


Abstract
Methods that permit controlled changes in the expression of genes are important tools for biological and medical research, and for biotechnological applications. Conventional methods are directed at individually changing each gene, its regulatory elements or its mRNA's translation rate. We demonstrate that the CRISPR-associated DNA-binding Cascade complex can be used for efficient, long-lasting and programmable gene silencing. When Cascade is targeted to a promoter sequence the transcription of the downstream gene is inhibited, resulting in dramatically reduced expression. The specificity of Cascade binding is provided by the integral crRNA component, which is easily designed to target virtually any stretch of DNA. Cascade targeted to the ORF sequence of the gene can also silence expression, albeit at lower efficiency. The system can be used to silence plasmid and chromosome targets, simultaneously target several genes and is active in different bacterial species and strains. The findings described here are an addition to the expanding range of CRISPR-based technologies and may be adapted to additional organisms and cell systems.
INTRODUCTION
Rapid modulation of gene expression is an essential feature of bacterial adaptive responses to changing environments. Gene expression can be regulated on both transcriptional and post-transcriptional level, often in a multi-layer fashion. Regulation by activators and repressors that alter transcription initiation is a well-studied phenomenon. The activity of protein factors is often affected by their expression levels by modifications, such as phosphorylation, and by binding of various co-factors (1). Once formed, the mRNA is subject to a variety of post-transcriptional regulatory mechanisms including those that regulate premature transcription termination, translation initiation and mRNA stability. With the discovery of riboregulators and riboswitches it has become clear that RNA structures can regulate gene expression. It is, however, interesting to note that most instances of regulation involving RNA operate at the post-transcriptional level (2) and that there are no known natural examples of RNA-mediated control of promoter activity or transcription initiation in bacteria.
Genetic engineering has allowed exploitation of naturally occurring regulatory mechanisms for both applied use and basic science. Artificial transcription factors have also been created; by fusing DNA-binding protein domains with regulatory domains transcription of specific genes can be controlled (3). Other approaches have involved manipulation of the sequence specificity of a given DNA binding protein, such as transcription-activator-like-effector or zinc finger proteins to direct transcription regulation (4,5). However, engineering of sequence specificity of proteins is a complex and time-consuming process which reduces applicability for simultaneous targeting of multiple genes (6). A programmable, simple and generic method for transcription control is a desirable tool for analysis of any cell and organism. Its effect would be comparable to that of RNAi-based silencing systems in eukaryotes, but it should be noted that these act on the post-transcriptional level.
Promising candidates for easy and efficient gene regulation are the RNA-guided DNA-binding components of CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) systems, an adaptive immune system in bacteria and archaea that provides protection from invading viruses and mobile elements. A CRISPR-Cas system is characterized by two features: the CRISPR locus and a set of CRISPR-associated (cas) genes. Both components need to be expressed for defense against invasive genetic elements. For an in-depth review, see, e.g. van der Oost et al. (7).
The CRISPR loci contain short repeated sequences separated by short unique spacers usually acquired from phages or other mobile genetic elements (8–10). The CRISPR is transcribed and the RNA is processed by Cas proteins into CRISPR RNA (crRNA) (11–13); crRNAs use sequence complementarity to guide Cas proteins to target DNA for degradation (14–17), although other functions have also been described (18).
CRISPR-Cas systems are present in about 45% of bacteria and 83% of archaea, and they are currently divided into three major types (I, II and III) and further subtypes (19,20). We focus our work on the Escherichia coli (E. coli) type I-E system, where Cas proteins and crRNA form a complex called Cascade (11). Cascade is a surveillance unit that upon detection of target DNA recruits Cas3 to destroy the target. Targeting requires the presence of a cognate protospacer-adjacent motif (PAM) which is a short sequence (3 bp in the case of Cascade) flanking the protospacer (11,16,21). Complexes similar to Cascade are also found in other type I and type III CRISPR-Cas systems (7,22,23, and references therein).
Type II CRISPR-Cas systems have already been adapted for biotechnological applications, mainly as a tool for genome editing (24,25), but also for gene regulation where a catalytically inactive ‘dead’ Cas9 (dCas9) was used. The dCas9 containing a small guide RNA (sgRNA) was shown to reduce transcription when targeted to a cognate promoter or the open reading frame (ORF) in E. coli and other organisms (26,27). Transcription activation or repression can also be achieved by fusing dCas9 with either a transcriptional activation domain or a repressor (28–30).
Here, we exploit the DNA-binding ability of the Cascade protein-RNA complex to design a programmable system for targeted silencing of gene expression, adding to the emerging CRISPR-Cas toolbox for genetic modifications and gene regulation. We express the components of Cascade in the absence of the Cas3 nuclease, which allows stable target binding without inducing its cleavage. We show that co-expression of a minimal CRISPR array carrying spacers designed to target a promoter result in effective silencing of the downstream gene. Repression could also be achieved by targeting coding regions of a gene, although at lower efficiency. As the system can be made inducible and can be targeted to virtually any region of DNA, it holds the potential as a general silencing tool for any gene of interest in tractable bacteria, archaea and eukaryotes.
MATERIALS AND METHODS
Strains, plasmids and culture media
All experiments in E. coli were performed in Δcas3 genetic background, except BL21 strains that naturally lack cas3 genes. The kanamycin cassette from JW2731(2) Δcas3, a Keio collection strain (31), was removed by Flp recombinase expressed from a temperature-sensitive plasmid, pCP20, as previously described (32). P1vir lysate grown on BL21AI was used, in a transduction protocol previously described (33), to introduce arabinose-inducible T7 RNA polymerase gene into this strain. This strain was designated MLS367. For routine growth, E. coli strains BL21DE3 (34), BL21AI (Invitrogen), MLS367 and Salmonella typhimurium LT2 strain DA6192 were cultured in Luria Bertani (LB) medium and grown with aeration at 37°C. When necessary, the media was supplemented with kanamycin (50 μg/ml), ampicillin (100 μg/ml), streptomycin (50 μg/ml), tetracycline (25 μg/ml) and chloramphenicol (15 μg/ml). Chromosomal markers were not selected for during growth in liquid cultures.
The plasmid pEH9 was used as silencing target and carries a venus gfp gene preceded by a PLtetO-1 promoter (35). It has a 158-bp polymerase chain reaction (PCR)-amplified DNA fragment containing the 5′ UTR and first 8 codons of the ompA gene inserted into the NsiI and NheI sites of the plasmid pXG10 (36). To compare targeting of a plasmid and targeting of the chromosome, the gfp gene, PLtetO-1 promoter and resistance marker from pEH9 were amplified by PCR using primer LA085 and LA086 (see Supplementary Table S1) and introduced into the E. coli BW25113 chromosome in the Lambda attB site (position 807747–807769 on the BW25113 chromosome) using Lambda red recombination (37). After verification of insertion, the construct was moved into MLS367 by P1 transduction, this strain was designated MLS541.
To test the simultaneous silencing of multiple targets, the mTagBfp2 gene under the control of the constitutive promoter J23101 (Genbank KM018299) was inserted by P1 transduction in the galK locus of MLS367. The chloramphenicol selection marker was removed by Flp recombinase expressed from pCP20. To construct the final strain containing both reporters, the gfp construct (above) was moved to the chromosome of this strain by P1 transduction, this strain was designated MLS561.
Cascade was expressed from pWUR400 (11). For expression of crRNA, minimal CRISPR arrays containing the transcribed 53 bp of the leader sequence from the CRISPR1 array of E. coli K12 (38) and a spacer flanked by two repeats were synthesized and subcloned by Life technologies. The CRISPRs were cloned into the EcoRI and XbaI sites of pZE12Luc (35), thereby removing the luciferase gene and placing the CRISPR cassette downstream of the IPTG-inducible promoter. For demonstration of silencing in S. typhimurium LT2 strain DA6192 cultures were transformed with pWUR400, pEH9 and CRISPR expressing plasmids. For details of the target sequences, CRISPR arrays and PAMs used, see Supplementary Table S2.
Measurement of bacterial growth and fluorescence
For E. coli fluorescence measurements on population level, overnight cultures grown in LB were diluted 1:100 in 10 ml of LB medium supplemented with required antibiotics, and 0.2% arabinose and 0.1 mM IPTG to induce CRISPR and Cas protein expression. Cultures were grown in flasks with aeration at 37°C. OD600 and fluorescence were acquired in exponential phase, 3 h after inoculation, or in stationary phase, 9 h after inoculation. Measurements were made on 100 μl of culture in black Corning flat-bottomed plates with an Infinite M200 Pro microplate reader (Tecan). S. typhimurium was analyzed as E. coli, except that modified M9-CA medium was used (1× M9 salts, 0.1 mM CaCl2, 2 mM MgSO4, 10 μg/ml thiamine hydrochloride and 1% (w/v) casamino acids, supplemented with 0.4% glycerol and antibiotics as required) and a Infinite 200 microplate reader (Tecan) was used for fluorescence and OD600 measurements. Note that 0.2% arabinose and 0.1 mM IPTG were added to the media to induce crRNA and Cas protein expression. For investigation of E. coli silencing over time, overnight cultures were diluted 1:100 in 100 μl of LB with inducers and required antibiotics and transferred to a 96-well plate. Fluorescence and OD600 were measured in the microplate reader every 5 min. For fluorescence measurements the following wavelengths were used: 480 nm for Gfp excitation, 520 nm for Gfp emission, 399 nm for Bfp excitation and 456 nm for Bfp emission. Fluorescence readings were normalized to OD and corrected for background fluorescence by subtracting normalized fluorescence from a strain without fluorescent reporters. Relative fluorescence is calculated as the ratio between the tested cells and cells expressing non-targeting crRNA.
Flow cytometry
To investigate silencing in individual cells, cultures were diluted in modified M9-CA medium (above) and ampicillin (50 μg/ml), streptomycin (50 μg/ml) and chloramphenicol (25 μg/ml) when required. Samples were taken after 4 h of growth at 37°C; the cells were pelleted and resuspended in phosphate buffered saline before analysis in a BD LSR II flow cytometer using FACS Diva 6.0 software (BD Biosciences). Excitation wavelength was 488 nm and a 525/50 bandpass filter was used for detection; ∼10 000 events were recorded per sample. For data handling FlowJo 7.6.5 software (FlowJo) was used.
Total RNA isolation and northern blots
Total RNA was extracted by hot phenol method (39) from cultures grown as described for flow cytometry (above). The RNA was DNase I-treated (Thermo Scientific) according to manufacturer's instructions. 1 μg of total RNA was separated on a denaturing agarose gel (1.5% agarose, 1× MOPS, 25 mM guanidine thiocyanate) and transferred to a Hybond-N+ membrane (GE Healthcare Life Sciences) by overnight capillary transfer in 20× SSC. The RNA was ultraviolet-cross-linked two times at 150 mJ/cm2. Digoxigenin (DIG) was used for detection (Roche). A DIG labeled RNA probe was used for detection of full-length gfp transcripts while DIG end-labeled DNA oligonucleotide probes were used for detection of 5S rRNA and partial gfp transcripts. DIG-labeled RNA probe was hybridized to the membrane in high sodium dodecyl sulphate (SDS) buffer (7% SDS, 50% formamide, 5× SSC, 2% blocking reagent, 0.1% N-lauroylsarcosine, 50 mM NaPO4) at 68°C. For DNA probes, the hybridization temperature used is indicated in Supplementary Table S1. Hybridization, washing, blocking, antibody binding and chemiluminescence detection were done according to DIG manufacturers's instructions. RNA probe was constructed by PCR-amplifying the probe sequence from pEH9 using primers that included the T7 promoter (Supplementary Table S1). The PCR-product was treated with fast digest DpnI (Thermo Scientific) to remove the template plasmid and used as template for in vitro T7 transcription using DIG RNA labeling mix (Roche) containing DIG-labeled uridine triphosphates (UTPs). The probe was treated with DNase I (Thermo Scientific) according to manufacturer's instructions before use. Oligonucleotide probes were 3′-labeled using the DIG oligonucleotide 3′-end labeling kit (Roche) according to manufacturer's instructions. Sequences of oligonucleotides and probes are provided in Supplementary Table S1.
RESULTS
Design of silencing system
To test our hypothesis of Cascade-based transcription silencing (Figure 1), we used an E. coli cas3-knockout strain, where we overexpressed Cascade from an IPTG-inducible T7 promoter (11) together with a CRISPR array targeting a gfp reporter gene. The gfp target was encoded on the chromosome or on a low-copy plasmid (pEH9), constitutively expressed from a PLtetO-1 promoter, which allowed quantitative fluorescence-based read-out of changes in gene expression. Minimal CRISPR arrays consisting of the transcribed leader (32) and two 29-bp repeats flanking a 32-bp spacer were designed to produce crRNAs that guide Cascade to various elements of the PLtetO-1 promoter or gfp ORF. Pairs of spacers were designed to recognize the opposite strands of DNA at approximately the same location to investigate strand bias (Figure 2A). CRISPR arrays were expressed from plasmids using a PLlacO-1 promoter. For analysis of simultaneous silencing of multiple targets we constructed a strain carrying constitutively expressed gfp and bfp genes on the chromosome. To test the system's functionality in other species, we transferred the same Cascade, gfp target and CRISPR plasmids used in E. coli to S. typhimurium.
Figure 1.
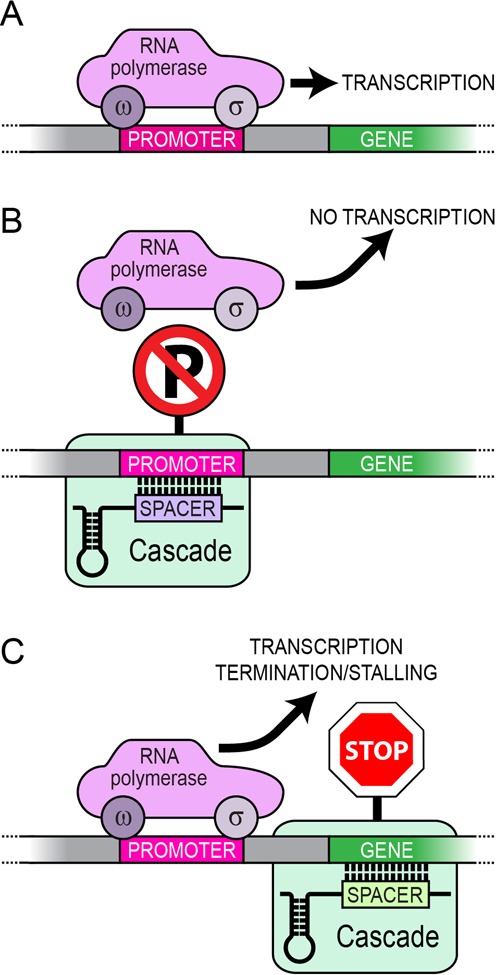
Model of Cascade-mediated silencing system. A crRNA co-expressed with Cascade proteins form the Cascade RNA-protein complex. Efficient binding of Cascade to DNA is directed by base pairing of crRNA to a cognate sequence in the target DNA, which also requires an adjacent PAM motif. (A) Aided by the sigma factor, the RNA polymerase initiates transcription of a gene and elongation produces the mRNA. (B) When Cascade is targeted to the promoter it may interfere with transcription initiation. (C) When Cascade is targeted to a region within the ORF, it may affect transcription elongation.
Figure 2.
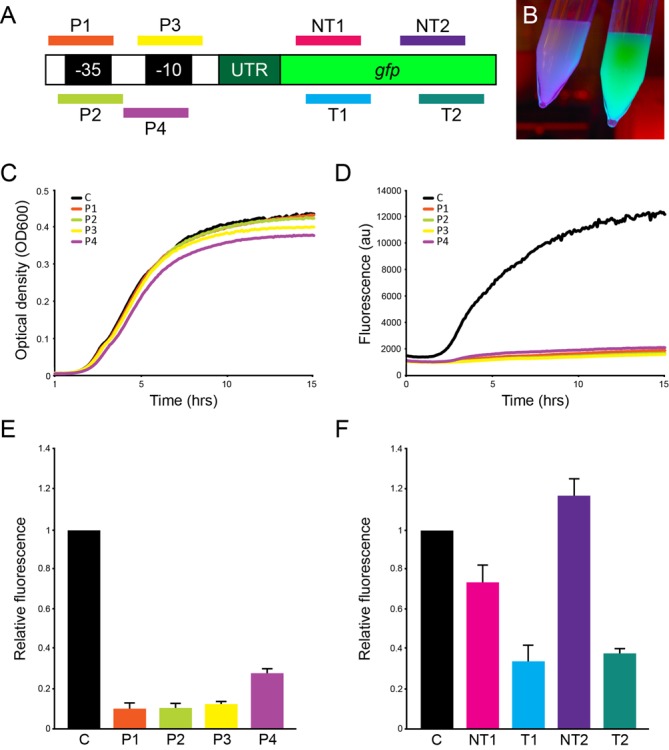
Cascade-mediated control of transcription. (A) Schematic drawing showing the positions of protospacers in the PLtetO-1 promoter and the gfp ORF (not drawn to scale). (B) E. coli cells expressing Cascade with P3 crRNA display Gfp silencing (left), compared to cells expressing Cascade with non-targeting crRNA (right). (C) Optical density analysis of cultures grown in 96-well plates demonstrates little difference in growth between strains expressing Cascade targeted to different regions of the PLtetO-1 promoter and strain expressing non-targeting Cascade (indicated by ‘C’). (D) Gfp fluorescence of cultures grown in 96-well plates expressing Cascade guided to different regions of the PLtetO-1 promoter is dramatically lower than of cultures expressing non-targeting Cascade, demonstrating efficient silencing of gfp. (E) Relative Gfp fluorescence of cultures expressing Cascade guided to different parts of the promoter 3 h after induction compared to cultures expressing Cascade and non-targeting crRNA. (F) Relative Gfp fluorescence of cultures expressing Cascade guided to different part of the gfp ORF 3 h after induction compared to cultures expressing Cascade and non-targeting crRNA. Target is plasmid encoded in all experiments. Error bars represent SD.
Cascade targeted to promoter silences expression
All CRISPR-expressing strains exhibited comparable growth but Gfp fluorescence was dramatically reduced in all strains that carried spacers against the PLtetO-1 promoter compared to a control with non-targeting spacer (Figure 2B–E). Silencing was effective on both plasmid and chromosome targets, with similar efficiency (Figure 3A). Gfp repression was evident very early during growth, but we used a time point 3 h post-induction, in exponential growth phase, to quantify change in fluorescence (Figure 2E). With gfp on a plasmid, P2 and P3 crRNA targeting the -35 and -10 element, respectively, decreased the fluorescence the most, up to about 50-fold. Silencing required both Cascade and a targeting crRNA, demonstrating that silencing was not due to any activity of the crRNA on its own, or the overexpression of the Cas proteins (Supplementary Figure S1). Overall, these results show that Cascade binding to promoter elements results in highly effective transcription silencing. Analysis of individual cells expressing P1 and P2 crRNA by flow cytometry demonstrates symmetric fluorescence distribution, suggesting that all cells displayed approximately similar level of silencing (Figure 4A and B and Supplementary Figure S2). We hypothesize that Cascade stably bound to the promoter blocks RNA polymerase access, which in turn prevents transcription.
Figure 3.
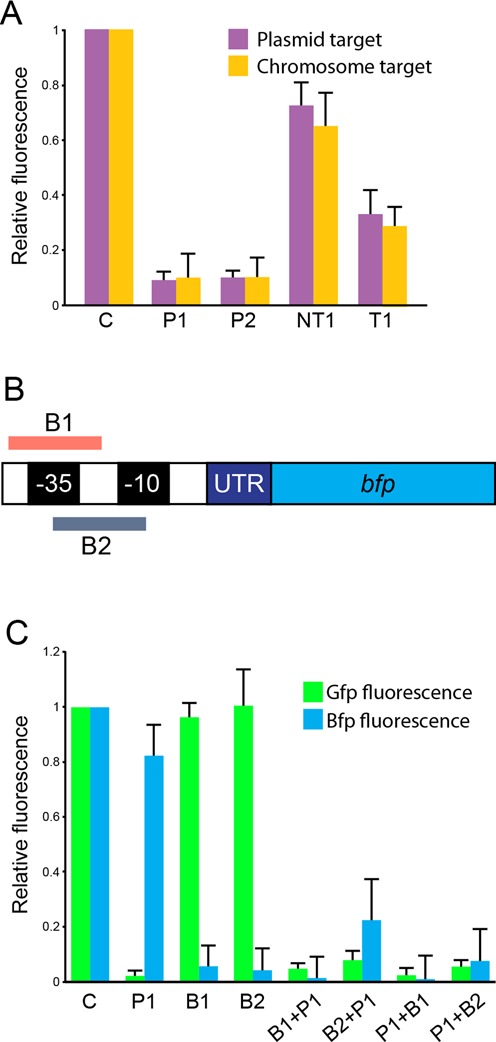
Transcription silencing of chromosomal, plasmid and multiple targets. (A) Comparison of relative fluorescence 3 h after induction when target is on a plasmid or on the chromosome. (B) Schematic drawing showing the positions of protospacers in the J23101 promoter (not drawn to scale). (C) Relative Bfp and Gfp fluorescence when expressing different CRISPRs targeting bfp, gfp or both 9 h after induction. Order of spacers in double-spacer CRISPRs is indicated. Error bars represent SD. ‘C’ indicates strain expressing Cascade and non-targeting CRISPR.
Figure 4.
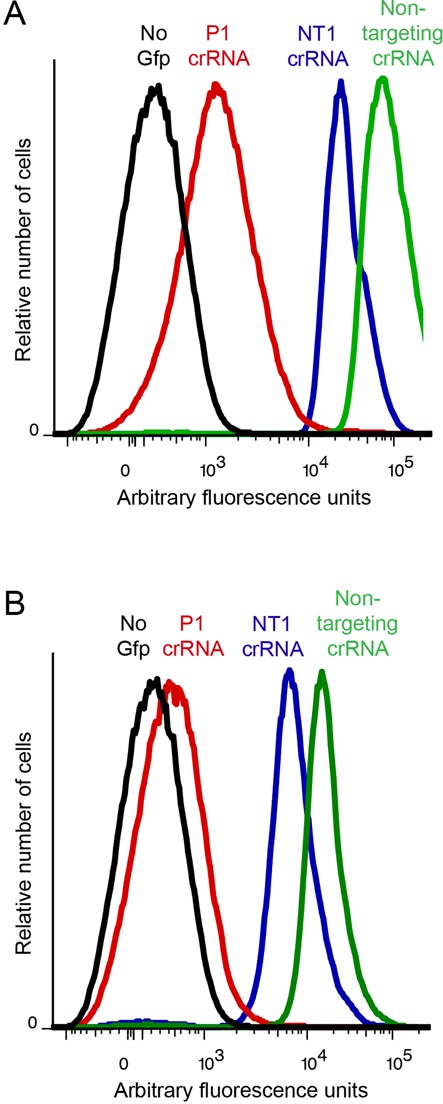
Single-cell analysis of silencing. Flow cytometry analysis of Gfp fluorescence in individual cells when gfp is on plasmid (A) or chromosome (B) and targeted by P1 or NT1 crRNA. Fluorescence of cells with non-targeting crRNA and cells lacking gfp are shown for comparison.
Cascade targeted to ORF also silences expression
We then designed spacers that targeted different regions within the gfp ORF to test whether Cascade binding can interfere with an elongating RNA polymerase. Again, pairs of ORF-targeting spacers were designed to bind the same location, on either the template or non-template strand (Figure 2A), and Gfp fluorescence was quantified 3 h post-induction (Figure 2F). With a plasmid target, the most efficient silencing when targeting the ORF was achieved with the T1 and T2 crRNA which both gave about 5-fold reduction of fluorescence. T1 and T2 target the template strand 269–301 and 437–469 bp, respectively, downstream of the transcription start site. NT1 gave only slight reduction of fluorescence and NT2 crRNA had no silencing effect (Figure 2F). Like promoter targets, ORF targets on chromosome and plasmid were silenced to same degree (Figure 3A). Our results indicate that Cascade targeted to the template strand of the ORF gives better silencing than when targeted to the non-template strand (Figure 2F), but that neither is as efficient as promoter targeting. Flow cytometry analysis revealed that cells expressing Cascade and NT1 or T1 crRNA displayed reduced fluorescence from both plasmid- and chromosome-encoded Gfp compared to non-targeting control, but again not to the same degree as promoter-targeting spacers (Figure 4A and B and Supplementary Figure S2). In contrast to when the promoter was targeted, the fluorescence distribution was not symmetrical when the ORF was targeted, particularly for plasmid-encoded Gfp (Figure 4A and Supplementary Figure S2A). The level of silencing thus appears to be different in individual cells, but it should be noted that very few cells displayed fluorescence equal to the non-silenced control.
Cascade-mediated silencing is long lasting
Since the in vivo nature of Cascade-DNA interaction is not known, we investigated the efficiency of our silencing system over time in the presence of inducers. Measurement of relative fluorescence from 3 to 23 h after induction with promoter-targeting crRNA demonstrated robust gfp silencing over time (Figure 5). For cultures expressing NT1, T1 or T2 crRNAs targeting the ORF gfp remained silenced over time (Supplementary Figure S3), while NT2 expressing cells did not show any silencing, even with prolonged induction.
Figure 5.
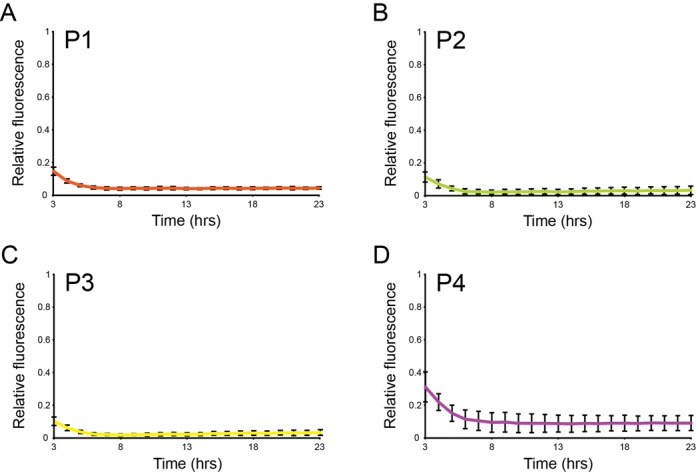
Stability of silencing over time. (A–D) Gfp fluorescence of cells expressing Cascade guided to indicated regions of the PLtetO-1 promoter relative to cells expressing Cascade and non-targeting crRNA. Fluorescence was monitored in 96-well plates 3–23 h after induction. Error bars represent SD.
Silencing of multiple targets
To determine if it is possible to use Cascade to silence several targets simultaneously, we constructed a strain carrying both gfp and bfp on the chromosome in a cas3 knockout background. In this strain we expressed plasmid-encoded Cascade and CRISPRs containing spacers targeting the bfp and/or the gfp promoter. The bfp spacer pair was designed to target both strands of the promoter (Figure 3B). Spacers targeting gfp or bfp promoters were only active on their intended targets and, when encoded in same CRISPR array, gfp and bfp expression were silenced simultaneously. Silencing was effective independent of spacer order in the CRISPR array (Figure 3C). A time point in stationary phase (9 h after inoculation) was chosen for quantification as small fluctuations in the measurements during exponential phase had large impact on normalized Bfp fluorescence values. However, the time course experiments show stable silencing over time (Supplementary Figure S4).
Cascade-based silencing is effective in different strains and species
To demonstrate the applicability of Cascade-mediated silencing in strains other than E. coli K-12, we tested our system in BL21AI and BL21DE3, two E. coli B strains that are distinctly different from K-12 (40). Silencing of Gfp expression was demonstrated in BL21AI with P1 (12-fold at most) and P2 (15-fold at most) targeting PLtetO-1 promoter (Figure 2A and Supplementary Figure S5). Similar results were obtained with BL21DE3 (data not shown). Transfer of Cascade, CRISPR and target plasmid to S. typhimurium LT2 demonstrate on average 8-fold silencing with P1 and 12-fold with P2 after 4 h of induction (Supplementary Figure S6) but longer induction resulted in more silencing (data not shown). The results demonstrate that Cascade-based transcription silencing is applicable in different strains and species but that the efficiency of silencing may differ.
Cascade silencing is caused by transcription interference
To assess if silencing is, as hypothesized, caused by transcription interference (and not by post-transcriptional effects), we measured gfp mRNA levels using northern blots and a probe binding downstream of the crRNA target sites, thereby detecting full-length gfp transcripts. Plasmid-derived gfp transcripts were non-detectable in cells expressing P1–4 crRNA, in a Cascade-dependent manner, but observed in cells expressing non-targeting crRNA (Figure 6A). T1, T2 or NT1 crRNA caused a reduction in gfp mRNA levels, but not as efficiently as P1–4 (Figure 6B), and NT2 did not reduce gfp transcripts. All results were consistent with fluorescence data (Figure 2E and F). A probe upstream of ORF target sites revealed that T1, T2 and NT1 crRNA gave rise to partial gfp transcripts of sizes corresponding to transcription termination at the position of the protospacer (Supplementary Figure S7), further indicating interference with transcription.
Figure 6.
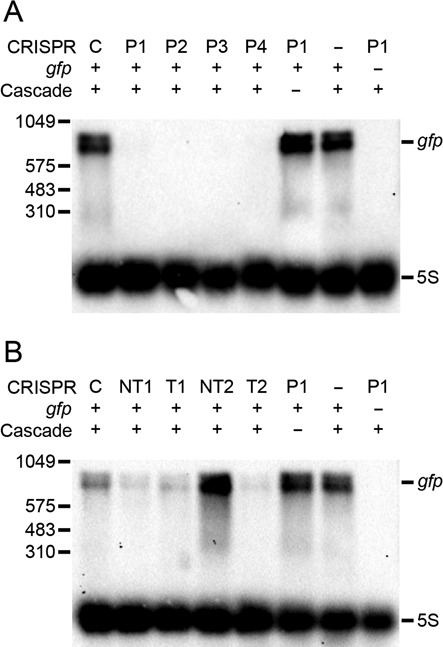
Cascade binding interferes with transcription. Northern blot analysis of full-length gfp transcript abundance during Cascade-crRNA expression. The crRNAs used are indicated above the blot; +/- indicate presence/absence of gfp, Cascade or crRNA. 5S rRNA was used as loading control. (A) No gfp transcripts are detected when promoter-targeting crRNA and Cascade are expressed. (B) Cells expressing Cascade and NT1, T1 or T2 crRNA have decreased gfp transcript levels compared to cells expressing Cascade and non-targeting crRNA (indicated by ‘C’) while NT2 provides no reduction, in accordance with fluorescence data (Figure 2).
DISCUSSION
In this report, we used the sequence-specific DNA binding activity by the crRNA-guided Cascade complex as a simple and efficient method for targeted transcription silencing. The crRNA component guides Cascade to complementary target DNA containing an adjacent PAM motif. By changing the spacer sequence in the crRNA, Cascade can be directed to bind virtually any chosen stretch of DNA. Binding of Cascade to target DNA is strong, specific and virtually permanent, with very low Kd values (13± 1.4 nM, (16); 8 ± 4 nM, (41)). Cascade remains bound to DNA until released by Cas3‐mediated DNA degradation (16). In absence of functional Cas3, Cascade could therefore be used to bind regulatory DNA elements, such as promoters, enhancers or operators, to prevent access of RNA polymerase, activators or repressors, respectively, thereby causing increase or decrease in transcript levels. In extension, any DNA-interacting element could potentially be affected by Cascade binding to its target site. Using E. coli and S. typhimurium as model systems, we show that Cascade targeted to promoter elements gives highly efficient expression silencing, determined both on transcript and protein level. Cascade silencing is specific and efficient, the silencing effect is stable over time and independent of growth phase. Cascade can be used to silence both chromosome and plasmid targets, as well as to silence several targets simultaneously. Silencing is also achieved when Cascade is targeted to the ORF albeit, for the spacers tested here, at lower efficiency than when targeting the promoter. Flow cytometry analysis of individual cells reveal that when targeting the promoter, all cells are uniformly silenced to a high degree, but when targeting the ORF, cells display some variation in silencing, especially when the target is on a multi-copy plasmid. We speculate that actively transcribing RNA polymerase occasionally dislodges or by-passes DNA-bound Cascade and that this explain the cell-to-cell variation in expression. It is also possible that an RNA polymerase stalls as it reaches Cascade-bound DNA and then proceeds when Cascade stochastically dissociates from DNA. Further investigations are required to elucidate the mechanistic details of the silencing process.
Since the inactivation of cas3 is the only requirement for turning a type I CRISPR-Cas system into a transcription regulator, this proof of principle raises the interesting question if this function may be found occurring naturally.
Cascade-based silencing is analogous to silencing based on dCas9 and the choice of method will be determined by practical considerations in each experimental set-up, such as off-target effects, expression efficiency, PAM availability and compatibility with genetic background. Off-target effects, activity on unintended targets, are important to minimize. The more specific the binding is, the smaller is the risk of off-target effects (42 and references therein). The silencing method based on catalytically inactive Cas9 from Streptococcus pyogenes requires 20 bp spacers and an NGG PAM at the 3′-end (26,27). Qi et al. (26) observed repression with a guide RNA possessing only 12 nt homology to the protospacer, a finding reproduced by Bikard et al. (27). In a recent report, Kuscu et al. (43) determined the off-target binding sites for sgRNA-dCas9 complexes by ChIP-seq. Their results suggest that a dCas9 can bind a higher number of off-target sites than previously anticipated. Their findings also suggest that the total number of mismatches at dCas9 binding sites can be as high as 10, and as many as nine of the mismatches can be consecutive in the PAM-distal region. A perfect match of 10 bases in the PAM-proximal region of the sgRNA guiding sequence is sufficient to mediate dCas9 binding to DNA. Cascade utilizes a 32-bp long spacer and an adjacent 3-bp PAM motif for target recognition. Perfect match is required for the PAM and the seven-nucleotide seed region, but up to five mismatches are tolerated in the protospacer outside the seed region (44), effectively reducing the recognition region to 30 bp. Since this is a much longer sequence match than the 12–23 bp required by Cas9, it is conceivable that Cascade produces less off-target effects than dCas9.
Qi et al. (26) achieved the best repression with dCas9 that targeted the non-template strand of the ORF close to the promoter, and repression efficiency decreased dramatically with increasing distance of the protospacer from the ORF start site. This suggests that distance of the protospacer from the start site is an influential parameter for dCas9. Further, when targeting a promoter on the chromosome, not all spacers gave repression (27). These observations suggest that dCas9-mediated gene silencing is affected by several parameters, not all of which are fully understood. Cascade directed to the template strand of the ORF gave slightly better silencing than when targeted to the non-template strand, but due to the limited number of spacers tested we cannot rule out that that the differences are caused by the efficiency of the individual spacers. Target binding by Cascade proceeds via formation of an R-loop (45). A stronger inhibition of RNA polymerase when Cascade is bound to the template strand (compared to when bound to the non-template strand) can be rationalized as RNA polymerase primarily needs access to the template strand for elongation.
Our system uses an unperturbed Cascade complex which, unlike systems based on engineered dCas9, allows for the use of endogenous cellular components for efficient programmable silencing in cells where Cas3 is absent, inactive or silenced. The larger size of Cascade compared to Cas9 may also be an additional advantage when interfering with RNA polymerase-DNA interaction. Cascade also uses a different set of PAMs than Cas9 and may have a different range of species and strains where it can be used. However, in-depth comparative studies are required to exactly determine the advantages and disadvantages of different programmable silencing methods.
Design of new CRISPRs is simple and cost-effective compared to constructing gene knockouts and, by using inducible CRISPR-Cas systems, essential genes can be targeted, which obviates the need for time-consuming construction of thermo-sensitive or other conditional mutants. By designing spacers against standard promoters used in common cloning and expression vectors, the same system can be used for many applications. By using different CRISPRs under different inducible promoters, where each CRISPR target different gene(s), it is possible to use the system as a silencing switch. Tuning of target expression may be achieved by regulating the expression of the CRISPR array or Cascade. The method could also be developed to activate genes, e.g. by targeting operators.
In conclusion, we have demonstrated specific gene silencing using Cascade. We have used E. coli and S. typhimurium for proof of concept, but this method should be applicable to any gene in any organism, the requirement being the presence of a 3-bp PAM in the vicinity of the promoter and the ability of Cascade to assemble and access target DNA. The Cascade-mediated transcriptional silencing demonstrated in this work is a powerful new addition to the rapidly emerging CRISPR-Cas toolbox for genetic modifications and gene regulation, expanding the versatility and applicability of CRISPR-Cas tools.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Erik Holmqvist for sharing plasmids, and the group of Gerhart Wagner for technical assistance and valuable discussion. We thank Erik Gullberg for sharing strains and for important insights and Gargi Bindal for assistance with Salmonella experiments. We thank the SciLifeLab BioVis facility for assistance with flow cytometry analysis.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
Present addresses:
Devashish Rath, Molecular Biology Division, Bhabha Atomic Research Centre, Mumbai 400 085, Maharashtra, India.
Praneeth Reddy Devulapally, Max Planck Institute for Molecular Genetics, 14195 Berlin, Germany.
FUNDING
Swedish Research Council; Wenner-Gren foundations; Royal Academy of Sciences; Uppsala RNA Research Center. Funding for open access charge: Swedish Research Council; Uppsala RNA Research Center (URRC).
Conflict of interest statement. The authors have submitted a provisional patent application that includes the findings presented in the manuscript.
REFERENCES
- 1.Browning D.F., Busby S.J. The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2004;2:57–65. doi: 10.1038/nrmicro787. [DOI] [PubMed] [Google Scholar]
- 2.Winkler W.C., Breaker R.R. Regulation of bacterial gene expression by riboswitches. Annu. Rev. Microbiol. 2005;59:487–517. doi: 10.1146/annurev.micro.59.030804.121336. [DOI] [PubMed] [Google Scholar]
- 3.Ausländer S., Fussenegger M. From gene switches to mammalian designer cells: present and future prospects. Trends Biotechnol. 2013;31:155–168. doi: 10.1016/j.tibtech.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 4.Beerli R.R., Segal D.J., Dreier B., Barbas C.F., 3rd Toward controlling gene expression at will: specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc. Natl. Acad. Sci. U.S.A. 1998;95:14628–14633. doi: 10.1073/pnas.95.25.14628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cong L., Zhou R., Kuo Y.C., Cunniff M., Zhang F. Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nat. Commun. 2012;3:968. doi: 10.1038/ncomms1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klug A. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu. Rev. Biochem. 2010;7:213–231. doi: 10.1146/annurev-biochem-010909-095056. [DOI] [PubMed] [Google Scholar]
- 7.Van der Oost J., Westra E.R., Jackson R.N., Wiedenheft B. Unravelling the structural and mechanistic basis of CRISPR-Cas systems. Nat. Rev. Microbiol. 2014;12:479–492. doi: 10.1038/nrmicro3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolotin A., Quinquis B., Sorokin A., Ehrlich S.D. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005;151:2551–2561. doi: 10.1099/mic.0.28048-0. [DOI] [PubMed] [Google Scholar]
- 9.Mojica F.J., Díez-Villaseñor C., García-Martínez J., Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005;60:174–182. doi: 10.1007/s00239-004-0046-3. [DOI] [PubMed] [Google Scholar]
- 10.Pourcel C., Salvignol G., Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology. 2005;151:653–663. doi: 10.1099/mic.0.27437-0. [DOI] [PubMed] [Google Scholar]
- 11.Brouns S.J., Jore M.M., Lundgren M., Westra E.R., Slijkhuis R.J., Snijders A.P., Dickman M.J., Makarova K.S., Koonin E.V., van der Oost J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carte J., Wang R., Li H., Terns R.M., Terns M.P. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 2008;22:3489–3496. doi: 10.1101/gad.1742908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haurwitz R.E., Jinek M., Wiedenheft B., Zhou K., Doudna J.A. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science. 2010;329:1355–1358. doi: 10.1126/science.1192272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gasiunas G., Barrangou R., Horvath P., Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl Acad. Sci. U.S.A. 2012;109:2579–2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Westra E.R., van Erp P.B., Kunne T., Wong S.P., Staals R.H., Seegers C.L., Bollen S., Jore M.M., Semenova E., Severinov K., et al. CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by Cascade and Cas3. Mol. Cell. 2012;46:595–605. doi: 10.1016/j.molcel.2012.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garneau J.E., Dupuis M.E., Villion M., Romero D.A., Barrangou R., Boyaval P., Fremaux C., Horvath P., Magadan A.H., Moineau S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67–71. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- 18.Sampson T.R., Saroj S.D., Llewellyn A.C., Tzeng Y.L., Weiss D.S. A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature. 2013;497:254–257. doi: 10.1038/nature12048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makarova K.S., Haft D.H., Barrangou R., Brouns S.J., Charpentier E., Horvath P., Moineau S., Mojica F.J., Wolf Y.I., Yakunin A.F., et al. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 2011;9:467–477. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grissa I., Vergnaud G., Pourcel C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics. 2007;8:172. doi: 10.1186/1471-2105-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mojica F.J., Díez-Villaseñor C., García-Martínez J., Almendros C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155:733–740. doi: 10.1099/mic.0.023960-0. [DOI] [PubMed] [Google Scholar]
- 22.Wiedenheft B., van Duijn E., Bultema J.B., Waghmare S.P., Zhou K., Barendregt A., Westphal W., Heck A.J., Boekema E.J., Dickman M.J., et al. RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proc. Natl Acad. Sci. U.S.A. 2011;108:10092–10097. doi: 10.1073/pnas.1102716108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heidrich N., Vogel J. Same same but different: new structural insight into CRISPR-Cas complexes. Mol. Cell. 2013;52:4–7. doi: 10.1016/j.molcel.2013.09.023. [DOI] [PubMed] [Google Scholar]
- 24.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qi L.S., Larson M.H., Gilbert L.A., Doudna J.A., Weissman J.S., Arkin A.P., Lim W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bikard D., Jiang W., Samai P., Hochschild A., Zhang F., Marraffini L.A. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013;41:7429–7437. doi: 10.1093/nar/gkt520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng A.W., Wang H., Yang H., Shi L., Katz Y., Theunissen T.W., Rangarajan S., Shivalila C.S., Dadon D.B., Jaenisch R. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013;23:1163–1171. doi: 10.1038/cr.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A., et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farzadfard F., Perli S.D., Lu T.K. Tunable and multifunctional eukaryotic transcription factors based on CRISPR/Cas. ACS Synth. Biol. 2013;2:604–613. doi: 10.1021/sb400081r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., Datsenko K.A., Tomita M., Wanner B.L., Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2006;2 doi: 10.1038/msb4100050. 2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cherepanov P.P., Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158:9–14. doi: 10.1016/0378-1119(95)00193-a. [DOI] [PubMed] [Google Scholar]
- 33.Yosef I., Goren M.G., Kiro R., Edgar R., Qimron U. High-temperature protein G is essential for activity of the Escherichia coli clustered regularly interspaced short palindromic repeats (CRISPR)/Cas system. Proc. Natl Acad. Sci. U.S.A. 2011;108:20136–20141. doi: 10.1073/pnas.1113519108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Studier F.W., Moffatt B.A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- 35.Lutz R., Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997;25:1203–1210. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Urban J.H., Vogel J. Translational control and target recognition by Escherichia coli small RNAs in vivo. Nucleic Acids Res. 2007;35:1018–1037. doi: 10.1093/nar/gkl1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Datsenko K.A., Wanner B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pougach K., Semenova E., Bogdanova E., Datsenko K.A., Djordjevic M., Wanner B.L., Severinov K. Transcription, processing and function of CRISPR cassettes in Escherichia coli. Mol. Micro. 2010;77:1367–1379. doi: 10.1111/j.1365-2958.2010.07265.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blomberg P., Wagner E.G., Nordström K. Control of replication of plasmid R1: the duplex between the antisense RNA, CopA, and its target, CopT, is processed specifically in vivo and in vitro by RNase III. EMBO J. 1990;9:2331–2340. doi: 10.1002/j.1460-2075.1990.tb07405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Studier F.W., Daegelen P., Lenski R.E., Maslov S., Kim J.F. Understanding the differences between Escherichia coli B strains REL606 and BL21(DE3) and comparison of the E. coli B and K-12 genomes. J. Mol. Biol. 2009;394:653–680. doi: 10.1016/j.jmb.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 41.Sashital D.G., Wiedenheft B., Doudna J.A. Mechanism of foreign DNA selection in a bacterial adaptive immune system. Mol. Cell. 2012;46:606–615. doi: 10.1016/j.molcel.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuscu C., Arslan S., Singh R., Thorpe J., Adli M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 2014;32:677–683. doi: 10.1038/nbt.2916. [DOI] [PubMed] [Google Scholar]
- 44.Semenova E., Jore M.M., Datsenko K.A., Semenova A., Westra E.R., Wanner B., van der Oost J., Brouns S.J., Severinov K. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl Acad. Sci. U.S.A. 2011;108:10098–10103. doi: 10.1073/pnas.1104144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jore M.M., Lundgren M., vanDuijn E., Bultema J.B., Westra E.R., Waghmare S.P., Wiedenheft B., Pul U., Wurm R, Wagner R., et al. Structural basis for CRISPR RNA-guided DNA recognition by Cascade. Nat. Struct. Mol. Biol. 2011;18:529–536. doi: 10.1038/nsmb.2019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.