


Abstract
Riboswitches are RNA elements that regulate gene expression in response to their ligand. Although these regulations are thought to be performed without any aid of other factors, recent studies suggested the participation of protein factors such as transcriptional termination factor Rho and RNase in some riboswitch regulations. However, to what extent these protein factors contribute to the regulation was unclear. Here, we studied the regulatory mechanism of the flavin mononucleotide (FMN) riboswitch of Corynebacterium glutamicum which controls the expression of downstream ribM gene. Our results showed that this riboswitch downregulates both ribM mRNA and RibM protein levels in FMN-rich cells. Analysis of mRNA stability and chromatin immunoprecipitation–real-time PCR analysis targeting RNA polymerase suggested the involvement of the mRNA degradation and premature transcriptional termination in this regulation, respectively. Simultaneous disruption of RNase E/G and Rho function completely abolished the regulation at the mRNA level. Also, the regulation at the protein level was largely diminished. However, some FMN-dependent regulation at the protein level remained, suggesting the presence of other minor regulatory mechanisms. Altogether, we demonstrated for the first time that two protein factors, Rho and RNase E/G, play a central role in the riboswitch-mediated gene expression control.
INTRODUCTION
Riboswitches are non-coding RNA elements that control gene expression in response to cellular conditions. They are typically composed of two functional domains, a ligand-binding aptamer and downstream expression platform, which is involved in the genetic control of associated genes (1,2). Riboswitches are classified by their specific ligands; more than 10 small molecule compounds ranging from amino acids to coenzymes are known to act as ligands. A ligand binding to the aptamer during and/or after transcription leads to the formation of the ‘on’ or ‘off’ conformation of the expression platform region. In the past decade, many riboswitches have been discovered, and their molecular mechanisms of gene regulation have been extensively studied (3). From these studies, two general mechanisms, control of the translation initiation by an anti-Shine–Dalgarno (SD) sequence and control of the transcription termination by an intrinsic terminator, have been revealed. In the case of control at the translational level, ligand binding typically promotes the sequestration of SD sequences by anti-SD sequences, thus inhibiting ribosome binding to SD sequences. Without a ligand, an anti-anti-SD sequence in the aptamer domain base-pairs with an anti-SD sequence, resulting in the liberation of the SD sequence and translation of the mRNA. In the transcription termination regulation of riboswitches, a ligand bound to the aptamer typically facilitates the formation of a transcriptional terminator stem in the expression platform and promotes premature transcription termination. In the absence of ligand, the formation of a termination stem is inhibited by an anti-terminator sequence in the aptamer domain, and transcription elongates into the protein-coding region.
In contrast to these canonical mechanisms, recent studies suggested the participation of protein factors, such as transcription termination factor Rho and RNase, in some riboswitch-based gene regulations (4,5). In both cases, these riboswitches are equipped with anti-SD sequences; therefore, ligand-dependent regulation is thought to be performed at both the transcriptional and translational levels. Thus the regulation pathways of riboswitches are more complicated than had been thought. The redundant regulation pathways in one riboswitch make it difficult to dissect the relative contribution from either pathway in physiological condition. Therefore, to what extent these protein factors contribute to the regulation is unclear. Also, in some putative riboswitches, a lack of cis-acting elements such as anti-SD sequences or the pair of intrinsic terminator and anti-terminator sequences suggests that protein factors like Rho and RNase are involved in the function of these riboswitches (3). However, regulatory mechanism of these riboswitches has not been elucidated and reports that suggest the participation of protein factors in riboswitch regulation are limited in γ-proteobacteria such as Escherichia coli and Salmonella enterica (4,5). It is unclear if functional interplay between riboswitches and protein factors is widely distributed.
Corynebacterium glutamicum is a high-GC gram-positive bacterium that is widely used for the industrial production of amino acids, and it is expected to be a versatile biocatalyst for biofuels and biochemicals (6–10). In addition, this non-pathogenic soil bacterium is of interest as a model organism for the Corynebacterineae, an Actinomycetales suborder that includes medically important pathogens such as Corynebacterium diphtheriae and Mycobacterium tuberculosis (11–13). With respect to the riboswitches of C. glutamicum, the existence of flavin mononucleotide (FMN), thiamin pyrophosphate, cobalamin, and S-adenosyl methionine riboswitches has been predicted by comparative genomics or RNA-Seq analysis (14–16). Because the ligand compounds of these riboswitches are used as cofactors for many enzymes, establishing the fundamental characteristics of the metabolic regulation of these cofactors is of great interest.
Previously, we demonstrated that the expression of the C. glutamicum ribM gene, which encodes riboflavin transporter, is regulated by the FMN riboswitch (17). However, an experimental evaluation of the mechanism has not been performed. Here, we report that the FMN riboswitch of C. glutamicum mainly controls ribM gene expression at the mRNA level by regulating the action of two trans-acting protein factors, Rho and RNase E/G. Also, we assessed the relative contributions from either regulation pathways in physiological condition. Although FMN riboswitch of C. glutamicum has been thought to be controlled at translational level by anti-SD sequence from comparative genomics study (15), our results demonstrated that the regulation at the mRNA level by Rho and RNase E/G comprises the basis of this riboswitch regulation. These findings revealed the physiological significance of regulation at the mRNA level by protein factors in riboswitch-mediated gene expression control. Also, our observations raise a question about the control of many other riboswitches that have been thought to be controlled by cis-elements.
MATERIALS AND METHODS
Media and growth conditions
E. coli was grown at 37°C in Luria-Bertani medium while C. glutamicum was grown aerobically at 33°C with shaking at 180 rpm. Media were supplemented with 50-μg ml−1 antibiotics when necessary. For all analyses, 10-ml overnight pre-cultures of C. glutamicum in A medium (18) was washed twice with 10-ml minimal medium (BTM medium) (19), and the cells were resuspended in BTM medium for subsequent analysis. For all analysis, pre-cultured cells were inoculated in 100-ml BTM medium supplemented with 1% glucose and a limited (0.1 μM) or sufficient (1 μM) concentration of riboflavin and grown to mid-log phase.
Bacterial strains and plasmids
Strains and plasmids used in this study are listed in Supplementary Table S1. Oligonucleotide primers used in this study are listed in Supplementary Tables S2 and S3. A detailed procedure of construction of strains and plasmids is described in the SI MATERIAL AND METHODS (Supplementary data).
Quantification of cytoplasmic FMN level
Cytoplasmic FMN levels were quantified as described previously (17).
Total RNA purification
After cultivation, cultures were mixed with an equal amount of RNAprotect Bacteria Reagent (Qiagen) and incubated for 5 min at room temperature. Cells were collected by centrifugation (5 min, 6000 × g, 25°C) and stored at −80°C. Cells were resuspended in 10-mg/ml lysozyme/TE buffer and incubated for 1.5 h at 37°C. Total RNA was purified using NucleoSpin® RNA (MACHEREY-NAGEL) according to the manufacturer's instructions. Purified RNA was further treated with DNaseI (TaKaRa), ethanol precipitated and suspended in water.
Northern blot analysis
A detailed procedure of northern blot analysis is described in the SI MATERIAL AND METHODS (Supplementary data). Briefly, digoxigenin (DIG)-labeled anti-sense RNA probes targeting the RFN element (RFN probe) and ribM coding region (CDS probe) were used to detect ribM mRNA. Sense strands of ribM RNA of different lengths (157, 245 and 950 bases) were synthesized by in vitro transcription and were used as an RNA molecular weight marker. Total RNA purified from cultured cells and synthesized sense-strand RNA were analyzed on a 1.25% denaturing formaldehyde agarose gel followed by blotting to nylon membrane. After the hybridization and washing, hybridized RNA probes were detected using anti-DIG-AP, Fab fragments, and CDP-star (Roche Applied Science).
Quantitative reverse transcription-polymerase chain reaction
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) analysis was performed as described previously (20). Total RNA (20 ng) was used as a template for analysis of the ribM, aceA and rneG genes, and 0.4 ng was used for analysis of the 16S rRNA to generate cDNA and for the subsequent PCR. Each qRT-PCR mixture (20 μl) contained 500 nM each primer, 10-μl Power SYBR green PCR Master Mix (Applied Biosystems) or FastStart SYBR Green Master (Roche Applied Science), 8 units RNase inhibitor and 5 units murine leukemia virus reverse transcriptase (Applied Biosystems). The primers used in these reactions are listed in Supplementary Table S3. Reactions were performed using the ABI 7500 Fast Real-Time PCR System (Applied Biosystems) with the following cycle parameters: one cycle of 50°C for 30 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 30 s. The 16S rRNA result was used as an internal control.
Quantitative analysis of RibM protein expression level
Relative expression level of RibM protein was measured by western blot analysis as described previously (17). Strains harboring a modified ribM gene encoding C-terminally FLAG-tagged RibM protein were used. Briefly, cells were disrupted by vigorous shaking with zirconia/silica beads (BIO SPEC). After centrifugation to eliminate intact cells and debris, the supernatant was collected as the cell-free extract. Total protein in the soluble fraction of the cell-free extract was determined by the Bio-Rad protein assay (Bio-Rad Laboratories) using bovine serum albumin as a standard. Cell-free extracts were analyzed on 12% polyacrylamide gels. After transfer to Immobilon-P polyvinylidene fluoride membranes (Millipore), FLAG-tagged proteins were detected with anti-FLAG M2 monoclonal antibody (Sigma). Peroxidase-coupled anti-mouse IgG antibody was used as secondary antibody, and chemiluminescence reagents (GE Healthcare) were used to develop the blots.
Semi-quantitative analysis of the relative RibM protein level was conducted using cell-free extract of strain TK05 cultured with 1-μM riboflavin for 9 h as a standard (17). A calibration curve was obtained using standard samples containing different amounts of total soluble protein. The calibration curve was used to estimate the RibM-FLAG expression content of samples. Relative expression levels of RibM protein were calculated by normalizing values to that obtained in −FMN cells of the ΔribA ribM-FLAG strain (TK05).
Analysis of mRNA stability by the Rifampicin chase assay
After cultivation to mid-log phase, 1-ml culture was mixed with an equal amount of RNAprotect Bacteria Reagent (Qiagen) for a reference sample (0 min). Immediately after the sampling of the reference sample, Rifampicin (Wako) was added at 200 μg/ml, and samples were taken at 1, 2, 3, 4 and 6 min for strains with an intact rneG gene and at 1, 2, 4 and 8 min for the Δrneg ΔribA ribM-FLAG strain (TK12). After incubation for 5–15 min at room temperature, cells were collected by centrifugation (5 min, 6000 × g, 25°C) and stored at −80°C. The purification of total RNA and qRT-PCR analysis was performed as described above.
Chromatin immunoprecipitation-qPCR analysis targeting RNA polymerase
The detailed chromatin immunoprecipitation (ChIP) procedure is described in the SI MATERIAL AND METHODS (Supplementary data). Briefly, C. glutamicum strains harboring the rpoC gene with a FLAG-tagged coding sequence were grown to mid-log phase. Formaldehyde was added to cross-link chromosomal DNA and RNA polymerase (RNAP) complexes. Cross-linking was quenched by the addition of glycine, and cells were then collected. Cell disruption and genomic DNA shearing were conducted using a Bioruptor UCD-250 (Cosmo Bio). After the preparation of the cell-free extract, an aliquot was stored for the purification of total genomic DNA sample. FLAG-tagged RNAP cross-linked with chromosomal DNA fragments was immunoprecipitated using anti-FLAG M2 agarose (Sigma). The total genomic DNA sample and ChIP DNA sample were purified from the cell-free extract and immunoprecipitated fraction, respectively.
Reference DNA (4 ng) and 4-μl ChIP DNA were used as a template for qPCR analysis. Each qPCR mixture (20 μl) contained 500 nM each primer and 10-μl FastStart SYBR Green Master (Roche Applied Science). The primers used in these reactions are listed in Supplementary Table S3. Reactions were performed using the ABI 7500 Fast Real-Time PCR System (Applied Biosystems) with the following cycle parameters: one cycle of 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 30 s. Cycle threshold values were obtained using automated settings and were used to calculate the concentration factor and relative RNAP occupancies (see the legend of Figure 3).
Figure 3.
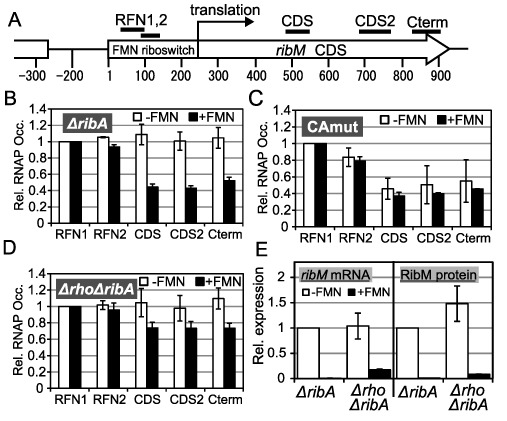
Involvement of premature transcription termination in the FMN riboswitch function. (A) Schematic of targets of ChIP-qPCR analysis. A chromosomal region around the ribM gene is represented. Regions coding the FMN riboswitch and ribM CDS are indicated as a box and an arrow, respectively. Numbers indicate the distance from the transcription start point of ribM, which is designated as ‘1’. Bars above the diagram are targets of ChIP-qPCR (RFN1; 26–107 nt, RFN2; 69–131 nt, CDS; 492–548 nt, CDS2; 692–790 nt, Cterm; 832–911 nt) (B–D) ChIP-qPCR analysis. Relative RNAP occupancies (Rel. RNAP Occ.) at each target region (horizontal axis) in −FMN (white bars) and +FMN (black bars) cells of the ΔribA rpoC-FLAG (TK13) (B), ΔribA CAmut rpoC-FLAG (TK14) (C) and Δrho ΔribA rpoC-FLAG (TK15) (D) strains. The values shown are means from at least three experiments and standard deviations. (E) Relative expression levels of ribM mRNA (left) and RibM protein (right) in −FMN cells (white bars) and +FMN cells (black bars) of the ΔribA ribM-FLAG (TK05) and Δrho ΔribA ribM-FLAG (TK17) strains. The strains are indicated along the horizontal axis (ΔribA, TK05; and Δrho ΔribA, TK17). The values shown are means from at least three experiments standard deviations.
Rho-function knock-down
The Rho-function knock-down system was developed as described in the Supplementary data. C. glutamicum strains carrying pCRC104 were grown to mid-log phase. During cultivation, the expression of mutant Rho protein was induced for 3 h by 1-mM Isopropyl β-D-1-thiogalactopyranoside (IPTG).
RESULTS
Expression of ribM mRNA is increased under FMN-depletion conditions
Previously, we demonstrated that the expression of RibM protein is upregulated under FMN-depleted conditions (17). Although translational regulation was predicted from comparative genomics study (15), a prediction of the secondary structure of the C. glutamicum FMN riboswitch suggested that a putative SD sequence forms a stem even in the ligand-free state (Figure 1A). This prompted us to examine whether the FMN riboswitch regulation is controlled by some mechanism other than SD sequestration. Therefore, we carried out northern blot analysis using RNA probes for the RFN element (an aptamer region of FMN riboswitch; RFN probe) or RibM protein-coding sequence (CDS probe) with RNA samples isolated from C. glutamicum strain TK02 (ΔribA) (Figure 1B). Because ribA gene encodes bifunctional GTP cyclohydrolase II/3,4-dihydroxy-2-butanone 4-phosphate synthase which catalyzes the first step of riboflavin synthesis, a ribA disruptant will not make FMN and it is therefore possible to better control cellular FMN levels in this mutant. The cultivation of ribA disruptant to mid-log phase in the presence of a limited (0.1 μM) concentration of riboflavin resulted in decreased cytoplasmic FMN levels without affecting growth. In contrast, supplementing riboflavin at a sufficient (1 μM) concentration rescued the cytoplasmic FMN level (Supplementary Table S4). Thus, in this study, ribA-deleted cells cultured with limited and sufficient riboflavin concentrations are called FMN-deprivation (−FMN) and FMN-rich (+FMN) cells, respectively. Interestingly, northern analysis showed that transcripts including the ribM coding region were markedly increased in −FMN cells (Figure 1B, filled arrowhead). This fragment is slightly longer than the control RNA and encompasses the nucleotides from the transcription start site to the C-terminus of the RibM protein-coding region (Figure 1B, lanes 3 and 9), indicating that the RNA includes the entire ribM CDS with an untranslated region. qRT-PCR was performed to compare the mRNA expression level using primers targeting the ribM coding region (Figure 1C and Supplementary Table S5). The expression of ribM mRNA increased ∼100-fold in −FMN cells. These results were consistent with the increase of RibM protein. Interestingly, in northern analysis, although we did not observe any signal with the CDS probe in +FMN cells (Figure 1B, lane 11), the RFN probe detected a short transcript that corresponds to the length of the RFN element (Figure 1B, lane 5, open arrowhead), indicating that the regulation of ribM gene expression is executed after transcription initiation.
Figure 1.
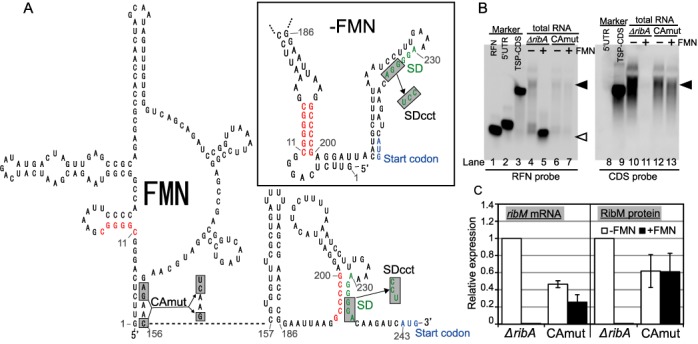
Predicted secondary structure of the FMN riboswitch and FMN-dependent regulation of ribM gene expression. (A) Prediction of the RNA secondary structure of the FMN riboswitch of the C. glutamicum ribM gene. The structures of the RFN element (1–156 nt) and expression platform (157–243 nt) in the presence of FMN were predicted based on reports from comparative genomics research (15) and RNA fold software (mfold version 3.2; http://www.frontend.bioinfo.rpi.edu/applications/mfold/cgibin/rna-form1.cgi), respectively. The structure in the absence of FMN (upper right box) was predicted by RNA fold software (mfold version 3.2). Nucleotides were numbered from the transcription start site, which is designated as ‘1’. The putative SD sequence and the translation start codon are indicated in green and blue, respectively. Putative anti-SD and anti-anti-SD sequences are indicated in red. Nucleotides that are mutated in the CAmut or SDcct strains are in gray boxes. (B) Northern blot analysis of ribM. Synthesized RNAs were used as a molecular weight marker (lanes 1–3, 8 and 9). RFN (156 bases), 5′ untranslated region (UTR) (243 bases) and TSP-CDS (935 bases) RNA markers were synthesized by T7 RNAP. Total RNAs from −FMN cells (lanes 4, 6, 10 and 12) and +FMN cells (lanes 5, 7, 11 and 13) of the ΔribA (lanes 4, 5, 10 and 11) and ΔribA CAmut (lanes 6, 7, 12 and 13) strains were applied. RNA probes targeting the RFN element (left panel) and CDS of ribM (right panel) were used. White arrowhead represents the transcript that corresponds to the length of the RFN element. Filled arrowhead represents the transcripts including the ribM coding region. (C) Relative expression levels of ribM mRNA (left) and RibM protein (right) in −FMN cells (white bars) and +FMN cells (black bars) of the ΔribA ribM-FLAG (TK05) and ΔribA CAmut ribM-FLAG (TK06) strains. The strains are indicated along the horizontal axis (ΔribA, TK05; and CAmut, TK06). The values shown are means from at least three experiments and standard deviations.
We previously found that mutations in the aptamer region of the FMN riboswitch lead to constitutive expression of RibM protein (17) and referred to these mutations as constitutive active mutation (CAmut). When the ΔribA CAmut strain (TK07) was analyzed, northern analysis showed the presence of similar signals between −FMN and +FMN cells; this pattern resembles that of −FMN cells of the intact FMN riboswitch strain (Figure 1B). Thus, regulation at the mRNA level was abolished by CAmut. The signal intensity was slightly decreased in the CAmut strain, and the decreased ribM mRNA level was also confirmed by qRT-PCR analysis (Figure 1C). The origin of this slight decrease is discussed below.
RNase E/G-dependent degradation of ribM mRNA controlled by the FMN riboswitch
Since the steady-state mRNA level is controlled by the balance between de novo synthesis and degradation, decreased mRNA level in +FMN cells suggests that synthesis and/or degradation efficiency is changed in response to cytoplasmic FMN level. To examine whether ribM mRNA degradation is enhanced in +FMN cells, ribM mRNA stability was measured in −FMN and +FMN cells of ΔribA ribM-FLAG strain (Figure 2A). As expected, the ribM mRNA decay was accelerated in +FMN cells. This suggests that the decrease of the steady-state ribM mRNA level in +FMN cells is attributed to the destabilization of ribM mRNA. To evaluate the role of RNase in the modulation of ribM mRNA stability, we attempted to disrupt the RNase gene. In E. coli, RNase E is a primary endoribonuclease in mRNA decay (21). In the C. glutamicum genome, there is one RNase E homolog (rneG), and its gene product, RNase E/G, has been reported to degrade mRNA (22). Therefore, we analyzed ribM mRNA stability in the rneG disruptant. The stability was markedly increased with the disruption of the rneG gene in +FMN conditions, while the effect of rneG disruption was very small in −FMN cells (Figure 2A). Consequently, FMN-dependent regulation of ribM mRNA stability was abolished in the rneG disruptant. The results in the ΔribA strain that neither the stability of known RNase E/G target (aceA mRNA) (22) nor the rneG mRNA level was modulated by cytoplasmic FMN level ruled out the possibility that RNase E/G activity was decreased in −FMN cells (Supplementary Figure S1). These results suggest that RNase E/G specifically degrades the FMN-bound form of ribM mRNA. Consistent with this, a 6-fold increase in the steady-state level of ribM mRNA was observed in rneG-deleted cells under +FMN conditions but not under −FMN conditions by qRT-PCR (Figure 2B and Supplementary Table S5). However, a large part of the FMN-dependent regulation of ribM mRNA remained. Indeed, in northern blot analysis, the signal in +FMN cells was undetectable even in the rneG disruptant (Figure 2C, lane 12). Unexpectedly, in contrast to the mRNA level, the RibM protein level in +FMN cells was not increased by rneG deletion. Instead, the RibM protein level in −FMN cells was slightly decreased with rneG deletion (Figure 2B). Thus, RNase E/G clearly takes part in the FMN riboswitch function. However, regulation at both the mRNA and protein levels remained in the rneG disruptant.
Figure 2.
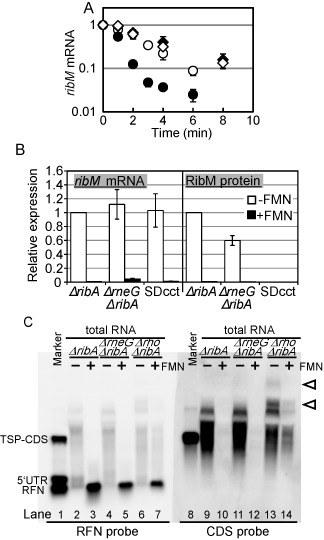
Involvement of RNase E/G in ribM mRNA stability regulation. (A) The ribM mRNA stability in +FMN cells (filled symbols) and −FMN cells (open symbols) of the ΔribA ribM-FLAG (circles) and ΔrneG ΔribA ribM-FLAG (diamonds) strains. The values shown are means from at least three experiments and standard deviations. (B) Relative expression levels of ribM mRNA (left) and RibM protein (right) in −FMN cells (white bars) and +FMN cells (black bars) of the ΔribA ribM-FLAG (TK05), ΔrneG ΔribA ribM-FLAG (TK09) and ΔribA SDcct ribM-FLAG (TK08) strains. The strains are indicated along the horizontal axis (ΔribA, TK05; ΔrneG ΔribA, TK09; and SDcct, TK08). The values shown are means from at least three experiments and standard deviations. (C) Northern blot analysis of ribM. Synthesized RNAs are used as a molecular weight marker (lanes 1 and 8). Total RNAs from −FMN cells (lanes 2, 4, 6, 9, 11 and 13) and +FMN cells (lane 3, 5, 7, 10, 12 and 14) of the ΔribA (lanes 2, 3, 9 and 10), ΔrneG ΔribA (lanes 4, 5, 11 and 12) and Δrho ΔribA (lanes 6, 7, 13 and14) strains were applied. Anti-sense RNA probes targeting the RFN element (left panel) and CDS of ribM (right panel) were used. Arrowheads represent the transcripts increased in the rho disruptant.
In E. coli, a translating ribosome lowers the accessibility of the RNA degradosome to the mRNA, resulting in increased mRNA stability (23). To examine whether the reduction in mRNA level in +FMN cells is a secondary effect of the decreased translation efficiency, a putative SD sequence of the ribM gene (AGGGGA) was mutated to AGGCCT to lower the ribosome affinity (SDcct mutant). As expected, the mutations markedly decreased the RibM protein level, which could not be detected even in −FMN cells (Figure 1C, less than 0.002-fold compared with the intact SD strain). However, in contrast to protein levels, ribM mRNA levels of the SDcct strain were nearly identical to those of the intact SD strain in both +FMN and −FMN cells (Figure 2B and Supplementary Table S5) and decay of ribM mRNA was only slightly accelerated by SD mutations under −FMN conditions (Supplementary Figure S2A). This slight difference in ribM mRNA stability is probably due to the stabilization effect by efficient translation which occurs only in intact SD strain. Absence of such difference in +FMN conditions suggests that translation is inhibited in +FMN cells of intact SD strain. These results imply the existence of FMN-dependent translation inhibition. In the CAmut strain, mRNA stability control was not observed, and the stability was almost the same as that of −FMN cells of the intact FMN riboswitch strain (Supplementary Figure S2B). These results are consistent with the hypothesis that the regulation of ribM mRNA stability depends on the FMN-dependent conformational change. Also, the result that CAmut did not destabilize ribM mRNA indicates that the decreased mRNA level in the CAmut strain (Figure 1B and C) is not due to the altered mRNA stability caused by the mutations.
Premature transcriptional termination occurs in +FMN cells
The results presented thus far imply the presence of ribM gene regulatory mechanisms other than mRNA degradation. In riboswitch-based gene regulation, the control of premature transcriptional termination is one of the general mechanisms. To test the possibility that premature transcriptional termination occurs in +FMN cells, we compared the transcription elongation efficiency of +FMN and −FMN cells. For this purpose, we measured the amount of ribM gene DNA fragment purified by ChIP with RNAP. Because the occupancy of RNAP on specific genome regions is correlated with the transcription efficiency of the region (24), this analysis enables us to clarify whether the transcription efficiency is modulated in ribM. Using the ΔribA rpoC-FLAG strain, DNA associated with RNAP in vivo was co-immunoprecipitated, and the amount of purified DNA was measured by quantitative PCR (qPCR). To monitor the local occupancy of RNAP within the ribM gene, five primer pairs were designed (Figure 3A). The results were normalized to the RFN1 region to compare the transcription elongation efficiency. In −FMN cells, RNAP occupancies were constant throughout the entire coding region of ribM, indicating that most transcription elongated to the end of the ribM gene, as expected. In contrast, in +FMN cells, RNAP occupancies in the coding region decreased to 40–60% that of the RFN1 region (Figure 3B). These results strongly indicate that premature transcription termination occurs selectively in +FMN cells. In the ΔribA CAmut rpoC-FLAG strain (TK14), we expected that RNAP occupancies in the coding region would not be decreased even in +FMN cells and that FMN-dependent regulation of RNAP occupancies would be abolished because CAmut fixes the conformation of the FMN riboswitch in the ‘on’ state. Unexpectedly, RNAP occupancies in the CDS decreased in −FMN cells of the CAmut strain. These results suggest that mutations in the RFN element may raise some termination sites or transcription pausing sites that enhance termination events, which leads to the decreased expression of ribM mRNA in CAmut strain (Figure 1C). However, we also observed FMN-dependent regulation of RNAP occupancies being abolished by CAmut (Figure 3C). These results suggest that the FMN-dependent response of RNAP elongation depends on an FMN-dependent conformational change of the FMN riboswitch.
Rho-factor contributes to the FMN riboswitch-mediated control of transcription elongation
To test the possibility that Rho-factor is involved in the premature transcriptional termination observed in ribM, we performed ChIP-qPCR analysis in the rho disruptant (Figure 3D). In −FMN cells, RNAP occupancies in the coding region were similar to those in the RFN1 region, as observed in the rho-intact strain (Figure 3D). However, in contrast to the rho-intact strain, deletion of the rho gene resulted in RNAP occupancies in the protein-coding region that were more than 70% of those of the RFN1 region in +FMN conditions (Figure 3D). Thus, the FMN-dependent decrease of RNAP occupancies was partially relieved by deletion of the rho gene. This result strongly indicates that Rho-factor contributes to the premature transcription termination of ribM in +FMN cells.
Then we assessed the effect of rho disruption on ribM gene expression. In −FMN cells of the rho gene disruptant, in addition to the ribM transcript detected in the ΔribA strain, longer transcripts were detected (Figure 2C, lanes 13 and 14, open arrowheads). This suggests that Rho-dependent transcription termination occurs at the end of the ribM gene and that the disruption of the rho gene diminished termination at the proper site. Interestingly, ribM transcripts were also detected in +FMN cells, indicating that expression of ribM mRNA was increased by rho disruption in +FMN conditions (Figure 2C, lane 14). Quantitative analysis showed that, in +FMN cells, disruption of the rho gene increased both the mRNA and protein levels more than 10-fold, while in −FMN cells, no such increase was observed (Figure 3E and Supplementary Table S5). To test the possibility that rho gene expression is increased in +FMN cells, leading to enhanced transcription termination, we examined the expression level of Rho protein (Supplementary Figure S3). Western blot analysis showed that the expression level of Rho protein decreased rather than increased in +FMN cells (Supplementary Figure S3, lanes 3 and 4). Although the cause of this decrease is unclear, this result rules out the possibility. Taken together, these results indicate that Rho selectively recognizes the FMN-bound form of the FMN riboswitch and terminates transcription.
Inhibition of Rho function in the ΔrneG strain completely abolished the FMN-dependent regulation of ribM mRNA
The results presented thus far indicated that both RNase E/G and Rho contribute to FMN riboswitch-dependent regulation of ribM gene expression. These observations prompted us to construct double deletion mutant of rneG and rho. However, we could not obtain such a strain, probably owing to a synthetic lethal effect. Therefore, we developed a Rho-function inhibition system in which Rho function is inhibited by overexpressing a dominant-negative mutant of Rho protein (see SI and Supplementary Figure S3). Although the effect of the knock-down of Rho function (RhoKD) on ribM gene expression is smaller to that of rho gene deletion (Figures 3E, 4B and Supplementary Table S5), signals in northern blot analysis of +FMN cells were detected in the RhoKD strain (Figure 4A, lanes 10 and 12). In accordance, qRT-PCR analysis showed that RhoKD had 10-fold increased ribM mRNA expression in +FMN cells but not in −FMN cells (Figure 4B and Supplementary Table S5). Similar effects were also observed in RibM protein expression levels. Then, we tested the effect of rneG gene deletion in the RhoKD background. Deletion of the rneG gene diminished the differences in the patterns of northern blot signals between +FMN and −FMN cells. Although the signal intensities seems slightly stronger in +FMN cells than in −FMN cells (ratio of signal intensity of whole lane was 1:1.5 (lane 13:lane 14)), qRT-PCR analysis showed that simultaneous disruption of the rneG gene and Rho function almost completely diminished the FMN-dependent regulation of ribM mRNA levels (Figure 4B). These results strongly indicate that FMN-dependent regulation of ribM mRNA was mainly controlled by RNase E/G and Rho. In addition to these observations, northern blot analysis revealed that a signal corresponding to the size of the ribM gene was decreased in the ΔrneG ΔribA RhoKD strain (Figure 4A, gray arrowhead). Instead, a strong signal was observed at ∼3 kb (Figure 4A, filled arrowhead). Although the signal intensity was weak, a band with similar mobility was observed in −FMN cells of the Δrho ΔribA strain (Figure 2C, lane 13). These results suggest that longer transcripts produced under Rho-function-deficient conditions were processed by RNase E/G. With respect to the short fragment corresponding to the size of the RFN element observed in +FMN cells (Figure 4A, open arrowhead); although the signal was markedly diminished by the simultaneous disruption of rneG and Rho function, a band with the same mobility was detected in the ΔrneG ΔribA RhoKD strain. Some exoribonucleases may work after FMN-dependent premature transcription termination by Rho and/or FMN-dependent cleavage by RNase E/G.
Figure 4.
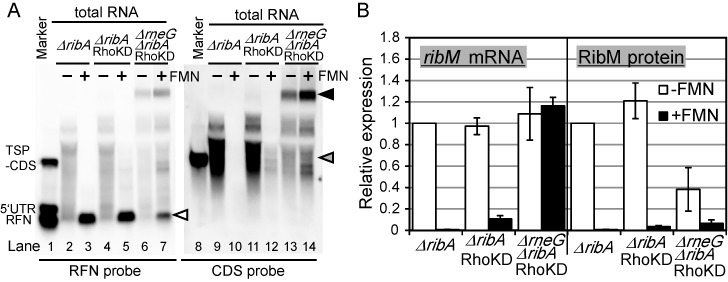
Effects of the double disruption of Rho function and the rneG gene on ribM expression. (A) Northern blot analysis of ribM. Synthesized RNAs were used as a molecular weight marker (lanes 1 and 8). Total RNAs from −FMN cells (lanes 2, 4, 6, 9, 11 and 13) and +FMN cells (lane 3, 5, 7, 10, 12 and 14) of the ΔribA with pCRB12iP (lanes 2, 3, 9 and 10), ΔribA with pCRB12iP-Rho (TriMut) (lanes 4, 5, 11 and 12) and ΔrneG ΔribA with pCRB12iP-Rho (TriMut) (lanes 6, 7, 13 and14) strains were applied. The strains are indicated above the lanes (ΔribA, ΔribA with pCRB12iP; ΔribA RhoKD, ΔribA with pCRB12iP-Rho (TriMut); and ΔrneG ΔribA RhoKD, ΔrneG ΔribA with pCRB12iP-Rho (TriMut)). Anti-sense RNA probes targeting the RFN element (left panel) and CDS of ribM (right panel) were used. White arrowhead represents the transcript that corresponds to the length of RFN element. Gray and black arrowheads represent the transcripts differentially expressed by deletion of the rneG gene in the background of RhoKD. (B) Relative expression levels of ribM mRNA (left) and RibM protein (right) in −FMN cells (white bars) and +FMN cells (black bars) of the ΔribA ribM-FLAG with pCRB12iP, ΔribA ribM-FLAG with pCRB12iP-Rho (Mut) and ΔrneG ΔribA ribM-FLAG with pCRB12iP-Rho (Mut) strains. The strains are indicated along the horizontal axis (ΔribA, ΔribA ribM-FLAG with pCRB12iP; ΔribA RhoKD, ΔribA ribM-FLAG with pCRB12iP-Rho (Mut); and ΔrneG ΔribA RhoKD, ΔrneG ΔribA ribM-FLAG with pCRB12iP-Rho (Mut)). The values shown are means from at least three experiments and standard deviations.
With respect to the RibM protein level in +FMN cells, as expected, deletion of the rneG gene increased RibM expression about 2-fold in the RhoKD background (Figure 4B and Supplementary Table S5). This result indicates that RNase E/G-dependent degradation of ribM mRNA contributed to the repression of RibM protein expression in +FMN cells. In contrast, in −FMN cells, deletion of the rneG gene in the RhoKD background decreased RibM protein expression to about one-third the level in rneG-intact cells (Figure 4B). This result is similar to that observed in strains harboring intact Rho function (Figure 2B). Considering that ribM mRNA levels in −FMN cells were not changed as a result of rneG deletion, the translational efficiency may be decreased in ΔrneG strains. Although further investigations are required to uncover the role of RNase E/G, differences in the RibM protein expression level in +FMN and −FMN conditions were considerably reduced with the simultaneous disruption of Rho function and the rneG gene, indicating these two factors plays a key role in the FMN riboswitch function in C. glutamicum. Besides these mechanisms regulating the mRNA level, minor FMN-dependent regulation of the RibM protein level remained even in the absence of regulation at the mRNA level, implying the existence of translational regulation of this riboswitch (Figure 4B).
DISCUSSION
With respect to the relationship between the translation efficiency and mRNA stability, many studies reported (23) that ribosome association with mRNA for translation prevents the mRNA from RNase attack, resulting in increased stability. Conversely, when translation is prematurely terminated by a non-sense mutation or amino-acid starvation, the untranslated region of RNA is degraded more rapidly than that being translated. Similar phenomena have been observed in some riboswitches that operate at the translational level. In the vitamin B12 riboswitch of the E. coli btuB gene, sequestration of the SD sequence occurs in response to its ligand, adenosylcobalamin (25). A precise analysis using a reporter system demonstrated that the inhibition of translation initiation by ligand binding is accompanied by a decrease in mRNA stability. Thus, ribosome-free RNA is subjected to ribonuclease-dependent degradation (25,26). A similar consequence was observed in the THI-box riboswitch of thiM (4,27). In both cases, it is thought that the modulation of mRNA stability is a secondary effect of the translational regulation.
In this study, we demonstrated that the FMN riboswitch of C. glutamicum ribM also decreased the stability of ribM mRNA in an FMN-dependent manner. However, it should be noted that this riboswitch is essentially different from that of btuB and thiM because the FMN-dependent regulation of ribM mRNA stability occurred even in strains with defective translation (Supplementary Figure S2A). Although the inhibition of translation slightly decreased the ribM mRNA stability in −FMN cells, the effect was too small to affect the steady-state level of ribM mRNA (Figure 2B). Analysis using an rneG disruptant showed that the deletion of rneG eliminated the FMN-dependent regulation of ribM mRNA stability, demonstrating the importance of RNase E/G in the regulation of mRNA stability (Figure 2A). From these observations, we conclude that the FMN riboswitch of C. glutamicum ribM directly controls the stability of mRNA in response to its ligand. This situation is similar to that of the lysine riboswitch of E. coli lysC. It has been recently reported that, in addition to translational regulation, the lysC riboswitch directly controls mRNA stability by altering the accessibility to the degradosome in response to lysine (4). Thus, the ribM and lysC riboswitches may utilize a similar mechanism to control mRNA stability. However, the effect of mRNA stability control on the regulation of the protein expression level is different between the ribM and lysC riboswitches. In the lysC riboswitch, the deletion of the RNase E cleavage site had only minor effects on the ligand-dependent regulation of translational fusion. In contrast, in the ribM riboswitch, the deletion of rneG in RhoKD conditions impaired the regulation not only of mRNA but also of RibM protein (Figure 4B). This result revealed the importance of ribM mRNA stability control by RNase E/G in the regulation of RibM protein expression.
The control of transcription termination is one of the general mechanisms of many of the riboswitches discovered to date. A previous comparative genomics study predicted that ∼40% of riboswitches analyzed use ligand-dependent sequestration of an intrinsic transcription terminator to regulate downstream gene expression (28). In addition to these predictions, the experimental demonstration of ligand-dependent transcription termination has also been reported for many riboswitches since their discovery (29–31). These analyses were performed using an in vitro transcription assay, and they demonstrated that ligand binding facilitates premature transcription termination. In this study, we performed RNAP-ChIP-qPCR analysis instead of in vitro transcription analysis to examine if premature transcription termination occurs in ribM (Figure 3B-D). Although this analysis cannot rule out the possibility that some factors other than the riboswitch or ligand are involved, this method has the advantage that the results obtained reflect the transcription efficiency in vivo. Using this analysis, we succeeded in demonstrating that the FMN riboswitch induces transcription termination in response to its ligand more directly than has been reported.
We also confirmed that Rho is involved in this termination regulation under +FMN conditions. The involvement of Rho-factor in the riboswitch function was suggested recently (5). Using in vitro transcription analysis, the authors clearly revealed that the magnesium riboswitch of S. enterica and the FMN riboswitch of E. coli control the Rho-dependent termination independent of translation. They also tested the effect of Rho inhibition on reporter gene expression in vivo using transcriptional fusion construct. However, the physiological significance of Rho-dependent termination on target gene expression control remains obscure because it is possible that these riboswitches primarily control translation initiation to regulate downstream gene expression (15,32). In the present study, we demonstrated that disrupting the rho gene increased the transcription elongation efficiency under +FMN conditions (Figure 3B and D). We also demonstrated that disruption of Rho function increased ribM mRNA level only under +FMN conditions (Figures 3E and 4B). Together with the result that Rho expression was not increased in +FMN cells (Supplementary Figure S3), these results indicate that Rho monitors the FMN-dependent conformational change in the FMN riboswitch and selectively terminates transcription in the presence of FMN. Furthermore, our results show that disruption of Rho function significantly increases the RibM protein level in addition to the ribM mRNA level. These results strongly indicate that Rho-dependent termination practically participates in FMN riboswitch-based ribM gene expression control. Taken together, this is the first assessment of the physiological significance of Rho-dependent termination in gene expression control by a riboswitch.
Although our results suggested that both RNase E/G and Rho monitor the FMN-dependent conformational change of FMN riboswitch, the precise locations of Rho-dependent termination site(s) and RNase E/G cleavage site(s) were not determined. Probably these sites are located in expression platform and would be exposed in the presence of FMN as reported in other riboswitches previously (4,5). Structure-probing assay together with in vitro transcription/cleavage assay using various mutant DNA templates or RNAs is required to determine the accurate site(s).
A previous comparative genomics study predicted that most riboswitches of bacterial groups other than Firmicutes and Fusobacterium are controlled at the translational level (28). However, recent findings in γ-proteobacteria that suggested the participation of protein factors such as RNase or transcription termination factor Rho in these riboswitch functions raise the question about the generalization of these situations (4,5). In this study, we demonstrated that the FMN riboswitch of C. glutamicum ribM mainly controls ribM mRNA by using Rho and RNase E/G to regulate RibM protein expression. This observation not only revealed that the interplay between the riboswitch and protein factors is not confined to γ-proteobacteria but also raised a question about the control of many other riboswitches that have been thought to be controlled by cis-elements. In addition, recent studies in Sinorhizobium meliloti (α-proteobacteria), Treponema denticola (spirochaeta) and Streptomyces griseus (actinobacteria) have shown that riboswitches lacking an obvious intrinsic transcription terminator sequence seem to be controlled at the mRNA level (33–35). Our observations suggest that these riboswitches would utilize some protein factors as the FMN riboswitch of C. glutamicum ribM. Taken together, the participation of protein factors in riboswitch function would be a widely distributed mechanism.
One interesting observation is that the FMN riboswitch of C. glutamicum ribM utilizes both Rho and RNase E/G for its regulation. This is the first example of a riboswitch that employs two trans-acting protein factors acting in mRNA metabolism. Both Rho and RNase E/G act in mRNA metabolism but in different pathways. Although FMN is a key effector, the use of these protein factors would render the FMN riboswitch responsive to physiological states other than the FMN concentration. For example, in E. coli, it is speculated that Rho activity is unlocked from inhibition by the RNA chaperone Hfq in response to amino acid starvation via RelA protein-dependent Hfq oligomerization (36–38). Although these observations were made in E. coli and C. glutamicum does not have a homolog of hfq, we can speculate that some signals cause Rho activity to be inhibited, leading to an increased expression of RibM protein in C. glutamicum. On the other hand, our results of the rho disruptant showed FMN-dependent regulation of ribM expression probably owing to RNase E/G and translational regulation (Figure 3E). Thus, redundancy in regulatory pathways might not only expand the diversity of effectors but also ensure the regulation under various growth conditions. Furthermore, it should be noted that the dynamic range of the ribM FMN riboswitch gene regulation is relatively large compared with other riboswitches (Supplementary Table S6). By using two factors that act in different pathways, this riboswitch has a relatively large dynamic range of regulation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: Scientific research fund of Research Institute of Innovative Technology for the Earth.
Conflict of interest statement. None declared.
REFERENCES
- 1.Roth A., Breaker R.R. The structural and functional diversity of metabolite-binding riboswitches. Annu. Rev. Biochem. 2009;78:305–334. doi: 10.1146/annurev.biochem.78.070507.135656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henkin T.M. Riboswitch RNAs: using RNA to sense cellular metabolism. Genes Dev. 2008;22:3383–3390. doi: 10.1101/gad.1747308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Serganov A., Nudler E. A decade of riboswitches. Cell. 2013;152:17–24. doi: 10.1016/j.cell.2012.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caron M.P., Bastet L., Lussier A., Simoneau-Roy M., Masse E., Lafontaine D.A. Dual-acting riboswitch control of translation initiation and mRNA decay. Proc. Natl Acad. Sci. U.S.A. 2012;109:E3444–E3453. doi: 10.1073/pnas.1214024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hollands K., Proshkin S., Sklyarova S., Epshtein V., Mironov A., Nudler E., Groisman E.A. Riboswitch control of Rho-dependent transcription termination. Proc. Natl Acad. Sci. U.S.A. 2012;109:5376–5381. doi: 10.1073/pnas.1112211109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hermann T. Industrial production of amino acids by coryneform bacteria. J. Biotechnol. 2003;104:155–172. doi: 10.1016/s0168-1656(03)00149-4. [DOI] [PubMed] [Google Scholar]
- 7.Inui M., Kawaguchi H., Murakami S., Vertès A.A., Yukawa H. Metabolic engineering of Corynebacterium glutamicum for fuel ethanol production under oxygen-deprivation conditions. J. Mol. Microbiol. Biotechnol. 2004;8:243–254. doi: 10.1159/000086705. [DOI] [PubMed] [Google Scholar]
- 8.Okino S., Noburyu R., Suda M., Jojima T., Inui M., Yukawa H. An efficient succinic acid production process in a metabolically engineered Corynebacterium glutamicum strain. Appl. Microbiol. Biotechnol. 2008;81:459–464. doi: 10.1007/s00253-008-1668-y. [DOI] [PubMed] [Google Scholar]
- 9.Inui M., Murakami S., Okino S., Kawaguchi H., Vertès A.A., Yukawa H. Metabolic analysis of Corynebacterium glutamicum during lactate and succinate productions under oxygen deprivation conditions. J. Mol. Microbiol. Biotechnol. 2004;7:182–196. doi: 10.1159/000079827. [DOI] [PubMed] [Google Scholar]
- 10.Smith K.M., Cho K.M., Liao J.C. Engineering Corynebacterium glutamicum for isobutanol production. Appl. Microbiol. Biotechnol. 2010;87:1045–1055. doi: 10.1007/s00253-010-2522-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baumbach J. CoryneRegNet 4.0—a reference database for corynebacterial gene regulatory networks. BMC Bioinformatics. 2007;8:429. doi: 10.1186/1471-2105-8-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brune I., Brinkrolf K., Kalinowski J., Puhler A., Tauch A. The individual and common repertoire of DNA-binding transcriptional regulators of Corynebacterium glutamicum, Corynebacterium efficiens, Corynebacterium diphtheriae and Corynebacterium jeikeium deduced from the complete genome sequences. BMC genomics. 2005;6:86. doi: 10.1186/1471-2164-6-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liebl W. Corynebacterium Taxonomy. In: Eggeling L, Bott M, editors. Handbook of Corynebacterium glutamicum. Boca Raton, FL: CRC Press; 2005. pp. 9–34. [Google Scholar]
- 14.Rodionov D.A., Vitreschak A.G., Mironov A.A., Gelfand M.S. Comparative genomics of thiamin biosynthesis in procaryotes. New genes and regulatory mechanisms. J. Biol. Chem. 2002;277:48949–48959. doi: 10.1074/jbc.M208965200. [DOI] [PubMed] [Google Scholar]
- 15.Vitreschak A.G., Rodionov D.A., Mironov A.A., Gelfand M.S. Regulation of riboflavin biosynthesis and transport genes in bacteria by transcriptional and translational attenuation. Nucleic Acids Res. 2002;30:3141–3151. doi: 10.1093/nar/gkf433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mentz A., Neshat A., Pfeifer-Sancar K., Puhler A., Ruckert C., Kalinowski J. Comprehensive discovery and characterization of small RNAs in Corynebacterium glutamicum ATCC 13032. BMC Genomics. 2013;14:714. doi: 10.1186/1471-2164-14-714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takemoto N., Tanaka Y., Inui M., Yukawa H. The physiological role of riboflavin transporter and involvement of FMN-riboswitch in its gene expression in Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 2014;98:4159–4168. doi: 10.1007/s00253-014-5570-5. [DOI] [PubMed] [Google Scholar]
- 18.Inui M., Suda M., Okino S., Nonaka H., Puskas L.G., Vertès A.A., Yukawa H. Transcriptional profiling of Corynebacterium glutamicum metabolism during organic acid production under oxygen deprivation conditions. Microbiology. 2007;153:2491–2504. doi: 10.1099/mic.0.2006/005587-0. [DOI] [PubMed] [Google Scholar]
- 19.Teramoto H., Suda M., Inui M., Yukawa H. Regulation of the expression of genes involved in NAD de novo biosynthesis in Corynebacterium glutamicum. Appl. Environ. Microbiol. 2010;76:5488–5495. doi: 10.1128/AEM.00906-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanaka Y., Teramoto H., Inui M., Yukawa H. Translation efficiency of antiterminator proteins is a determinant for the difference in glucose repression of two beta-glucoside phosphotransferase system gene clusters in Corynebacterium glutamicum R. J. Bacteriol. 2011;193:349–357. doi: 10.1128/JB.01123-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kushner S.R. mRNA decay in prokaryotes and eukaryotes: different approaches to a similar problem. IUBMB Life. 2004;56:585–594. doi: 10.1080/15216540400022441. [DOI] [PubMed] [Google Scholar]
- 22.Maeda T., Wachi M. 3′ untranslated region-dependent degradation of the aceA mRNA, encoding the glyoxylate cycle enzyme isocitrate lyase, by RNase E/G in Corynebacterium glutamicum. Appl. Environ. Microbiol. 2012;78:8753–8761. doi: 10.1128/AEM.02304-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deana A., Belasco J.G. Lost in translation: the influence of ribosomes on bacterial mRNA decay. Genes Dev. 2005;19:2526–2533. doi: 10.1101/gad.1348805. [DOI] [PubMed] [Google Scholar]
- 24.Uplekar S., Rougemont J., Cole S.T., Sala C. High-resolution transcriptome and genome-wide dynamics of RNA polymerase and NusA in Mycobacterium tuberculosis. Nucleic Acids Res. 2013;41:961–977. doi: 10.1093/nar/gks1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nou X., Kadner R.J. Adenosylcobalamin inhibits ribosome binding to btuB RNA. Proc. Natl Acad. Sci. U.S.A. 2000;97:7190–7195. doi: 10.1073/pnas.130013897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nou X., Kadner R.J. Coupled changes in translation and transcription during cobalamin-dependent regulation of btuB expression in Escherichia coli. J. Bacteriol. 1998;180:6719–6728. doi: 10.1128/jb.180.24.6719-6728.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ontiveros-Palacios N., Smith A.M., Grundy F.J., Soberon M., Henkin T.M., Miranda-Rios J. Molecular basis of gene regulation by the THI-box riboswitch. Mol. Microbiol. 2008;67:793–803. doi: 10.1111/j.1365-2958.2007.06088.x. [DOI] [PubMed] [Google Scholar]
- 28.Barrick J.E., Breaker R.R. The distributions, mechanisms, and structures of metabolite-binding riboswitches. Genome Biol. 2007;8:R239. doi: 10.1186/gb-2007-8-11-r239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winkler W.C., Cohen-Chalamish S., Breaker R.R. An mRNA structure that controls gene expression by binding FMN. Proc. Natl Acad. Sci. U.S.A. 2002;99:15908–15913. doi: 10.1073/pnas.212628899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Epshtein V., Mironov A.S., Nudler E. The riboswitch-mediated control of sulfur metabolism in bacteria. Proc. Natl Acad. Sci. U.S.A. 2003;100:5052–5056. doi: 10.1073/pnas.0531307100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sudarsan N., Wickiser J.K., Nakamura S., Ebert M.S., Breaker R.R. An mRNA structure in bacteria that controls gene expression by binding lysine. Genes Dev. 2003;17:2688–2697. doi: 10.1101/gad.1140003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Groisman E.A., Cromie M.J., Shi Y., Latifi T. A Mg2+-responding RNA that controls the expression of a Mg2+ transporter. Cold Spring Harb. Symp. Quant. Biol. 2006;71:251–258. doi: 10.1101/sqb.2006.71.005. [DOI] [PubMed] [Google Scholar]
- 33.Tezuka T., Ohnishi Y. Two glycine riboswitches activate the glycine cleavage system essential for glycine detoxification in Streptomyces griseus. J. Bacteriol. 2014;196:1369–1376. doi: 10.1128/JB.01480-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bian J., Shen H., Tu Y., Yu A., Li C. The riboswitch regulates a thiamine pyrophosphate ABC transporter of the oral spirochete Treponema denticola. J. Bacteriol. 2011;193:3912–3922. doi: 10.1128/JB.00386-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng J., Poduska B., Morton R.A., Finan T.M. An ABC-type cobalt transport system is essential for growth of Sinorhizobium meliloti at trace metal concentrations. J. Bacteriol. 2011;193:4405–4416. doi: 10.1128/JB.05045-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Argaman L., Elgrably-Weiss M., Hershko T., Vogel J., Altuvia S. RelA protein stimulates the activity of RyhB small RNA by acting on RNA-binding protein Hfq. Proc. Natl Acad. Sci. U.S.A. 2012;109:4621–4626. doi: 10.1073/pnas.1113113109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bossi L., Schwartz A., Guillemardet B., Boudvillain M., Figueroa-Bossi N. A role for Rho-dependent polarity in gene regulation by a noncoding small RNA. Genes Dev. 2012;26:1864–1873. doi: 10.1101/gad.195412.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rabhi M., Espeli O., Schwartz A., Cayrol B., Rahmouni A.R., Arluison V., Boudvillain M. The Sm-like RNA chaperone Hfq mediates transcription antitermination at Rho-dependent terminators. EMBO J. 2011;30:2805–2816. doi: 10.1038/emboj.2011.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.