


Abstract
Cockayne syndrome (CS) is a premature aging disorder characterized by photosensitivity, impaired development and multisystem progressive degeneration, and consists of two strict complementation groups, A and B. Using a yeast two-hybrid approach, we identified the 5′-3′ exonuclease SNM1A as one of four strong interacting partners of CSB. This direct interaction was confirmed using purified recombinant proteins—with CSB able to modulate the exonuclease activity of SNM1A on oligonucleotide substrates in vitro—and the two proteins were shown to exist in a common complex in human cell extracts. CSB and SNM1A were also found, using fluorescently tagged proteins in combination with confocal microscopy and laser microirradiation, to be recruited to localized trioxsalen-induced ICL damage in human cells, with accumulation being suppressed by transcription inhibition. Moreover, SNM1A recruitment was significantly reduced in CSB-deficient cells, suggesting coordination between the two proteins in vivo. CSB-deficient neural cells exhibited increased sensitivity to DNA crosslinking agents, particularly, in a non-cycling, differentiated state, as well as delayed ICL processing as revealed by a modified Comet assay and γ-H2AX foci persistence. The results indicate that CSB coordinates the resolution of ICLs, possibly in a transcription-associated repair mechanism involving SNM1A, and that defects in the process could contribute to the post-mitotic degenerative pathologies associated with CS.
INTRODUCTION
Cockayne syndrome (CS) is a rare recessive segmental progeria in which the patient exhibits increased sensitivity to ultraviolet (UV) light, developmental failings and severe multi-system progressive degeneration, including atrophy of the brain (1,2). The disorder is comprised of two major complementation groups, CSA (ERCC8) and CSB (ERCC6). The CSA protein consists of seven WD40 repeat motifs with a β-propeller architecture, and exists as part of a complex with DDB1, cullin 4A and Roc1 that exhibits ubiquitin ligase activity (3). CSB is a member of the SWI2/SNF2 family of DNA-dependent ATPases, and appears to operate in aspects of chromatin remodeling (4). Evidence suggests that CSA and CSB play critical roles in transcription regulation and DNA repair, particularly when these two processes intersect in a pathway termed transcription-coupled repair (5). The nature of the endogenous DNA damage that elicits a CS protein response, however, remains largely undefined, though presumably, at minimum, involves lesions that block transcription.
Reactive oxygen species are formed as by-products of mitochondrial oxidative phosphorylation or after exposure to many physical and chemical exogenous agents. These products can react with lipids, proteins and nucleic acids, contributing to aging and age-related disease by promoting the gradual accumulation of macromolecular damage and consequent cellular dysfunction or cell death. Oxidative DNA damage includes a wide variety of genomic alterations, such as non-bulky (e.g. 8-oxoguanine) and bulky (e.g. cyclopurine) base modifications, abasic sites, non-conventional single-strand breaks, protein-DNA adducts and intra-/interstrand crosslinks; all pose continuous mutagenic or cytotoxic threats to the cell (6). DNA interstrand crosslinks (ICLs) are covalent linkages between the opposing strands of the DNA helix and are formed by products of lipid peroxidation, such as malondialdehyde, as well as by widely used chemotherapeutic drugs (e.g. psoralen/UVA, mitomycin C (MMC), cisplatin, etc.). ICLs, although considered infrequent endogenous DNA modifications, are extremely toxic, because they present blocks to the replication and transcription machinery, leading to cell death or gross chromosome instability if not removed or resolved improperly. To combat the deleterious consequences of such lesions, cells have evolved a set of DNA repair mechanisms that excise the target damage and restore genome integrity and normal cellular operations (7).
Repair systems for ICLs have mostly been characterized in proliferating cells, where the corrective response is triggered by arrest of the replication fork in S-phase. This replication-dependent pathway requires nucleases, helicases, translesion synthesis DNA polymerases, Fanconi anemia (FA) proteins and homologous recombination factors to remove the crosslink and reinitiate DNA synthesis (8). While it is well-established that replication-independent ICL repair pathways operate in bacteria and yeast (9), recent evidence also suggests that ICLs can be resolved via a replication-independent pathway in post-mitotic cells, such as neurons, or outside of S-phase in proliferating vertebrate cells (10,11). The ICL repair response in the G1 cell cycle of replicating cells or in non-cycling cells is thought to involve steps of recognition, crosslink ‘unhooking’ (flanking incisions that release the ICL), translesion synthesis gap-filling and crosslink remnant removal, and appears to occur via both global genome and transcription-associated mechanisms. In global genome repair, the XPC-RAD23B nucleotide excision repair complex likely initiates the process, whereas in transcription-associated repair, the pathway is presumably triggered by arrest of a progressing RNA polymerase (RNAP), followed by recruitment of factors, such as CSA and CSB, to the damage site.
Previous work has indicated contributions of the CS proteins to ICL repair. For instance, deficiencies in CSA or CSB result in hypersensitivity of primary and SV40-transformed fibroblasts to cisplatin (12,13) and of mammalian cells in G0/G1 to MMC (14). In addition, CSB-deficient UV61 Chinese hamster ovary cells show reduced ICL unhooking in G1 following treatment with psoralen/UVA relative to wild-type cells (15). Moreover, reporter construct assays found reduced MMC ICL removal in CSB-deficient proliferating cells (16) and cisplatin ICLs in G0/G1 cells (14). The studies herein describe a novel physical and functional, coordinated interaction between CSB and the 5′ to 3′ exonuclease DNA crosslink repair 1A (DCLRE1A) protein—more commonly referred to as sensitive to nitrogen mustard 1A (SNM1A)—and reveal biological roles of CSB in a replication-independent, transcription-associated ICL repair response.
MATERIALS AND METHODS
Synthetic oligonucleotides
All oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA, USA) and are described in Supplementary Table S1.
Yeast two-hybrid screen
The yeast two-hybrid screen using full-length CSB as bait was carried out by Dualsystems Biotech, Zurich, Switzerland (see Supplementary Figure S1).
Recombinant proteins
Recombinant hemagglutinin-tagged full-length CSB (HA-CSB) and SNM1A nuclease fragment consisting of residues 608 to 1040 (ΔN-SNM1A) were purified as described (17,18).
Plasmid constructs
To generate N-terminal GFP tagged CSB or SNM1A, a serine- and glycine-rich linker sequence (19) was inserted into the C-terminal sequences of EGFP. In brief, the pEGFP-C1 vector (Clontech, Mountain View, CA, USA) was digested with BspEI and BglII, and subsequently ligated with linker 1 (linker Fw1 and linker Rv1) or linker 2 (linker Fw2 and linker Rv2). Polymerase chain reaction (PCR) was carried out using the Hercules II Fusion Enzyme according to the manufacturer's instruction (Agilent Technologies, Santa Clara, CA, USA). The CSB coding region was amplified using primers CSB XhoI-Fw and CSB BamHI-Rv, and the in-house cDNA template (20). The SNM1A coding region was amplified using the primers SNM1A XhoI-Fw and SNM1A BamHI-Rv, and a cDNA template obtained from Origene (SC322694; Rockville, MD, USA). All oligonucleotides above are described in Supplementary Table S1. The PCR products were digested accordingly and subcloned into the XhoI and BamHI restriction sites of pEGFP-C1 with linker 1 for CSB (pCSB-GFP) or linker 2 for SNM1A (pSNM1A-GFP). To create the N-terminal mCherry-tagged CSB expression construct (pCSB-mCherry), the CSB fragment was transferred from pCSB-GFP to the XhoI and BamHI restriction sites of pmCherry-C1 (Clontech). The nucleotide sequence of each cDNA was confirmed at the Johns Hopkins Sequencing Facility or Eurofins Genomics (Huntsville, AL, USA).
Protein interaction assay
Recombinant ΔN-SNM1A (500 ng) was incubated with or without HA-CSB (500 ng) in the presence of α-HA magnetic beads (Thermo Fisher Scientific, Rockford, IL, USA) in a 500 μl reaction containing 20 mM HEPES pH 7.9, 4 mM MgCl2, 0.05 mM ATP, 40 μg/ml bovine serum albumin (BSA) and 1 mM DTT at 4°C for 2 h. The beads and associated material were captured on a magnetic stand via a 1-min incubation, and washed three times with 20 mM HEPES pH 7.9, 4 mM MgCl2, 1 mM DTT and 0.1% Nonidet P-40. The bead-bound material was suspended in 2× sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) loading dye and incubated at 95°C for 5 min. Proteins were resolved on an 8% Tris-glycine-SDS polyacrylamide gel and detected using the Pierce Silver Stain Kit for Mass Spectrometry (Thermo Fisher Scientific).
Co-immunoprecipitation
Whole cell extracts were prepared from untreated HeLa cells, or where indicated, at 0.5 or 2 h after 6 μM trioxsalen/UVA treatment. In brief, cells were lysed in 20 mM Tris pH 7.5, 150 mM NaCl, 1% Triton-X-100, 1 mM ethylenediaminetetraacetic acid (EDTA) and complete protease inhibitor cocktail (Roche, Mannheim, Germany) with sonication, and insoluble material was removed by centrifugation at 14 000 × g for 10 min at 4°C. Prior to α-CSB immunoprecipitation, the soluble whole cell extract was pre-treated with protein A/G magnetic beads (Thermo Fisher Scientific) at 4°C for 2 h to remove non-specific protein binders. Extracts were then incubated with mouse α-CSB antibody (ab66598; Abcam, Cambridge, MA, USA) for 12 h, and the immunocomplexes were captured by protein A/G magnetic beads for 2 h at 4°C. The bead-bound material was washed five times, suspended in 2× SDS-PAGE loading dye and incubated at 95°C for 5 min. Proteins were resolved on a polyacrylamide gel and detected by western blotting.
Nuclease assay
The substrates used in the nuclease assays were as described (17), with the relevant oligonucleotides listed in Supplementary Table S1. To measure exonuclease activity, ΔN-SNM1A (0.35 ng, 0.8 nM) was mixed with 1 pmol (100 nM) of 3′-[32P]-labeled DNA substrate (in the presence of CSB where indicated) in 10 μl of 20 mM HEPES pH 7.9, 50 mM KCl, 10 mM MgCl2, 0.5 mM DTT, 0.05% Triton-X, 0.1 mg/ml BSA and 5% glycerol. Reactions were incubated at 37°C for 10 or 30 min for the 21-mer or 61-mer substrates, respectively, and stopped by adding 2 μl of 80% formamide/10 mM EDTA to each reaction and heating at 95°C for 5 min. Following separation on a 15% polyacrylamide/7 M urea denaturing gel, substrate and product bands were visualized by a Typoon Trio+ Variable Model Imager, and the signals were quantified using ImageQuant software (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA).
Real-time kinetic measurements
Real-time kinetic measurements were performed as described (21) using the 20-mer oligonucleotides listed in Supplementary Table S1. Reactions were carried out in black 384-well microplates, and measurements were made using a SpectraMax M2e fluorescent plate reader in fluorescent top read mode, with SoftMaxPro software (Molecular Devices, Sunnyvale, CA, USA) to control the settings. Reactions were performed in 15 μl of the above buffer with varying concentrations of DNA substrate (10, 25, 50, 100, 250, 500, 1000, 1500 and 2500 nM), 0.242 nM ΔN-SNM1A and 0.242 nM CSB (where indicated). Each reaction was started by the addition of ΔN-SNM1A, and the fluorescein emission spectra measured (excitation at 495 nm, emission at 525 nm and cutoff at 515 nm) with six readings taken at 7 s intervals for 6 min. The fluorescence intensity of each well was plotted against time, and the rate of increase was determined, plotted against substrate concentration and fitted to a Michaelis–Menten curve on Prism software (GraphPad Software, Inc., La Jolla, CA, USA) to determine KM and kcat.
Protein recruitment and retention
Targeted microirradiation was performed on a set-up involving a Nikon Eclipse TE2000-E microscope, a CSU10 confocal scanner unit (Yokogawa, Japan), and a NL100 nitrogen laser (Stanford Research Systems, Sunny Vale, CA, USA) adjusted with a MicroPoint ablation system (Photonics Instruments, St. Charles, IL, USA) to generate a wavelength of 365 nm (22). In brief, cells were precultured in 10 mm microwells, and then transfected with either pCSB-GFP or pSNM1A-GFP, or pCSB-mCherry and pSNM1A-GFP simultaneously, using the JetPrime reagent and cultured an additional 24 h before microirradiation. Thirty minutes prior to microirradiation, transfected cells were treated with 6 μM trioxsalen or not at all. A defined laser intensity sufficient to activate trioxsalen to generate ICLs, but not γH2AX foci on its own, was directed to a designated rectangular region of interest (5 × 20 pixels, 0.16 μm/pixel). Where indicated, 10 mM N-acetylcysteine (NAC) or 20 μM α-amanitin was added to the media 1 or 8 h, respectively, prior to microirradiation. The images were photographically recorded and analyzed using the Volocity software 6.3 (PerkinElmer, Waltham, MA, USA). Relative fluorescence intensity (RFI) was determined using the Volocity software, and reports the fluorescence signal intensity within a defined microirradiated region relative to a similarly defined region in an unirradiated (background) location within the nucleus. All images were acquired using identical gain, exposure, sensitivity and contrast settings.
Cell line construction and maintenance
HeLa cells were grown in normal culture media: high glucose Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum and 1% penicillin/streptomycin. SV40-transformed CS1AN cells stably transfected with either CSB (CS1AN-CSB) or empty vector (CS1AN-vector) were cultured as above, except with 400 μg/ml geneticin (23,24). All cell lines were grown in a cell culture incubator maintained at 5% CO2 and 37°C. Lentivirus generation and infections were performed essentially as described previously (23). A vector containing the following shRNA sequence against CSB was used: 5′-CCAGAAGCAAGACAGTGAATTCAAGAGATTCACTGTCTTGCTTCTGGTTTTT-3′ (Santa Cruz Biotechnology, Dallas, TX, USA). As a control, a plasmid containing a scrambled shRNA was used (Plasmid-A; Santa Cruz Biotechnology). To generate stable knockdown cell lines, SH-SY5Y cells were suspended in normal culture media containing virus and then incubated at 37°C for 2 h with gentle rocking every 15 min. The cells were then cultured as above for 2 days, and subsequently in media containing 6 μg/ml puromycin for 2 weeks, to select for shRNA vector containing clones. Puromycin-resistant colonies were subcloned by limited dilution, and periodically checked by standard western blotting to measure CSB expression. Stable cell lines were maintained in normal culture media with 3 μg/ml puromycin.
Western blotting
Western blots were performed using standard procedures and the following primary antibodies: goat anti-SNM1 (ab14805), rabbit anti-CSB (ab96089), mouse anti-β-actin (ab8226) purchased from Abcam, and mouse anti-GAPDH (MAB374; Millipore, Billerica, MA, USA). Secondary horseradish peroxidase-conjugated antibodies and ECL prime (GE Healthcare Bio-Sciences) or SuperSignal West Femto Chemiluminescent Substrate (Thermo Fisher Scientific) were used to visualize proteins on a ChemiDoc XRS+ system (Bio-Rad Laboratories, Hercules, CA, USA). Digitized images were obtained and quantified with ImageLab version 5.1 (Bio-Rad Laboratories), and processed using the Adobe Photoshop 8.0 software package (Adobe Systems, San Jose, CA, USA).
Differentiation of SH-SY5Y cells
Differentiation of SH-SY5Y cells was performed as described (25). In brief, 20–30% confluent SH-SY5Y cells were treated with 30 μM retinoic acid in normal culture media for 5 days. This medium was then replaced with serum-free DMEM with 10 ng/ml brain derived neurotrophic factor (Millipore), and differentiation was allowed to continue for another 5 days.
Cell viability
To determine UVC light sensitivity, 2 × 104 cycling or 1 × 106 non-cycling (differentiated) scramble shRNA and CSB shRNA SH-SY5Y cells were seeded into each well of a 6-well plate. After 2 days of incubation, culture media was removed, and the cells were washed once with phosphate buffered saline (PBS). The cells were then irradiated with UVC (254 nm) at 0, 2.5, 5, 7.5 or 10 J/m2, and fresh media was added to each well for further incubation for 5 days. To remain consistent with prior lab practices, cell counts were determined by hemocytometer analysis, and percent survival was plotted relative to the unirradiated samples. For sensitivity to angelicin and trioxsalen, we used the more efficient Cell Counting Kit-8 with the WST-8 reagent (Dojindo, Kumamoto, Japan). In brief, 2 × 103 cycling or 2 × 104 non-cycling cells were seeded into each well of a 96-well plate. After 1 day, cells were treated with the indicated concentration of trioxsalen or angelicin for 30 min, and then irradiated with UVA light using a Rayonet box (Southern New England Ultraviolet Company, Branford, CT, USA) as previously outlined (22). Cell viability was scored using a BioRad microplate spectrophotometer, with the number of viable cells proportional to the OD 450 nm reading. Survival rate at each dose was calculated relative to the OD 450 nm for the untreated (UVA alone) cells.
Single-cell gel electrophoresis
To monitor ICL unhooking, comet assays were performed under alkaline conditions as described (11,15,26), with minor modifications according to the Comet Assay Kit of Trevigen (Gaithersburg, MD, USA). In brief, cycling or non-cycling, scramble or CSB shRNA SH-SY5Y cells were treated in culture with 40 nM trioxsalen for 30 min and then exposed to 1.8 J/cm2 UVA, washed with PBS, scraped either immediately or 8 h after irradiation and suspended in cold PBS. Cells were embedded in soft agarose on glass slides and subjected to comet analysis. Images of more than 150 cells for each time point and treatment were obtained using a fluorescence microscope (Axiovert 200M) and Axiovision 4.9 software (Carl Zeiss, Oberkochen, Germany), and analyzed using the Comet Assay Software Project program to quantify tail moment (27). Statistical analysis was carried out using Graph Pad Prism software version 6.01.
Immunostaining for γ-H2AX
Non-cycling, scramble or CSB shRNA SH-SY5Y cells were treated in culture with 10 nM trioxsalen and UVA as above. Cells were subsequently fixed either immediately or at the indicated time point in 4% formaldehyde for 10 min at room temperature. Fixed cells were permeabilized with 0.5% Triton X-100, 1% BSA, 100 mM glycine and 0.2 mg/ml EDTA in PBS on ice for 10 min, and then digested with RNase A for 30 min at 37°C. Cells were blocked with Block ACE (DS Pharma. Biomedical Co., Osaka, Japan) in PBS for 1 h at room temperature. Immunofluorescence staining was carried out using an anti-γ-H2AX antibody (sc-101696; Santa Cruz Biotechnology), along with Alexa Fluor 647 goat anti-rabbit immunoglobulin G (IgG) (A-21244, Life Technologies, Grand Island, NY, USA). Nuclei were counterstained with 4′-diamino-2-phenylindole (DAPI) (H-1200, Vector Laboratories, Burlingame, CA, USA). Percentage of γ-H2AX positive cells (>10 foci per cell) was calculated from images obtained using the confocal microscope of more than 200 cells for each time point.
Statistical analysis
All results are expressed as the mean ± SD. Statistical analysis was performed using Prism software, and each method of statistical analysis is specified in the relevant figure legend. P < 0.05 was considered statistically significant.
RESULTS
Identification of an interaction between CSB and SNM1A
To identify molecular pathways that engage CSB, we conducted a yeast two-hybrid screen using a CSB-LexA DNA binding domain (DBD) fusion protein as bait against a normalized human universal cDNA library fused to the GAL4 activation domain (AD) (summarized in Supplementary Figure S1 and Supplementary Table S2). The studies utilized a yeast strain, NMY32, which was designed to harbor LexA operator sequences upstream of the HIS3 and ADE2 genes, as well as a LacZ reporter gene. Of the 2.5 × 106 co-transformants screened, 59 independent isolates grew on selective medium lacking histidine, indicative of an interaction between CSB-LexA(DBD) and the cDNA-GAL4(AD) fusion protein. To verify the putative interactions and to gauge their strength, two additional criteria were assessed: (i) induction of β-galactosidase activity via the lacZ gene and (ii) growth on minimal media lacking both histidine and adenine. Forty-six of the 59 initial candidates induced β-galactosidase activity when co-transformed with the CSB bait plasmid (Supplementary Figure S1B), and four of the clones showed robust growth when challenged with the more stringent (His−/Ade−) selection conditions (Supplementary Figure S1C). The four candidates strongly positive for all screening criteria were considered bona fide interactors of CSB and were as follows: (i) clone 28, deoxynucleotidyltransferase, terminal, interacting protein 2 (DNTTIP2); (ii) clone 44, DCLRE1A/SNM1A; (iii) clone 56, Leo1, Paf1/RNAP II complex component, homolog (S. cerevisiae) (LEO1); and (iv) clone 59, chromatin modifying protein 5 (CHMP5).
Characterization of the CSB-SNM1A physical association
Given the likely role that endogenous DNA damage plays in at least a subset of the pathologies associated with CS, we focus here on the CSB-SNM1A interaction, and the contribution of CSB to the resolution of DNA ICLs. SNM1A is a S. cerevisiae SNM1/PSO2 (sensitive to psoralen 2) homolog and a member of the β-CASP family of metallo-β-lactamase-fold-containing proteins that process nucleic acids and are related to polypeptides encoded by prokaryotic antibiotic resistance genes (28). Human SNM1A is a DNA 5′ to 3′ exonuclease with documented roles in ICL repair.
First, we used the yeast two-hybrid technology to determine the region of CSB that associates with SNM1A. In particular, we designed a bait construct that expresses the final 306 C-terminal residues of CSB (corresponding to amino acids 1187–1493) fused to the LexA(DBD). When co-transformed with the prey construct expressing SNM1A-GAL4(AD), these cells grew at least as well on selective medium as co-transformants harboring full-length CSB-LexA(DBD) and SNM1A-GAL4(AD), revealing the C-terminal portion of CSB as the interacting region with the exonuclease (Figure 1A). We note that unlike full-length CSB-LexA(DBD) or CSB C-terminal-LexA(DBD), neither p53-LexA(DBD) nor Lamin C-LexA(DBD) grew on selective medium when co-transformed with SNM1A-GAL4(AD) (Figure 1A).
Figure 1.
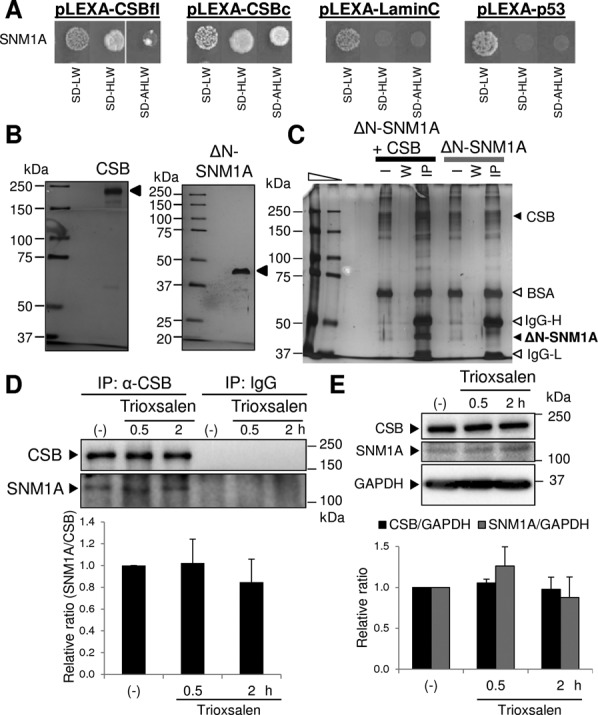
CSB and SNM1A interact. (A) Yeast two-hybrid interaction. The SNM1A-GAL4(AD) construct was cotransformed into NMY32 yeast with the indicated LexA(DBD) fusion construct (the pLEXA plasmids). The transformation mix was then spotted and allowed to grow on normal medium (SD-LW) or selection medium lacking histidine (SD-HLW) or histidine and adenine (SD-AHLW). Full-length CSB (CSBfl) and the 306 amino acid C-terminal CSB fragment (CSBc), as well as Lamin C and p53, were tested to determine interaction specificity. (B) Purified recombinant HA-tagged CSB and ΔN-SNM1A proteins. Purified recombinant HA-CSB (300 ng, left) and ΔN-SNM1A (300 ng, right) were loaded and separated on an 8% or 10% Tris–glycine–SDS-polyacrylamide gel, respectively. Proteins (designated by arrow) were then visualized using a Pierce Silver Stain Kit. Protein standards are indicated in kDa. (C) CSB physically interacts with the SNM1A nuclease fragment. ΔN-SNM1A (500 ng) was incubated with or without HA-CSB (500 ng) in the presence of α-HA magnetic beads. Bead-bound material was captured, washed and analyzed by silver staining after separation on an SDS-polyacrylamide gel. The positions of the CSB and SNM1A proteins are designated, as are the BSA, IgG-Heavy (H) and IgG-Light (L) bands intrinsic to the reaction mixture. I, 1/20th of the initial reaction mixture or input; W, material recovered from the third wash; IP, bead-bound, IP material. Protein standards are indicated in kDa. (D) Co-purification of endogenous CSB and SNM1A from human cell extracts. Whole cell extracts were prepared from HeLa cells, either without (-) or 0.5 or 2 h post-treatment with trioxsalen/UVA. Extracts were then incubated with α-CSB antibody or mouse IgG, and subsequently with protein A/G magnetic beads. Bead-bound IP material was captured, washed, extracted with SDS sampling buffer and subjected to western blot analysis. The positions of CSB and SNM1A are shown, as are the protein standards in kDa. Bar graph indicates the relative SNM1A:CSB ratio (average and standard deviation of three independent experiments), with the untreated (-) sample set as 1. (E) Expression of CSB and SNM1A is unchanged following trioxsalen/UVA treatment. Whole cell extracts were prepared from HeLa cells, either without (-) or 0.5 or 2 h post-treatment with trioxsalen/UVA. Extracts were immediately subjected to western blot analysis for CSB, SNM1A or GAPDH, as designated. Bar graph reports the relative expression for CSB/GAPDH or SNM1A/GAPDH, as determined from three independent experiments.
Second, we determined whether highly purified protein preparations of ΔN-SNM1A (residues 608–1040) and HA-CSB (Figure 1B) could be co-captured on α-HA beads. Indeed, ΔN-SNM1A was pulled down by α-HA resin on which HA-CSB had been immobilized (+ CSB, IP lane), but not by the α-HA resin alone, confirming a direct association between the two proteins (Figure 1C).
Third, we determined whether the CSB and SNM1A proteins exist in a common complex within human cells. Initially, we examined whether SNM1A could be captured by a transiently expressed, N-terminal HA-tagged and C-terminal His-tagged CSB protein (dtCSB) in HeLa cells. Following transfection of the vector control or pcDNA-dtCSB expression plasmid, whole cell extracts were prepared and incubated with α-HA beads. This strategy revealed that endogenous SNM1A is indeed retrieved from extracts containing dtCSB, but not the pcDNA control extracts (Supplementary Figure S2A). After optimization of an immunoprecipitation procedure using a mouse monoclonal antibody against CSB, we also found that SNM1A (∼11% of total) co-immunoprecipitated (IP) with endogenous CSB from HeLa whole cell extracts, while neither protein was captured by mouse IgG used as a negative control (Figure 1D, lanes labeled ‘(-)’). In addition, we found that the percentage of SNM1A retrieved in the anti-CSB immunoprecipitant was unchanged at 0.5 or 2 h post-treatment of HeLa cells with the DNA interstrand crosslinking agent, trioxsalen (+ UVA), suggesting that the interaction of the two proteins is not altered in response to ICL induction (Figure 1D, specified lanes). Finally, the overall level of CSB or SNM1A was not affected by trioxsalen/UVA treatment, at least under the experimental conditions explored and at the time points analyzed (Figure 1E); and while these two proteins may associate as part of a common complex in vivo, a deficiency in CSB did not notably affect the steady-state level of SNM1A, indicating that CSB is not a major stabilization factor for the exonuclease (Supplementary Figure S2B and C).
CSB modulates SNM1A exonuclease activity
We next examined whether the CSB-SNM1A interaction could regulate the in vitro biochemical functions of each protein. No significant effect of ΔN-SNM1A was observed on CSB DNA-dependent ATPase activity, its key enzymatic function (29), even when the nuclease fragment was at a 4-fold excess over CSB or when different relative DNA concentrations were employed (Supplementary Figure S3). Experiments, however, revealed that CSB could markedly stimulate the exonuclease activity of ΔN-SNM1A on simple 61-mer single-stranded oligonucleotides (single-stranded DNA is the preferred substrate of SNM1A (21)) and on blunt double-stranded DNA (Figure 2A). Notably, this stimulation was observed when CSB and SNM1A were present at an equimolar concentration (0.8 nM). Interestingly, although a similar pattern of increased DNA degradation was observed on shorter, 21-mer single- and double-stranded oligonucleotides at low CSB concentrations (Figure 2B), some inhibition—most pronounced with the double-stranded substrate—was observed at higher concentrations of CSB (>20 nM, a 25-fold excess over SNM1A), likely reflecting DNA binding and substrate occlusion by CSB.
Figure 2.
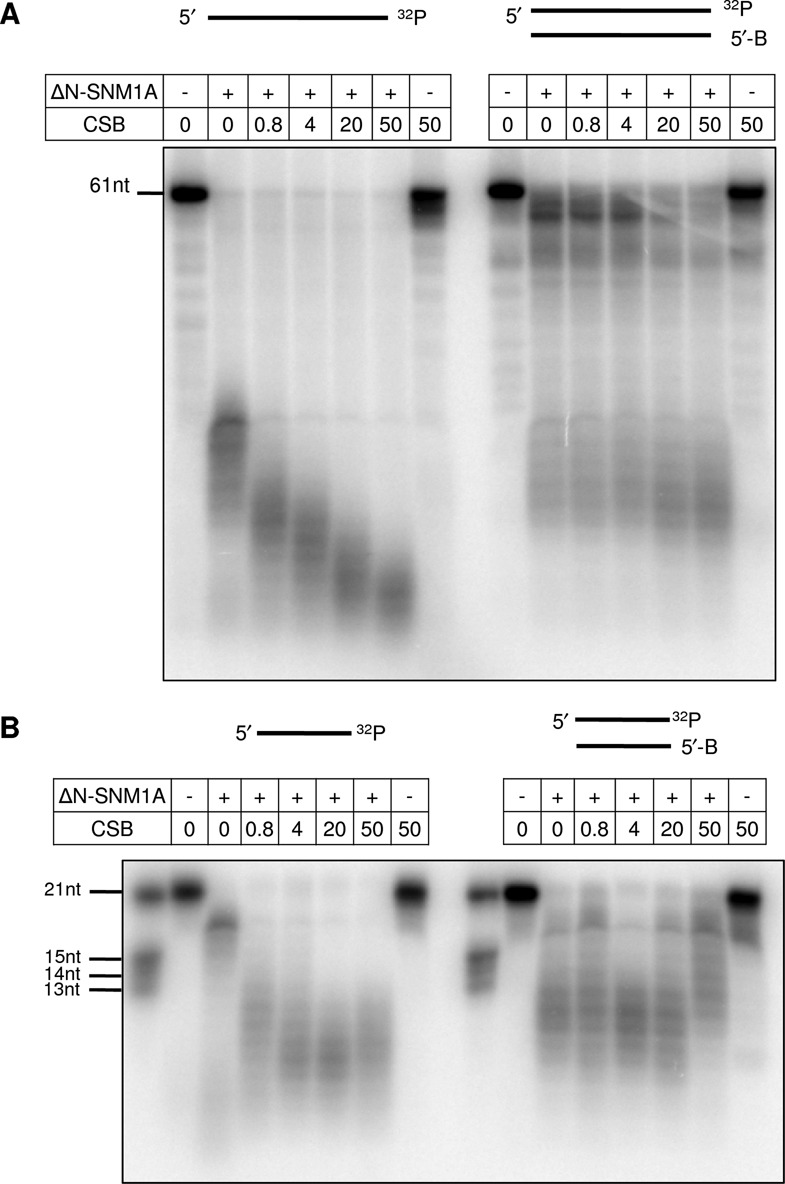
CSB modulates SNM1A exonuclease activity. Single-stranded or double-stranded 3′-end labeled 61-mer (A) or 21-mer (B) oligonucleotides were incubated with SNM1A (+; fixed concentration of 0.8 nM), and increasing concentrations of CSB where indicated (0.8–50 nM), for 30 min at 37°C. Reactions were resolved on a denaturing gel, and images were captured during phosphorimager analysis (shown is a representative gel from three independent experiments). Note that the non-labeled strand of the double-stranded substrate is blocked at its 5′-end with a biotin moiety (B) to prevent competing digestion of this strand.
To determine how CSB might stimulate SNM1A activity, we measured the kinetic parameters of SNM1A on single- and double-stranded 20-mer oligonucleotides either in the absence or in the presence of equimolar CSB using a previously described real-time fluorescence-based assay. These experiments revealed that the activation observed is due to an ∼2- to 4-fold lower KM (Table 1), indicating that CSB improves the affinity of SNM1A for DNA. Since single-stranded DNA is not stably bound by CSB and does not serve as a co-factor for its ATPase function (18), the stimulation of SNM1A appears to primarily arise from the physical interaction of CSB and not its reported DNA re-modeling activity (30,31).
Table 1. Effect of CSB on SNM1A kinetic parameters.
| Single-stranded DNA | Double-stranded DNA | |||||
|---|---|---|---|---|---|---|
| kcat (AU nM−1min−1) | KM (nM) | Catalytic efficiency | kcat (AU nM−1 min−1) | KM (nM) | Catalytic efficiency | |
| SNM1A | 57 ± 2.6 | 37 ± 8.3 | 1.5 ± 0.5 | 78 ± 5.9 | 123 ± 34 | 0.6 ± 0.3 |
| SNM1A + CSB* | 60 ± 2.8 | 20 ± 5.9 | 3.0 ± 1.4 | 74 ± 4.2 | 32 ± 9.2 | 2.4 ± 1.2 |
Real-time reaction assays were conducted using single- and double-stranded fluorescent oligonucleotide substrates, and kinetic constants were subsequently determined, as described in Materials and Methods. Values shown represent the average and standard deviation of best curve fits involving nine substrate concentrations and three data points at each concentration. AU, absorbance units; catalytic efficiency = kcat/KM; *, SNM1A and CSB were at equal molar concentrations.
CSB and SNM1A respond coordinately to site-specific DNA ICLs
To elucidate the possible role(s) of CSB and SNM1A in the ICL response, we employed fluorescently tagged fusion proteins in combination with confocal microscopy and laser microirradiation, which together permit the assessment of the reaction to targeted DNA damage. Following transfection of pCSB-GFP into HeLa cells, we determined the recruitment and retention dynamics of the CSB protein to site-specific oxidative DNA damage (laser alone) or DNA ICLs (trioxsalen + laser); trioxsalen is a psoralen derivative that produces a high ratio of ICLs:monoadducts following UVA activation (32). While we observed no response of the GFP tag alone to either DNA damage scenario (Supplementary Figure S4), we found CSB-GFP to respond similarly to either oxidative damage or ICLs, although recruitment to the latter was slightly more rapid and robust (Figure 3A and B). Notably, the response of CSB to laser alone was suppressed by the anti-oxidant NAC, whereas the recruitment to trioxsalen ICLs was not (Figure 3A and B). These data indicate that: (i) laser alone generated mainly oxidative DNA damage and (ii) CSB accumulation at trioxsalen-induced DNA ICLs was specific and distinct. Focusing on the response to crosslinks, we next determined whether transcriptional status would alter CSB recruitment. As shown in Figure 3A and C, pre-treating cells with α-amanitin, an RNAP II inhibitor, greatly reduced CSB-GFP accumulation at site-specific trioxsalen ICLs, indicating that active transcription was a prerequisite for robust CSB recruitment to ICLs and consistent with a transcription-associated repair response.
Figure 3.
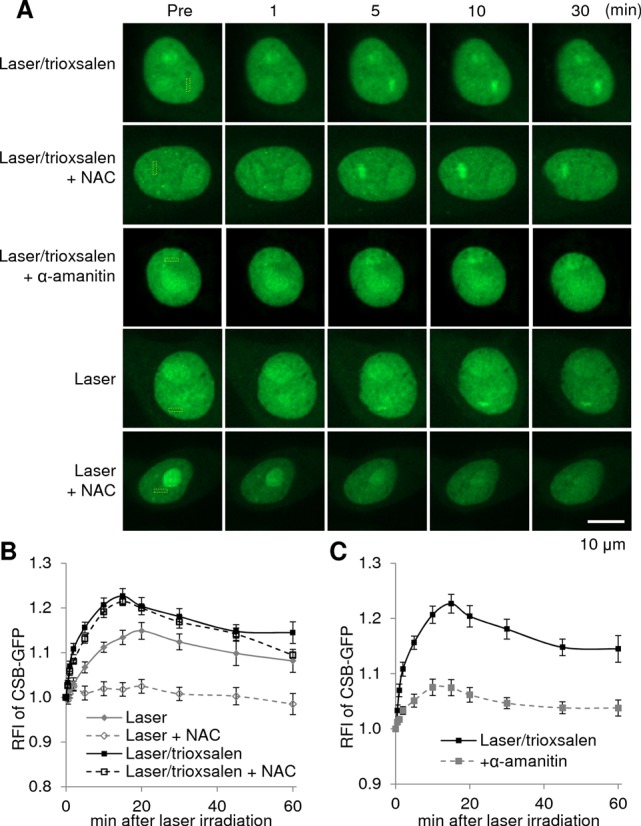
Response of CSB to site-specific DNA damage. (A) CSB recruitment and retention at site-specific DNA damage. pCSB-GFP was transfected into HeLa cells, and the indicated region (yellow box) was subsequently laser irradiated under the conditions specified. In particular, cells were grown in the presence of 6 μM trioxsalen, 10 mM NAC and/or 20 μM α-amanitin as indicated. Shown are representative images of unirradiated cells (Pre), and the CSB response at 1, 5, 10 and 30 min post-laser irradiation. (B) Quantification of the CSB response to laser (oxidative DNA damage) or laser/trioxsalen (ICLs), with or without the anti-oxidant NAC. (C) Quantification of the CSB response to laser/trioxsalen, with or without the RNAP II inhibitor, α-amanitin. The graphs shown in panels B and C report the RFI of CSB-GFP at the microirradiated area relative to unirradiated (background) parts of the nucleus. Each data point is derived from at least 12 independent cells, respectively, from three independent experiments. Error bars indicate SEM.
Studies using a GFP-tagged SNM1A fusion protein (SNM1A-GFP) revealed that the nuclease displayed similar recruitment kinetics as CSB to trioxsalen ICLs, peaking at around 15 min post-laser exposure, yet exhibited a more sustained persistence at the DNA damage site (Supplementary Figure S5A; Figure 4A). As seen for CSB, the SNM1A-GFP response was greatly suppressed by α-amanitin, consistent with a role for transcription in protein recruitment. When these two proteins were co-expressed in HeLa cells, with CSB tagged with mCherry and SNM1A with GFP, we observed co-localization specifically at sites of DNA ICLs, supporting the notion that these two proteins operate in a common DNA damage response (Figure 4B). Moreover, SNM1A accumulation at DNA crosslinks was significantly reduced in CSB-deficient CS1AN-vector complemented cells relative to the corrected CS1AN-CSB control line (Supplementary Figure S5B; Figure 4C), indicating that CSB coordinates at least a component of SNM1A recruitment.
Figure 4.
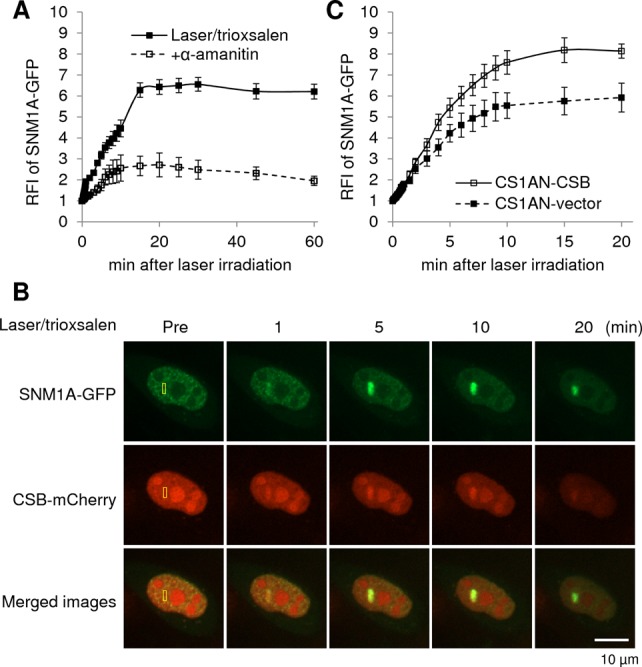
Response of SNM1A to site-specific DNA ICLs. (A) Quantification of the SNM1A response to laser/trioxsalen, and the effects of α-amanitin on protein recruitment/retention. pSNM1A-GFP was transfected into HeLa cells, which were pre-treated with 6 μM trioxsalen ± 20 μM α-amanitin as indicated, before targeted laser irradiation. The graph shows the RFI of SNM1A-GFP at the microirradiated area relative to unirradiated (background) parts of the nucleus. Each data point is derived from at least 16 independent cells from two independent experiments. Error bars indicate SEM. See Supplementary Figure S5A for representative images. (B) SNM1A and CSB co-localize to sites of DNA ICLs in live human cells. HeLa cells were co-transfected with pSNM1A-GFP and pCSB-mCherry, and co-expressing cells were identified 20–24 h post-transfection. Cells were then microirradiated at the indicated region (yellow box) under the conditions specified. Images of a representative single live cell are shown, along with the merge. (C) CSB coordinates the SNM1A response to DNA ICLs. CSB-deficient (CS1AN-vector) or CSB complemented (CS1AN-CSB) cells were transfected with pSNM1A-GFP and treated with 6 μM trioxsalen for 30 min prior to targeted microirradiation. See panel A for information about the graph, and Supplementary Figure S5B for representative images.
CSB modulates cellular resistance to DNA crosslinking agents
To gain further insight into the contribution of CSB to ICL repair in cycling (replicating) and non-cycling (non-replicating) cells, we stably knocked-down CSB protein expression by ≥80% in the neuroblastoma cell line SH-SY5Y (Figure 5A), which can be selectively terminally differentiated into a neuronal-like state in culture. We then measured DNA-damaging agent sensitivity of control (scramble shRNA) and CSB-deficient (CSB shRNA) cells in both the non-differentiated, cycling state and the differentiated, non-cycling state. Initial experiments revealed a hypersensitivity of CSB shRNA SH-SY5Y cells to UVC irradiation in both cycling and non-cycling situations, confirming CSB deficiency and, in the latter case, indicating a role for the protein in a replication-independent UV DNA damage repair response (Figure 5B). As observed with UVC, CSB knockdown resulted in hypersensitivity of SH-SY5Y cells to angelicin/UVA (a trioxsalen analog able to form only DNA monoadducts), regardless of cycling status (Figure 5C), consistent with CSB participating in transcription-associated repair of bulky DNA lesions independent of DNA replication. Notably, in contrast to the sensitivity profile seen for UVC or angelicin, trioxsalen/UVA sensitivity was most pronounced in CSB-deficient non-cycling cells in comparison to the cycling cells (Figure 5D), indicating a more critical replication-independent, CSB-dependent, ICL repair process. Consistent with these observations, CSB-deficient, CS1AN-vector patient fibroblasts, in comparison to corrected CS1AN-CSB cells, were hypersensitive to angelicin/UVA regardless of replication status, but only significantly sensitive to trioxsalen/UVA when arrested (i.e. non-replicating) by serum starvation (Supplementary Figure S6). Finally, in keeping with CSB functioning prominently in the repair of DNA crosslink damage in non-cycling cells, differentiated CSB shRNA SH-SY5Y neural cells displayed a significant hypersensitivity to the crosslinking agents cisplatin and MMC, whereas non-differentiated CSB shRNA cells did not (Supplementary Figure S7).
Figure 5.
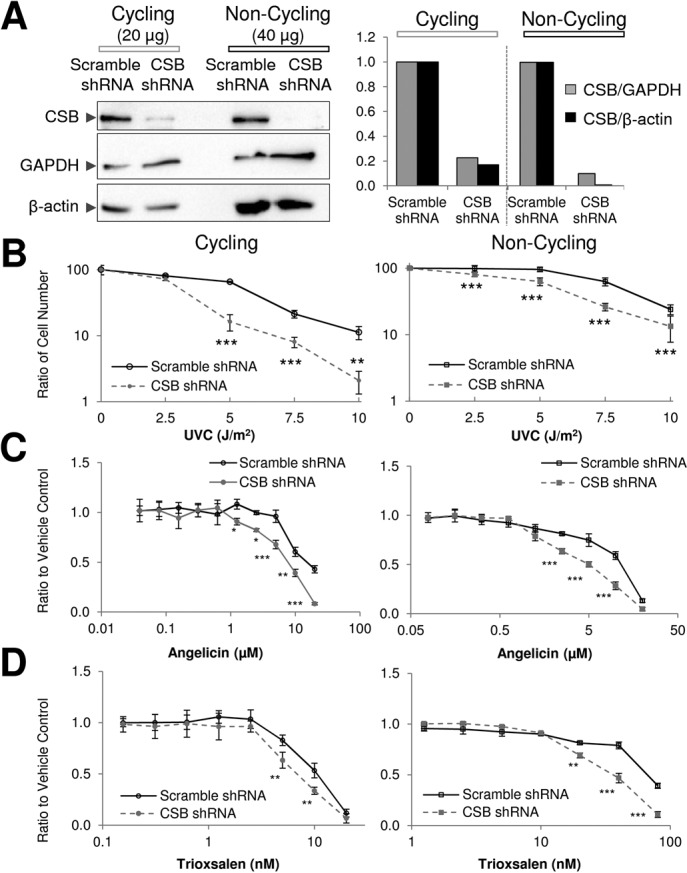
Sensitivity of cycling and non-cycling CSB-deficient SH-SY5Y cells to DNA-damaging agents. (A) Expression of CSB protein in cycling and non-cycling scramble shRNA or CSB shRNA SH-SY5Y cells. CSB protein level was determined in whole cell extracts (μg amount designated) by western blotting (left), in comparison to GAPDH and β-actin. The relative ratio of CSB to the indicated standard is shown (right; from a representative gel), with the scramble control line being set as 1. (B) UV sensitivity of cycling and non-cycling scramble shRNA and CSB shRNA SH-SY5Y cells. Cell viability was quantified after UVC irradiation (0, 2.5, 5, 7.5 or 10 J/m2) by hemocytometer analysis. Reported is the ratio of the cell count determined at the indicated UVC dose in comparison to the untreated control sample. (C) Viability of cycling and non-cycling scramble shRNA and CSB shRNA SH-SY5Y cells treated with angelicin (cycling cells, 0.04–20 μM; non-cycling cells, 0.08–20 μM) and UVA. Viability was measured using a WST-8 assay 72 h after treatment. Results are presented as the ratio of the value obtained at the indicated concentration of angelicin relative to that of the untreated (UVA only) cells. (D) Viability of cycling and non-cycling scramble shRNA and CSB shRNA SH-SY5Y cells treated with trioxsalen (cycling cells, 0.15–20 nM; non-cycling cells, 1.25–80 nM) and UVA. See panel C for further details. The data in panels B–D represent the mean ± SD from three independent experiments. The statistical analysis of each graph was performed by two-way ANOVA followed by the Bonferroni post hoc test (*P < 0.05, **P < 0.005, ***P < 0.0001).
CSB promotes ICL unhooking
Exploiting the capacity to terminally differentiate SH-SY5Y cells, we more thoroughly characterized the molecular involvement of CSB in ICL repair in the absence of active replication. Specifically, we treated scramble shRNA and CSB shRNA non-cycling SH-SY5Y neural cells in culture with trioxsalen/UVA, and measured the induction and repair (unhooking) of DNA ICLs at the single-cell level by the alkaline comet assay. ICLs prevent migration of DNA during the electrophoresis process, and thus, the formation of a comet tail, at least until the covalent crosslink is degraded or unhooked. As seen in Figure 6A (no treatment), CSB deficiency alone does not affect the overall steady-state level of strand breaks or alkaline sensitive sites in the genome. Moreover, the initial level of DNA ICL induction appears to be identical in the control and CSB knockdown cells, as evidenced by the lack of comet tail (Figure 6A, trioxsalen/UVA, 0 h). CSB-deficient cells, however, recover their tail moment less efficiently than the control cells at 8 h after treatment (P < 0.0001), indicative of failed ICL unhooking.
Figure 6.
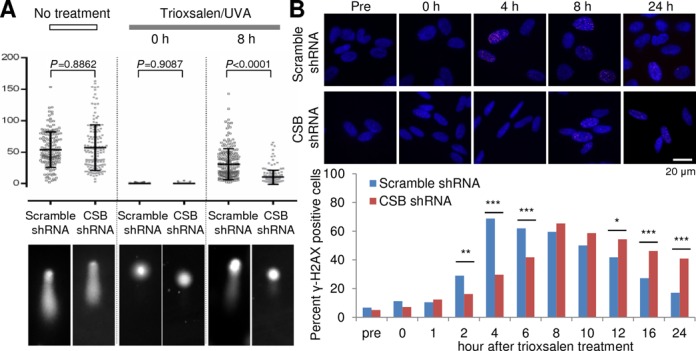
CSB promotes DNA ICL processing. (A) Unhooking of DNA ICLs in non-cycling scramble shRNA and CSB shRNA SH-SY5Y cells. Designated cells received no treatment or were treated with trioxsalen/UVA and fixed immediately (0 h) or 8 h later. The box plots (above) show the tail moments determined from analysis of more than 150 nuclear DNAs from at least three different experiments. Error bars indicate SD, and P-values were determined by statistical analysis using the Mann–Whitney U-test. Representative Comet images are shown (below) for the indicated treatments, cells and time points. (B) γ-H2AX foci formation and disappearance following trioxsalen/UVA treatment of non-cycling scramble shRNA or CSB shRNA SH-SY5Y cells. (Top) Representative images of γ-H2AX staining (red) and DAPI (blue) at the indicated time after trioxsalen/UVA treatment are shown. (Bottom) The percentage of γ-H2AX positive cells (>10 foci per cell) was quantified from more than 200 cells of two different preparations. Statistical significance between the values obtained for scramble shRNA and CSB shRNA SH-SY5Y cells was determined using Fisher's exact test and Bonferroni correction. *P < 0.05, **P < 0.01, ***P < 0.001.
To further characterize DNA ICL processing in non-cycling CSB-deficient cells, we monitored the formation and clearance of phosphorylated histone H2AX (γ-H2AX) foci in differentiated scramble shRNA and CSB shRNA SH-SY5Y neural cells. γ-H2AX foci are most commonly used to assess the response to DNA double-strand breaks, but can be used more broadly to evaluate for perturbations in DNA integrity (33). As shown in Figure 6B, the percentage of non-cycling, scramble knockdown cells positive for γ-H2AX staining (i.e. cells with >10 foci) increases up to 4 h post-treatment with trioxsalen/UVA, and then decreases back to approximately the pre-treatment level around 24 h. In the CSB-deficient cells, however, the percentage of γH2AX positive cells did not peak until 8 h after treatment and remained well above the untreated background to at least 24 h post-damage induction (Figure 6B). These results support the comet data indicating that replication-independent processing of DNA ICLs is perturbed in CSB-deficient mammalian cells.
DISCUSSION
Prior cellular work indicated a role for the CSB protein in the repair of DNA ICLs (14,15). Using a yeast two-hybrid approach, we identified SNM1A, a 5′ to 3′ exonuclease with documented roles in ICL repair (28,34), as one of four strong interacting partners of CSB, revealing a mechanism by which CSB could facilitate ICL resolution. Interestingly, recent observations indicate that inherited mutations in the XPF (ERCC4) and ERCC1 genes, which encode proteins that constitute a nuclease complex that functions in both nucleotide excision repair and ICL repair, are associated with clinical features that resemble the premature aging disorder CS (35). One individual deficient in XPF exhibited clinical phenotypes seen not only in CS, but also in xeroderma pigmentosum (XP) and FA, the latter of which is diagnosed by cellular sensitivity to DNA crosslinking agents. In addition, an ERCC1-/Δ mutant mouse model reveals strong functional, regulatory and histopathological parallels between accelerated aging—driven by a DNA repair defect—and normal aging (36). Thus, there appears to be a convergence of independent scientific evidence that supports a role for endogenous DNA lesions, such as ICLs, in the development of disease and the aging process.
Our two-hybrid experiments revealed that the 306 C-terminal residues of CSB can associate with SNM1A, and our biochemical studies uncovered that the nuclease portion of SNM1A was sufficient for the direct physical association between the two proteins. In addition, we observed that CSB and SNM1A can exist in a common protein complex within human cell extracts, and can co-localize to induced DNA ICLs in live cells. These data are consist with a prior study that found SNM1A to be one of the proteins that associates with tagged CSB in mouse FM3A cell extracts using tandem affinity purification followed by mass spectrometry analysis (37). Thus, the existence of these two proteins in a functional complex in vivo appears to be conserved among mammals.
Our in vitro studies indicate that CSB can stimulate the SNM1A exonuclease activity on single- and double-stranded oligonucleotides by enhancing substrate affinity, i.e. by lowering the KM. Coupled with the finding that SNM1A recruitment to ICLs is decreased in CSB-deficient cells, our data suggest that a main role for the interaction is for CSB to coordinate efficient assembly of SNM1A at sites of DNA damage. Since both CSB and SNM1A accumulation at ICLs was markedly suppressed by α-amanitin, this coordinated response appears to be influenced by active RNA transcription machinery. Thus, it would appear that upon RNAP arrest at a blocking damage, CSB is called upon to organize the necessary proteins to process the substrate lesion (38). In a situation involving an ICL, we propose that SNM1A is recruited by CSB to facilitate crosslink unhooking following 5′ incision by the ERCC1/XPF nuclease complex (see model in Supplementary Figure S8). Future studies will need to employ more comprehensive reconstitution systems using synthetic ICL-containing DNA substrates and cell biology strategies to fully delineate the molecular details of the CSB-directed ICL unhooking mechanism, which besides SNM1A, could involve the nucleases SLX1, FAN1, MUS81 and/or XPF (39).
While the ICL repair mechanism outlined in Supplementary Figure S8 could occur in both cycling (primarily in the G1 phase) and non-cycling cells, the viability studies reported herein indicate that the role of transcription-associated repair of ICLs is greater when the major repair pathway, i.e. replication-dependent homologous recombination, is presumably not operational. Conversely, it would appear that monoadducts, generated by either UVC or angelicin/UVA, are repaired with equal importance in cycling and non-cycling cells by classic transcription-coupled nucleotide excision repair, since both DNA-damaging agents produced a similar toxicity profile in CSB-deficient cells independent of replication status. It should be emphasized that while CSB is likely restricted to transcription-associated repair of ICLs, SNM1A appears to have roles in replication-dependent crosslink resolution as well (17,40). Notably, SNM1A has been found to interact via a PCNA-interaction protein box and ubiquitin binding zinc finger with the replication factor PCNA in a RAD18-monoubiquitinated manner in response to DNA damage (41), reflective of a broader role for SNM1A in DNA repair.
The other three strong interactors of CSB identified in the yeast two-hybrid screen included DNTTIP2 (also known as TdIF2), LEO1 and CHMP5. While these interactions need to be confirmed by additional means, the overall results of the screen support the hypothesis that CSB functions most prominently in pathways related to DNA repair and gene regulation. Based in part on the studies presented herein, we propose a model whereby CSB, in a replication-independent, transcription-associated mechanism, coordinates key nucleases, such as SNM1A, to carry out ICL unhooking, repair and transcription restart. Defects in this process likely contribute to the pathologies of CS, particularly those that involve the loss of post-mitotic cells.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Drs. Morten Scheibye-Knudsen and Robert Brosh (NIA) for their critical reading of the manuscript, Dr. Manikandan Paramasivam (NIA) for his helpful advice on the confocal microscopy and laser microirradiation studies and Dr. Chris Morrell (Loyola University Maryland) for assistance with statistical analysis.
FUNDING
Intramural Research Program at the NIH, National Institute on Aging; Luke O'Brien Foundation (DMWIII); MRC [to P.J.M. and O.G.]; Cancer Research UK [to P.J.M.]; National Science Scholarship from the Singaporean Agency for Science, Technology, and Research (A*STAR) [to S.Y.L.]. Funding for open access charge: Intramural Research Program at the NIH, National Institute on Aging.
Conflict of interest statement. None declared.
REFERENCES
- 1.Laugel V. Cockayne syndrome: the expanding clinical and mutational spectrum. Mech. Ageing Dev. 2013;134:161–170. doi: 10.1016/j.mad.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 2.Lanzafame M., Vaz B., Nardo T., Botta E., Orioli D., Stefanini M. From laboratory tests to functional characterisation of Cockayne syndrome. Mech. Ageing Dev. 2013;134:171–179. doi: 10.1016/j.mad.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 3.Saijo M. The role of Cockayne syndrome group A (CSA) protein in transcription-coupled nucleotide excision repair. Mech. Ageing Dev. 2013;134:196–201. doi: 10.1016/j.mad.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 4.Lake R.J., Fan H.Y. Structure, function and regulation of CSB: a multi-talented gymnast. Mech. Ageing Dev. 2013;134:202–211. doi: 10.1016/j.mad.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vermeulen W., Fousteri M. Mammalian transcription-coupled excision repair. Cold Spring Harb. Perspect. Biol. 2013;5:a012625. doi: 10.1101/cshperspect.a012625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berquist B.R., Wilson D.M., 3rd Pathways for repairing and tolerating the spectrum of oxidative DNA lesions. Cancer Lett. 2012;327:61–72. doi: 10.1016/j.canlet.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muniandy P.A., Liu J., Majumdar A., Liu S.T., Seidman M.M. DNA interstrand crosslink repair in mammalian cells: step by step. Crit. Rev. Biochem. Mol. Biol. 2010;45:23–49. doi: 10.3109/10409230903501819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deans A.J., West S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer. 2011;11:467–480. doi: 10.1038/nrc3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McHugh P.J., Sarkar S. DNA interstrand cross-link repair in the cell cycle: a critical role for polymerase zeta in G1 phase. Cell Cycle. 2006;5:1044–1047. doi: 10.4161/cc.5.10.2763. [DOI] [PubMed] [Google Scholar]
- 10.Williams H.L., Gottesman M.E., Gautier J. The differences between ICL repair during and outside of S phase. Trends Biochem. Sci. 2013;38:386–393. doi: 10.1016/j.tibs.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muniandy P.A., Thapa D., Thazhathveetil A.K., Liu S.T., Seidman M.M. Repair of laser-localized DNA interstrand cross-links in G1 phase mammalian cells. J. Biol. Chem. 2009;284:27908–27917. doi: 10.1074/jbc.M109.029025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furuta T., Ueda T., Aune G., Sarasin A., Kraemer K.H., Pommier Y. Transcription-coupled nucleotide excision repair as a determinant of cisplatin sensitivity of human cells. Cancer Res. 2002;62:4899–4902. [PubMed] [Google Scholar]
- 13.McKay B.C., Becerril C., Ljungman M. P53 plays a protective role against UV- and cisplatin-induced apoptosis in transcription-coupled repair proficient fibroblasts. Oncogene. 2001;20:6805–6808. doi: 10.1038/sj.onc.1204901. [DOI] [PubMed] [Google Scholar]
- 14.Enoiu M., Jiricny J., Scharer O.D. Repair of cisplatin-induced DNA interstrand crosslinks by a replication-independent pathway involving transcription-coupled repair and translesion synthesis. Nucleic Acids Res. 2012;40:8953–8964. doi: 10.1093/nar/gks670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards S., Liu S.T., Majumdar A., Liu J.L., Nairn R.S., Bernier M., Maher V., Seidman M.M. Triplex targeted genomic crosslinks enter separable deletion and base substitution pathways. Nucleic Acids Res. 2005;33:5382–5393. doi: 10.1093/nar/gki851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng H., Wang X., Warren A.J., Legerski R.J., Nairn R.S., Hamilton J.W., Li L. Nucleotide excision repair- and polymerase eta-mediated error-prone removal of mitomycin C interstrand cross-links. Mol. Cell. Biol. 2003;23:754–761. doi: 10.1128/MCB.23.2.754-761.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang A.T., Sengerova B., Cattell E., Inagawa T., Hartley J.M., Kiakos K., Burgess-Brown N.A., Swift L.P., Enzlin J.H., Schofield C.J., et al. Human SNM1A and XPF-ERCC1 collaborate to initiate DNA interstrand cross-link repair. Genes Dev. 2011;25:1859–1870. doi: 10.1101/gad.15699211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berquist B.R., Wilson D.M., 3rd Nucleic acid binding activity of human Cockayne syndrome B protein and identification of Ca(2+) as a novel metal cofactor. J. Mol. Biol. 2009;391:820–832. doi: 10.1016/j.jmb.2009.06.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimozono S., Miyawaki A. Engineering FRET constructs using CFP and YFP. Methods Cell. Biol. 2008;85:381–393. doi: 10.1016/S0091-679X(08)85016-9. [DOI] [PubMed] [Google Scholar]
- 20.Christiansen M., Stevnsner T., Modin C., Martensen P.M., Brosh R.M., Jr, Bohr V.A. Functional consequences of mutations in the conserved SF2 motifs and post-translational phosphorylation of the CSB protein. Nucleic Acids Res. 2003;31:963–973. doi: 10.1093/nar/gkg164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sengerova B., Allerston C.K., Abu M., Lee S.Y., Hartley J., Kiakos K., Schofield C.J., Hartley J.A., Gileadi O., McHugh P.J. Characterization of the human SNM1A and SNM1B/Apollo DNA repair exonucleases. J. Biol. Chem. 2012;287:26254–26267. doi: 10.1074/jbc.M112.367243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McNeill D.R., Paramasivam M., Baldwin J., Huang J., Vyjayanti V.N., Seidman M.M., Wilson D.M., 3rd NEIL1 responds and binds to psoralen-induced DNA interstrand crosslinks. J. Biol. Chem. 2013;288:12426–12436. doi: 10.1074/jbc.M113.456087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scheibye-Knudsen M., Ramamoorthy M., Sykora P., Maynard S., Lin P.C., Minor R.K., Wilson D.M., 3rd, Cooper M., Spencer R., de Cabo R., et al. Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J. Exp. Med. 2012;209:855–869. doi: 10.1084/jem.20111721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berquist B.R., Canugovi C., Sykora P., Wilson D.M., 3rd, Bohr V.A. Human Cockayne syndrome B protein reciprocally communicates with mitochondrial proteins and promotes transcriptional elongation. Nucleic Acids Res. 2012;40:8392–8405. doi: 10.1093/nar/gks565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kulkarni A., McNeill D.R., Gleichmann M., Mattson M.P., Wilson D.M., 3rd XRCC1 protects against the lethality of induced oxidative DNA damage in nondividing neural cells. Nucleic Acids Res. 2008;36:5111–5121. doi: 10.1093/nar/gkn480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramamoorthy M., May A., Tadokoro T., Popuri V., Seidman M.M., Croteau D.L., Bohr V.A. The RecQ helicase RECQL5 participates in psoralen-induced interstrand cross-link repair. Carcinogenesis. 2013;34:2218–2230. doi: 10.1093/carcin/bgt183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konca K., Lankoff A., Banasik A., Lisowska H., Kuszewski T., Gozdz S., Koza Z., Wojcik A. A cross-platform public domain PC image-analysis program for the comet assay. Mutat. Res. 2003;534:15–20. doi: 10.1016/s1383-5718(02)00251-6. [DOI] [PubMed] [Google Scholar]
- 28.Cattell E., Sengerova B., McHugh P.J. The SNM1/Pso2 family of ICL repair nucleases: from yeast to man. Environ. Mol. Mutagen. 2010;51:635–645. doi: 10.1002/em.20556. [DOI] [PubMed] [Google Scholar]
- 29.Citterio E., Rademakers S., van der Horst G.T., van Gool A.J., Hoeijmakers J.H., Vermeulen W. Biochemical and biological characterization of wild-type and ATPase-deficient Cockayne syndrome B repair protein. J. Biol. Chem. 1998;273:11844–11851. doi: 10.1074/jbc.273.19.11844. [DOI] [PubMed] [Google Scholar]
- 30.Beerens N., Hoeijmakers J.H., Kanaar R., Vermeulen W., Wyman C. The CSB protein actively wraps DNA. J. Biol. Chem. 2005;280:4722–4729. doi: 10.1074/jbc.M409147200. [DOI] [PubMed] [Google Scholar]
- 31.Citterio E., Van Den Boom V., Schnitzler G., Kanaar R., Bonte E., Kingston R.E., Hoeijmakers J.H., Vermeulen W. ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol. Cell. Biol. 2000;20:7643–7653. doi: 10.1128/mcb.20.20.7643-7653.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chatterjee P.K., Cantor C.R. Photochemical production of psoralen - DNA monoadducts capable of subsequent photocrosslinking. Nucleic Acids Res. 1978;5:3619–3633. doi: 10.1093/nar/5.10.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cleaver J.E. gammaH2Ax: biomarker of damage or functional participant in DNA repair “all that glitters is not gold!”. Photochem. Photobiol. 2011;87:1230–1239. doi: 10.1111/j.1751-1097.2011.00995.x. [DOI] [PubMed] [Google Scholar]
- 34.Yan Y., Akhter S., Zhang X., Legerski R. The multifunctional SNM1 gene family: not just nucleases. Future Oncol. 2010;6:1015–1029. doi: 10.2217/fon.10.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kashiyama K., Nakazawa Y., Pilz D.T., Guo C., Shimada M., Sasaki K., Fawcett H., Wing J.F., Lewin S.O., Carr L., et al. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am. J. Hum. Genet. 2013;92:807–819. doi: 10.1016/j.ajhg.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Niedernhofer L.J., Garinis G.A., Raams A., Lalai A.S., Robinson A.R., Appeldoorn E., Odijk H., Oostendorp R., Ahmad A., van Leeuwen W., et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444:1038–1043. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- 37.Xie W., Ling T., Zhou Y., Feng W., Zhu Q., Stunnenberg H.G., Grummt I., Tao W. The chromatin remodeling complex NuRD establishes the poised state of rRNA genes characterized by bivalent histone modifications and altered nucleosome positions. Proc. Natl. Acad. Sci. U.S.A. 2012;109:8161–8166. doi: 10.1073/pnas.1201262109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spivak G., Ganesan A.K. The complex choreography of transcription-coupled repair. DNA Repair (Amst) 2014;19:64–70. doi: 10.1016/j.dnarep.2014.03.025. [DOI] [PubMed] [Google Scholar]
- 39.Zhang J., Walter J.C. Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair (Amst) 2014;19:135–142. doi: 10.1016/j.dnarep.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sengerova B., Wang A.T., McHugh P.J. Orchestrating the nucleases involved in DNA interstrand cross-link (ICL) repair. Cell Cycle. 2011;10:3999–4008. doi: 10.4161/cc.10.23.18385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang K., Moldovan G.L., D'Andrea A.D. RAD18-dependent recruitment of SNM1A to DNA repair complexes by a ubiquitin-binding zinc finger. J. Biol. Chem. 2010;285:19085–19091. doi: 10.1074/jbc.M109.100032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.