


Abstract
DNA replication initiation is mediated across all domains of life by initiator proteins oligomerizing at replication origins. Recently, it was shown that initiators can directly bind single-stranded DNA (ssDNA) and thus might enhance origin melting. In this study, we used single-molecule fluorescence assays to probe the ssDNA binding mechanism of the replication initiator DnaA. Our experiments revealed that DnaA forms a dynamic filament on ssDNA in 3′ to 5′ directionality in the presence of ATP and analogs. After nucleation with a three-monomer seed, monomers dynamically assemble and disassemble one monomer at a time at the 5′ end, each monomer binding three nucleotides of ssDNA. The addition of adjacent double-stranded DnaA binding sites stabilized the DnaA filament on ssDNA. Our results extend the current models of origin melting via DnaA ssDNA interaction.
INTRODUCTION
Initiation of DNA replication is an essential step in cell proliferation of all organisms. Defects in the start of replication can be correlated to DNA damage as well as genetic instability (1–3). DNA replication is initiated across all domains of life by so-called initiator proteins interacting with replication origin sites and coordinating the replisome assembly (4–6). For bacterial chromosomes, DnaA is the initiator protein, and Orc1 and Cdc6 are eukaryotic homologs (1,6–8). Typical bacterial chromosomes contain a single replication origin (oriC) that consists of two functional segments. The first one is a duplex DNA binding region, which contains several conserved repeats, so-called DnaA boxes that are recognized by DnaA and lead to DnaA assembly. The second segment is an AT-rich DNA-unwinding element (DUE), in which strands separate to allow replisome assembly (1,9). In Escherichia coli, strong DnaA boxes (regions R1, R2 and R4) are recognized and bound by ATP-DnaA or ADP-DnaA, while other intermediate (R3 and R5) and low (I sites and τ sites) affinity sites are preferentially occupied by ATP-DnaA protomers (1,10–14). Cooperative, adenosine triphosphate (ATP)-dependent assembly of DnaA monomers on the duplex DNA binding sites results in the formation of a nucleoprotein complex and induces with a yet unknown mechanism of the melting of the DUE (6,13).
Previous studies suggest two mechanisms, based either on torsional strain of double-stranded DNA (dsDNA) or DnaA binding to single-stranded DNA (ssDNA) (1). In the first mechanism, the DnaA–oriC complex adopts a positive wrap that may destabilize the origin of replication by introducing torsional strain into the DUE through compensatory negative supercoiling, which is known to facilitate DNA melting (15,16). The second mechanism assumes, ATP-DnaA assists DUE melting through a direct interaction between the AAA+ domain of DnaA and single-stranded segments within the DUE capturing breathing of the DUE (6,9,17). A combination of both mechanisms could increase the efficiency of the melting process.
In Aquifex aeolicus, the helicase loader DnaC, a close paralog of DnaA, was shown to interact with DnaA and, thus, it was hypothesized that DnaC docks to ATP-DnaA and loads one hexameric helicase DnaB on the top strand of the single-stranded DUE. Loading of the other hexameric helicase on the bottom strand of the DUE is supposed to be mediated through the direct interaction between DnaA and DnaB (18,19). These highly coordinated helicase loading mechanisms require a well-coordinated DnaA assembly. In the presence of ATP, E. coli DnaA shows a preference for binding to the T-rich instead of the A-rich strand in the DUE, potentially an indication that DnaA has a preferential binding directionality 3′ to 5′ (9,17,20). Recently, a truncated A. aeolicus DnaA, a close homolog to E. coli DnaA, was co-crystallized with ssDNA and showed that DnaA binds to ssDNA in nucleotide triplets and thereby stretches the ssDNA filament (6). In combination with biochemical assays, the binding of DnaA to ssDNA has been observed; however, the ssDNA binding directionality of DnaA filament formation remains unknown.
In this study, we investigated the DnaA assembly dynamics on ssDNA. Using single-molecule Förster Resonance Energy Transfer (smFRET) and Protein Induced Fluorescence Enhancement (smPIFE), we found that DnaA assembles in filaments on ssDNA in the 3′ to 5′ direction one monomer at a time. In agreement with the structural data, we found that a DnaA monomer binds to three nucleotides (6). The DnaA filaments are highly dynamic with a kon ≈ 0.1 s−1μM−1 and a koff ≈ 0.5 s−1. The initial filament requires a nucleation seed of approximately three DnaA monomers binding simultaneously within our time resolution. Finally, we found that DnaA boxes stabilize the assembly of DnaA on ssDNA and thus allow a longer filament length. Our findings of the ssDNA binding mechanism of DnaA open a new view to a better understanding of how the origin of replication can be melted and kept open via the dynamic interaction between DnaA and ssDNA.
MATERIALS AND METHODS
Expression and purification of DnaA
The A. aeolicus initiator DnaA (residues 77–399, AaDnaA) was polymerase chain reaction amplified from the genome and cloned into E. coli expression vector pET-28a, generating a C-terminal hexahistidine-tagged construct. The construct was confirmed by sequencing. AaDnaA was overexpressed in E. coli BL21-RIL cells and purified by affinity chromatography. For further purification, the His-trap elution was purified on a Superdex 75 size exclusion column in size exclusion buffer (50-mM HEPES/KOH, pH 7.5, 500-mM KCl, 10-mM MgAc, 1-mM DTT and 2-mM ethylenediaminetetraacetic acid), concentrated, flash frozen and stored at −80°C.
DNA constructs
ssDNA molecules were purchased from PURIMEX, eurofins mwg, biomers, or IBA. The DNA molecules with biotin or dye modification at the ends are labeled during the DNA synthesis. Cy3 or Cy5 (NHS-ester from GE Healthcare) were attached to Amino-C6-dT base, avoiding DNA backbone modifications to reduce the disturbance on DnaA–DNA interactions, followed by purification on a reverse phase C18 column (Phenomenex) with high-performance liquid chromatography. The sequences of all DNA strands are listed in Supplementary Table S3.
Reaction conditions
Biotinylated DNA was immobilized on a polyethylene glycol (PEG)-passivated quartz surface via biotin-NeutrAvidin interaction. 10–100-pM DNA molecules were immobilized and observed in the standard imaging buffer: 20-mM Tris-HCl, 100-mM NaCl, 10-mM MgCl2 in a saturated aged Trolox-solution (6-hydroxy-2, 5, 7, 8-teramethylchroman-2-carboxylic acid) at pH 8 with an oxygen scavenging system (1-mg/ml glucose oxidase, 0.8% (w/v) D-glucose, 1-kU/ml catalase). 1-mM ATP or analogs were used in all experiments if not noted otherwise.
Single-molecule experiments
A custom-built prism-type total internal reflection microscope was used for single-molecule data acquisition using a custom-written LabVIEW software as described previously (21).
Single-molecule acceptor and donor time traces were extracted from recorded movies using a custom-written MATLAB script. The apparent FRET efficiency Eapp was calculated by E = IA / (IA+ID). Background fluorescence was subtracted from both intensities ID and IA for each trace individually by setting the intensity value of bleached fluorophores to zero. Leakage β = 0.088 from donor emission into the acceptor channel was corrected as IA = IA,0 − β*ID. Underlying transitions of DnaA assembly and disassembly were identified using hidden Markov modeling with the software package vbFRET (http://vbfret.sourceforge.net/) for MATLAB (22). For the PIFE analysis, HaMMy (http://bio.physics.illinois.edu/) was used (23).
Transition density plots (TDPs) were used to merge the information from individual single molecules. TDPs allow the identification of regularly appearing transitions between FRET efficiencies, so-called FRET states (23). A peak height of N in the heat map at a position (Ei, Ej) correlates to N transitions from FRET Ei to Ej. These peaks can be identified, separated and assigned to population transitions or so-called islands. Transition rates were extracted from TDPs as described previously (23).
RESULTS
DnaA filament formation on ssDNA
We probed the ssDNA binding kinetics of a truncated DnaA variant using an assay based on smFRET (24). A partial duplex DNA with a 3′-(dT)21 ssDNA tail (pdT21) was immobilized on a PEG-passivated surface via biotin–NeutrAvidin interaction. The donor fluorophore Cy3 was attached to the 3′ end of the unstructured poly-dT tail, while the acceptor fluorophore Cy5 was attached to the end of the duplex stem, which does not contain a known DnaA binding site (Figure 1A). A typical intensity time trace of the donor and acceptor attached to a single pdT21 sample without DnaA is shown in the top part of Figure 1B. The FRET efficiency of the pdT21 DNA is approximately EFRET = 0.55 in the experiment buffer (see the Materials and Methods section) (Figure 1B bottom and Figure 1D, blue). Addition of DnaA in the presence of ATP is expected to lead to an assembly of DnaA monomers to ssDNA and, by extension, to a conformationally restricted ssDNA with an increased end-to-end distance. This would result in a reduced average FRET efficiency. In a first set of experiments, we incubated 5-μM DnaA and 1-mM ATP analog ADP·BeF3 for 5 min with the immobilized pdT21 substrate and observed afterwards large fluctuations in FRET traces (Figure 1C) (6). FRET efficiencies dropped to lower values (EFRET ≈ 0.3) in a stepwise fashion and increased to the original FRET efficiencies repetitively until photophysical bleaching of the donor or acceptor fluorophore occurred (arrow in Figure 1C). These fluctuations were observed for DnaA with ATP and a large variety of ATP analogs (ATPγS, AMPPNP, AMPPCP and ADP·BeF3), unlike previously reported for RecA (see the Supplementary data and Supplementary Figure S1). Without ATP analogs or only ADP present, the fluctuations in the FRET efficiencies were not observed (Supplementary Figure S1). The FRET efficiencies in the presence of DnaA with ADP·BeF3 showed brief excursions to FRET efficiencies EFRET ≈ 0.8, which most likely originated from known influence of proteins on fluorophore properties (25–27). The FRET efficiency histogram in the presence of DnaA and ADP·BeF3 was significantly broadened showing a heterogeneous population (Figure 1D, green). This result together with the traces showing high FRET fluctuations (Figure 1C) suggests a highly dynamic rearrangement of DnaA molecules on the ssDNA filament. In additional experiments, we verified that DnaA did not melt the partial duplex DNA stem (see the Supplementary data and Supplementary Figure S2). We hypothesize that the origin of these large fluctuations is the attachment and detachment of DnaA monomers to form a DnaA-decorated ssDNA.
Figure 1.
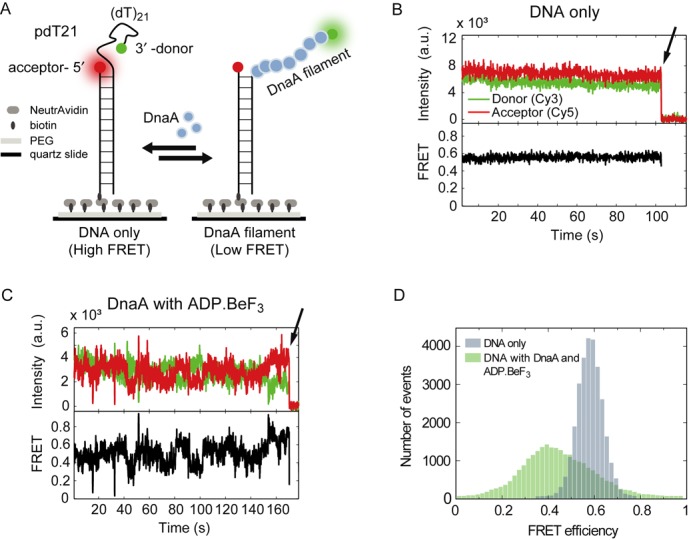
DnaA binds dynamically to ssDNA. (A) Schematic representation of a partial duplex DNA (pdT21) immobilized on a PEG-passivated surface via specific biotin-NeutrAvidin interaction. Stepwise DnaA assembly stretches ssDNA and separates the donor and acceptor fluorophore. (B) A representative time trace of a donor (green) and acceptor (red) FRET pair of the bare DNA construct at 100-ms time resolution. The FRET efficiency was calculated of the donor and acceptor intensities. Donor bleaching is highlighted by the arrow. (C) A representative time trace of pdT21 with donor and acceptor fluorophores (green and red, respectively) in the presence of 5-μM DnaA and 1-mM ADP·BeF3. FRET efficiencies are broadly distributed. Donor bleaching is highlighted by the arrow. (D) FRET histograms of DNA without initiator DnaA (blue) and in the presence of 5-μM DnaA with 1-mM ADP·BeF3 (green). (40 molecules for each).
DnaA assembles into a filament in 3′ to 5′ direction on ssDNA
While smFRET probes DnaA monomer binding to ssDNA, it does not yield information about the polarity of binding, i.e. 3′ to 5′ or 5′ to 3′ directionality. In order to study the directionality of DnaA assembly, we used smPIFE. PIFE is a photophysical phenomenon that leads to an increased emission intensity of a fluorophore upon protein binding in its vicinity with basepair sensitivity (25–27). We used two nearly identical DNA substrates with Cy3 at the 3′ end of the ssDNA tail or Cy3 labeled at the 5′ of the ssDNA (Figure 2A and D). In order to observe the Cy3 fluorescence intensity enhancement in real time, we performed flow experiments. Immobilized DNA molecules were imaged continuously while a 5-μM DnaA, 1-mM ADP·BeF3 solution was rapidly (<1 s) injected into the flow chamber. Shortly after the injection, the majority of DNA molecules with the Cy3 label at the 3′ end of the ssDNA showed a single-step increase in its intensity (Figure 2B and C). Conversely, when the DNA sample with Cy3 labeled at the 5′ end was used, a multi-step enhancement of the Cy3 intensity with 2.4 steps on average was observed (Figure 2E and F). Each step showed less fluorescence enhancement than the one step process of the previous sample indicating a stepwise closer proximity of protein to the fluorophore. Control experiments using the DNA substrate with a Cy3 at a free 5′ tail and a Cy3 labeling 3 nt away from the ds–ssDNA junction also showed a fluorescence enhancement of on average 2.2 and 2.1 steps, respectively (see Supplementary Figure S3A–D). This indicates that proteins bind stepwise closer and closer to the fluorophore at the 5′ end. In contrast, the one step enhancement for the first DNA substrate (Cy3 labeled at the 3′ end) illustrates that the DnaA filament does not grow toward the 3′ end direction. Moving the reporter fluorophore to the 3′ end of the ssDNA tail close to the ds–ssDNA junction further resulted in a majority of single-step fluorescence enhancement (Supplementary Figure S3E and F) supporting that the DnaA filament on ssDNA grows toward the 5′ end. We conclude that DnaA monomers assemble into a filament on ssDNA in the 3′ to 5′ directionality (Figure 2G). The highest intensity state of the 3′-labeled DNA construct had a significantly longer lifetime in comparison to the 5′-labeled state, suggesting that the 5′ end of the DnaA filament is the growing and shrinking end of the DnaA filament.
Figure 2.
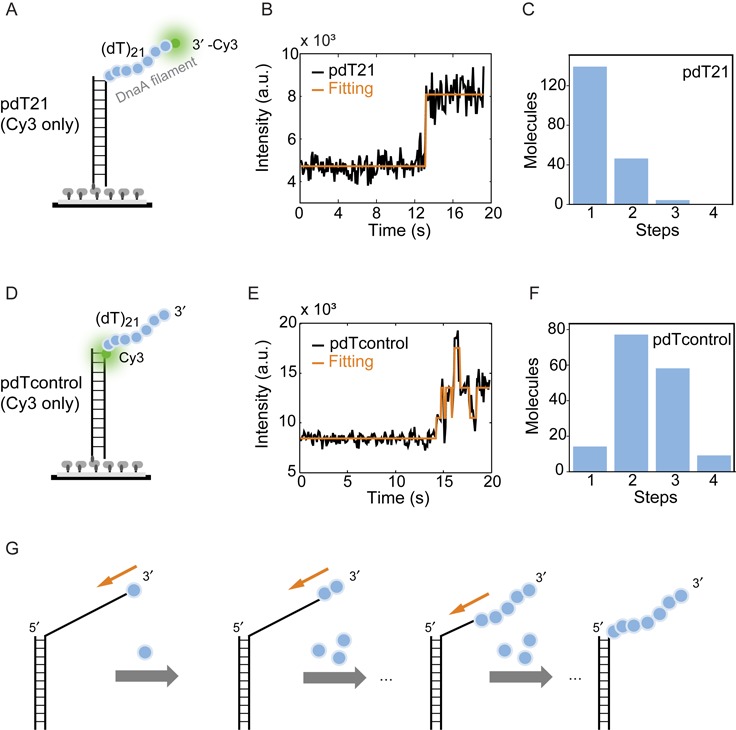
DnaA assembles in 3′ to 5′ directionality. (A) Schematic representation of the partial duplex DNA substrate used in smPIFE experiment with Cy3 labeled at the 3′ end of ssDNA tail. (B) A representative time trace of pdT21 substrate with a single-step enhancement in Cy3 intensity (black) after adding 5-μM DnaA and 1-mM ADP·BeF3. The transition is identified by eHaMMy (orange). (C) Number of steps identified for the pdT21-PIFE experiment. The majority of traces exhibited a one state behavior. (D) The partial duplex DNA substrate used in smPIFE as pdTcontrol with Cy3 labeled at the 5′ end of the ssDNA. (E) A representative time trace of pdTcontrol (Cy3 only) substrate shows multi-steps enhancement in Cy3 intensity (black) after adding 5-μM DnaA and 1-mM ADP·BeF3. The states are identified by eHaMMy (orange). (F) The histogram for pdTcontrol (Cy3 only) shows the majority number of steps from the initial intensity state to the highest intensity state is 2.4. (G) Schematic model of DnaA assembly in a 3′ to 5′ directionality.
DnaA assembly and disassembly dynamics on ssDNA
Individual DnaA molecules are known to bind three nucleotides (6), therefore we would expect a maximum of seven DnaA monomers bound to the pdT21 substrate and thus eight different FRET states are possible but unlikely to be resolved due to a limited signal-to-noise ratio.
To further study the assembly and disassembly dynamics of DnaA, we designed a set of high-resolution (HR) DNA FRET substrates. Figure 3A illustrates the HR DNA substrate with a longer ssDNA tail, to ensure the observation of 5′ end kinetics. Donor and acceptor fluorophores were placed in closer proximity to increase the sensitivity for DnaA monomer binding. We designed three different DNA templates with distances of 13, 10 or 7 dT nucleotides between donor and acceptor fluorophore (HR13, HR10 and HR7, respectively). Single-molecule time traces for HR13 in the presence of 5-μM DnaA with 1-mM ADP·BeF3 showed FRET fluctuations, in this case with distinct FRET states due to the improved resolution (Figure 3B). We identified these levels using hidden Markov models (HMMs) (23,28) (Figure 3B bottom). The analysis of 134 FRET time traces (1666 transitions) was summarized in a two-dimensional histogram, a TDP (Figure 3C, left) (24). Here, six distinct islands can be identified, three representing binding transitions of DnaA monomers from high FRET to low FRET efficiencies, three representing DnaA dissociation from low FRET to high FRET efficiencies. Most transitions (80%) occur between neighboring states along a diagonal, indicating that the DnaA filament grows or shrinks by one unit at a time. Transitions on the lower diagonal can be related to binding of DnaA monomers elongating the DnaA filament in the 5′ direction resulting in three FRET transitions from a FRET efficiency of 0.67 to 0.55, 0.55 to 0.45 and 0.45 to 0.35 with EFRET = 0.67 corresponding to the protein-free ssDNA construct (Supplementary Figure S4A). Transitions on the upper diagonal are dissociation events with average FRET values changes from 0.35 to 0.45, 0.45 to 0.55 and 0.55 to 0.67. In total, four distinct FRET populations were observed (Figure 3C). According to a recent crystal structure, each protein monomer is supposed to bind to three nucleotides (6). For the HR13 substrate with 13 nucleotides between donor and acceptor fluorophores, we would expect five FRET states corresponding to 0, 1, 2, 3 and 4 bound DnaA monomers. It could be that the missing state is only very transiently populated, potentially due to the unstable binding of the last monomer. Alternatively, the binding site is larger than three nucleotides, e.g. four nucleotides, which would result in four distinct binding states for the HR13 substrate. We tested these hypotheses by reducing the potential binding sites for DnaA monomers between the donor and acceptor fluorophores by three or six nucleotides with the HR10 and HR7 substrates. In this case, we would expect for the HR10 substrate a maximum of four distinct states and for the HR7 three distinct states if three nucleotides are bound by a DnaA monomer. Single-molecule FRET transitions for HR10 (n = 250, 1334 transitions) were analyzed using HMMs and the TDP for the HR10 substrate showed four FRET states (0.72, 0.62, 0.52 and 0.46), although the lowest FRET state was only rarely populated (Figure 3D and Supplementary Figure S4B, left). Analysis of the HR7 (n = 68, 236 transitions) substrate yielded three FRET states (0.76, 0.66 and 0.59), again the lowest FRET state only rarely populated (Figure 3E and Supplementary Figure S4B, right). Identifying in HR10 and HR7 the rarely populated lowest FRET state eliminates the possibility that DnaA binds to four instead of three nucleotides, but rather suggests that in the HR13 substrate the lowest FRET state if being populated is too short-lived to be detected within the experiment. The transition rates (kon(obs) and koff) for the highly populated states of HR substrates are determined between 0.4 and 0.6 s−1 (Supplementary Table S2). There is no trend of transition rates (increasing or decreasing) throughout the assembly stages, indicating these transition rates are independent of the position at which DnaA monomers bind. The ratio koff/kon(obs) increases throughout the assembly stages, consistent with models for equilibrium polymers of biomolecules with a limited polymer length.
Figure 3.
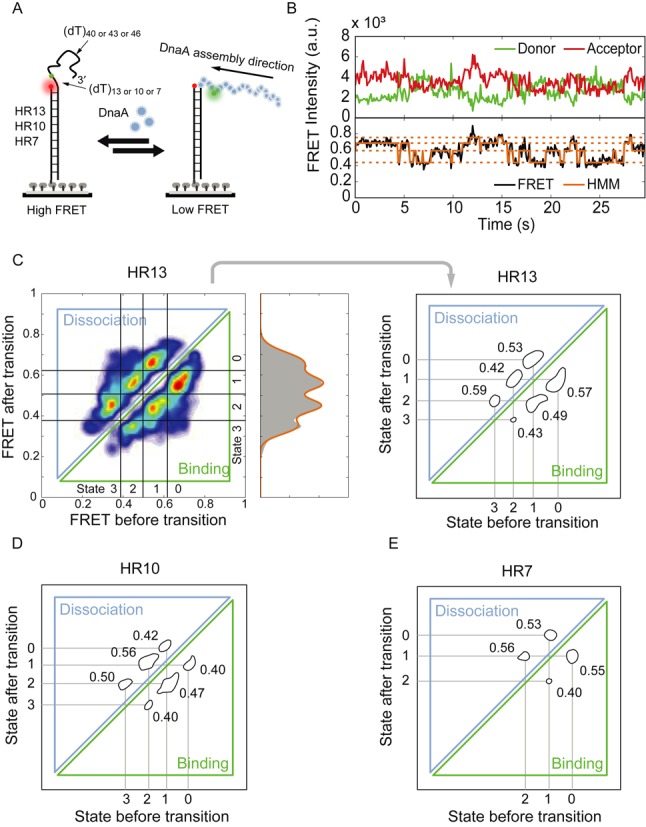
DnaA assembles and disassembles monomer by monomer. (A) High-resolution DNA substrates for DnaA binding. The distances between donor and acceptor are varied from 13, 10, to 7 poly-dT for the substrates named HR13, HR10 and HR7, respectively. (B) A representative time trace of HR13 in the presence of 5-μM DnaA and 1-mM ADP·BeF3. The FRET trace (black) was fitted by hidden Markov modeling (orange) to extract FRET populations and kinetic information. In this case, four FRET states were assigned. (C) The transition density plot (TDP) for HR13. Left: The heat-map representation illustrates islands for the monomer assembly and disassembly. Right: The island plot is generated by thresholding the heat-map. Four different FRET states (peaks) with the FRET efficiency values of 0.67, 0.55, 0.45 and 0.35 are identified. The transition rates per second of each transition are represented next to each island. (D) The TDP for HR10 shows three prominent FRET efficiency states and a fourth weak FRET efficiency state with EFRET = 0.72, 0.62, 0.52 and 0.42, respectively. The transition rates per second of each transition are shown next to the islands. (E) The TDP for HR7 shows two distinct FRET efficiency states and a third weak FRET efficiency state with EFRET = 0.76, 0.66 and 0.59, respectively. The transition rates per second of each transition are shown next to the islands.
The DnaA nucleation seed is composed of three monomers
While the 5′ end of the DnaA filament appeared highly dynamic with frequent transitions between different states (Figure 3), the PIFE experiments indicated a less dynamic 3′ end of the DnaA filament that remained bound (Figure 2B). This nucleation-like situation was studied using a FRET substrate based on HR13 but with opposing directionality (OP13), i.e. a long 5′ ssDNA tail and 13 nucleotides between donor and acceptor at the 3′ end (Figure 4A).
Figure 4.
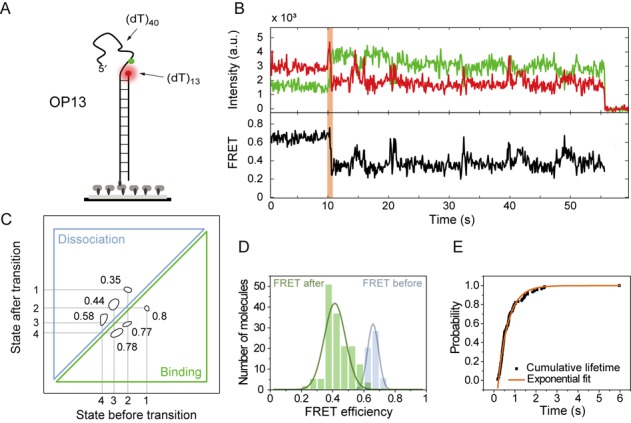
DnaA filament nucleation is comprised of two to three monomers. (A) Schematic representation of the OP13 substrate with opposite polarity to HR13. (B) A representative time trace of the flow experiment for OP13 in the presence of 5-μM DnaA and 1-mM ADP·BeF3. The initial FRET efficiency drop and, by extension, the initial DnaA binding is highlighted in orange. (C) The TDP for OP13 (n = 168) shows four different states with the FRET values of 0.6, 0.51, 0.43 and 0.33. The majority of transitions occur in the low FRET efficiency region with the rates indicated. (D) The FRET histograms show a EFRET = 0.67 before DnaA binding (blue) and EFRET = 0.42 after DnaA binding (green). (E) Cumulative distribution of the transition time (black squares, n = 90 molecules). The data are well described with a single exponential function and the characteristic transition time is determined as 0.48 ± 0.01 s (orange line).
In order to observe the first protein binding steps we performed flow experiments, with immobilized DNA molecules being imaged continuously while at t = ∼10 s a 5-μM DnaA, 1-mM ADP·BeF3 solution was rapidly injected into the flow chamber (Figure 4B). The first FRET efficiency change reflects the initial binding event of DnaA to the ssDNA and could thus potentially yield information about the filament nucleation. The FRET efficiency change in the first detected binding event was analyzed and the duration tΔE of the first binding event (n = 90 traces) characterized. The initial FRET level drop was from Ebefore = 0.67 to Eafter = 0.42 indicating the binding of several monomers at a time (Figure 4D). The cumulative transition time distribution could be well reproduced by a single-exponential function with a characteristic time of tΔE = 0.48 ± 0.01 s (Figure 4E). A single-exponential time distribution indicates a first-order reaction without any hidden steps (29,30). Therefore we conclude that the first binding event represents the binding of several monomers within the time resolution forming a nucleation seed.
After the first binding event, distinct binding and unbinding events like for the HR13 substrate were observed (Figure 4B). An analysis with HMM revealed four distinct states (0.6, 0.51, 0.43, 0.33) shown in the TDP (Figure 4C and Supplementary Figure S4D); however, the highest FRET state corresponding to DNA without protein (EFRET = 0.7) was missing indicating a very stable bound first monomer at the 3′ end, probably stabilized by the duplex junction (Supplementary Figure S4C). The average FRET level Eafter = 0.42 after the first binding corresponded to three bound DnaA monomers. This implies that the DnaA filament is formed by a nucleus of three DnaA monomers. We cannot rule out a dimer nucleus and potentially also a monomer nucleus, indicated by the asymmetry in the FRET efficiency distribution after the initial binding (Figure 4D, green), but the majority of the data exhibited a trimer nucleus.
DnaA boxes enhance the assembly of DnaA on ssDNA
The replication origin is composed of the dsDNA DnaA boxes and the DNA unwinding element (DUE) (9). DnaA boxes are key elements to position DnaA proteins at the origin of replication and serve as an anchor for binding DnaA close to the AT-rich region (17). We expanded the HR13 DNA substrate by a dsDNA section at the 3′ end of the ssDNA containing two (HR13–2box) or five (HR13–5box) consecutive DnaA boxes from the A. aeolicus genome (Figure 5A and Supplementary Table S3) to study the effects of DnaA boxes on the DnaA filament on ssDNA. The HR13–2box substrate was incubated with 5-μM DnaA and 1-mM ADP·BeF3 and FRET efficiency fluctuations similar to the HR13 substrate were observed. HMM analysis revealed five distinct FRET states compared to four states for the HR13 substrate (Figure 5B and Supplementary Figure S4F, left). The additional state appeared at a lower FRET level implying that the ssDNA is further stretched and a fourth DnaA monomer is binding more stable.
Figure 5.
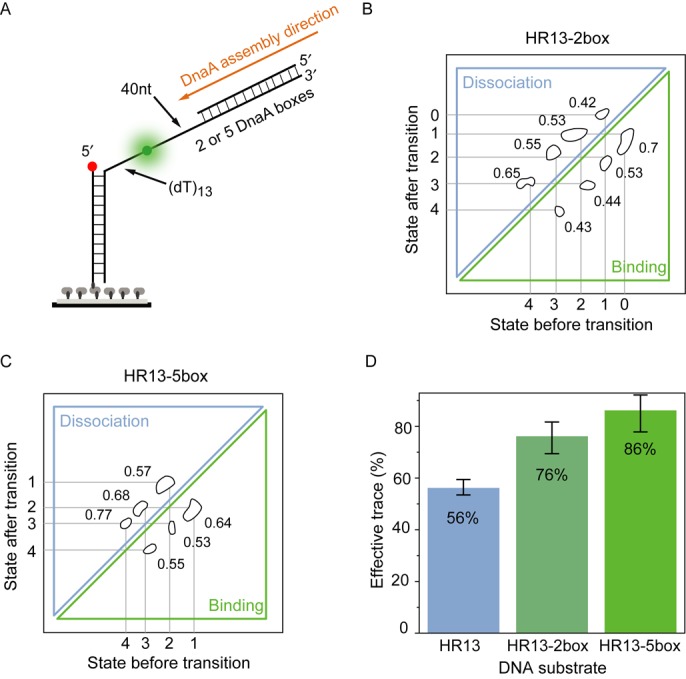
DnaA boxes enhance DnaA filament formation. (A) High-resolution HR13 substrate with two (HR13–2box) or five (HR13–5box) dsDNA DnaA binding boxes at the 3′ end of the ssDNA tail. (B) The TDP for HR13–2box shows five FRET different states with the FRET values of 0.75, 0.67, 0.57, 0.45 and 0.34 in the presence of 1-mM ADP·BeF3 and 5-μM DnaA. Compared to HR13, a lower FRET efficiency state is found. The transition rates per second are denoted besides the transition islands. (C) The TDP for HR13–5box shows four different states with the FRET values of 0.61, 0.52, 0.46 and 0.36. Compared to HR13–2box, the highest FRET state is missing. The transition rates per second of each transition are shown in the plot. (D) The ratio of effective traces with FRET fluctuations for different DNA substrates in the presence of 5-μM DnaA and 1-mM ADP·BeF3. Error bars represent the standard error. DnaA boxes increase the number of observed traces per field of view.
Extending the dsDNA region to five DnaA boxes, we observed in the FRET traces and the TDP only four distinct FRET states (Figure 5C and Supplementary Figure S4F, right). Interestingly, the missing fifth FRET level is the highest FRET efficiency level. The HR13–5box sample without DnaA in solution showed EFRET = 0.68 (Supplementary Figure S4E). The five DnaA boxes enhance DnaA filament formation on ssDNA significantly and the filament rarely shrinks more than four monomers at the 3′ end. It is also important to note that the ratio of single-molecule traces exhibiting several FRET levels versus single-molecule traces without detectable protein binding was significantly increased by the addition of DnaA boxes at the 3′ end of the filament. While for the HR13 substrate ∼56% of the traces showed FRET fluctuations, the HR13–2box and HR13–5box substrates yielded 76 and 86% of ‘active’ single molecules, respectively (Figure 5D). This observation supports that DnaA boxes enhance ssDNA binding activities of DnaA. The association rates (kon(obs)) compared to the HR13 increased by 20 and 30% in the presence of two or five DnaA boxes, respectively (Supplementary Table S2).
DISCUSSION
In this work, we used smFRET and smPIFE to investigate the DnaA assembly dynamics on ssDNA. The truncated DnaA variant based on a recent crystal structure was deficient in domain I, which is known to mediate protein–protein interactions such as DnaA–DnaB or DnaA–DiaA, but does not affect the ssDNA binding ability of DnaA (6,31). The experimental design of unstructured ssDNA mimics a partially melted dsDNA, which is known to temporarily occur and postulated to be enhanced by DnaA binding to DnaA boxes (32). We found that DnaA forms filaments on ssDNA with a directionality growing from 3′ to 5′. Unlike RecA, DnaA still exhibited a dynamic binding and unbinding to and from ssDNA instead of forming a static filament in the presence of non-hydrolyzable ATP analogs. However, in the absence of nucleotide triphosphates or analogs, we did not observe binding activity of DnaA to ssDNA, indicating that ATP or its analogs modulated DnaA affinities to ssDNA. These observations are consistent with results from a previous study, which determined similar dissociation constants of A. aeolicus DnaA to ssDNA in the presence of ATP, ADP·BeF3, ATPγS or AMPPNP (13). It is important to note that ATP will be hydrolyzed during experiments leading to an inactive DnaA–ADP complex (15).
Up to now, it had been unclear whether DnaA forms a directed filament on ssDNA. Considering the location of the DnaA binding boxes and the DUE within the oriC, the indication of DnaA binding to T-rich strand of the DUE (9,17), a DnaA assembly in the 3′ to 5′ direction was considered favorable. Here, our data directly show for the first time that the DnaA filament on ssDNA assembles in the 3′ to 5′ directionality with one monomer at a time.
Interestingly, a DnaA filament formed only with a nucleation seed of two to three monomers. After nucleation we observed a highly dynamic 5′ end of the DnaA filament potentially limiting the length of the DnaA filament on ssDNA. In contrast, the 3′ end of the filament did not show any significant growing or shrinking dynamics. This directionality might be important to direct the melting and replisome assembly to the DUE region.
Furthermore, DnaA binding boxes next to the ssDNA region enhanced filament formation. The association rates (kon(obs)) of HR13 and the HR-box substrates in the presence of two or five DnaA boxes are increased by 20 and 30%, respectively (Supplementary Table S2). At the same time, the unbinding rate remained unchanged leading to an increased length of the DnaA filament. This observation supports previous models suggesting a continuous filament of DnaA ranging from the DnaA boxes to the DUE region binding dsDNA and ssDNA, respectively (13,17) (Supplementary Figure S5A). We cannot exclude the model by Katayama et al. in which DnaA assembly occurs at the DnaA boxes and in the presence of IHF, the DNA bends and allows the identical DnaA monomers to bind also to single-stranded DUE regions (9). This model would also lead to a 3′ to 5′ assembly direction; however, it appears very unlikely that this filament grows and shrinks by individual monomers with similar kinetics, since the geometry would imply a more cooperative binding of several monomers (Supplementary Figure S5B).
In conclusion, our data extend the currently existing models (13) of origin melting by intermediate steps (Figure 6C and D). Typically, the models describe that DnaA recognizes the dsDNA binding sites and preassembles (Figure 6A and B) to melt the origin of replication and stabilize the DNA bubble—also by ssDNA interactions (Figure 6E). Taking into account our direct observation of the very dynamic DnaA filament assembly, spontaneous DNA bubble formation near the DnaA binding sites could be stabilized by DnaA, which itself is stabilized through the adjacent dsDNA-bound DnaA (Figure 6C). The directionality of the DnaA filament growth would lead to a subsequent DNA bubble growth by capturing transiently melted DNA to extend the single-stranded origin of replication state for subsequent replisome assembly (Figure 6D). We anticipate further single-molecule studies with DnaA can yield a more detailed mechanistic understanding of the structure–function relationship in origin melting and the structural organization of the origin during the melting process.
Figure 6.
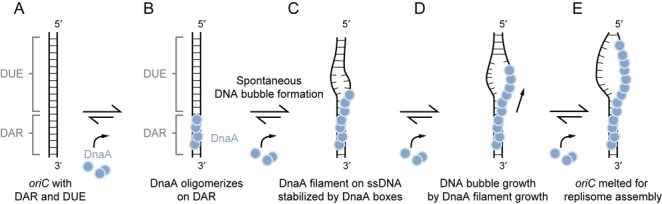
A proposed model illustrates the origin melting via direct DnaA–ssDUE interaction. (A,B) DnaA recognizes and oligomerizes on the dsDNA assembly region. (C) Spontaneous DNA melting occurs via DnaA–ssDNA interaction. The DnaA filament formation on ssDUE is stabilized by the adjacent dsDNA-bound DnaA. (D) Continuous filament growth promotes replication origin melting and (E) allows the replisome assembly.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors thank all group members of the Schlierf group, in particular Steffen Hahn for the software development in LabVIEW, as well as Alvin Thomas and Weilin Lin from Dr Yixin Zhang's group for discussions. We thank Dr Harald Huber (Universität Regensburg, Germany) for providing Aquifex aeolicus. Comments on the manuscript by Dr Karl Duderstadt are highly appreciated.
FUNDING
German Federal Ministry of Education and Research BMBF [03Z2EN11 to M.S.]; German Research Foundation in the framework of the Dresden International Graduate School for Biomedicine and Bioengineering [GS97 to H.-M.C., in part]. Funding for open access charge: German Federal Ministry of Education and Research [03Z2EN11 to M.S.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Costa A., Hood I.V., Berger J.M. Mechanisms for initiating cellular DNA replication. Annu. Rev. Biochem. 2013;82:25–54. doi: 10.1146/annurev-biochem-052610-094414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arias E.E., Walter J.C. Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells. Genes Dev. 2007;21:497–518. doi: 10.1101/gad.1508907. [DOI] [PubMed] [Google Scholar]
- 3.Sutera V.A., Lovett S.T. The role of replication initiation control in promoting survival of replication fork damage. Mol. Microbiol. 2006;60:229–239. doi: 10.1111/j.1365-2958.2006.05093.x. [DOI] [PubMed] [Google Scholar]
- 4.Leonard A.C., Mechali M. DNA replication origins. Cold Spring Harb. Perspect. Biol. 2013;5:a010116. doi: 10.1101/cshperspect.a010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stillman B. Origin recognition and the chromosome cycle. FEBS Lett. 2005;579:877–884. doi: 10.1016/j.febslet.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 6.Duderstadt K.E., Chuang K., Berger J.M. DNA stretching by bacterial initiators promotes replication origin opening. Nature. 2011;478:209–213. doi: 10.1038/nature10455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mott M.L., Berger J.M. DNA replication initiation: mechanisms and regulation in bacteria. Nat. Rev. Microbiol. 2007;5:343–354. doi: 10.1038/nrmicro1640. [DOI] [PubMed] [Google Scholar]
- 8.Iyer L.M., Leipe D.D., Koonin E.V., Aravind L. Evolutionary history and higher order classification of AAA plus ATPases. J. Struct. Biol. 2004;146:11–31. doi: 10.1016/j.jsb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 9.Ozaki S., Katayama T. Highly organized DnaA-oriC complexes recruit the single-stranded DNA for replication initiation. Nucleic Acids Res. 2012;40:1648–1665. doi: 10.1093/nar/gkr832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGarry K.C., Ryan V.T., Grimwade J.E., Leonard A.C. Two discriminatory binding sites in the Escherichia coli replication origin are required for DNA strand opening by initiator DnaA-ATP. Proc. Natl. Acad. Sci. U.S.A. 2004;101:2811–2816. doi: 10.1073/pnas.0400340101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leonard A.C., Grimwade J.E. Regulation of DnaA assembly and activity: taking directions from the genome. Annu. Rev. Microbiol. 2011;65:19–35. doi: 10.1146/annurev-micro-090110-102934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawakami H., Keyamura K., Katayama T. Formation of an ATP-DnaA-specific initiation complex requires DnaA arginine 285, a conserved motif in the AAA plus protein family. J. Biol. Chem. 2005;280:27420–27430. doi: 10.1074/jbc.M502764200. [DOI] [PubMed] [Google Scholar]
- 13.Duderstadt K.E., Mott M.L., Crisona N.J., Chuang K., Yang H., Berger J.M. Origin remodeling and opening in bacteria rely on distinct assembly states of the DnaA initiator. J. Biol. Chem. 2010;285:28229–28239. doi: 10.1074/jbc.M110.147975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ryan V.T., Grimwade J.E., Camara J.E., Crooke E., Leonard A.C. Escherichia coli prereplication complex assembly is regulated by dynamic interplay among Fis, IHF and DnaA. Mol. Microbiol. 2004;51:1347–1359. doi: 10.1046/j.1365-2958.2003.03906.x. [DOI] [PubMed] [Google Scholar]
- 15.Erzberger J.P., Mott M.L., Berger J.M. Structural basis for ATP-dependent DnaA assembly and replication-origin remodeling. Nat. Struct. Mol. Biol. 2006;13:676–683. doi: 10.1038/nsmb1115. [DOI] [PubMed] [Google Scholar]
- 16.Zorman S., Seitz H., Sclavi B., Strick T.R. Topological characterization of the DnaA-oriC complex using single-molecule nanomanipuation. Nucleic Acids Res. 2012;40:7375–7383. doi: 10.1093/nar/gks371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Speck C., Messer W. Mechanism of origin unwinding: sequential binding of DnaA to double- and single-stranded DNA. EMBO J. 2001;20:1469–1476. doi: 10.1093/emboj/20.6.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soultanas P. Loading mechanisms of ring helicases at replication origins. Mol. Microbiol. 2012;84:6–16. doi: 10.1111/j.1365-2958.2012.08012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mott M.L., Erzberger J.P., Coons M.M., Berger J.M. Structural synergy and molecular crosstalk between bacterial helicase loaders and replication initiators. Cell. 2008;135:623–634. doi: 10.1016/j.cell.2008.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weigel C., Seitz H. Strand-specific loading of DnaB helicase by DnaA to a substrate mimicking unwound oriC. Mol. Microbiol. 2002;46:1149–1156. doi: 10.1046/j.1365-2958.2002.03232.x. [DOI] [PubMed] [Google Scholar]
- 21.Swoboda M., Henig J., Cheng H.-M., Brugger D., Haltrich D., Plumere N., Schlierf M. Enzymatic oxygen scavenging for photostability without pH drop in single-molecule experiments. ACS Nano. 2012;6:6364–6369. doi: 10.1021/nn301895c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bronson J.E., Fei J., Hofman J.M., Gonzalez R.L., Jr, Wiggins C.H. Learning rates and states from biophysical time series: a Bayesian approach to model selection and single-molecule FRET data. Biophys. J. 2009;97:3196–3205. doi: 10.1016/j.bpj.2009.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKinney S.A., Joo C., Ha T. Analysis of single-molecule FRET trajectories using hidden Markov modeling. Biophys. J. 2006;91:1941–1951. doi: 10.1529/biophysj.106.082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joo C., McKinney S.A., Nakamura M., Rasnik I., Myong S., Ha T. Real-time observation of RecA filament dynamics with single monomer resolution. Cell. 2006;126:515–527. doi: 10.1016/j.cell.2006.06.042. [DOI] [PubMed] [Google Scholar]
- 25.Luo G., Wang M., Konigsberg W.H., Xie X.S. Single-molecule and ensemble fluorescence assays for a functionally important conformational change in T7 DNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 2007;104:12610–12615. doi: 10.1073/pnas.0700920104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hwang H., Kim H., Myong S. Protein induced fluorescence enhancement as a single molecule assay with short distance sensitivity. Proc. Natl. Acad. Sci. U.S.A. 2011;108:7414–7418. doi: 10.1073/pnas.1017672108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hwang H., Myong S. Protein induced fluorescence enhancement (PIFE) for probing protein-nucleic acid interactions. Chem. Soc. Rev. 2014;43:1221–1229. doi: 10.1039/c3cs60201j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blanco M., Walter N.G. Analysis of complex single-molecule FRET timetrajectories. In: Walter NG, editor. Methods in Enzymology, Vol 472: Single Molecule Tools, Pt A: Fluorescence Based Approaches. Vol. 472. San Diego: Elsevier Academic Press; 2010. pp. 153–178. [Google Scholar]
- 29.Park J., Myong S., Niedziela-Majka A., Lee K.S., Yu J., Lohman T.M., Ha T. PcrA helicase dismantles RecA filaments by reeling in DNA in uniform steps. Cell. 2010;142:544–555. doi: 10.1016/j.cell.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Floyd D.L., Harrison S.C., van Oijen A.M. Analysis of kinetic intermediates in single-particle dwell-time distributions. Biophys. J. 2010;99:360–366. doi: 10.1016/j.bpj.2010.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ozaki S., Katayama T. DnaA structure, function, and dynamics in the initiation at the chromosomal origin. Plasmid. 2009;62:71–82. doi: 10.1016/j.plasmid.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 32.Duderstadt K.E., Berger J.M. A structural framework for replication origin opening by AAA plus initiation factors. Curr. Opin. Struct. Biol. 2013;23:144–153. doi: 10.1016/j.sbi.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.