


Abstract
Escherichia coli has served as the archetypal organism on which the overwhelming majority of biochemical characterizations of bacterial RNA polymerase (RNAP) have been focused; the properties of E. coli RNAP have been accepted as generally representative for all bacterial RNAPs. Here, we directly compare the initiation properties of a mycobacterial transcription system with E. coli RNAP on two different promoters. The detailed characterizations include abortive transcription assays, RNAP/promoter complex stability assays and DNAse I and KMnO4 footprinting. Based on footprinting, we find that promoter complexes formed by E. coli and mycobacterial RNAPs use very similar protein/DNA interactions and generate the same transcription bubbles. However, we find that the open promoter complexes formed by E. coli RNAP on the two promoters tested are highly stable and essentially irreversible (with lifetimes much greater than 1 h), while the open promoter complexes on the same two promoters formed by mycobacterial RNAP are very unstable (lifetimes of about 2 min or less) and readily reversible. We show here that CarD, an essential mycobacterial transcription activator that is not found in E. coli, stabilizes the mycobacterial RNAP/open promoter complexes considerably by preventing transcription bubble collapse.
INTRODUCTION
Tuberculosis, caused by infection with the bacterium Mycobacterium tuberculosis (Mtb), continues to pose a major health problem, particularly due to the increase in multidrug resistant strains (WHO Global tuberculosis report 2013 http://www.who.int/tb/publications/global_report/en/). RNA polymerase (RNAP), the enzyme responsible for all transcription in bacteria, is the target for the antibiotic rifampicin, a first line therapeutic treatment for tuberculosis (1), and is thus an attractive target for the development of new drugs.
The catalytic core of the bacterial RNAP, comprising five subunits (α2, β, β′ and ω), is competent for RNA synthesis (2). Promoter-specific transcription initiation, however, requires a promoter specificity factor, σ, which binds the core to form the holoenzyme (3,4). Escherichia coli (Eco) has served as the archetypal organism on which the overwhelming majority of biochemical characterizations of bacterial RNAP have been focused. The properties of Eco RNAP have been accepted as generally representative for all bacterial RNAPs.
The availability of high-resolution X-ray crystal structures of Thermus aquaticus (Taq) and Thermus thermophilus (Tth) RNAPs (5–7) has prompted biochemical characterization of these enzymes (8–13). Studies of transcription initiation by RNAPs from gram-positive organisms are sparse but include characterizations of RNAPs purified endogenously from Bacillus subtilis (14), M. smegmatis (15) and Mtb (16).
CarD [also called CdnL in Myxococcus xanthus; (17)] was identified as a direct RNAP binding protein that is an essential regulator of ribosomal RNA (rRNA) transcription in Mtb (18). CarD, although widely distributed across many eubacteria phyla, is not found in Eco (18,19). Loss of CarD is lethal for Mtb in culture and during infection of mice. Depletion of CarD results in sensitivity to killing by oxidative stress, starvation, DNA damage and changes in the mRNA levels of hundreds of genes. A combination of in vivo and in vitro approaches established that CarD is a global regulator that activates transcription by stimulating the formation of the RNAP/open promoter complex (19). The X-ray crystal structure of Tth CarD, combined with detailed structural and functional analyses, led to the proposal that CarD functions by forming protein/protein and protein/DNA interactions with a DNA structure uniquely presented by the open promoter complex (RPo)—the splayed minor groove at the double-/single-stranded DNA junction at the upstream edge of the transcription bubble.
Here, we directly compare the initiation properties of a mycobacterial transcription system with Eco RNAP on two different promoters. The detailed characterizations include abortive transcription assays, RPo stability assays and DNAse I and KMnO4 footprinting. We find that the tested open promoter complexes formed by Eco RNAP are highly stable and essentially irreversible, while the same open promoter complexes formed by mycobacterial RNAP are very unstable and readily reversible (in equilibrium with RNAP and free promoter or other intermediates on the promoter melting pathway). The transcription activator CarD stabilizes the mycobacterial RNAP/open promoter complexes considerably by preventing collapse of the transcription bubble, thereby compensating for the enzyme's relatively feeble activity on aMtb rRNA promoter.
MATERIALS AND METHODS
Protein purification
Eco core RNAP was overexpressed and purified from Eco BL21(DE3) cells co-transformed with pGEMABC (encoding Eco RNAP rpoA, rpoB and rpoC; Addgene plasmid 45398) and pACYCDuet-1_Ec_rpoZ (encoding rpoZ) as described (20). Eco σ70 was overexpressed and purified as described (21). M. bovis (Mbo) core RNAP and σA were overexpressed and purified using methods modified from Czyz et al. (22). Briefly, the Mbo core RNAP subunits were co-overexpressed in Eco pRARE2 (Novagen) cells overnight at room temperature for ∼16 h after induction with 0.1 mM isopropyl-beta-D-thiogalactopyranoside (IPTG). Cell pellets were lysed with a continuous flow French press (Avestin) and the clarified cell lysate was treated by polyethyleneimine (PEI) precipitation to remove nucleic acids. Proteins eluted from the PEI pellet were then purified by Ni2+—affinity chromatography and the eluted sample concentrated by centrifugal filtration (VivaSpin) and further purified by size exclusion chromatography. Buffers and detailed methods are as described in detail in Twist et al. (23). Mbo/Mtb σA was overexpressed in Eco pRARE2 cells and purified as previously described (22). Mbo/Mtb CarD was overexpressed from Eco BL21(DE3) as previously described for Tth CarD (19).
Transcription assays
An AC50 [also called -35con; (24)] promoter DNA fragment (−152 to +72) was polymerase chain reaction (PCR) amplified from plasmid pAC50 (22). Promoter DNA (−60 to +15) of rrnAP3 from Mtb (25) was synthesized (GenScript) and placed into the pUC57 plasmid to generate pUC57-AP3. Fragment −86 to +70 of pUC57-AP3 was also PCR amplified. Both promoter DNA fragments were subsequently subjected to agarose electrophoresis and gel purified (Qiagen). These promoter fragments (AC50 and AP3) served as templates for all biochemical assays except where described otherwise. Artificial transcription bubble and double-stranded templates of AC50 (−60 to +20) were synthesized and gel purified (IDT). Purified oligos were then annealed and used as templates for transcription as described in Figure 7.
Figure 7.
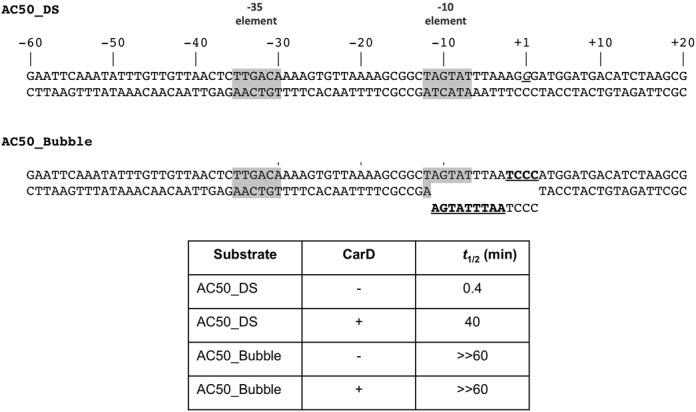
A non-complementary transcription bubble rescues short half-life of Mbo RPo on the AC50 promoter and renders CarD redundant. The AC50 double-stranded promoter (from −60 to +20) was synthesized and used as a template for abortive transcription assays (AC50-DS). In AC50-bubble, non-complementary mismatches (underlined) were introduced from −11 to +2, generating a non-collapsible transcription bubble (AC50_Bubble). Half-life assays were performed and RPo half-lives calculated as described in Figure 3B.
Proteins used for the in vitro transcription assays were diluted into 1× transcription buffer [10 mM Tris-HCl, pH 8.0, 10–150 mM KGlu (unless otherwise noted), 1 mM MgCl2, 0.1 mM DTT, 50 μg/ml bovine serum albumin]. Reactions (20 μl) were carried out in a 37°C water bath with proteins using the following protocol: core RNAP (200 nM) and σA (1 μM) were combined and incubated at 37°C for 5 min to form holoenzyme. CarD (2 μM, when used) was then added to holoenzyme and incubated for an additional 5 min. Next, promoter DNA (10 nM) was added and RPO was allowed to form for 15 min at 37°C. Abortive transcription was initiated by the addition of an NTP mix containing the initiating dinucleotide (250 μM, GpU for AP3, GpG for AC50; TriLINK), the next NTP (α-32P labeled, UTP for AP3, ATP for AC50; 1.25 μCi, with 50 μM of the same unlabeled NTP) and FC-bubble competitor DNA (2 μM, see below). After 10 min, transcription was quenched by the addition of 2× stop buffer (8 M Urea, 0.5× TBE, 0.05% Bromophenol blue, 0.05% Xylene cyanol). Reactions were heated at 95°C for 1 min and immediately loaded on a 23% polyacrylamide gel (19:1 acrylamide:bis-acrylamide). Abortive products were visualized by exposing the gel on a phosphorimager plate overnight and digitized using a Typhoon phosphorimager. Data were quantified using Image J (26).
DNase I footprinting
Promoter DNAs with 5′-end-labeled template strand was prepared by PCR amplification using a 5′-[32P]-end-labeled PCR primer. The resulting PCR products were then gel purified as described above. DNaseI (New England Biolabs) was diluted to 500 U/μl and kept on ice. Reactions (25 μl) were carried out in a 37°C water bath and in 1× transcription buffer. Core RNAP (400 nM) and σA (2 μM) were incubated for 5 min to form holoenzyme. CarD (4 μM), when used, was added to the holoenzyme and incubated for another 5 min, followed by the addition of the 32P-labeled promoter DNA (200 fmol). Formation of RPo was allowed to proceed for 15 min and competitor added for times indicated (Figure 5). DNase I (500 U) was then added to the mixture and the reaction incubated for an additional 2 min. The reactions were quenched by the addition of 100 μl of 0.5 M phenol, 75 μl of sodium acetate (0.3 M) and ethylenediaminetetraacetic acid (10 mM final). The DNA was recovered in the aqueous layer, ethanol precipitated and washed. The air-dried pellet was resuspended in 2× loading buffer, heated at 95°C for 1 min before being immediately loaded on an 8% polyacrylamide (19:1 acrylamide:bis-acrylamide) 8M urea gel. The gel was visualized as described above.
Figure 5.
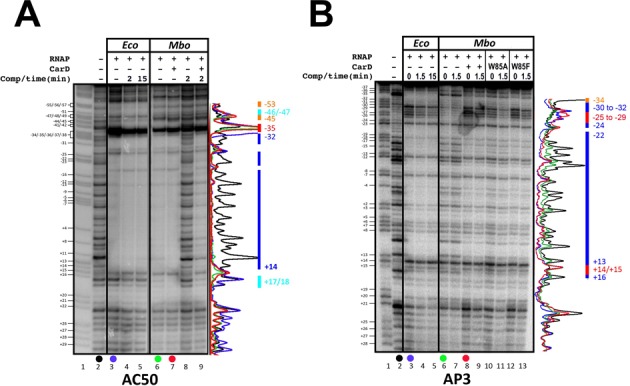
DNAse I footprints (template strand) of Eco and Mbo (±CarD) RNAPs on the AP3 and AC50 promoters. In each panel (A and B), lane 1 shows the AG sequencing ladder (assignments shown on the left), lane 2 shows DNAse I cleavage in the absence of any proteins. DNAse I footprints are shown without competitor trap, and with competitor trap incubation prior to cleavage (times as indicated). The colored bars on the right denote the footprint characteristics (blue, DNAse I protection for both Eco and Mbo RNAPs; red, DNAse I hypersensitivity for both RNAPs; orange, protection by Eco RNAP but not Mbo RNAP; cyan, protection by Mbo RNAP but not Eco RNAP). Densitometric traces provided on the right illustrate the protection profiles. Colors of each trace correspond to samples indicated by the colored dots below the gel lanes. (A) AP3 promoter: protection by Mbo RNAP alone is not as apparent as with CarD, therefore, the blue bars only represent protection by Eco RNAP and Mbo RNAP + CarD. (B) AC50 promoter.
KMnO4 footprinting
Open complexes were formed on 5′-[32P]-labeled (template strand) promoter DNA as described above for DNAse I footprinting. Potassium permanganate (KMnO4) was added to a final concentration of 2 mM, incubated for 2 min, then the reactions were quenched by the addition of 25 μl of stop buffer (1 M β-mercaptoethanol, 1.5 M sodium acetate). The DNA was precipitated with 200 μl EtOH, pelleted, then washed with 100% EtOH to remove all traces of KMnO4. The DNA pellet was then resuspended in 100 μl of piperidine (1 M) and incubated at 90°C for 30 min to cleave the DNA at modified thymine residues. After cleavage, the DNA was precipitated with 100% EtOH, pelleted, washed, air-dried, re-suspended in 2× loading buffer and subjected to electrophoresis and visualized as described for the DNase I assays.
RESULTS
Mycobacterial transcription system
Recombinant Mbo core RNAP was generated by co-overexpression and in vivo assembly of the RNAP subunits (α, β, β′, ω) in host Eco cells and purified to homogeneity using a revised purification procedure (22) (Supplementary Figure S1). The Mbo housekeeping promoter specificity factor, σA, was also overexpressed in Eco and purified as described (22). The Mbo holoenzyme is identical to that of the pathogen Mtb with the exception of one amino acid (Mbo β’P69 is R69 in Mtb). CarD is identical between Mbo and Mtb. Our analyses focused on two promoters, Mtb rrnA-P3 (Mtb AP3), a promoter of the Mtb rrnA operon (25), and AC50, based on -35con of Gaal et al. (24) (Figure 1). The AP3 promoter has a nearly consensus −35 element but a non-optimal 18 base-pair (bp) spacer between the −10 and −35 elements (Figure 1). The AC50 promoter harbors an optimized −35 element and an optimal 17 bp −10/−35 spacer. On AC50 at saturating concentration of RNAP, Mbo RNAP holoenzyme showed similar levels of transcription activity as Eco RNAP, indicating comparable activity ((22); Figures 2 and 3).
Figure 1.
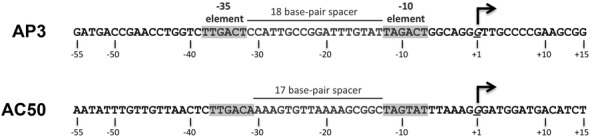
Sequences of the Mtb AP3 [Mtb rrnA–P3; (25)] and AC50 [-35con of (24)] promoters.
Figure 2.
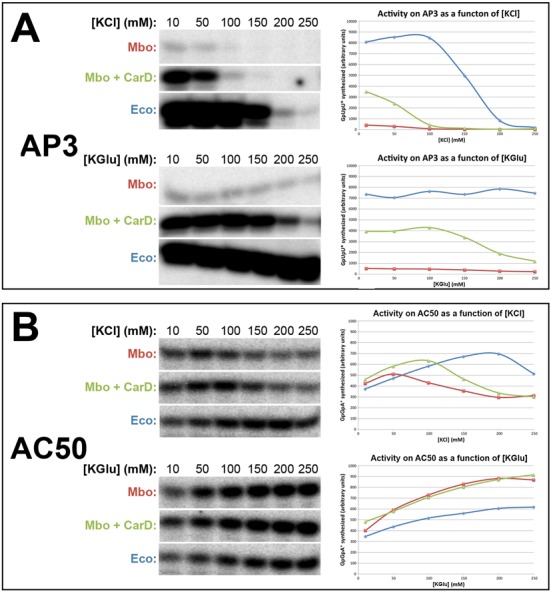
Dependence of Mbo and Eco RNAP transcription activity on salt concentration for both the AP3 and AC50 promoters. Single round abortive initiation assays measured GpUpU (AP3) or GpGpA (AC50) production. On the left, [32P]-labeled abortive transcript production was monitored by polyacrylamide gel electrophoresis and autoradiography. On the right, transcript production was quantified by phosphorimagery and plotted versus [KCl] (top) or [KGlu] (bottom) concentration (10–250 mM). On the plots, Mbo RNAP (alone) is shown in red, Mbo RNAP + CarD in green, Eco RNAP in blue. (A) AP3 promoter. (B) AC50 promoter.
Figure 3.
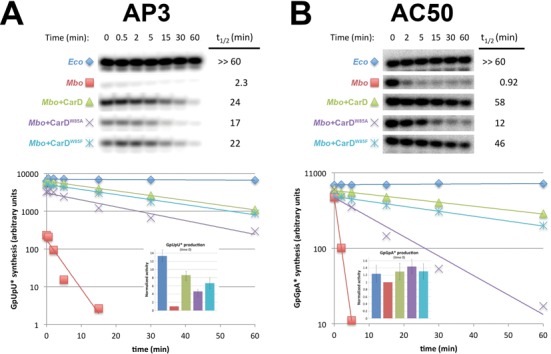
Lifetimes of promoter complexes measured by abortive transcription. On the top, [32P]-labeled abortive transcript production at times after addition of a large excess of competitor promoter DNA trap was monitored by polyacrylamide gel electophoresis and autoradiography. On the bottom, transcript production was quantified by phosphorimagery and plotted. The lines indicate single-exponential decay curves fit to the data points. The decay half-lives (t1/2) calculated from the fits are shown to the right of the gel images. The insets show histograms denoting transcription activity at time 0 (before incubation with competitor trap DNA). (A) AP3 promoter: assays were performed in transcription buffer (see Materials and Methods) with 10 mM KGlu. (B) AC50 promoter: assays were performed in transcription buffer (see Materials and Methods) with 150 mM KGlu.
Salt sensitivity of Mbo RNAP promoter complexes
Single round abortive initiation assays were used to compare the activity of Eco and Mbo RNAPs as a function of [KCl] and [K-glutamate] ([KGlu]) on both the AP3 and AC50 promoters. Although KCl or NaCl are typically used in in vitro transcription studies, in Eco cells, [Cl−] is always very low. The primary anion is glutamate, which varies dramatically in concentration (30–260 mM) depending on the osmolarity of the surrounding medium (27). Even higher levels of intracellular glutamate have been estimated for Mtb (28). Previous studies found that moderate concentrations of KGlu significantly stabilized Eco RNAP interactions with some promoters compared to KCl or NaCl (29). In particular, transcription from the Eco rrnB promoter was particularly sensitive to Cl− but tolerated high concentrations of KGlu. However, the effect was promoter specific. For instance, the lac UV5 promoter maintained the same activity in both low and high concentrations of KCl and KGlu (30).
On the AP3 promoter, Eco RNAP showed high activity in KCl up to 100 mM which then dropped off rapidly at increasing [KCl] (Figure 2A; half-maximal activity at ∼160 mM [KCl]). By contrast, activity of Mbo RNAP was more than an order of magnitude less and was essentially completely absent at 100 mM KCl and above (half-maximal activity at ∼60 mM [KCl]). Saturating amounts of CarD boosted Mbo RNAP transcription almost 9-fold but did not alter the KCl-sensitivity (half-maximal activity at ∼60 mM [KCl]).
In low [KGlu] (≤50 mM) on AP3, both Eco and Mbo (±CarD) RNAPs showed very similar activites as in low [KCl]. However, both RNAPs were much less sensitive to increasing [KGlu]. Eco RNAP activity was completely resistant to increasing [KGlu] up to the highest [KGlu] tested, 250 mM (Figure 2A). Mbo RNAP, both with and without CarD, was also more resistant to increasing [KGlu] (half-maximal activity at ∼200 mM [KGlu]).
Like on the AP3 promoter, the activity of Eco RNAP on AC50 was much less sensitive to increasing [KCl] (≥150 mM) than Mbo RNAP (with or without CarD; Figure 2B). The similarity of each enzyme's activity on AP3 as a function of [salt] ended there, however. Unlike on the AP3 promoter, at low [KCl] (≤50 mM), both Eco and Mbo activities were similar to each other, and Mbo activity was not dependent on CarD. In KGlu, the activities of both Eco and Mbo RNAPs increased with increasing [KGlu] (up to 250 mM). For all subsequent assays (both Eco and Mbo RNAPs on both AP3 and AC50 promoters), transcription conditions used are 10 mM (for AP3) or 150 mM (for AC50) [Kglu], as noted.
Mbo RNAP promoter open complexes are extremely unstable compared to Eco RNAP, a phenotype partially rescued by CarD
We tested the lifetime of competitor-resistant RNAP/promoter complexes using the abortive initiation assay in KGlu. For these and other assays where promoter complexes were challenged with competitor, we chose not to use heparin, which actively dissociates RNAP from promoters (31). Instead, we designed a competitive promoter trap comprising the optimized full-con promoter sequence (24) but also with a non-complementary transcription bubble to afford rapid and irreversible RNAP binding (FC-bubble, Supplementary Figure S2A). Control experiments demonstrated that the FC-bubble is an extremely effective competitor (Supplementary Figure S2B).
With Mbo RNAP on both promoters, we observed biphasic decay with a rapidly decaying component (t1/2 ∼2 min) with a decay rate strongly dependent on the presence of CarD, and a very slow component (t1/2 ∼5 h) that did not seem to depend on CarD (Supplementary Figure S3). On AC50, the slow-decaying component could be essentially eliminated by increasing [KGlu]. The experiments shown in Figure 3B were obtained in 150 mM [KGlu], giving rise to single-exponential decay kinetics. For AP3, Mbo RNAP transcription activity in the absence of CarD was extremely weak and only decreased with higher [KGlu] (Figure 2A). We therefore chose to perform the AP3 promoter lifetime experiments at 10 mM KGlu and removed the slow-decaying component from the data shown in Figure 3A (see Supplementary Figure S3). Under these conditions, the slow-decaying component accounted for less than 10% of the transcription activity of Mbo RNAP with CarD, and ∼50% of the (extremely weak) activity of Mbo RNAP without CarD (Supplementary Figure S3).
On the AP3 promoter, Eco RNAP showed high transcription activity that was essentially completely competitor resistant over the 60 min assay time (Figure 3A). By contrast, Mbo RNAP showed very weak transcription that decayed rapidly (t1/2 = 2.3 min). The presence of saturating amounts of CarD stimulated Mbo RNAP transcription by almost an order of magnitude (consistent with previous results) to nearly the same level as Eco RNAP (at time 0). CarD also dramatically stabilized the Mbo RNAP promoter complexes, increasing the t1/2 by more than 10-fold (Figure 3A).
Substitution of conserved CarD W85 to A (CarDW85A) showed that this residue is critical for optimal transcription of the rRNA promoters in both Tth and Mtb (19). We tested both CarDW85A and a more conservatively substituted mutant, CarDW85F, in our transcription assays. Consistent with previous results, CarDW85A showed a roughly 2-fold reduction in transcription activation (54% of the transcription activity compared to wt-CarD at time 0) but was still able to stabilize the Mbo RNAP promoter complexes to nearly the same level as wt-CarD (Figure 3A). CarDW85F showed near wild-type activity when compared to CarDW85A (77% versus 54% shown by CarDW85A of the transcription activity compared to wt-CarD at time 0).
On the AC50 promoter, Eco RNAP also showed high transcription activity that was essentially completely competitor resistant over the 60 min assay time (Figure 3B). Mbo RNAP also showed strong transcription activity (69% of Eco RNAP activity), but this activity decayed extremely rapidly upon competitor challenge (t1/2 = 0.92 min). Saturating amounts of CarD had only a slight effect on Mbo RNAP transcription at time 0 (1.3-fold activation) but, like on AP3, dramatically stabilized the complexes to dissociation (63-fold increase in t1/2 compared to no CarD). The CarDW85A mutant had little effect on Mbo RNAP transcription at time 0, and stabilized the promoter complexes at an intermediate level (13-fold increase in t1/2 compared to no CarD). As with the AP3 promoter, CarDW85F was able to stabilize the open complex on AC50 at levels similar to wt-CarD (Figure 3B).
In summary, under these assay conditions, Eco RNAP formed exceedingly stable promoter complexes on both the nearly optimal AC50 and the non-optimal AP3 promoters (t1/2's >> 60 min), while promoter complexes of Mbo RNAP were highly unstable (t1/2 = 0.92 and 2.3 min, respectively). CarD had two major effects on Mbo RNAP initiation, to activate the overall level of transcription (nearly 9-fold on AP3, but only slightly on AC50) and to stabilize the otherwise highly unstable Mbo RNAP promoter complexes (on both promoters). The CarDW85A substitution caused a partial loss of transcription activation (on AP3) activity as well as a partial defect in promoter complex stabilization (on both promoters) while CarDW85F showed stabilization similar to that of wt-CarD.
The suboptimal 18 bp −10/−35 spacer is not the origin of the weak transcription activity on the AP3 promoter
The Mtb AP3 promoter has a non-optimal 18-bp −10/−35 spacer (Figure 1). Nevertheless, Eco RNAP transcribes the AP3 promoter well and forms very stable promoter complexes (Figure 3A). On the other hand, Mbo RNAP shows very weak activity in the absence of CarD that is very sensitive to [KCl] (Figure 2A), and forms unstable promoter complexes (Figure 3A). To test whether Mbo RNAP was particularly sensitive to the non-optimal 18-bp spacer, we examined two engineered AP3 derivatives with optimal 17-bp spacers. In AP3Δ23, we generated a 17-bp spacer by deleting the bp at −23 (Figure 4A). In AP3(AC50sp), we swapped the 18-bp AP3 spacer for the 17-bp AC50 spacer (Figure 4A). Single round abortive initiation assays were performed on these promoters using RNAP from Eco and Mtb (±CarD) (Figure 4B). None of the promoter alterations had a significant effect on the efficiency of transcription by either RNAP. Thus, the poor transcription of the AP3 promoter by Mbo RNAP (in the absence of CarD) is not due to the non-optimal, 18-bp spacer.
Figure 4.
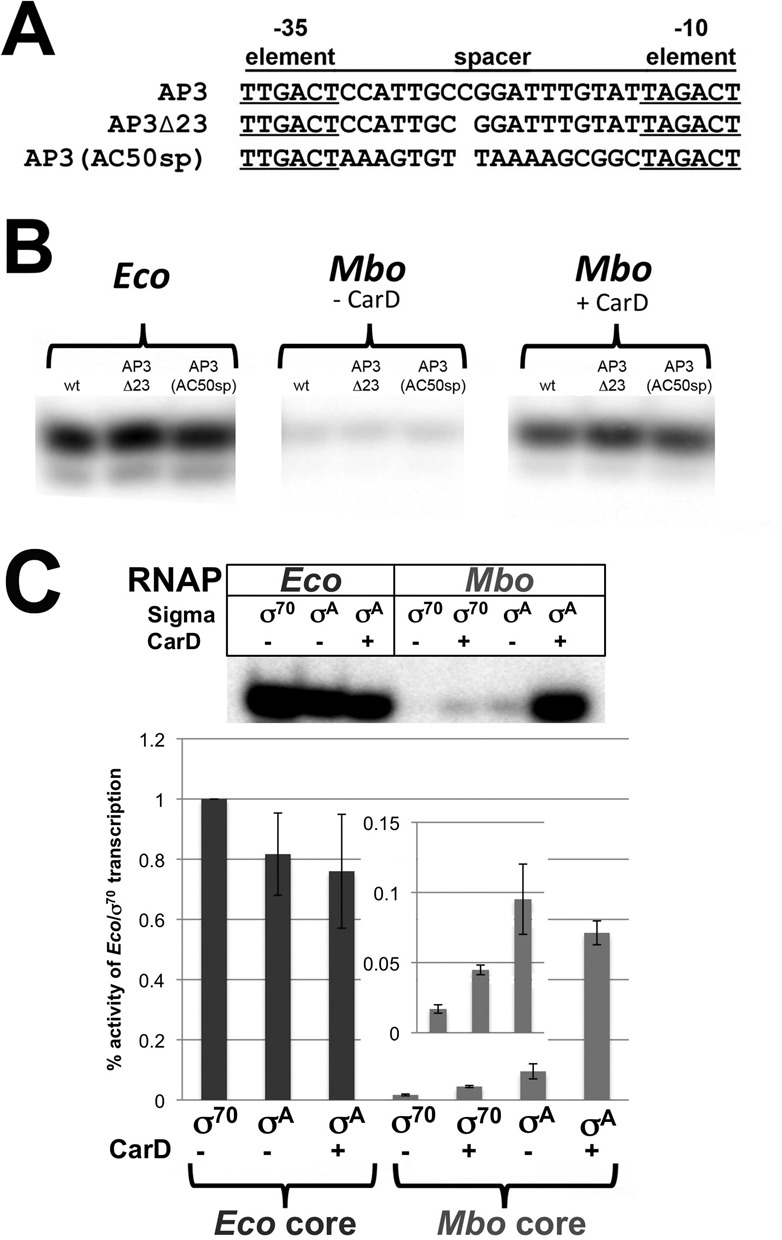
Weak activity of Mbo RNAP on the AP3 promoter is not due to the suboptimal 18-bp −10/-35 spacer of AP3 nor Mbo σA. (A) Sequences of AP3 (18-bp −10/-35 spacer, top) and spacer mutant promoters. AP3Δ23 has a deletion of the −23 bp, giving AP3Δ23 a 17-bp −10/-35 spacer. AP3(AC50sp) has the optimal 17-bp spacer of AC50 (blue) swapped for the AP3 spacer. (B) Single round abortive initiation activity of RNAPs on wt AP3, AP3Δ23 and AP3(AC50sp) was determined in transcription buffer as described (Figure 2 and Materials and Methods) with 10 mM KGlu. Gels show transcription initiation products (GpUpU* synthesis). (C) Single round abortive initiation assays were performed as described in (B) with hybrid holoenzymes. Eco core RNAP was mixed with either Eco σ70 or Mbo σA and assayed for activity on the AP3 promoter. The reverse experiment was also performed with Mbo core RNAP mixed with Eco σ70 or Mbo σA. CarD was also tested for effects on transcription where indicated. Graphs below represent relative activities of hybrid holoenzymes normalized to Eco-core/σ70 holoenzyme. The inset shows a magnification of Mbo-core/Eco σ70, Mbo-core/Eco σ70 + CarD and Mbo-core/Mbo σA to better visualize the weak activity (<0.1% that of Eco-core/σ70).
Mbo σA is not responsible for the weak transcription activity on the AP3 promoter
Mbo σA is very similar to Eco σ70 and the residues contacting the −10 and −35 promoter regions are highly conserved (32,33). In contrast, the non-conserved regions (NCR), inserted within domain 2 of housekeeping σ factors (34), share virtually no homology. Therefore, we tested whether Mbo σA was the cause of the instability and weak activity of the Mbo holoenzyme by comparing the transcriptional activity of Eco core RNAP with Eco σ70 (Eco-core/σ70) to Eco core RNAP with Mbo σA (Eco-core/σA) on the AP3 promoter. Mbo σA was able to direct transcription of AP3 by Eco core RNAP almost as well as σ70, suggesting that the weak activity of Mbo holoenzyme does not originate from σA (Figure 4C). As expected, CarD was unable to activate Eco-core/σ70 (data not shown) nor Eco-core/σA holoenzymes (we have no evidence that CarD interacts with Eco RNAP).
Previous studies found that interaction between Eco σ70 and Mbo core RNAP was not detectable by native gel electrophoresis (22). Therefore, as expected, Eco σ70 directed only very weak transcription of AP3 by the Mbo core RNAP (Figure 4C). One possible explanation for the weak Eco σ70/Mbo RNAP interaction is that the relatively large NCR of Eco σ70 (247 residues, compared to the 32 residue Mbo σA NCR) could clash with lineage-specific insert 2 of the mycobacterial RNAP β′ subunit (35), a ∼130-residue insert located near the σ NCR that is not present in Eco. CarD very weakly activates Mbo-core RNAP/Eco σ70 (Figure 4C).
DNase I footprinting
Occupancy of the promoter by RNAP protects the DNA from DNAse I cleavage, usually over a range from about −55 to +20 (36–39). DNAse I binds DNA in the minor groove, widening it and bending the DNA away from itself toward the major groove (40). Thus, RNAP-mediated DNA distortion resulting in exposed, widened minor grooves can also give rise to sites of DNAse I hypersensitivity. We examined promoter complexes of Eco and Mbo RNAPs on both the AP3 and AC50 promoters by DNase I footprinting (Figure 5).
On the AP3 promoter, Eco RNAP produced a strong DNAse I footprint, protecting the promoter DNA completely from ∼−22 to +13. Additional regions of protection and hypersensitivity extended upstream to −34 (Figure 5A, lanes 3–5). By contrast, Mbo RNAP (without CarD) failed to protect most of the DNA from DNAse I cleavage. Evidence of Mbo RNAP interaction with the DNA was apparent through the presence of some hypersensitive sites, such as at −34 and +14/+15 (Figure 5A, lanes 6–7). Addition of CarD conferred partial DNA protection (Figure 5A, lanes 8–9). The CarDW85 substitutions (W85A, W85F) produced footprints similar to wild-type CarD (Figure 5A, lanes 10–13). The DNAse I footprint due to Eco RNAP binding was resistant to competitor (Figure 5A, lanes 4–5), as was Mbo RNAP in the presence of CarD (Figure 5A, lanes 9, 11, 13).
On the AC50 promoter, Eco and Mbo RNAPs produced strong DNAse I footprints that were very similar to each other. Some minor differences in the footprints were observed upstream of the −35 element (−45 to −53), and the Mbo RNAP footprint extended slightly further downstream (+17/+18) than the Eco RNAP footprint (Figure 5B, lanes 3–5 compared to lanes 6–7). Addition of CarD had no effect whatsoever on the Mbo RNAP footprint (Figure 5B, compare lanes 6 and 7). This is not surprising since the region of DNA expected to be contacted by CarD [just upstream of the −10 element; (19)] is already completely protected from DNAse I cleavage by Mbo RNAP alone. The Mbo RNAP footprint was very sensitive to the presence of competitor DNA. CarD substantially stabilized the Mbo RNAP complex to competitor (Figure 5B, lanes 8–9).
KMnO4 footprinting
KMnO4 reacts with unstacked thymine (T) bases, and the modified T's can be subsequently detected by strand cleavage. This approach can thus be used to probe transcription bubble formation in RPo (39,41). We used KMnO4 footprinting (template strand) to examine the transcription bubble formed by Eco and Mbo RNAPs, and to examine the effect of CarD on Mbo RNAP transcription bubble formation.
The transcription bubble formed by Eco RNAP holoenzyme at promoters, as measured by KMnO4 footprinting, typically extends from about −11 to +2 (41). On the AP3 promoter, the transcription bubble formed by Eco RNAP results in strong KMnO4 reactivity of template strand T's at −11, −9 and −3, the only T's present within the typical transcription bubble range (Figure 6A, B, lane 2). Mbo RNAP, without CarD, formed a barely detectable transcription bubble (Figure 6B, lane 6). Transcription bubble formation by Mbo RNAP was stimulated dramatically by CarD (Figure 6B, lane 10). The extent of the transcription bubbles formed by Mbo and Eco RNAPs was identical, as far as could be determined.
Figure 6.
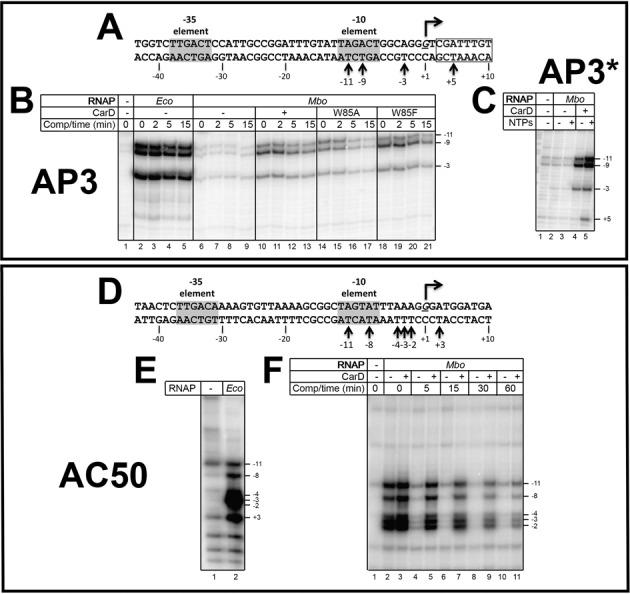
KMnO4 footprints (template strand) of Eco and Mbo (±CarD) RNAPs on the AP3 and AC50 promoters. (A) Sequence of the AP3 promoter, except the altered sequence (+3 and downstream) of AP3* is boxed (gives rise to the template-strand T at +5 which is absent in AP3, see Figure 1). Template strand (bottom) thymidines rendered KMnO4 reactive by RNAP are denoted. (B) KMnO4 footprints. Lane 1, no protein added. (C) Effect of adding initiating NTPs (GpU ribonucleotide dimer and CTP) on the KMnO4 footprint of Mbo RNAP on the AP3* promoter. (D) Sequence of the AC50 promoter. Template strand (bottom) thymidines rendered KMnO4 reactive by RNAP are denoted. (E) KMnO4 footprint of Eco RNAP on the AC50 promoter. (F) KMnO4 footprints of Mbo RNAP (±CarD as indicated) on the AC50 promoter.
Addition of NTPs has been shown to extend DNAse I protection of the rrnB P1 promoter downstream of the transcription bubble and to increase the intensity of KMnO4-reactive thymines (42). Therefore, we tested whether addition of NTPs would stimulate and possibly extend bubble formation on AP3 by Mbo RNAP. In order to detect if the bubble would be extended by the addition of NTPs, we used an alternative construct of AP3 (AP3*) where the sequence from +3 and downstream was altered, generating a template-strand T at +5 (Figure 6A). Addition of a dinucleotide primer (GpU) and CTP supported the abortive synthesis of GpUpC (Figure 2A) but did not enhance bubble formation by Mbo RNAP without CarD (Figure 6C, lanes 2 and 3). In the presence of CarD KMnO4 reactivity was slightly enhanced upon the addition of the initiating nucleotides. In addition, the bubble extended downstream to +5 (Figure 6C, lanes 4 and 5), most likely due to ‘scrunching’ during abortive synthesis (43,44).
The CarDW85A mutant stimulated bubble formation somewhat less than wt-CarD (Figure 6B, compare lanes 10 and 14), and upon challenge with competitor, dissociated more rapidly. The CarDW85F mutant stimulated bubble formation similar to wild-type CarD (Figure 6B, lanes 18–21).
On AC50, RPo formation by Eco RNAP induced KMnO4 reactivity at all template-strand T's between −11 and +3 (−11, −8, −4, −3, −2, +3; Figure 6D and E). Mbo RNAP without CarD induced a very similar footprint except the template strand T at +3 was relatively much less reactive (Figure 6F, lane 2). With CarD, KmnO4 reactivity was slightly stimulated, but the pattern of reactivity was unaltered (Figure 6F, lanes 2 and 3). The striking effect of CarD was seen after challenging the complexes with competitor, where the promoter complexes were dramatically stabilized over time by CarD (Figure 6F).
CarD stabilizes RPo by preventing transcription bubble collapse
CarD stabilizes Mbo RNAP open complexes on the AC50 promoter template (spanning −152 to +72), increasing the half-life in the presence of competitor more than 60-fold (Figure 3B). We hypothesized that prevention of transcription bubble collapse (reannealing) could explain the dramatic effect of CarD on RPo stability. To test this hypothesis, we determined the effect of CarD on Mbo holoenzyme stability on a synthetic promoter template based on the AC50 sequence (spanning −60 to +20; Figure 7, AC50_DS) and compared it with the exact same synthetic template but with a non-complementary transcription bubble (from −11 to +2; Figure 7, AC50_Bubble) unable to collapse. The non-complementary bubble was generated by altering the template strand sequence from −11 to −3 [thus maintaining the −10 element and discriminator sequences on the non-template strand (33,45,46)] and the non-template strand from −2 to +2 (thus maintaining the same initially transcribed sequence). Half-life assays were performed as described for Figure 3. On the AC50_DS template, CarD increased the half-life 100-fold (Figure 7), consistent with our results on the full AC50 double-stranded template (Figure 3B). However, on the AC50_Bubble template, Mbo holoenzyme behaved much like Eco holoenzyme, with no detectable dissociation over the 60-min experiment (half-life >> 60 min), and addition of CarD had no effect. We conclude that the very short half-life of Mbo RPo on AC50 (Figure 3B) is due, at least in large part, to collapse of the transcription bubble, which generates the closed promoter complex (RPc) at the expense of RPo. RPc is in rapid equilibrium with RNAP and promoter DNA in solution, and in the presence of the full-con promoter trap competitor DNA, transcription competent RPo is rapidly depleted. The stabilization of Mbo RPo by CarD can be largely attributed to the inhibition of transcription bubble collapse by CarD.
DISCUSSION
In this study, we used a mycobacterial transcription system (Mbo core RNAP, Mbo/Mtb σA and Mbo/Mtb CarD—essentially identical to a complete Mtb transcription system) that shows transcription activity on the optimized AC50 promoter (Figure 1) essentially equal to or better than the very well established Eco transcription system (Figure 2B). We thus propose that differences in behavior we observed between the Mbo and Eco RNAPs can be attributed to mechanistic differences in enzyme function and not due to suboptimal RNAP preparations or conditions.
Our studies revealed some similarities between the two RNAPs. These include:
Both the Mbo and Eco RNAPs gave rise to nearly identical Dnase I footprints on each promoter (Figure 5), indicating similar physical protein/DNA interactions.
The RNAPs gave rise to nearly identical KMnO4 footprints on each promoter (Figure 6), indicating very similar transcription bubbles in the open complexes.
Despite these similarities listed above, the mechanistic differences we have observed between the two RNAPs are profound, and include:
Weak transcription activity of Mbo RNAP on the native Mtb AP3 promoter (more than an order of magnitude less than Eco RNAP, Figure 2A). This finding was echoed by the absence of a robust Dnase I footprint for Mbo RNAP on AP3 (Figure 5A, lane 6) and the absence of a KMnO4 footprint for Mbo RNAP on AP3 (Figure 6A, lane 6). The weak transcription activity of Mbo RNAP holoenzyme on AP3 is a property of the core RNAP and not Mbo σA (Figure 4C).
High sensitivity of the Mbo RNAP to Cl−, particularly on AP3 (1/2-maximal activity on AP3 at about 60 mM KCl, compared to 160 mM KCl for Eco RNAP, Figure 2A).
Very unstable Mbo RNAP open promoter complexes on both promoters (t1/2 of 2.3 min on AP3 and less than 1 min on AC50), while Eco RNAP formed essentially irreversible promoter complexes on both promoters (Figure 3). This finding was echoed in the rapid disappearance of the Dnase I and KMnO4 footprints for Mbo RNAP upon competitor challenge (Figure 5B, lanes 8 and 9; Figure 6F, lanes 2, 4, 6, 8, 10).
These significant differences between the Mbo and Eco RNAPs were, to a significant extent, rescued by the presence of the transcription activator CarD:
The weak Mbo RNAP transcription activity on AP3 was boosted by CarD nearly 10-fold (Figure 2A).
The very unstable promoter complexes formed by Mbo RNAP on both promoters were dramatically stabilized by CarD (t1/2 increased more than 10-fold for AP3, more than 60-fold for AC50; Figure 3). This was also reflected in the behavior of the Dnase I and KMnO4 footprints (Figures 5 and 6).
Remarkably, despite these profound effects of CarD on the stability of the Mbo RNAP promoter complexes, the presence of CarD stimulated the strength of the DNase I and KMnO4 footprints on the AC50 promoter (where the footprints in the absence of CarD could be compared) as expected, but did not alter the structure of the footprints (Figure 5B, compare lanes 6 and 7; Figure 6F, compare lanes 2 and 3). On the AP3 promoter, a mutant of CarD that altered the effectiveness of CarD as a transcription activator (CarDW85A) also did not alter the structure of the footprints (Figure 5A, lane 10; Figure 6B, lane 14).
The in vitro transcription conditions used here, which included a large excess of RNAP over promoter DNA and therefore favored RPo formation and transcription activity, were chosen to allow detection of the very weak transcription activity of Mbo RNAP on AP3. Such conditions explain how the Mbo RNAP promoter complexes on the optimized AC50 promoter can have such a rapid dissociation rate (short t1/2) but still show transcription levels similar to Eco RNAP (Figure 2B). Even in these strongly favorable conditions, however, AP3 transcription by Mbo RNAP without CarD was very weak (Figure 2A). These observations suggest that Mbo RNAP has a very fast on-rate on AC50, while on AP3 the on-rate is slow. On AC50, at high RNAP concentration favoring the formation of RPo, transcription activity is high (in the absence of competitor promoter trap DNA) despite the high off-rate, even in the absence of CarD (Figure 3B). Eliminating the forward reaction by adding competitor promoter trap DNA reveals the rapid dissociation from AC50 and the effect of CarD in stabilizing the AC50 RPo to dissociation (Figure 3B). AP3 activity, on the other hand, may be limited by a slow on-rate: the slow on-rate combined with the fast off-rate in the absence of CarD would yield very low levels of RPo and thus weak transcription activity. CarD stimulates AP3 activity by dramatically slowing the off-rate (Figure 3A), allowing the build-up of increased levels of RPo (we cannot rule out that CarD also affects the on-rate).
The most striking distinction between the AC50 and AP3 promoters is that AC50 harbors an optimal 17-bp −10/−35 spacer, while AP3 harbors a suboptimal 18-bp spacer (Figure 1). Nevertheless, the promoter spacer mutagenesis and swapping experiments demonstrate that the suboptimal −10/−35 spacer of AP3 is not the source of its inability to support transcription by the Mbo RNAP (Figure 4). Mbo RNAP transcription activity on several different mutant AP3 promoters with optimal 17-bp −10/−35 spacers [AP3Δ23 and AP3(AC50sp), Figure 4A] was just as weak as the wild-type AP3 promoter (Figure 4B). Thus, the characteristics of the AP3 promoter that give rise to such feeble Mbo RNAP transcription activity remain unknown. More extensive swapping of promoter regions, such as the discriminator (45,46), as well as targeted promoter mutagenesis, will be required to understand this property. Understanding the characteristics of the AP3 promoter that give rise to such weak Mbo RNAP transcription activity is important since it will shed light on mechanistic differences between the Mbo and Eco RNAPs, since Eco RNAP shows robust transcription activity on AP3.
Our current analysis allows us to extend the model for CarD function (19). The results establish that CarD dramatically stabilizes Mbo RNAP open promoter complexes (Figure 3) but does not alter the RNAP/promoter interactions as revealed by DNase I footprinting (Figure 5), and does not alter the transcription bubble as revealed by KMnO4 footprinting (Figure 6). Engineering a non-complementary transcription bubble in the AC50 promoter extended the half-life of the Mbo RPo to values similar to that of Eco and made the function of CarD redundant (Figures 3 and 7). These findings support a model whereby CarD forms favorable interactions with the upstream edge of the pre-formed transcription bubble, stabilizing the bubble against collapse. This implies that bubble collapse is a major contributor to the instability of Mbo RNAP promoter complexes. However, we do note that our findings neither support nor contradict models for CarD function that posit allosteric effects on RNAP/promoter interactions (47). We also note that the CarD mechanism may be more complex and could affect other steps in the pathway of RPo formation.
Depletion of CarD in Msm cells results in increased levels of 16S rRNA, leading to the initial proposal that CarD acts as a repressor of rRNA transcription (18). This was coupled to the finding that overexpression of CarD amino acids 1–104 could rescue the phenotype of ΔDksA in Eco cells, since DksA negatively regulates rRNA transcription in vivo (48). Our results here clearly confirm that CarD functions in vitro as a transcription activator, as was also shown previously (19). CarD is an essential global regulator in mycobacteria; it is found on the Msm chromosome at essentially all σA promoters (19). Its depletion leads to sensitivity to multiple cellular stresses (18). We suggest that depletion of CarD leads to indirect, pleiotropic effects that ultimately result in an increase of 16S rRNA levels (19).
The CarD/DNA interaction involves a universally conserved Trp (Mbo CarDW85) that is predicted to insert into the distorted minor groove at the upstream edge of the −10 element (19). The CarDW85A substitution was previously shown to be defective in transcription activation (19). We show here that CarDW85A is also defective in stabilizing Mbo RNAP promoter complexes. The t1/2 for CarDW85A on AP3 is 71% of the wild-type CarD, while on AC50 it is only 21% (Figure 3). Nevertheless, CarDW85A still retains significant capacity to stabilize Mbo RNAP promoter compexes, indicating that other CarD/promoter DNA interactions must contribute to CarD function. CarDW85F exhibited transcription activation and promoter complex stabilization function similar to that of wild-type CarD, indicating that a bulky, hydrophobic (and likely aromatic) residue inserted into the upstream edge of the transcription bubble is sufficient for nearly full CarD function.
Eco has served as the archetypical organism on which the overwhelming majority of biochemical characterization of RNAP has been focused, and the properties of this RNAP have been assumed to be generally representative for all bacterial RNAPs. Our extensive comparisons of the properties of Eco and Mbo RNAPs at the same in vitro transcription conditions and on the same promoters demonstrate that some of these assumptions are not valid. Dnase I footprinting studies indicate that physical RNAP/promoter interactions are very similar between the two RNAPs, and KMnO4 footprinting indicates that the transcription bubble formed by both RNAPs at promoters is essentially identical. However, Eco RNAP is characterized by very stable open promoter complexes when double-stranded DNA is used as a competitor, with half-lives ranging between ∼10 and 100s of minutes. Even the Eco rrnB P1 promoter, the regulation of which is dependent on the instability of its open complexes (49), has a reported half-life of 20–58 min when double-stranded promoter DNA is used as a competitor (50,51). In particular, for promoters that are close to optimum (close to consensus −10 and −35 elements, optimal 17-bp −10/−35 spacer), Eco RNAP typically forms RPo irreversibly (52). This is borne out in our study, where Eco RNAP forms essentially irreversible open promoter complexes on both the optimized AC50 promoter and even on the suboptimal AP3 promoter (Figure 3). This is decidedly not the case for Mbo RNAP, which forms exceedingly short-lived promoter complexes on both AP3 and even on the optimized AC50 promoter (Figure 3). Thus, the assumption of RPo irreversibility for Mbo RNAP, even on optimal promoters, is not valid. Investigations of other RNAPs from non-Eco sources, such as B. subtilis (14), Tth (8), T. aquaticus (12) and M. smegmatis (15), have also noted characteristically unstable promoter complexes compared with Eco. The possibility that the properties of Eco RNAP may not be representative of most bacterial RNAPs, but rather that these properties make Eco RNAP an outlier, needs to be considered. It is interesting to note that CarD is found in Bacillus, Thermus and Mycobacterium species, where purified RNAPs have been found to generate relatively unstable open promoter complexes, but not in Eco, where RNAP generally forms exceedingly stable open complexes (19). Our finding that the Mbo σ factor is not the source of the observed RPo instability (Figure 4C) suggests that the instability comes from a property of the core RNAP. In general, important structural features of the bacterial RNAPs are highly conserved, but RNAPs from different bacterial lineages can differ substantially due to the presence/absence of so-called lineage-specific insertions (35). For example, previous studies found that deletion of 188-residues inserted in the middle of the Eco β′ Trigger-Loop [β′In6 according to the nomenclature of (35); SI3 according to the nomenclature of (53)] decreased the stability of promoter complexes by 10-fold (53). In RNAPs from Bacillus, Thermus and Mycobacterium species (which form unstable open complexes), β′In6 is absent (35), suggesting a correlation.
ChIP-seq experiments in M. smegmatis established that CarD is a global regulator, being present at essentially all promoter regions in the genome, suggesting that CarD is not a promoter-specific regulator (19). We have shown here that unstable promoter complexes seem to be a general property of Mbo RNAP, even on an optimized promoter like AC50 (Figure 3B). This suggests that CarD may be thought of as a general transcription factor that functions to prevent transcription bubble collapse, helping to compensate for the otherwise rapidly dissociating RNAP/promoter complexes throughout the genome.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Elizabeth Hubin, Rachel Mooney, Robert Landick, Wilma Ross, Rick Gourse and Christina Stallings for sharing plasmids, methodological advice and discussion. We also thank Ruth Saecker for important discussions.
Footnotes
Present addresses:
Elizabeth Davis, The University of Minnesota School of Medicine, 420 Delaware St. SE, Minneapolis, MN 55455, USA.
Katherine Leon, Department of Biochemistry and Molecular Biology, University of Chicago, 929 East 57th Street, GCIS W219, Chicago, IL 60637, USA.
FUNDING
The Rockefeller University. Funding for open access charge: The Rockefeller University.
Conflict of interest statement. None declared.
REFERENCES
- 1.Floss H.G., Yu T.-W. Rifamycin-mode of action, resistance, and biosynthesis. Chem. Rev. 2005;105:621–632. doi: 10.1021/cr030112j. [DOI] [PubMed] [Google Scholar]
- 2.Burgess R.R. Separation and characterization of the subunits of ribonucleic acid polymerase. J. Biol. Chem. 1969;244:6168–6176. [PubMed] [Google Scholar]
- 3.Burgess R.R., Travers A.A., Dunn J.J., Bautz E.K. Factor stimulating transcription by RNA polymerase. Nature. 1969;221:43–46. doi: 10.1038/221043a0. [DOI] [PubMed] [Google Scholar]
- 4.Burgess R.R., Travers A.A. Escherichia coli RNA polymerase: purification, subunit structure, and factor requirements. Fed. Proc. 1970;29:1164–1169. [PubMed] [Google Scholar]
- 5.Zhang G., Campbell E.A., Minakhin L., Richter C., Severinov K., Darst S.A. Crystal structure of Thermus aquaticus core RNA polymerase at 3.3 A resolution. Cell. 1999;98:811–824. doi: 10.1016/s0092-8674(00)81515-9. [DOI] [PubMed] [Google Scholar]
- 6.Murakami K.S., Masuda S., Campbell E.A., Muzzin O., Darst S.A. Structural basis of transcription initiation: an RNA polymerase holoenzyme-DNA complex. Science. 2002;296:1285–1290. doi: 10.1126/science.1069595. [DOI] [PubMed] [Google Scholar]
- 7.Vassylyev D.G., Sekine S.-I., Laptenko O., Lee J., Vassylyeva M.N., Borukhov S., Yokoyama S. Crystal structure of a bacterial RNA polymerase holoenzyme at 2.6 Å resolution. Nature. 2002;417:712–719. doi: 10.1038/nature752. [DOI] [PubMed] [Google Scholar]
- 8.Xue Y., Hogan B.P., Erie D.A. Purification and initial characterization of RNA polymerase from Thermus thermophilus strain HB8. Biochemistry. 2000;39:14356–14362. doi: 10.1021/bi0012538. [DOI] [PubMed] [Google Scholar]
- 9.Hogan B.P., Hartsch T., Erie D.A. Transcript cleavage by Thermus thermophilus RNA polymerase. Effects of GreA and anti-GreA factors. J. Biol. Chem. 2002;277:967–975. doi: 10.1074/jbc.M108737200. [DOI] [PubMed] [Google Scholar]
- 10.Kashkina E., Anikin M., Tahirov T.H., Kochetkov S.N., Vassylyev D.G., Temiakov D. Elongation complexes of Thermus thermophilus RNA polymerase that possess distinct translocation conformations. Nucl Acids Res. 2006;34:4036–4045. doi: 10.1093/nar/gkl559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mekler V., Minakhin L., Kuznedelov K., Mukhamedyarov D., Severinov K. RNA polymerase-promoter interactions determining different stability of the Escherichia coli and Thermus aquaticus transcription initiation complexes. Nucl Acids Res. 2012;40:11352–11362. doi: 10.1093/nar/gks973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miropolskaya N., Ignatov A., Bass I., Zhilina E., Pupov D., Kulbachinskiy A. Distinct functions of regions 1.1 and 1.2 of RNA polymerase subunits from Escherichia coli and Thermus aquaticus in transcription initiation. J. Biol. Chem. 2012;287:23779–23789. doi: 10.1074/jbc.M112.363242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schroeder L.A., deHaseth P.L. Mechanistic differences in promoter DNA melting by Thermus aquaticus and Escherichia coli RNA polymerases. J. Biol. Chem. 2005;280:17422–17429. doi: 10.1074/jbc.M501281200. [DOI] [PubMed] [Google Scholar]
- 14.Whipple F.W., Sonenshein A.L. Mechanism of initiation of transcription by Bacillus subtilis RNA polymerase at several promoters. J. Mol. Biol. 1992;223:399–414. doi: 10.1016/0022-2836(92)90660-c. [DOI] [PubMed] [Google Scholar]
- 15.China A., Tare P., Nagaraja V. Comparison of promoter-specific events during transcription initiation in mycobacteria. Microbiology. 2010;156:1942–1952. doi: 10.1099/mic.0.038620-0. [DOI] [PubMed] [Google Scholar]
- 16.Tare P., China A., Nagaraja V. Distinct and contrasting transcription initiation patterns at Mycobacterium tuberculosis promoters. PLoS ONE. 2012;7:e43900. doi: 10.1371/journal.pone.0043900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia-Moreno D., Abellon-Ruiz J., Garcia-Heras F., Murillo F.J., Padmanabhan S., Elias-Arnanz M. CdnL, a member of the large CarD-like family of bacterial proteins, is vital for Myxococcus xanthus and differs functionally from the global transcriptional regulator CarD. Nucleic Acids Res. 2010;38:4586–4598. doi: 10.1093/nar/gkq214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stallings C.L., Stephanou N.C., Chu L., Hochschild A., Nickels B.E., Glickman M.S. CarD is an essential regulator of rRNA transcription required for mycobacterium tuberculosis persistence. Cell. 2009;138:146–159. doi: 10.1016/j.cell.2009.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Srivastava D.B., Leon K., Osmundson J., Garner A.L., Weiss L.A., Westblade L.F., Glickman M.S., Landick R., Darst S.A., Stallings C.L., et al. Structure and function of CarD, an essential mycobacterial transcription factor. Proc. Natl. Acad. Sci. 2013;110:12619–12624. doi: 10.1073/pnas.1308270110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murakami K.S. The X-ray crystal structure of Escherichia coli RNA polymerase Sigma70 holoenzyme. J. Biol. Chem. 2013;288:9126–9134. doi: 10.1074/jbc.M112.430900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bae B., Davis E., Brown D., Campbell E.A., Wigneshweraraj S.R., Darst S.A. Phage T7 Gp2 inhibition of Escherichia coli RNA polymerase involves misappropriation of σ70 domain 1.1. Proc. Natl. Acad. Sci. 2013;110:19772–19777. doi: 10.1073/pnas.1314576110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Czyz A., Mooney R.A., Iaconi A., Landick R. Mycobacterial RNA polymerase requires a U-tract at intrinsic terminators and is aided by NusG at suboptimal terminators. MBio. 2014;5:e00931. doi: 10.1128/mBio.00931-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Twist K.-A., Husnain S.I., Franke J.D., Jain D., Campbell E.A., Nickels B.E., Thomas M.S., Darst S.A., Westblade L.F. A novel method for the production of in vivo-assembled, recombinant Escherichia coli RNA polymerase lacking the α C-terminal domain. Protein Sci. 2011;20:986–995. doi: 10.1002/pro.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaal T., Ross W., Estrem S.T., Nguyen L.H., Burgess R.R., Gourse R.L. Promoter recognition and discrimination by EsigmaS RNA polymerase. Mol. Microbiol. 2001;42:939–954. doi: 10.1046/j.1365-2958.2001.02703.x. [DOI] [PubMed] [Google Scholar]
- 25.Gonzalez-y-Merchand J.A., Colston M.J., Cox R.A. The rRNA operons of Mycobacterium smegmatis and Mycobacterium tuberculosis: comparison of promoter elements and of neighbouring upstream genes. Microbiology (Reading, Engl.) 1996;142 (Pt 3):667–674. doi: 10.1099/13500872-142-3-667. [DOI] [PubMed] [Google Scholar]
- 26.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cayley S., Lewis B.A., Guttman H.J., Record M.T., Jr Characterization of the cytoplasm of Escherichia coli K-12 as a function of external osmolarity. Implications for protein-DNA interactions in vivo. J. Mol. Biol. 1991;222:281–300. doi: 10.1016/0022-2836(91)90212-o. [DOI] [PubMed] [Google Scholar]
- 28.Tian J., Bryk R., Itoh M., Suematsu M., Nathan C. Variant tricarboxylic acid cycle in Mycobacterium tuberculosis: identification of alpha-ketoglutarate decarboxylase. Proc. Natl. Acad. Sci. 2005;102:10670–10675. doi: 10.1073/pnas.0501605102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leirmo S., Harrison C., Cayley D.S., Burgess R.R., Record M.T., Jr Replacement of potassium chloride by potassium glutamate dramatically enhances protein-DNA interactions in vitro. Biochemistry. 1987;26:2095–2101. doi: 10.1021/bi00382a006. [DOI] [PubMed] [Google Scholar]
- 30.Gralla J.D., Vargas D.R. Potassium glutamate as a transcriptional inhibitor during bacterial osmoregulation. EMBO J. 2006;25:1515–1521. doi: 10.1038/sj.emboj.7601041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dayton C.J., Prosen D.E., Parker K.L., Cech C.L. Kinetic measurements of Escherichia coli RNA polymerase association with bacteriophage T7 early promoters. J. Biol. Chem. 1984;259:1616–1621. [PubMed] [Google Scholar]
- 32.Campbell E.A., Muzzin O., Chlenov M., Sun J.L., Olson C.A., Weinman O., Trester-Zedlitz M.L., Darst S.A. Structure of the bacterial RNA polymerase promoter specificity sigma subunit. Mol. Cell. 2002;9:527–539. doi: 10.1016/s1097-2765(02)00470-7. [DOI] [PubMed] [Google Scholar]
- 33.Feklistov A., Darst S.A. Structural basis for promoter -10 element recognition by the bacterial RNA polymerase σ subunit. Cell. 2011;147:1257–1269. doi: 10.1016/j.cell.2011.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lonetto M., Gribskov M., Gross C.A. The sigma 70 family: sequence conservation and evolutionary relationships. J. Bacteriol. 1992;174:3843–3849. doi: 10.1128/jb.174.12.3843-3849.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lane W.J., Darst S.A. Molecular evolution of multisubunit RNA polymerases: sequence analysis. J. Mol. Biol. 2010;395:671–685. doi: 10.1016/j.jmb.2009.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carpousis A.J., Gralla J.D. Interaction of RNA polymerase with lacUV5 promoter DNA during mRNA initiation and elongation. Footprinting, methylation, and rifampicin-sensitivity changes accompanying transcription initiation. J. Mol. Biol. 1985;183:165–177. doi: 10.1016/0022-2836(85)90210-4. [DOI] [PubMed] [Google Scholar]
- 37.Ozoline O.N., Tsyganov M.A. Structure of open promoter complexes with Escherichia coli RNA polymerase as revealed by the DNase I footprinting technique: compilation analysis. Nucleic Acids Res. 1995;23:4533–4541. doi: 10.1093/nar/23.22.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Craig M.L., Suh W.C., Record M.T., Jr HO. and DNase I probing of E sigma 70 RNA polymerase–lambda PR promoter open complexes: Mg2+ binding and its structural consequences at the transcription start site. Biochemistry. 1995;34:15624–15632. doi: 10.1021/bi00048a004. [DOI] [PubMed] [Google Scholar]
- 39.Ross W., Gourse R.L. Analysis of RNA polymerase-promoter complex formation. Methods. 2009;47:13–24. doi: 10.1016/j.ymeth.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weston S.A., Lahm A., Suck D. X-ray structure of the DNase I-d(GGTATACC)2 complex at 2.3 A resolution. J. Mol. Biol. 1992;226:1237–1256. doi: 10.1016/0022-2836(92)91064-v. [DOI] [PubMed] [Google Scholar]
- 41.Sasse-Dwight S., Gralla J.D. Footprinting protein-DNA complexes in vivo. Meth. Enzymol. 1991;208:146–168. doi: 10.1016/0076-6879(91)08012-7. [DOI] [PubMed] [Google Scholar]
- 42.Rutherford S.T., Villers C.L., Lee J.-H., Ross W., Gourse R.L. Allosteric control of Escherichia coli rRNA promoter complexes by DksA. Genes Dev. 2009;23:236–248. doi: 10.1101/gad.1745409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kapanidis A.N., Margeat E., Ho S.O., Kortkhonjia E., Weiss S., Ebright R.H. Initial transcription by RNA polymerase proceeds through a DNA-scrunching mechanism. Science. 2006;314:1144–1147. doi: 10.1126/science.1131399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Revyakin A., Liu C., Ebright R.H., Strick T.R. Abortive initiation and productive initiation by RNA polymerase involve DNA scrunching. Science. 2006;314:1139–1143. doi: 10.1126/science.1131398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feklistov A., Barinova N., Sevostyanova A., Heyduk E., Bass I., Vvedenskaya I., Kuznedelov K., Merkienė E., Stavrovskaya E., Klimašauskas S., et al. A basal promoter element recognized by free RNA polymerase σ subunit determines promoter recognition by RNA polymerase holoenzyme. Mol. Cell. 2006;23:97–107. doi: 10.1016/j.molcel.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 46.Haugen S.P., Berkmen M.B., Ross W., Gaal T., Ward C., Gourse R.L. rRNA promoter regulation by nonoptimal binding of σ region 1.2: an additional recognition element for RNA polymerase. Cell. 2006;125:1069–1082. doi: 10.1016/j.cell.2006.04.034. [DOI] [PubMed] [Google Scholar]
- 47.Gulten G., Sacchettini J.C. Structure of the Mtb CarD/RNAP β-lobes complex reveals the molecular basis of interaction and presents a distinct DNA-binding domain for Mtb CarD. Structure. 2013;21:1859–1869. doi: 10.1016/j.str.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paul B.J., Barker M.M., Ross W., Schneider D.A., Webb C., Foster J.W., Gourse R.L. DksA: a critical component of the transcription initiation machinery that potentiates the regulation of rRNA promoters by ppGpp and the initiating NTP. Cell. 2004;118:311–322. doi: 10.1016/j.cell.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 49.Paul B.J., Ross W., Gaal T., Gourse R.L. rRNA transcription in Escherichia coli. Annu. Rev. Genet. 2004;38:749–770. doi: 10.1146/annurev.genet.38.072902.091347. [DOI] [PubMed] [Google Scholar]
- 50.Barker M.M. Regulation without protein transcription factors: instrinsic properties of Escherichia coli promoters that lead to their regulation. Ph.D. thesis. 2013 University of Wisconsin-Madison. [Google Scholar]
- 51.Rutherford S.T., Lemke J.J., Vrentas C.E., Gaal T., Ross W., Gourse R.L. Effects of DksA, GreA, and GreB on transcription initiation: insights into the mechanisms of factors that bind in the secondary channel of RNA polymerase. J. Mol. Biol. 2007;366:1243–1257. doi: 10.1016/j.jmb.2006.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saecker R.M., Record M.T., Jr, deHaseth P.L. Mechanism of bacterial transcription initiation: RNA polymerase - promoter binding, isomerization to initiation-competent open complexes, and initiation of RNA synthesis. J. Mol. Biol. 2011;412:754–771. doi: 10.1016/j.jmb.2011.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Artsimovitch I. Co-overexpression of Escherichia coli RNA polymerase subunits allows isolation and analysis of mutant enzymes lacking lineage-specific sequence insertions. J. Biol. Chem. 2003;278:12344–12355. doi: 10.1074/jbc.M211214200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.