT1) after introduction into human cells; (ii) test whether introducing mNEAT1 into cells using snoVectors would constrain it to the nucleus and whether the overexpressed mNEAT1 would assemble into paraspeckle-like structures; and (iii) compare the expression pattern of mNEAT1 and the truncated mNEAT1 RNA fragments when expressed from our snoVector or from a commonly used vector.
Figure 2.
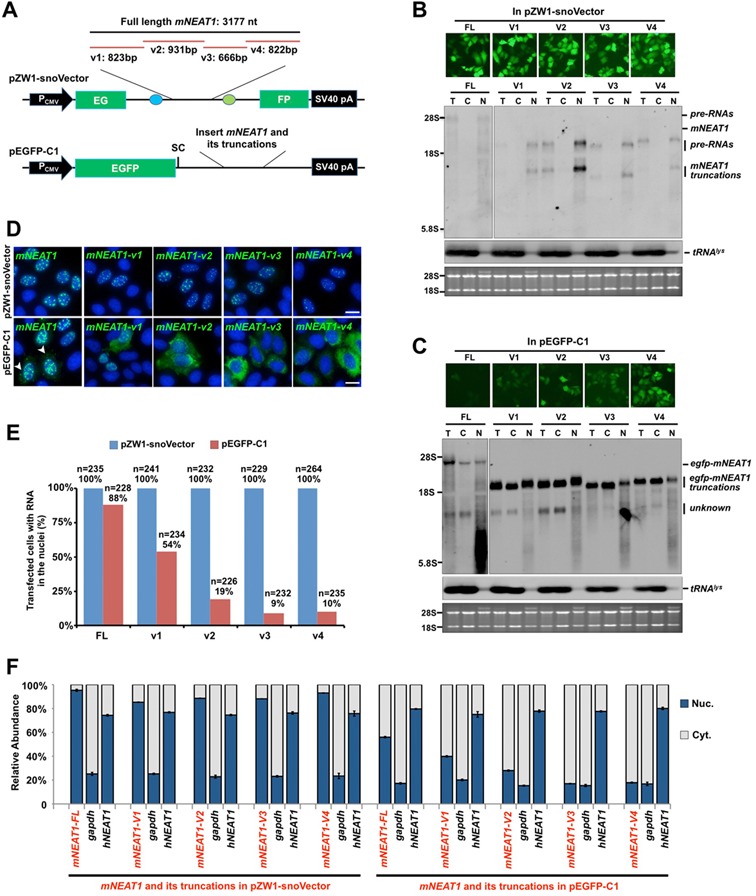
snoVectors express RNAs in the nucleus. (A) A schematic drawing to show full-length (FL) mNEAT1 RNA and different fragments of it (9) inserted into either the sno-lncRNA region in pZW1-snoVector (top) or into the UTR region of pEGFP-C1, which was engineered with a stop codon immediately downstream of the EGFP ORF (Bottom). (B) mNEAT1 RNA and different fragments of it expressed from pZW1-snoVector are retained in the nucleus. Top, HeLa cells were transfected with indicated plasmids individually and fluorescence pictures were taken 36 h after transfection. Bottom, total RNAs and fractionated nuclear and cytoplasmic RNAs were collected from the same batch of transfected HeLa cells as shown above, and then resolved on an agarose gel. Transcripts of mNEAT1 RNA and different fragments of it were probed with a Dig-labeled antisense SNORD116-13; tRNAlys was used as marker for nuclear/cytoplasmic RNA isolation. Equal amounts of total, cytoplasmic and nuclear RNAs were loaded onto an agarose gel and rRNAs were used as the loading control. T, total RNAs; C, cytoplasmic RNAs; N, nuclear RNAs. (C) mNEAT1 RNA and different fragments of it expressed from pEGFP-C1 are at least partially exported to the cytoplasm. Top, HeLa cells were transfected individually with the indicated plasmids. Total RNAs and fractionated nuclear and cytoplasmic RNAs were collected 36 h after transfection, and then resolved on an agarose gel. Transcripts of mNEAT1 RNA and different fragments of it were probed with Dig-labeled egfp. See (B) for details. (D) RNAs expressed from pZW1-snoVector are absolutely retained in the nucleus, while those from pEGFP-C1 are not. Top, HeLa cells were transfected with each indicated plasmid in a pZW1-snoVector for 36 h. RNA in situ hybridizations were performed with Dig-labeled probes for mNEAT1 RNA and different fragments of it. Bottom, HeLa cells were transfected with each indicated plasmid in a pEGFP-C1 vector for 36 h. RNA in situ hybridizations were performed with Dig-labeled egfp. Representative images are shown for each transfection. White arrow heads represent the cytoplasmic signals of egfp-mNEAT1-FL in cytoplasm in transfected HeLa cells. All nuclei were counterstained with DAPI. Scale bars 10 μm. (E) Subcellular distribution of transfected RNAs from different expression vectors. Quantitative analysis of the data from experiments shown in (D) is presented. More than 200 transfected cells were recorded randomly by confocal microscopy following each different transfection, and the percentage of each distinct nuclear/cytoplasmic localization pattern of RNAs transcribed from pZW1-snoVector or pEGFP-C1 was recorded. Note that mNEAT1 RNA and different fragments of it expressed from pZW1-snoVector are retained in the nucleus, while those from pEGFP-C1 are not. (F) mNEAT1 RNA and different fragments of it expressed from pZW1-snoVector are predominately retained in the nucleus. Nuclear and cytoplasmic RNAs extracted from equal numbers of transfected HeLa cells under each indicated condition were assayed by RT-qPCR. The cytoplasmic distributed gapdh and the nuclear retained endogenous hNEAT1 were used as markers to indicate a qualified cytoplasmic and nuclear fractionation under each transfection condition. Error bars were calculated from three replicates. In (B), (C) and (D), assays were repeated and the same results were obtained. Also see supplemental information Figure S2.
Northern blotting showed all inserts in the snoVector were expressed in HeLa cells (Figure 2B) and in PA-1 cells (data not shown). Similar to the reported endogenous PWS region sno-lncRNAs (7), the snoRNA-ended mNEAT1 fragments are very stable (Supplementary Figure S2). Importantly, their accumulation was almost exclusively nuclear as revealed by RNA fractionation followed by NB (Figure 2B). However, when the same mNEAT1 RNA and its truncations were expressed from the pEGFP-C1 vector, much of the product RNA was exported to the cytoplasm (Figure 2C). These results were confirmed by RNA in situ hybridization in individual transfected cells (Figure 2D and E). When expressed from the snoVector, all mNEAT1 sequences localized to punctate locations within the nucleus. When expressed from pEGFP-C1, only the full-length mNEAT1 transcripts showed similar nuclear punctate staining, with about 10% transfected cells also showing cytoplasmic signals (Figure 2D, bottom left); more strikingly, the cytoplasmic staining was prominent in most transfected cells for all truncated constructs and was especially prominent for some of the constructs (Figure 2D and E). The same observations were further confirmed by RT-qPCR as shown in Figure 2F. These results indicate that fragments of some nuclear retained RNAs, when expressed from a typical vector, may inappropriately localize to the cytoplasm, while those expressed from the snoVector do not. Interestingly, it was previously reported that the v1 and v4 fragments of mNEAT1 interacted with paraspeckle protein p54nrb (9), presumably resulting in the nuclear retention of mNEAT1. Our data revealed that when these fragments were expressed from the pEGFP-C1 vector, the majority of the transcripts were exported to the cytoplasm (Figure 2C–F), suggesting that the common expression vectors designed for mRNA expression may be inappropriate for the expression of these nuclear RNAs.
mNEAT1 and its fragments expressed from snoVectors retain their endogenous characteristics in the nucleus
While mNEAT1 RNA and its fragments were expressed in the nucleus after snoVector delivery (Figure 2), did these molecules associate with nuclear proteins that are known to co-localize with NEAT1 and which are important for its localization to paraspeckles? NEAT1 is known to be associated with p54nrb in the nucleus (Supplementary Figure S3A) (3,12–15). Following transfection, HeLa cells were stained for epitope-tagged p54nrb and simultaneously subjected to RNA in situ hybridization to localize mNEAT1 transcripts. When expressed from the snoVector, full-length mNEAT1 is not only almost exclusively nuclear, but most of the transfected nuclear RNA (66%) co-localizes with the epitope-tagged p54nrb (Figure 3A and Supplementary Figure S4A), and with the endogenous p54nrb with a similar ratio (61%) (Supplementary Figure S3B). This is largely true also for the full-length mNEAT1 expression from pEGFP-C1, although lower co-localization was seen with p54nrb in the nucleus (49%), compared to the snoVector expressed mNEAT1 (Figure 3A and Supplementary Figure S4A). However, expression of mNEAT1 fragments showed much more dramatic differences when the two different expression vectors were used. For example, fragment v1 of mNEAT1 was nuclear and mostly associated with p54nrb when expressed from the snoVector, but almost half of the v1 expressed from pEGFP-C1 was cytoplasmic and almost none of such v1 fragment was associated with p54nrb (Figure 3B and Supplementary Figure S4B). Fragments v2, v3 and v4 of mNEAT1 were likewise nuclear and co-localized with p54nrb when expressed from the snoVector, but mostly cytoplasmic when expressed from the more traditional vector (Figure 3C–E; Supplementary Figure S4C–S4E). These results further suggest that the common expression vector is inappropriate for the expression of these nuclear RNAs, while the snoVector can efficiently deliver such RNAs to the nucleus.
Figure 3.
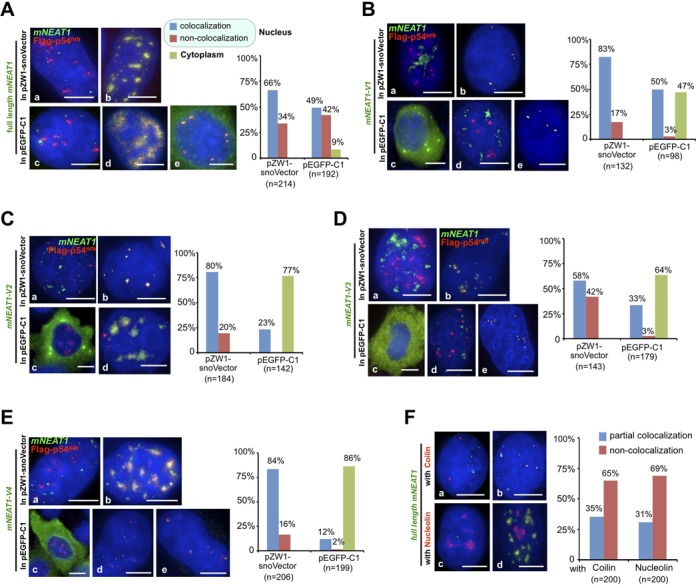
mNEAT1 and its fragments expressed from snoVectors largely retain their endogenous characteristics in the nucleus. (A) The subcellular localization of mNEAT1 RNA from a pZW1-snoVector or a pEGFP-C1 vector. Left, HeLa cells were co-transfected with Flag-p54nrb and pZW1-snoVector-mNEAT1-FL or Flag-p54nrb and pEGFP-C1-mNEAT1-FL, followed by co-staining of Flag-p54nrb (red) and mNEAT1 RNA (green). Representative images of all observed subcellular localization patterns of mNEAT1 expressed from these two vectors are shown (a-e). All nuclei were counterstained with DAPI. Scale bars 5 μm in all panels. Right, Quantitative analysis of each observed subcellular localization pattern of snoRNA-ended mNEAT1 or egfp-mNEAT1 counted from randomly selected microscopic fields. Note that the majority of expressed snoRNA-ended mNEAT1 RNA co-localizes with Flag-p54nrb. (B-E) The subcellular localization of the expressed fragments of mNEAT1 RNA from either pZW1-snoVectors or pEGFP-C1 vectors. Representative images of all observed subcellular localization patterns (cytoplasmic localization; co-localization with p54nrb in the nucleus; nonco-localization with p54nrb in the nucleus) of expressed mNEAT1 fragments from these two vectors are shown. See (A) for details. Note that fragments of mNEAT1 expressed from snoVectors are predominately retained in the nucleus and co-localize with Flag-p54nrb. (F) The majority of the snoVector expressed mNEAT1 RNAs do not localize to Cajal bodies or nucleoli. Left, HeLa cells were transfected with pZW1-snoVector-mNEAT1-FL, followed by co-staining of Coilin (red) or Nucleolin (red) with mNEAT1 RNA (green). Representative images of all observed subcellular localization patterns (partial co-localization or nonco-localization) of snoRNA-ended mNEAT1 with Colin or Nucleolin are shown (a-d). Right, Quantitative analysis of each observed subcellular co-localization pattern of snoRNA-ended mNEAT1 with Colin or Nucleolin counted from randomly selected microscopic fields. In all panels, assays were repeated and the same results were obtained. Also see supplemental information Figures S3 and S4.
mNEAT1 expressed from snoVector is functional
We further examined whether mNEAT1 expressed from snoVector is functional. First, as the endogenous NEAT1 does not localize to Cajal bodies (Supplementary Figure S3A) and as it has been reported for other sno-lncRNAs (7), mNEAT1 expressed from the snoVector largely does not co-localize with nucleoli or Cajal bodies, as most snoRNPs do (Figure 3F, Supplementary Figure S3A and S4F), suggesting that RNA expressed from the snoVector likely retains characteristics of the expressed RNA itself (Figures 2 and 3, Supplementary Figures S3 and S4). Second, in a HeLa cell line stably expressing snoRNA-ended mNEAT1, we found that such RNA products largely localize to their genomically integrated snoVector (Supplementary Figure S3C). This is in agreement with the recent report that Men ϵ/β (NEAT1) is essential to initiate the de novo assembly of paraspeckles at the NEAT1 transcription site (3,12). Third, we found that mNEAT1 expressed from snoVector significantly increased paraspeckle number (Figure 4A) as well as the size of individual paraspeckles (Figure 4B). This observation is in agreement with the previous report that overexpression of the 3.7 kb long NEAT1 in pcDNA3.1 could induce both the number and the size of paraspeckles in both NIH3T3 and HeLa cells (12). Finally, paraspeckles have been functionally implicated in the nuclear retention of mRNAs containing inverted repeated sequences (such as inverted repeated Alu elements, IRAlus, in human) in their 3’-UTRs (3,11,15). We found that the majority (>94%) of transfected mcherry mRNAs containing IRAlus, but not mcherry mRNAs lacking IRAlus, are retained at the snoRNA-ended mNEAT1 assembled paraspeckles (Figure 4C). Taken together, these results strongly suggest that mNEAT1 produced from snoVector acts properly and that snoVector represents yet another type of new vector that can be used to express a functional nuclear lncRNA.
snoVectors can lead to the nuclear retention of different types of RNA sequences
The above studies were carried out with sequences from mNEAT1, a lncRNA that is normally nuclear retained. Can snoVectors be used to express and induce the nuclear retention of other sequences, including not only those that never leave the nucleus but also those that are normally exported to the cytoplasm? We chose two intronic sequences, two coding regions and one other lncRNA (Supplementary Figure S5) to further study the ability of snoVectors to deliver RNA sequences to the nucleus. Figure 5A and C show that when expressed from a snoVector, two introns from gene ANKRD52 (10) and the nuclear lncRNA HOTAIR (4,5) are all strictly retained in the nucleus. When the same sequences were inserted into pEGFP-C1, in each case there was significant cytoplasmic localization (Figure 5B and C). These observations were further confirmed by RNA in situ hybridization in individual cells (Supplementary Figures S6A–S6C). It should also be noted that as the RNA FISH probe also detected the abundant nuclear retained pre-egfp-HOTAIR (Figure 5B, upper bands), the percentage (62%) of the actual fraction of processed egfp-HOTAIR localized in the nucleus (Supplementary Figure S6C) might actually be even lower.
Figure 5.
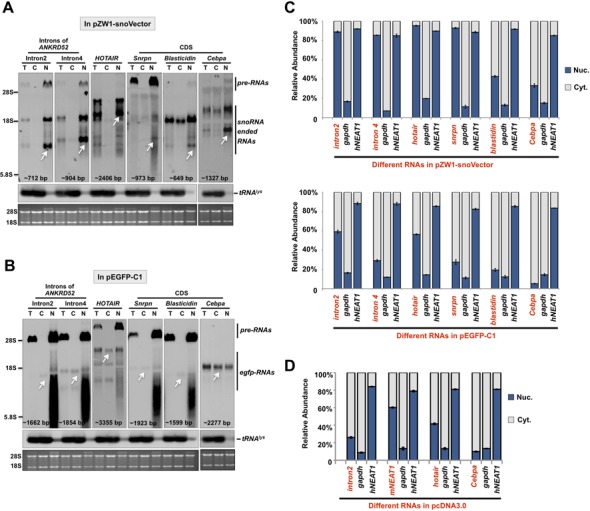
snoVectors can lead to the nuclear retention of different types of RNA sequences. (A) Different types of RNAs, including intron sequences, a lncRNA and mRNAs, expressed from pZW1-snoVector are retained in the nucleus. Total RNAs and fractionated nuclear and cytoplasmic RNAs were collected from HeLa cells transfected with the indicated plasmids individually and then resolved on an agarose gel. Transcripts of different types of expressed RNAs (indicated by white arrows) were probed with Dig-labeled antisense SNORD116-13 and their predicted sizes are labeled at the bottom of each NB. tRNAlys was used as markers for nuclear/cytoplasmic RNA isolation. Equal amounts of total, cytoplasmic and nuclear RNAs were loaded onto an agarose gel and rRNAs were used as the loading control. (B) Different types of RNAs, including intron sequences, a lncRNA and mRNAs, expressed from pEGFP-C1 are found in both the nucleus and in the cytoplasm. Total RNAs and fractionated nuclear and cytoplasmic RNAs were collected from HeLa cells transfected with the indicated plasmids individually and then resolved on an agarose gel. Transcripts of different types of expressed RNAs (indicated by white arrows) were probed with Dig-labeled egfp and their predicted sizes were labeled in the bottom of each NB. See (A) for details. (C) Different types of RNAs expressed from pZW1-snoVector are retained in the nucleus, while those expressed from pEGFP-C1 are found in both the nucleus and the cytoplasm. (D) Different types of RNAs expressed from pcDNA3.0 vector are found in both the nucleus and the cytoplasm. In (A) and (B), white arrows indicate the expected RNAs expressed from different vectors. Assays were repeated and the same results were obtained. In (C) and (D), nuclear and cytoplasmic RNAs extracted from equal numbers of transfected HeLa cells under each indicated condition were assayed by RT-qPCR. The cytoplasmic distributed gapdh and the nuclear retained endogenous hNEAT1 were used as markers to indicate a qualified cytoplasmic and nuclear fractionation under each transfection condition. Error bars were calculated from three replicates. Also see supplemental information Figures S5–S6.
To exclude the possibility that the egfp mRNA-related processing may facilitate the cytoplasmic export of expressed RNAs when pEGFP-C1 was used, we also cloned the same intron2 from gene ANKRD52 (10) and the lncRNAs HOTAIR and mNEAT1, into pcDNA3.0, which does not express any tag sequences. By nuclear and cytoplasmic RNA fractionation from transfected HeLa cells followed by Northern blotting, we confirmed that in all cases, significant levels of the intronic sequence and the lncRNAs HOTAIR and mNEAT1 were found in the cytoplasm (Figure 5D). These results again suggest that common expression vectors are inappropriate for the expression of nuclear RNAs.
Coding sequences are usually exported efficiently to the cytoplasm when expressed from commercially available vectors. We then expressed three coding regions from genes of Blasticidin, Cebpa, and Snrpn from pEGFP-C1 or pcDNA3.0 vectors in HeLa cells. These mRNAs were largely localized to the cytoplasm as expected (Figure 5B and C). However, when these coding regions were assayed using the snoVector, we found that the snoVector expressed RNAs were enhanced their nuclear retention in the nucleus (Figure 5A and C).
In addition, we explored other possible applications of the snoVector in the study of gene expression. We observed that when a snoVector insert contained a sequence containing the let-7g pre-miRNA, this sequence was not only expressed (Figure 6A), but also properly processed to generate let-7g (Figure 6B). This demonstrates that snoVectors can be used to express miRNAs, which are processed from nuclear precursors. Finally, we have used a snoVector for the selection of a stable cell line that expresses an RNA of interest in the nucleus by using Kan/Neo resistance and by the examination of fluorescent EGFP (Figure 2A). Such a stable cell line expresses the snoRNA ended fragment v3 of mNEAT1 persistently, shown by strong Northern blot signal from the nuclei at the passage 20 (P20, Figure 6C), as well as the nuclear localization of these snoRNA ended mNEAT1 fragments at similar expression levels in individual cells at P20 and P30 (Figure 6D).
Figure 6.
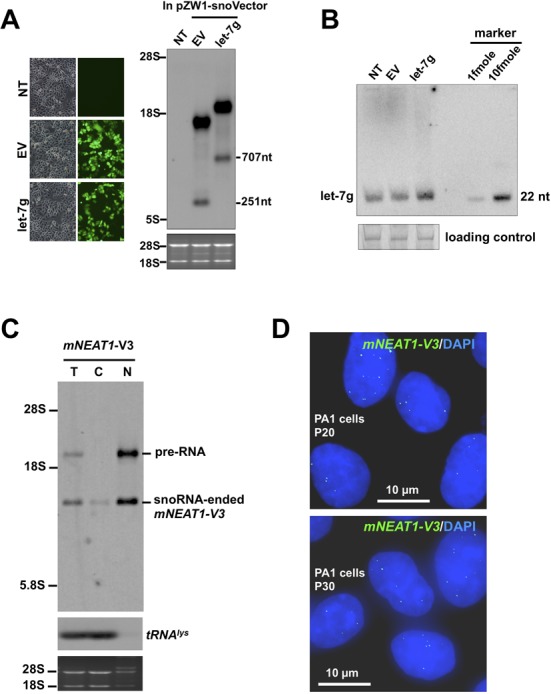
Application of snoVectors to miRNA expression and the selection of stable cell lines. (A) Expression of pre-miRNA-let-7g from the snoVector. Left, transfection of pZW1-snoVectors and the processing of transcribed pre-RNAs from transfected plasmids were indicated by EGFP fluorescence intensity. Right, NB revealed that pre-let-7g is expressed from pZW1-snoVector with total RNAs collected from HeLa cells transfected with the indicated plasmid. Transcripts of expressed RNAs were probed with Dig-labeled antisense SNORD116-13. NT, no transfection; EV, transfection with an empty vector. (B) Let-7g was produced from the pZW1-snoVector transfection revealed in (A). The production of let-7g was measured by NB with a [γ-32P] ATP labeled DNA probe complementary to the mature let-7g. The loading control is 5.8S rRNA. (C) Stable cell line selection with Kan/Neo resistance. Note that snoRNA-ended mNEAT1-v3 was stably produced in PA1 cells as revealed by NB and was predominately retained in the nucleus at the passage 20 (P20) after obtained the stable cells. (D) The stable cell line expressing snoRNA-ended mNEAT1-v3 (green, by RNA ISH) at similar levels in individual cells at different passages P20 (top) and P30 (bottom).
DISCUSSION
Recent studies have revealed that many regulatory RNAs are produced and matured in the nucleus. Very often such RNAs never leave the nucleus for their regulatory functions. However, commonly used vectors that were designed to express mRNAs have not been designed or optimized for the study of nuclear RNAs.
We have described here a strategy and a vector system to express sequences of interest in mammalian cells. This vector system ensures expressed RNAs accumulate only in the nucleus (Figures 2–5, Supplementary Figures S3, S4 and S6) and mNEAT1 acts properly in the nucleus (Figure 4). Importantly, such expression should be of use in a number of additional applications. First, the ability to ensure nuclear retention should be of value in studies interrogating the function of lncRNAs, or of fragments of these RNAs. While some studies have successfully investigated lncRNA function when expressed from standard vectors, as shown here, when expressed from a common vector, many transcripts, including intronic sequences and nuclear retained lncRNAs, are largely exported to the cytoplasm. In that compartment such RNAs may associate with different proteins than in the nucleus, or may exert unwanted or unexpected secondary biological effects.
Second, using snoVectors researchers should be able to express in the nucleus large amounts of RNAs with binding sites for specific nuclear factors such as splicing regulators. Thus, engineered sno-lncRNAs containing repeated binding motifs of interest can be used as sinks or sponges to titrate or locally concentrate proteins of interest. In fact, the endogenous sno-lncRNAs from the PWS deletion locus were shown to sequester the splicing regulator Fox2, and to alter Fox2-regulated alternative splicing patterns (7). Similarly, it is possible that sno-lncRNAs can be used as nuclear sponges for RNAs including miRNAs, pre-miRNAs or pri-miRNAs (Figure 5C). Another use might be as antisense RNAs to longer targets, or as tools to express double strand RNAs in the nucleus without the possibility of export to the cytoplasm and the activation of cytoplasmic dsRNA response pathways. Finally, snoVectors might be of use for the expression of RNAs that will be used for the affinity capture of associated nuclear proteins.
Our results suggest that snoVectors are promising for the expression of nuclear retained lncRNAs. However, we note that there are about 100 nucleotides derived from snoRNAs added to each end of the desired RNAs when expressed from the snoVectors. This may result in the unwanted recruitment of snoRNA associated proteins to sno-ncRNAs, and thus interfering with the function of the lncRNA of interest under certain circumstances. However, we have previously demonstrated the endogenous sno-lncRNAs from PWS deletion locus do not function as snoRNAs but instead to act as regulators of pre-RNA splicing (7). In addition, a number of transfected sno-lncRNAs investigated in this study are unlikely localize to nucleoli or Cajal bodies (Figure 3F). These snoRNA-ended-mNEAT1 and its fragments are largely associated paraspeckle protein, suggesting that snoVector expressed mNEAT1 RNAs could retain their normal functions. (Figure 3, Supplementary Figures S3 and S4). Importantly, mNEAT1 expressed from snoVector could increase both the number and the size of paraspeckles (Figure 4A and B) and such snoRNA-ended mNEAT1 associated paraspeckles are able to retain mRNAs containing IRAlus, a known role of paraspeckles (3,11,15). Thus, we conclude that addition of snoRNAs may not be able to override the normal function of the expressed nuclear lncRNAs, in particularly, if the expressed nuclear lncRNA acts in-trans. For those nuclear lncRNAs act in-cis, the recently developed genome editing technologies, such as TALENs (transcriptionactivator-like effector nucleases), have provided an excellent way to achieve gain-of-function study by manipulating their transcription and termination regulation in-cis. (17). Taken together, although RNAs expressed by the snoVector may not be able to allow the recapitulation of every function of all endogenous counterparts, snoVectors should be nevertheless prove useful in many conditions where nuclear RNA function is studied or where export to the cytoplasm needs to be avoided.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We are grateful to Z. Wang for the pZW1 plasmid, and Y. Zhang for providing pcDNA3.0-introns.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
FUNDING
NIH, USA [R01 HD072418 to G.G.C.]; CAS, China [2012OHTP08 to L.Y.]; CAS [XDA01010206, 2012SSTP01 to L.-L.Chen]; NSFC, China [31271376, 31322018 to L.-L.Chen]. Funding for open access charge: Chinese Academy of Sciences [XDA01010206]; NSFC, China [31322018].
Conflict of interest statement. None declared.
REFERENCES
- 1.Ulitsky I., Bartel D.P. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26–46. doi: 10.1016/j.cell.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Batista P.J., Chang H.Y. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298–1307. doi: 10.1016/j.cell.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mao Y.S., Sunwoo H., Zhang B., Spector D.L. Direct visualization of the co-transcriptional assembly of a nuclear body by noncoding RNAs. Nat. Cell Biol. 2011;13:95–101. doi: 10.1038/ncb2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rinn J.L., Kertesz M., Wang J.K., Squazzo S.L., Xu X., Brugmann S.A., Goodnough L.H., Helms J.A., Farnham P.J., Segal E., et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129:1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsai M.C., Manor O., Wan Y., Mosammaparast N., Wang J.K., Lan F., Shi Y., Segal E., Chang H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yap K.L., Li S., Munoz-Cabello A.M., Raguz S., Zeng L., Mujtaba S., Gil J., Walsh M.J., Zhou M.M. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell. 2010;38:662–674. doi: 10.1016/j.molcel.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yin Q.F., Yang L., Zhang Y., Xiang J.F., Wu Y.W., Carmichael G.G., Chen L.L. Long noncoding RNAs with snoRNA ends. Mol. Cell. 2012;48:219–230. doi: 10.1016/j.molcel.2012.07.033. [DOI] [PubMed] [Google Scholar]
- 8.Wang Z., Rolish M.E., Yeo G., Tung V., Mawson M., Burge C.B. Systematic identification and analysis of exonic splicing silencers. Cell. 2004;119:831–845. doi: 10.1016/j.cell.2004.11.010. [DOI] [PubMed] [Google Scholar]
- 9.Murthy U.M., Rangarajan P.N. Identification of protein interaction regions of VINC/NEAT1/Men epsilon RNA. FEBS Lett. 2010;584:1531–1535. doi: 10.1016/j.febslet.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y., Zhang X.O., Chen T., Xiang J.F., Yin Q.F., Xing Y.H., Zhu S., Yang L., Chen L.L. Circular intronic long noncoding RNAs. Mol. Cell. 2013;51:792–806. doi: 10.1016/j.molcel.2013.08.017. [DOI] [PubMed] [Google Scholar]
- 11.Chen L.L., DeCerbo J.N., Carmichael G.G. Alu element-mediated gene silencing. EMBO J. 2008;27:1694–1705. doi: 10.1038/emboj.2008.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clemson C.M., Hutchinson J.N., Sara S.A., Ensminger A.W., Fox A.H., Chess A., Lawrence J.B. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell. 2009;33:717–726. doi: 10.1016/j.molcel.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sunwoo H., Dinger M.E., Wilusz J.E., Amaral P.P., Mattick J.S., Spector D.L. MEN epsilon/beta nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res. 2009;19:347–359. doi: 10.1101/gr.087775.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sasaki Y.T., Ideue T., Sano M., Mituyama T., Hirose T. MENepsilon/beta noncoding RNAs are essential for structural integrity of nuclear paraspeckles. Proc. Natl Acad. Sci. U.S.A. 2009;106:2525–2530. doi: 10.1073/pnas.0807899106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen L.L., Carmichael G.G. Altered nuclear retention of mRNAs containing inverted repeats in human embryonic stem cells: functional role of a nuclear noncoding RNA. Mol. Cell. 2009;35:467–478. doi: 10.1016/j.molcel.2009.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hutchinson J.N., Ensminger A.W., Clemson C.M., Lynch C.R., Lawrence J.B., Chess A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics. 2007;8:39. doi: 10.1186/1471-2164-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiang J.F., Yin Q.F., Chen T., Zhang Y., Zhang X.O., Wu Z., Zhang S., Wang H.B., Ge J., Lu X., et al. Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 2014;24:513–531. doi: 10.1038/cr.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.