


Abstract
Serine/threonine kinase 11 (STK11, also known as LKB1) functions as a tumor suppressor in many human cancers. However, paradoxically loss of LKB1 in mouse embryonic fibroblast results in resistance to oncogene-induced transformation. Therefore, it is unclear why loss of LKB1 leads to increased predisposition to develop a wide variety of cancers. Here, we show that LKB1 protects cells from genotoxic stress. Cells lacking LKB1 display increased sensitivity to irradiation, accumulates more DNA double-strand breaks, display defective homology-directed DNA repair (HDR) and exhibit increased mutation rate, compared with that of LKB1-expressing cells. Conversely, the ectopic expression of LKB1 in cells lacking LKB1 protects them against genotoxic stress-induced DNA damage and prevents the accumulation of mutations. We find that LKB1 post-transcriptionally stimulates HDR gene BRCA1 expression by inhibiting the cytoplasmic localization of the RNA-binding protein, HU antigen R, in an AMP kinase-dependent manner and stabilizes BRCA1 mRNA. Cells lacking BRCA1 similar to the cell lacking LKB1 display increased genomic instability and ectopic expression of BRCA1 rescues LKB1 loss-induced sensitivity to genotoxic stress. Collectively, our results demonstrate that LKB1 is a crucial regulator of genome integrity and reveal a novel mechanism for LKB1-mediated tumor suppression with direct therapeutic implications for cancer prevention.
INTRODUCTION
Cancer cells differ from normal cells in many aspects, which are collectively dubbed as the hallmarks of cancer (1). To acquire these hallmarks, cancer cells undergo multiple genetic and epigenetic alterations (1). Among these, the inactivation of tumor suppressor genes (TSGs) due to genetic deletion, mutations or epigenetic gene silencing is frequently observed in human cancers (1–4). Loss of TSGs plays an important role in several aspects of cancer, including cancer initiation and metastatic progression (5,6).
Serine/threonine kinase 11 (STK11, commonly known as liver kinase B1 [LKB1]) was identified as a gene responsible for the Peutz-Jeghers Syndrome (PJS) (7,8). PJS is a rare autosomal dominant disease that is characterized by mucocutaneous pigmentation and benign hamartomatous polyps in gastrointestinal tracts (9). PJS patients display an increased predisposition to malignant tumors in multiple tissues (10–12). Notably, over 93% of PJS patients develop malignant tumors by the average age of 43 (13). Similar to PJS patients, LKB1 knockout mice are predisposed to cancer, particularly of the gastrointestinal tract (14–17). Furthermore, recent studies have discovered LKB1-inactivating mutations in multiple sporadic cancers, particularly of the lung and at a lower frequency in the pancreas and skin (18–21). Collectively, these studies suggest that LKB1 plays an important role as a TSG in many human malignancies.
As a tumor suppressor, LKB1 phosphorylates its target substrates and subsequently regulates their activities (22). LKB1 is activated through its interaction with the sterile 20 (STE20)-related kinase adaptor (STRAD) pseudokinase and mouse protein-25 (MO25) (23,24). In addition to activating STRAD, MO25 retains LKB1 in the cytoplasm, where it exerts cell cycle regulatory functions (25). Adenosine monophosphate-activated protein kinase (AMPK), which functions as a sensor of cellular energy changes, is one of the best-characterized substrates of LKB1. The reduction in cellular adenosine triphosphate levels activates AMPK. LKB1 phosphorylates and activates AMPK (26–28), which then activates TSC1/TSC2 and inhibits the oncogenic mTOR signaling pathway (22,29).
Here, we show that LKB1 preserves genome integrity by stimulating the expression of BRCA1. Our results identify a new role for LKB1 in mediating the DNA damage response (DDR) and DNA repair and suggest that the LKB1-mediated DDR pathway may be targeted for cancer prevention.
MATERIALS AND METHOD
Cell culture, plasmids and luciferase assay
HCT116, H1299, MCF7, SKMEL-28 and immortalized human diploid fibroblasts were obtained from American Type Culture Collection (ATCC) and A549 and H460 cells were obtained from the National Cancer Institute and grown as recommended. LKB1 wild-type and knockout mouse embryonic fibroblasts (MEFs) were obtained from Dr Boyi Gan (MD Anderson Cancer Center). LKB1 knockout were generated from LKB1 L/L, RosaCreERT2 MEFs as described previously (30). The BRCA1 mammalian expression construct was a kind gift from Steve Elledge (Harvard Medical School), and the BRCA1 reporter-luciferase reporter construct was a kind gift from Stephen Weiss (University of Michigan) (31). U2OS-DRGFP cells were a kind gift from Maria Jasin (Memorial Sloan Kettering Cancer Center). FLAG-LKB1 and FLAG-LKB1 KD was a kind gift from Lewis Cantley (Harvard Medical School). The luciferase assay was performed using the dual-luciferase reporter assay kit (Promega). Renilla luciferase was used as an internal control for normalizing transfection differences in the luciferase assay.
Transfections, shRNAs, preparation of retroviral and lentiviral particles, immunoblot analysis and cell fractionation
LKB1, BRCA1, HuR and control non-specific (NS) short-hairpin RNAs (shRNA)s were obtained from OpenBiosystems. Supplementary Table S1 shows the product IDs for all shRNAs. Lentiviral particles were prepared by co-transfecting the shRNA plasmids and lentiviral packaging plasmids, pSPAX2 and pMD2.G, into 293T cells using Effectene (Qiagen) and following the protocol at the Broad Institute's website (http://www.broadinstitute.org/rnai/public/resources/protocols). Retroviral particles were prepared as described previously (32). Immunoblot analysis was performed as described previously (33). Nuclear and cytoplasmic fractions were prepared as described previously (33). Protein concentrations were estimated using the Bradford Protein Estimation Kit (Bio-Rad), according to the manufacturer's instructions. The information of the antibodies and inhibitors used in this study is provided in Supplementary Table S1.
Gamma irradiation, γH2AX immunofluorescence, HuR immunofluorescence and flow cytometry analyses
Cells were gamma-irradiated at various doses and time points, as indicated in the figures. For γH2AX immunofluorescence 5 × 104 cells were plated onto multi-well tissue culture slides (Nalgene) and gamma-irradiated at 2 Gray dose. At different time points, cells were fixed with 3.7% paraformaldehyde (PFA) at room temperature for 10 min, after which the PFA was gently removed, and cells were washed three times with 1X phosphate-buffered saline (PBS). Afterward, cells were permeabilized with 0.5% Triton X-100 in PBS for 10 min and blocked with 1% bovine serum albumin (BSA) for 30 min at room temperature. Cells were then incubated with the γH2AX antibody (1:300; Cell Signaling) in 1% BSA for 2 h at 37°C and washed three times with 1X PBS, after which they were incubated with the anti-mouse Alexa 488 antibody (1:600; Life Technologies) for 1.5 h at 37°C. Following the incubation with the secondary antibody, cells were washed twice with 1X PBS, and the nuclei were stained with 0.01-mg/ml 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich) for 5 min. Images for γH2AX immunofluorescence were collected using the Olympus IX-71 inverted fluorescence microscope, and the percentages of cells with more than 10 γ-H2AX foci per cell in 40X magnification were counted in 10 different fields in biological triplicate and plotted at different time points as shown and indicated in the related figures and figure legends.
HuR immunofluorescence was performed as described above for γH2AX immunofluorescence using HuR-specific antibody listed in Supplementary Table S1. For flow cytometry analyses, cells were synchronized in G2/M phase by nocodazole arrest as described previously (34). Cells were stained with propidium iodide and cell cycle distributions were determined by fluorescence-activated cell sorting analysis. Quantitation of the fraction of cells in different cell cycle phases was done using FlowJoTM software. The number of cells in G1 (2n DNA), S phase (>2n <4n DNA) and G2/M (4n DNA) were quantified and percent fraction of cells in each phase was calculated. Doublets and cell aggregates were removed by plotting FL2A against FL2W and by gating for single-cell populations.
Cell viability analysis, clonogenic assay, drug treatment, spontaneous mutagenesis assay and Homologous recombination (HR) repair assay
To measure cell viability after irradiation, 0.2 × 106 cells were plated in 6-well plates and gamma-irradiated at different doses as indicated in the figures. At 48 h after irradiation, cells were harvested, mixed with an equal volume of trypan blue solution (Life Technologies) and counted using Countess (Life Technologies). The relative viability was plotted in reference to untreated cells.
The clonogenic ability of the cells stably expressing control or LKB1 shRNAs was measured in unirradiated and gamma-irradiated conditions. For clonogenic assay, 2 × 105 cells were seeded in a 6-well plate and 48 h after 2 Gy dose of gamma irradiation 5 × 103 cells were re-seeded in another 6-well plate. As an unirradiated control, 5 × 103 cells were seeded in a 6-well plate. After 2 weeks of plating, colonies were fixed with a fixing solution containing 50% methanol and 10% acetic acid and then stained with 0.05% crystal-violet (Sigma-Aldrich). The relative number of colonies was calculated by normalizing the average colony number of the triplicates carrying indicated shRNAs against those carrying NS shRNA.
To measure the cell viability HCT116 cells were treated with various concentrations of adriamycin and etoposide as indicated in the related figures for 48 h. The cell viability was determined by trypan blue exclusion assay as described above. The source of adriamycin and etoposide is listed in Supplementary Table S1.
The spontaneous mutagenesis assays were performed as described previously (35). Briefly, cells were seeded at a density of 1 × 104 cells/well in 48-well plates and grown in the presence of 1.5-μg/ml 6-thioguanine (Sigma-Aldrich) for 4 weeks. The mutation rate was calculated as the ratio of the number of 6-thioguanine-resistant colonies to the total number of cells that were seeded and normalized for plating efficiency. HR repair assay was performed in U2OS-DRGFP cells expressing an NS shRNA or shRNAs targeting LKB1 as described previously (36).
RNA isolation, Real-time-quantitative PCR (RT-qPCR) analysis and mRNA half-life measurement
Total RNA was extracted using TRIzol (Life technologies), according to the manufacturer's instructions. Total RNA was purified using RNAeasy mini columns (Qiagen). First-strand cDNA synthesis was performed using the ProtoScript M-MuLV First-Strand cDNA Synthesis Kit (New England Biolabs), and qPCR was performed using the Power SYBR Green PCR Master Mix (Applied Biosystems). The primers used for qPCR analysis are listed in Supplementary Table S1. Actin mRNA was used to normalize RT-qPCR data. For BRCA1 mRNA half-life measurement we performed actinomycin D chase in HCT116, SKMEL-28 and H1299 cells expressing shRNAs or cDNAs as presented in the related figures. Cells were irradiated at 20 gray and the total RNA was prepared at 0, 3, 6 and 12 h after irradiation and actinomycin D treatment (5 μM), and BRCA1 and actin mRNA expression was analyzed. The expression of BRCA1 mRNA at each time point was plotted in reference to mRNA samples at 0 h.
Statistical analysis
All experiments were performed at least three times in triplicate, and the data are expressed as mean ± standard error of the mean (SEM). The student's t-test for two-tailed distribution with unequal variance was performed in Microsoft Excel to derive the P-values.
RESULTS
LKB1 is necessary and sufficient for protecting cells from genotoxic stress
Mouse embryonic fibroblasts (MEFs) that harbor the genetic deletion of both alleles of LKB1 paradoxically display marked resistance to cellular transformation by oncogenes, compared with MEFs that have intact LKB1 loci (14). Therefore, why loss of LKB1 causes increase in cancer incidence remains unclear.
A common feature of many human malignancies is genome instability, which is proposed to drive cancer initiation and progression (37). To investigate whether LKB1 can protect the human genome from genotoxic stress, we used shRNAs to knockdown the expression of LKB1 in a variety of human cell lines of different tissue origin (Supplementary Figure S1A) and analyzed the sensitivity of these cells to gamma irradiation. We find that the shRNA-mediated knockdown of LKB1 sensitizes various human cell lines of different tissue origin to gamma irradiation (Figure 1A). To further confirm that the loss of LKB1 leads to increased sensitivity to gamma irradiation, we performed clonogenic assays. In complete agreement with our short-term survival assays, we find that the loss of LKB1 leads to increased sensitivity to gamma irradiation (Figure 1C and Supplementary Figures S2A). LKB1 knockdown was also able to sensitize the cells to other DNA damaging chemotherapeutic agents such as adriamycin and etoposide (Supplementary Figure S3). Conversely, the ectopic expression of LKB1 in LKB1-deficient cancer cell lines A549 and H460 protected them against gamma irradiation-induced genotoxic stress and decreased sensitivity to gamma irradiation, while a kinase-dead mutant of LKB1 failed to do so (Figure 1B and D and Supplementary Figures S1B and S2B). Collectively, these results show that LKB1 protects cells from genotoxic stress in a kinase activity-dependent manner.
Figure 1.
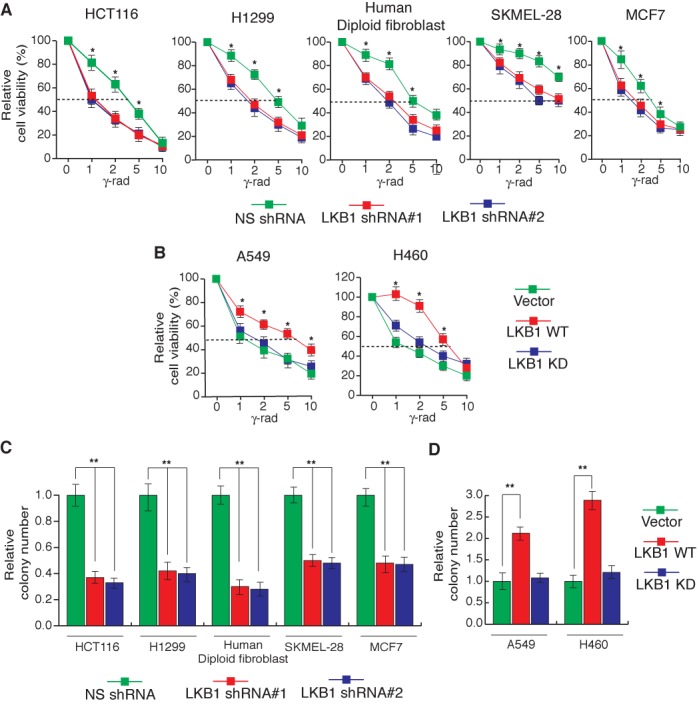
Loss of LKB1 expression causes increased sensitivity to DNA damage. (A) Indicated cell lines expressing LKB1 short-hairpin RNAs (shRNAs) or a non-specific (NS) shRNA were gamma-irradiated at the indicated doses. At 48 h post-irradiation, cell viability was measured by the trypan blue exclusion assay. The percentages of cell viability relative to corresponding unirradiated cells are plotted. (B) Indicated cell lines ectopically expressing wild-type LKB1 (LKB1 WT), kinase-dead mutant of LKB1 (LKB1 KD) or an empty vector were gamma-irradiated at the indicated doses. At 48 h post-irradiation, live cells were counted by the trypan blue exclusion assay. The percentages of cell viability relative to corresponding unirradiated cells are plotted. (C) Indicated cell lines expressing LKB1 shRNAs or a non-silencing (NS) shRNA were gamma-irradiated (2G) and plated on 6-well plates. Relative colony numbers 2 weeks after gamma irradiation in comparison to NS shRNA and normalized to unirradiated cells are presented. (D) Indicated cell lines expressing wild-type LKB1 (LKB1 WT), kinase-dead mutant LKB1 (LKB1 KD) or an empty vector were gamma-irradiated at 2G and plated on 6-well plates. Relative colony numbers 2 weeks after gamma irradiation in comparison to empty vector and normalized to unirradiated cells are presented. Error bars show standard error mean (SEM). (*P < 0.01; **P < 0.001).
Cells lacking LKB1 display increased DNA double-strand breaks and enhanced mutation rates
To determine the cause for increased sensitivity to gamma irradiation, we examined whether LKB1 loss leads to defects in DNA repair and the accumulation of DNA double-strand breaks, which can consequentially increase the sensitivity to genotoxic stress. To determine this, we knocked down the LKB1 expression in various human cancer cell lines and assessed for the DNA damage-induced formation of γ-H2AX foci before and after irradiating the cells with gamma irradiation. γ-H2AX is a marker of DNA double-strand breaks and can be used to determine the extent of DNA double-strand breaks (38). Remarkably, the shRNA-mediated knockdown of LKB1 expression significantly increased γH2AX foci formation (Figure 2A and Supplementary Figures S4A and S5A), indicating that the cells lacking LKB1 accumulate more DNA double-strand breaks. Notably, the ectopic expression of LKB1 in cancer cell lines that lack endogenous LKB1 protected these cells from gamma irradiation-induced DNA double-strand breaks, while a kinase-dead mutant of LKB1 or an empty vector failed to have any effect (Figure 2B and Supplementary Figure S4B and S5C). These results further support the role of LKB1 in protecting the human genome from genotoxic stress.
Figure 2.
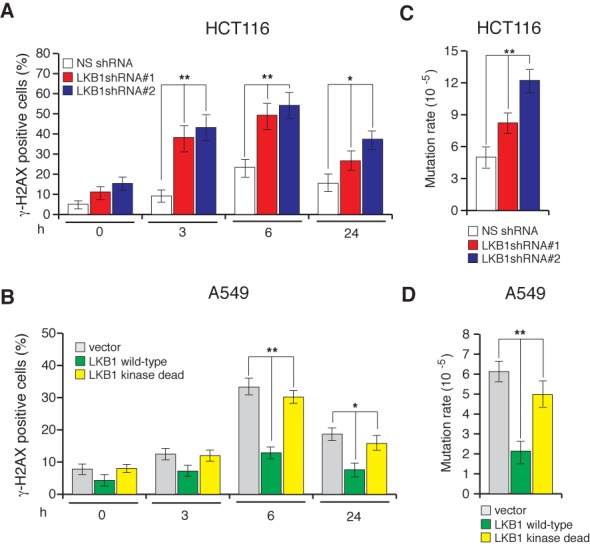
Loss of LKB1 expression increases DNA double strand breaks and mutation rates. (A) HCT116 cells expressing the indicated shRNAs or (B) A549 cells ectopically expressing LKB1 WT, LKB1 KD or an empty vector were gamma-irradiated (2G) and stained for γH2AX at indicated time points post-irradiation. Percentages of γH2AX-positive cells are plotted at the indicated time points. (C) Spontaneous mutation of HPRT in HCT116 cells expressing the indicated shRNAs. The mutation rates under the indicated conditions are plotted. (D) Spontaneous mutation of HPRT in A549 cells expressing the indicated constructs. The mutation rates under the indicated conditions are plotted. Error bars show standard error mean (SEM). (*P < 0.01; **P < 0.001).
Because the increased accumulation of DNA double-strand breaks was observed in cells lacking LKB1, we assessed whether this increase may affect genome integrity. As a measure of genome integrity, we monitored the spontaneous mutagenesis rate in LKB1-depleted cells using the hypoxanthine-guanine phosphoribosyltransferase (HPRT)-forward mutation assay. The HPRT assay is based on the spontaneous mutagenesis of the HGPRT locus, which, when mutated, allows cells to grow in the presence of 6-thioguanine. Under normal circumstances, 6-thioguanine blocks DNA replication and induces cytotoxicity (39). Notably, LKB1 shRNA-expressing cells had significantly higher mutation rates than that of NS shRNA-expressing cells (Figure 2C and Supplementary Figure S5B). On the contrary, the ectopic expression of LKB1 in cancer cells that lack LKB1 caused reduction in the mutation rate, while no reduction in mutation rate was observed when the kinase-dead mutant of LKB1 was expressed (Figure 2D and Supplementary Figure S5D). These results further confirm that LKB1 in kinase activity-dependent manner protects DNA from genotoxic stress by preventing the accumulation of DNA double-strand breaks and subsequently inhibiting the accumulation of deleterious mutations that may increase the likelihood of neoplastic transformation.
Cells lacking LKB1 display defective homology-directed DNA repair
Our results show that the cells lacking LKB1 display increased accumulation of double-stand DNA breaks and increased mutagenesis rate. Homology-directed DNA repair pathway has been shown to repair the DNA without error. Therefore, we asked if the cells that express LKB1 shRNA are defective in homology-directed DNA repair pathway. To do so, we used previously described U2OS cell line derivative U2OS-DRGFP cells that can be used to determine the homology-directed DNA repair by transfecting the nuclease I-Sce1 and measuring Green Fluorescent Protein (GFP) by flow cytometry analyses (36). To assess the effect of LKB1 on homology-directed DNA repair, we generated U2OS-DRGFP cell lines that either expressed an NS shRNA or shRNAs against LKB1 (Figure 3A). These cells were then transfected with the nuclease I-Sce1 and analyzed for GFP positive cells by flow cytometry analyses at 48 h post transfection. The results presented in Figure 3B show that cells expressing LKB1 shRNA display significant reduction in GFP-positive population compared to the cells that express an NS shRNA. Collectively, these results show that loss of LKB1 causes reduced homology-directed DNA repair that consequentially increases mutation rate.
Figure 3.
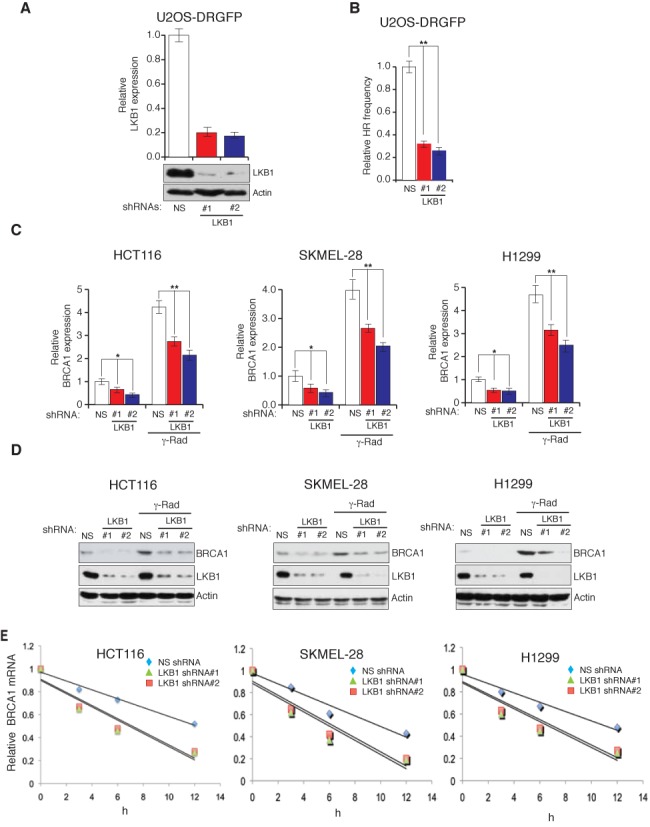
LKB1 regulates BRCA1 mRNA stability. (A) U2OS-DRGFP cell lines stably expressing a non-specific (NS) shRNA or LKB1 shRNAs were analyzed for the expression of LKB1 transcript and protein by RT-qPCR (top) and immunoblot (bottom), respectively. (B) U2OS-DRGFP cell expressing indicated shRNAs was analyzed for HR frequency by monitoring for GFP expression after 48 h of I-Sce1 transfection. Relative HR frequencies under indicated conditions are plotted. (C) The mRNA level of BRCA1 was measured by RT-qPCR in cells expressing LKB1 shRNAs or a non-specific (NS) shRNA that were either unirradiated or gamma-irradiated (20G). Actin expression was analyzed as the internal control. (D) The protein level of BRCA1 was measured by immunoblot analysis in cells expressing LKB1 shRNAs or the NS shRNA that were either unirradiated or gamma-irradiated (20G). Actin was measured as loading control. (E) The half-life of BRCA1 mRNA was measured at indicated time points after irradiation (20 Gray) and treatment with the transcription blocker, actinomycin D (5 μM), in cell lines that either expressed LKB1 shRNA or the non-specific shRNA. The relative BRCA1 mRNA abundance was normalized to actin at the indicated time points and is plotted. The BRCA1 half-lives were significantly different in cells expression NS shRNAs compared to the cells expressing LKB1 shRNAs (P < 0.01). Error bars show standard error mean (SEM). (*P < 0.01; **P < 0.001).
LKB1 stimulates BRCA1 expression by regulating BRCA1 mRNA stability
To further understand the mechanism by which LKB1 mediates genome integrity, we analyzed the expression of genes that were previously implicated in the regulation of homology-directed DNA repair (Supplementary Table S1). In comparison with NS shRNA-expressing cells, shRNA-mediated LKB1 knockdown significantly reduced the mRNA and protein levels of BRCA1 and also prevented the gamma irradiation-induced accumulation of BRCA1 (Figure 3C and D), while other HDR genes did not alter significantly (Supplementary Figure S6). To determine if LKB1 regulates BRCA1 at the transcriptional level, we tested the effect of LKB1 on BRCA1 promoter activity by transiently transfecting the BRCA1 promoter-luciferase reporter construct in cells expressing shRNAs against LKB1 or an NS shRNA. We observed no significant differences in BRCA1 promoter-luciferase reporter activity with or without gamma irradiation (Supplementary Figure S8A). Similarly, the ectopic expression of LKB1 did not affect BRCA1 promoter-luciferase reporter activity (Supplementary Figure S8B). These results indicated that LKB1 may possibly regulate the abundance of BRCA1 mRNA via a post-transcriptional mechanism. Therefore, we analyzed the half-life of BRCA1 mRNA by treating cells with the transcriptional blocker, actinomycin D. We find that the shRNA-mediated knockdown of LKB1 expression substantially reduces the half-life of BRCA1 mRNA, confirming that LKB1 promotes BRCA1 mRNA stability (Figure 3E and Supplementary Figure S7). In further support of these results, we find that ectopic expression of wild-type LKB1 in A549 cells enhanced the BRCA1 mRNA level and increased its half-life (Supplementary Figure S9A and B).
LKB1 stimulates BRCA1 expression independent of cell cycle stage
BRCA1 expression is highest at S and G2/M phases of cell cycle and lowest during the G1 phase of cell cycle in mammalian cells (40). Therefore, we asked if the observed decrease in BRCA1 expression following shRNA-mediated LKB1 knockdown is caused due to the effect of LKB1 loss on cell cycle progression. To determine this, we synchronized the cells in G2/M phase of cell cycle by treating them with nocodazole (Figure 4A) and the nocodazole synchronized cells that either expressed NS shRNA or shRNAs against LKB1 were checked for BRCA1 expression. We find that loss of LKB1 expression in both unsynchronized and synchronized cells leads to decrease in BRCA1 levels (Figure 4B). Collectively, these results show that the ability of LKB1 to stimulate BRCA1 expression is independent of cell cycle stage.
Figure 4.
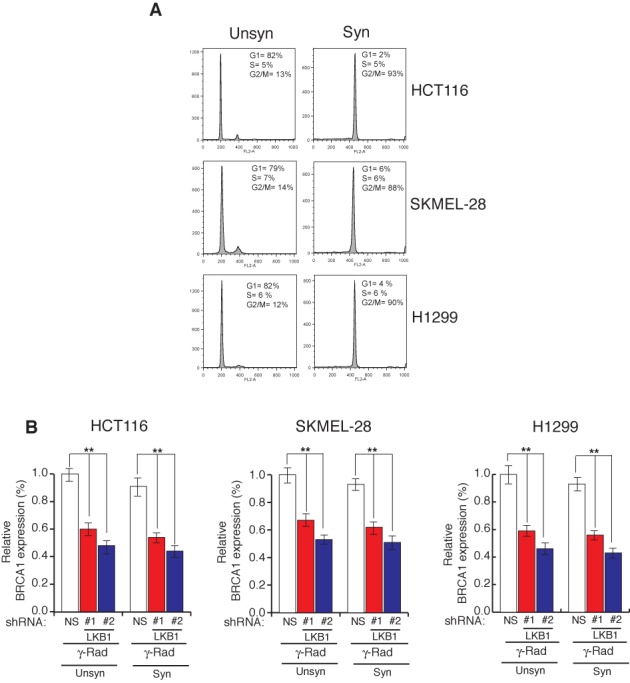
LKB1 regulates BRCA1 expression independent of cell cycle and cells lacking LKB1 show reduced homology-directed DNA repair. Indicated cell lines were treated with 200 ng/ml of nocodazole for 16 h for synchronization in G2/M phase. (A) Histograms and relative percentage of indicated cell cycle stages for unsynchronized and synchronized cells for indicated cell lines are shown. (B) Indicated cell lines expressing LKB1 shRNAs or a non-specific (NS) shRNA were gamma irradiated (20G) and either unsynchronized or synchronized using nocodazole. BRCA1 mRNA level was measured by RT-qPCR. Relative BRCA1 expression in relation to unsynchronized cells expressing NS shRNA is plotted. Error bars show standard error mean (SEM). (**P < 0.001).
LKB1 regulates BRCA1 by inhibiting the cytoplasmic localization of HuR via AMPK
Several mechanisms regarding the regulation of mRNA stability have been identified and proposed. Importantly, the association of RNA-binding proteins to the 3′-UTR (Untranslated region) of specific mRNAs has been shown to regulate mRNA abundance and translation (41,42). Interestingly, an RNA binding protein Hu Antigen R (HuR) has been shown to bind to the 3′-UTR of BRCA1 mRNA (43). HuR is an important prognostic marker in BRCA1-mutant breast cancers (44). The nuclear-cytoplasmic shuttling of HuR is the central mechanism by which its function is regulated (45,46), and it is predominately localized to the cytoplasm in various cancers, where it binds to different mRNAs to regulate their stability and/or translation and promote cancer (45,46). The cytoplasmic expression of HuR is associated with increased invasiveness and poor prognosis in many cancers (45,46), and its localization to the cytoplasm is blocked by AMPK (47–50). Therefore, we investigated whether LKB1 regulates BRCA1 stability by inhibiting the cytoplasmic localization of HuR in an AMPK-dependent manner. We find that the cells expressing shRNAs against LKB1 show a significantly higher accumulation of HuR in the cytoplasm, compared with cells expressing an NS shRNA (Figure 5A and Supplementary Figures S8C and S10). Notably, treatment of the LKB1 shRNA-expressing cells with the AMPK agonist, 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR), reduced the cytoplasmic levels of HuR and stabilized BRCA1 mRNA (Figure 5B and Supplementary Figure S8D). Similar to AICAR treatment, ectopic expression of wild-type LKB1 in A549 reduced cytoplasmic levels of HuR compared to the A549 cells expressing LKB1 kinase dead mutant or empty vector (Supplementary Figure S9C). Furthermore, BRCA1 expression was analyzed in cells in which the expression of both LKB1 and HuR was simultaneously knocked down using shRNAs (Supplementary Figure S11A). The shRNA-mediated knockdown of HuR expression in cells simultaneously expressing LKB1 shRNA restored BRCA1 levels (Figure 5C and Supplementary Figure S12), counteracted the sensitivity of cells to gamma irradiation (Figure 6A and Supplementary Figure S11B), reduced the number of γH2AX foci (Figure 6B and Supplementary Figures S11C and S13) and lowered the mutation rate (Figure 6C and Supplementary Figure S11D). Collectively, these results show that, in the absence of LKB1, HuR localizes to the cytoplasm in an AMPK-dependent manner, where it targets BRCA1 mRNA for degradation.
Figure 5.
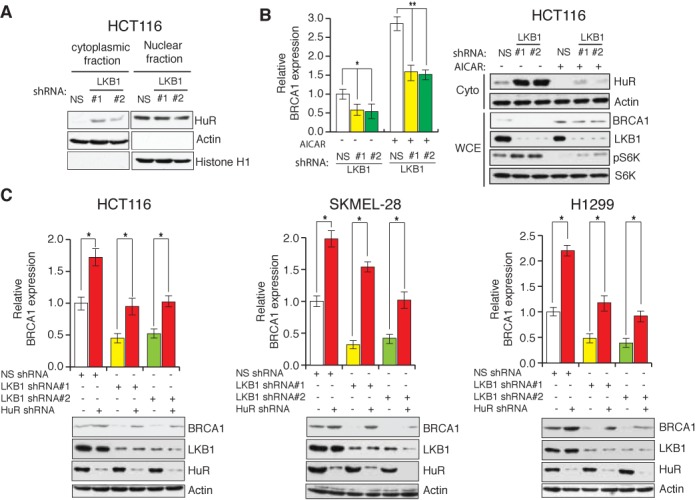
LKB1 stimulates BRCA1 expression by regulating the cytoplasmic localization of HuR. (A) Nuclear and cytoplasmic fractions were generated from HCT116 cells expressing LKB1 shRNAs or a non-specific (NS) shRNA, and the indicated proteins in each of the fractions were analyzed by immunoblotting. (B) HCT116 cells expressing LKB1 shRNAs or NS shRNA were left untreated or treated with AICAR (1.5 mM for 24 h). (Left) BRCA1 mRNA levels were measured by RT-qPCR analysis and (right) HuR protein and actin was analyzed in the cytoplasmic fractions (Cyto) and the other indicated proteins were probed in whole-cell extracts (WCE) by immunoblot analysis. (C) Cell lines expressing LKB1 shRNAs, HuR shRNA or the NS shRNA were analyzed for BRCA1 mRNA and indicated protein levels via RT-qPCR (top) or immunoblot analysis (bottom), respectively. Error bars show standard error mean (SEM). (*P < 0.01; **P < 0.001).
Figure 6.
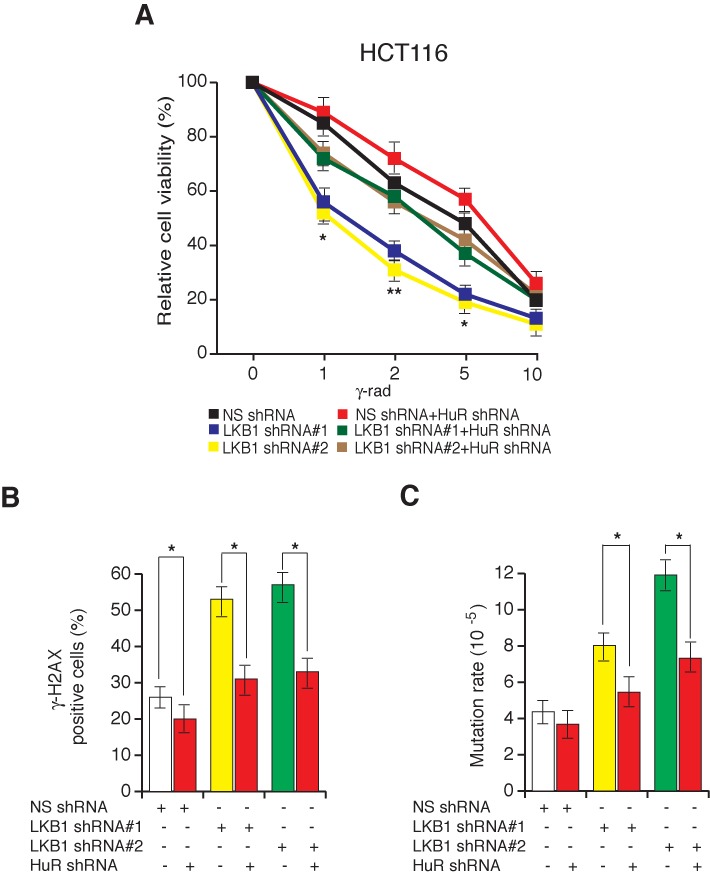
Simultaneous shRNA-mediated knockdown of LKB1 and HuR partly restores genome integrity. (A) HCT116 cells expressing the indicated shRNAs were gamma-irradiated at indicated doses, and cell viability was measured by the trypan blue exclusion assay 48 h post-irradiation. The cell viability relative to the unirradiated control is plotted. (B) HCT116 cell lines expressing the indicated shRNAs were gamma-irradiated (2G). At 6 h post-irradiation, cells were stained for γH2AX. Percentages of γH2AX-positive cells are plotted. (C) Spontaneous mutation of HPRT was measured in HCT116 cells expressing the indicated shRNAs. Mutation rates under the indicated conditions are plotted. Error bars show standard error mean (SEM). (*P < 0.01; **P < 0.001).
Ectopic expression of BRCA1 is sufficient to overcome the loss of the LKB1-mediated sensitization to genotoxic stress
Finally, we asked whether increased sensitivity of cells expressing LKB1 shRNA to genotoxic stress is due to the ability of LKB1 to regulate BRCA1 expression. To determine this, we first depleted the BRCA1 mRNA using shRNAs in HCT116 cells (Supplementary Figure S14A). We observed that cells expressing BRCA1 shRNAs were more sensitive to genotoxic stress (Supplementary Figure S14B), displayed increased DNA double-strand breaks (Supplementary Figure S14C) and accumulated more mutations (Supplementary Figure S14D), compared with that of NS shRNA-expressing cells. These results indicate that BRCA1 loss phenocopies the loss of LKB1. To conclusively determine the role of BRCA1 in mediating the genoprotective effect of LKB1, we knocked down the expression of LKB1 in HCT116 cells and then ectopically expressed BRCA1 (Supplementary Figure S15). Remarkably, the re-expression of BRCA1 largely rescued the sensitivity to genotoxic stress (Figure 7A and B and Supplementary Figure S16), reduced the number of DNA double-strand breaks (Figure 7C and Supplementary Figure S17) and lowered the mutation rate (Figure 7D) caused by the shRNA-mediated inhibition of LKB1 expression. Similar results were obtained by using LKB1 knockout MEFs (Supplementary Figure S18A and B). Collectively, these results confirm that LKB1 regulates BRCA1 mRNA stability to protect cells from the harmful effects of genotoxic stress and promotes genome stability.
Figure 7.
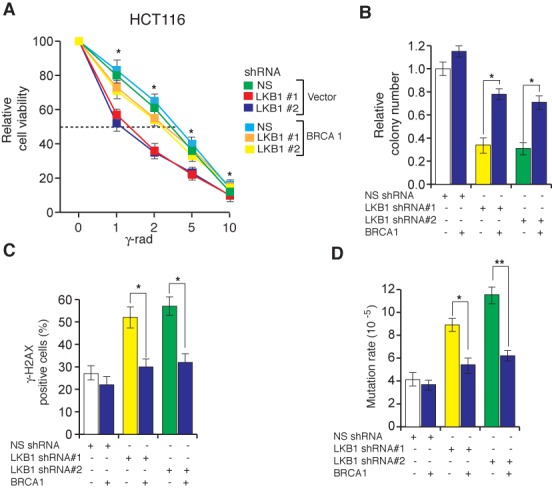
Ectopic expression of BRCA1 counteracts the LKB1 loss-mediated sensitivity to genotoxic stress and mutagenesis. (A) HCT116 cells with indicated shRNAs expressing either empty vector or BRCA1 were gamma-irradiated at the indicated doses. At 48 h post-irradiation, cell viability was measured by the trypan blue exclusion assay. Relative cell viability is plotted in reference to unirradiated cells. (B) HCT116 cell lines expressing LKB1 shRNAs or a non-silencing (NS) shRNA with or without BRCA1 were gamma irradiated (2G) and plated on 6-well plates. Relative colony numbers 2 weeks after gamma irradiation in comparison to NS shRNA are presented. (C) HCT116 cell lines expressing LKB1 shRNAs or a non-silencing (NS) shRNA with or without BRCA1 were gamma-irradiated (2G) and stained for γH2AX 6 h post-irradiation. Percentages of γH2AX positive cells are plotted. (D) Spontaneous mutation of HPRT in HCT116 cells expressing the indicated constructs. The mutation rates under the indicated conditions are plotted. Error bars show standard error mean (SEM). (*P < 0.01; **P < 0.001).
DISCUSSION
In this report, we identify a novel mechanism of LKB1-mediated tumor suppression. An overview of our results is presented in Figure 8 and discussed below. First, we find that LKB1 is necessary for protecting cells from genotoxic stress-induced genome instability. Furthermore, LKB1 stimulates BRCA1 expression by preventing the cytoplasmic localization of the RNA-binding protein, HuR. Finally, we document that the ectopic expression of BRCA1 protects cells from LKB1 loss-induced genome instability. Collectively, these findings demonstrate that LKB1 preserve genome stability by stimulating BRCA1 expression. These results also provide an attractive model to explain LKB1-loss-mediated increased predisposition to cancer, because LKB1 loss leads to increased accumulation of possible carcinogenic mutations in the genome.
Figure 8.
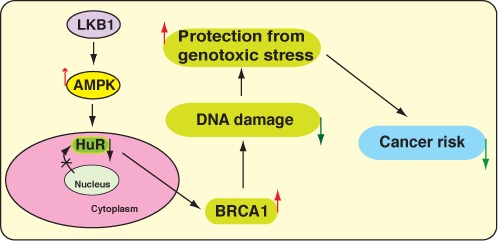
Model of the LKB1-mediated regulation of genome integrity. LKB1 prevents the cytoplasmic localization of the RNA-binding protein, HuR, in an AMPK-dependent manner. This, in turn, stimulates BRCA1 expression, preserves genome instability and subsequently prevents cancer.
LKB1 protects cells from genotoxic stress
The DDR pathway is a complex genetic pathway that is activated when a cell encounters genotoxic stress. A suitable DDR to genotoxic stimuli is essential for maintaining genome integrity, preventing neoplastic transformation and maintaining disease-free survival. In response to DNA damage, LKB1 is phosphorylated at threonine 366 by ATM kinase (51). Furthermore, LKB1 is associated with damaged DNA under certain scenarios and is implicated in the regulation of non-homologous end-joining repair (52). We find that the loss of LKB1 expression sensitizes cells to genotoxic stress and increases genomic instability, as observed by increased mutagenesis. In agreement with our results, a previous study showed that LKB1 deficiency sensitizes mice to a chemical mutagen, 7,12-dimethylbenz(a)anthracene-induced squamous cell carcinomas of the skin and the lung (53).
Regulation of BRCA1 expression by LKB1
BRCA1 is a TSG, and mutations in the BRCA1 gene are known to increase the susceptibility to develop many cancers, including that of the breast, ovary, pancreas and prostate (54–57). BRCA1 is a RING finger E3 ubiquitin ligase that regulates a wide variety of DNA-repair functions (55). LKB1 stimulates BRCA1 expression by regulating the cytoplasmic localization of HuR. Overall, these results for the first time reveal a previously undocumented role of LKB1 in the regulation of DDR by stimulating BRCA1 expression. A previous study has shown that gamma irradiation affects the ability of HuR to bind to its target genes (58). However, there are many differences between this study and the experiments described here. First, previous study was performed in LKB1 wild-type cells in the context of CHK2. Second, we observed increased cytoplasmic localization of HuR upon loss of LKB1 expression and contrary when AMPK is activated pharmacologically by AICAR or when wild-type LKB1 was ectopically expressed. These results show that the major mode of regulation of HuR activity in context of LKB1 is the regulation of the HuR cellular localization. Third, the goal of our studies is not to determine how BRCA1 is stabilized by gamma irradiation and what role HuR plays in this process, but rather to understand how LKB1 regulates HuR function, which, in turn, affects the stability of BRCA1 mRNA. Therefore, for our studies the comparison is between cells that either express or lack LKB1. Collectively, our results show that upon loss of LKB1, HuR is localized to the cytoplasm and downregulates BRCA1 mRNA levels.
Implication of BRCA1 regulation in LKB1-mediated tumor suppression
The loss of LKB1 causes marked resistance to oncogene-induced transformation (14). This finding is paradoxical, because LKB1 is known to be a TSG. An explanation for this observation could be that the introduction of oncogenes, many of which can increase proliferation and DNA damage, may compromise the survival of LKB1-deficient cells due to the increased number of DNA double-strand breaks. Indeed, we find that LKB1 mediates its tumor suppressor activity by regulating the response to genotoxic stress, and inhibition of LKB1 increases the accumulation of DNA double-strand breaks. Our findings provide an alternative model of LKB1-mediated tumor suppression and explain why LKB1 loss may cause resistance to oncogene-induced transformation.
A recent study has identified WEE1 as a HuR target and indicated that it might play an important role in determining the response to DNA damage in pancreatic cancer cells (59). Based on these studies, in the future it will be interesting to comprehensively analyze the role of other HuR targets beyond BRCA1 to determine their role in mediating LKB1-loss-induced sensitization to DNA damage. Additionally, since HuR only partly rescues LKB1-loss-induced sensitization to DNA damage, it is possible that HuR-independent mechanisms exist and will constitute a future direction for further studies.
Therapeutic implications of the LKB1-mediated regulation of the DDR
Genotoxic stress, such as exposure to irradiation (e.g. ultraviolet, ionizing radiation, etc.), is the major environmental factor that causes cancer. Therefore, strategies that would protect cells from genotoxic stress may yield new treatments for preventing cancer. In many scenarios, agents that enhance the protection from genotoxic stress can prevent or delay cancer initiation. Therefore, we anticipate that the use of agents that can enhance LKB1 activity by directly increasing its kinase activity or other downstream effectors, such as AMPK agonists (e.g. metformin), may protect the cells from neoplastic transformation. Finally, our results provide an alternative explanation for why AMPK agonists exert anticancer activities.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Ryan Jensen for useful discussion.
Authors Contributions: R.G. and N.W. designed the experiments. R.G. performed the majority of the experiments with the help of A.Y.L. P.M.G. provided reagents. R.G. and N.W. analyzed and interpreted the data, and R.G. and N.W. co-wrote the manuscript. All authors commented on the manuscript.
FUNDING
Young Investigator Awards from the National Lung Cancer Partnership [to N.W.]; Uniting Against Lung Cancer; International Association for the Study of Lung Cancer; Melanoma Research Alliance, Melanoma Research Foundation; American Lung Association's Biomedical Research Grant; Seed Grant from Hirshberg Foundation for Pancreatic Cancer Research; National Institutes of Health [RO1ES005775 to P.M.G.]. Funding for open access charge: Young Investigator Awards from the National Lung Cancer Partnership [to N.W.]; Uniting Against Lung Cancer; International Association for the Study of Lung Cancer; Melanoma Research Alliance, Melanoma Research Foundation; American Lung Association's Biomedical Research Grant; National Institutes of Health [RO1ES005775 to P.M.G.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D., Weinberg R.A. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B., Papadopoulos N., Velculescu V.E., Zhou S., Diaz L.A., Jr, Kinzler K.W. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones P.A., Baylin S.B. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fearon E.R., Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 6.Lowe S.W., Cepero E., Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–315. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- 7.Hemminki A., Tomlinson I., Markie D., Jarvinen H., Sistonen P., Bjorkqvist A.M., Knuutila S., Salovaara R., Bodmer W., Shibata D., et al. Localization of a susceptibility locus for Peutz-Jeghers syndrome to 19p using comparative genomic hybridization and targeted linkage analysis. Nat. Genet. 1997;15:87–90. doi: 10.1038/ng0197-87. [DOI] [PubMed] [Google Scholar]
- 8.Hemminki A., Markie D., Tomlinson I., Avizienyte E., Roth S., Loukola A., Bignell G., Warren W., Aminoff M., Hoglund P., et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 9.Jeghers H., Mc K.V., Katz K.H. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N. Engl. J. Med. 1949;241:993. doi: 10.1056/NEJM194912222412501. illust; passim. [DOI] [PubMed] [Google Scholar]
- 10.Hemminki A. The molecular basis and clinical aspects of Peutz-Jeghers syndrome. Cell. Mol. Life Sci. 1999;55:735–750. doi: 10.1007/s000180050329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tomlinson I.P., Houlston R.S. Peutz-Jeghers syndrome. J. Med. Genet. 1997;34:1007–1011. doi: 10.1136/jmg.34.12.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Westerman A.M., Entius M.M., de Baar E., Boor P.P., Koole R., van Velthuysen M.L., Offerhaus G.J., Lindhout D., de Rooij F.W., Wilson J.H. Peutz-Jeghers syndrome: 78-year follow-up of the original family. Lancet. 1999;353:1211–1215. doi: 10.1016/s0140-6736(98)08018-0. [DOI] [PubMed] [Google Scholar]
- 13.Giardiello F.M., Brensinger J.D., Tersmette A.C., Goodman S.N., Petersen G.M., Booker S.V., Cruz-Correa M., Offerhaus J.A. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119:1447–1453. doi: 10.1053/gast.2000.20228. [DOI] [PubMed] [Google Scholar]
- 14.Bardeesy N., Sinha M., Hezel A.F., Signoretti S., Hathaway N.A., Sharpless N.E., Loda M., Carrasco D.R., DePinho R.A. Loss of the Lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature. 2002;419:162–167. doi: 10.1038/nature01045. [DOI] [PubMed] [Google Scholar]
- 15.Jishage K., Nezu J., Kawase Y., Iwata T., Watanabe M., Miyoshi A., Ose A., Habu K., Kake T., Kamada N., et al. Role of Lkb1, the causative gene of Peutz-Jegher's syndrome, in embryogenesis and polyposis. Proc. Natl. Acad. Sci. U.S.A. 2002;99:8903–8908. doi: 10.1073/pnas.122254599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyoshi H., Nakau M., Ishikawa T.O., Seldin M.F., Oshima M., Taketo M.M. Gastrointestinal hamartomatous polyposis in Lkb1 heterozygous knockout mice. Cancer Res. 2002;62:2261–2266. [PubMed] [Google Scholar]
- 17.Rossi D.J., Ylikorkala A., Korsisaari N., Salovaara R., Luukko K., Launonen V., Henkemeyer M., Ristimaki A., Aaltonen L.A., Makela T.P. Induction of cyclooxygenase-2 in a mouse model of Peutz-Jeghers polyposis. Proc. Natl. Acad. Sci. U.S.A. 2002;99:12327–12332. doi: 10.1073/pnas.192301399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanchez-Cespedes M., Parrella P., Esteller M., Nomoto S., Trink B., Engles J.M., Westra W.H., Herman J.G., Sidransky D. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002;62:3659–3662. [PubMed] [Google Scholar]
- 19.Rowan A., Bataille V., MacKie R., Healy E., Bicknell D., Bodmer W., Tomlinson I. Somatic mutations in the Peutz-Jeghers (LKB1/STKII) gene in sporadic malignant melanomas. J. Invest. Dermatol. 1999;112:509–511. doi: 10.1046/j.1523-1747.1999.00551.x. [DOI] [PubMed] [Google Scholar]
- 20.Guldberg P., thor Straten P., Ahrenkiel V., Seremet T., Kirkin A.F., Zeuthen J. Somatic mutation of the Peutz-Jeghers syndrome gene, LKB1/STK11, in malignant melanoma. Oncogene. 1999;18:1777–1780. doi: 10.1038/sj.onc.1202486. [DOI] [PubMed] [Google Scholar]
- 21.Su G.H., Hruban R.H., Bansal R.K., Bova G.S., Tang D.J., Shekher M.C., Westerman A.M., Entius M.M., Goggins M., Yeo C.J., et al. Germline and somatic mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers. Am. J. Pathol. 1999;154:1835–1840. doi: 10.1016/S0002-9440(10)65440-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alessi D.R., Sakamoto K., Bayascas J.R. LKB1-dependent signaling pathways. Annu. Rev. Biochem. 2006;75:137–163. doi: 10.1146/annurev.biochem.75.103004.142702. [DOI] [PubMed] [Google Scholar]
- 23.Baas A.F., Boudeau J., Sapkota G.P., Smit L., Medema R., Morrice N.A., Alessi D.R., Clevers H.C. Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. EMBO J. 2003;22:3062–3072. doi: 10.1093/emboj/cdg292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boudeau J., Baas A.F., Deak M., Morrice N.A., Kieloch A., Schutkowski M., Prescott A.R., Clevers H.C., Alessi D.R. MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 2003;22:5102–5114. doi: 10.1093/emboj/cdg490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tiainen M., Vaahtomeri K., Ylikorkala A., Makela T.P. Growth arrest by the LKB1 tumor suppressor: induction of p21(WAF1/CIP1) Hum. Mol. Genet. 2002;11:1497–1504. doi: 10.1093/hmg/11.13.1497. [DOI] [PubMed] [Google Scholar]
- 26.Woods A., Johnstone S.R., Dickerson K., Leiper F.C., Fryer L.G., Neumann D., Schlattner U., Wallimann T., Carlson M., Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 27.Hawley S.A., Boudeau J., Reid J.L., Mustard K.J., Udd L., Makela T.P., Alessi D.R., Hardie D.G. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shaw R.J., Kosmatka M., Bardeesy N., Hurley R.L., Witters L.A., DePinho R.A., Cantley L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. U.S.A. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corradetti M.N., Inoki K., Bardeesy N., DePinho R.A., Guan K.L. Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004;18:1533–1538. doi: 10.1101/gad.1199104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gan B., Hu J., Jiang S., Liu Y., Sahin E., Zhuang L., Fletcher-Sananikone E., Colla S., Wang Y.A., Chin L., et al. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature. 2010;468:701–704. doi: 10.1038/nature09595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu Z.Q., Li X.Y., Hu C.Y., Ford M., Kleer C.G., Weiss S.J. Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc. Natl. Acad. Sci. U.S.A. 2012;109:16654–16659. doi: 10.1073/pnas.1205822109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wajapeyee N., Serra R.W., Zhu X., Mahalingam M., Green M.R. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132:363–374. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santra M.K., Wajapeyee N., Green M.R. F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature. 2009;459:722–725. doi: 10.1038/nature08011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whitfield M.L., Sherlock G., Saldanha A.J., Murray J.I., Ball C.A., Alexander K.E., Matese J.C., Perou C.M., Hurt M.M., Brown P.O., et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell. 2002;13:1977–2000. doi: 10.1091/mbc.02-02-0030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fang M., Xia F., Mahalingam M., Virbasius C.M., Wajapeyee N., Green M.R. MEN1 is a melanoma tumor suppressor that preserves genomic integrity by stimulating transcription of genes that promote homologous recombination-directed DNA repair. Mol. Cell. Biol. 2013;33:2635–2647. doi: 10.1128/MCB.00167-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakanishi K., Cavallo F., Brunet E., Jasin M. Homologous recombination assay for interstrand cross-link repair. Methods Mol. Biol. 2011;745:283–291. doi: 10.1007/978-1-61779-129-1_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Negrini S., Gorgoulis V.G., Halazonetis T.D. Genomic instability—an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 38.Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S., Bonner W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 39.Yan T., Berry S.E., Desai A.B., Kinsella T.J. DNA mismatch repair (MMR) mediates 6-thioguanine genotoxicity by introducing single-strand breaks to signal a G2-M arrest in MMR-proficient RKO cells. Clin. Cancer Res. 2003;9:2327–2334. [PubMed] [Google Scholar]
- 40.Ruffner H., Verma I.M. BRCA1 is a cell cycle-regulated nuclear phosphoprotein. Proc. Natl. Acad. Sci. U.S.A. 1997;94:7138–7143. doi: 10.1073/pnas.94.14.7138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pullmann R., Jr, Kim H.H., Abdelmohsen K., Lal A., Martindale J.L., Yang X., Gorospe M. Analysis of turnover and translation regulatory RNA-binding protein expression through binding to cognate mRNAs. Mol. Cell. Biol. 2007;27:6265–6278. doi: 10.1128/MCB.00500-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hogan D.J., Riordan D.P., Gerber A.P., Herschlag D., Brown P.O. Diverse RNA-binding proteins interact with functionally related sets of RNAs, suggesting an extensive regulatory system. PLoS Biol. 2008;6:e255. doi: 10.1371/journal.pbio.0060255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saunus J.M., French J.D., Edwards S.L., Beveridge D.J., Hatchell E.C., Wagner S.A., Stein S.R., Davidson A., Simpson K.J., Francis G.D., et al. Posttranscriptional regulation of the breast cancer susceptibility gene BRCA1 by the RNA binding protein HuR. Cancer Res. 2008;68:9469–9478. doi: 10.1158/0008-5472.CAN-08-1159. [DOI] [PubMed] [Google Scholar]
- 44.Heinonen M., Fagerholm R., Aaltonen K., Kilpivaara O., Aittomaki K., Blomqvist C., Heikkila P., Haglund C., Nevanlinna H., Ristimaki A. Prognostic role of HuR in hereditary breast cancer. Clin. Cancer Res. 2007;13:6959–6963. doi: 10.1158/1078-0432.CCR-07-1432. [DOI] [PubMed] [Google Scholar]
- 45.Srikantan S., Gorospe M. HuR function in disease. Front. Biosci. (Landmark Ed) 2012;17:189–205. doi: 10.2741/3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J., Guo Y., Chu H., Guan Y., Bi J., Wang B. Multiple functions of the RNA-binding protein HuR in cancer progression, treatment responses and prognosis. Int. J. Mol. Sci. 2013;14:10015–10041. doi: 10.3390/ijms140510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang W., Fan J., Yang X., Furer-Galban S., Lopez de Silanes I., von Kobbe C., Guo J., Georas S.N., Foufelle F., Hardie D.G., et al. AMP-activated kinase regulates cytoplasmic HuR. Mol. Cell. Biol. 2002;22:3425–3436. doi: 10.1128/MCB.22.10.3425-3436.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang W., Yang X., Lopez de Silanes I., Carling D., Gorospe M. Increased AMP:ATP ratio and AMP-activated protein kinase activity during cellular senescence linked to reduced HuR function. J. Biol. Chem. 2003;278:27016–27023. doi: 10.1074/jbc.M300318200. [DOI] [PubMed] [Google Scholar]
- 49.Wang W., Yang X., Kawai T., Lopez de Silanes I., Mazan-Mamczarz K., Chen P., Chook Y.M., Quensel C., Kohler M., Gorospe M. AMP-activated protein kinase-regulated phosphorylation and acetylation of importin alpha1: involvement in the nuclear import of RNA-binding protein HuR. J. Biol. Chem. 2004;279:48376–48388. doi: 10.1074/jbc.M409014200. [DOI] [PubMed] [Google Scholar]
- 50.Martinez-Chantar M.L., Vazquez-Chantada M., Garnacho M., Latasa M.U., Varela-Rey M., Dotor J., Santamaria M., Martinez-Cruz L.A., Parada L.A., Lu S.C., et al. S-adenosylmethionine regulates cytoplasmic HuR via AMP-activated kinase. Gastroenterology. 2006;131:223–232. doi: 10.1053/j.gastro.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 51.Sapkota G.P., Deak M., Kieloch A., Morrice N., Goodarzi A.A., Smythe C., Shiloh Y., Lees-Miller S.P., Alessi D.R. Ionizing radiation induces ataxia telangiectasia mutated kinase (ATM)-mediated phosphorylation of LKB1/STK11 at Thr-366. Biochem. J. 2002;368:507–516. doi: 10.1042/BJ20021284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ui A., Ogiwara H., Nakajima S., Kanno S., Watanabe R., Harata M., Okayama H., Harris C.C., Yokota J., Yasui A. Possible involvement of LKB1-AMPK signaling in non-homologous end joining. Oncogene. 2013;33:1640–1648. doi: 10.1038/onc.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gurumurthy S., Hezel A.F., Sahin E., Berger J.H., Bosenberg M.W., Bardeesy N. LKB1 deficiency sensitizes mice to carcinogen-induced tumorigenesis. Cancer Res. 2008;68:55–63. doi: 10.1158/0008-5472.CAN-07-3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li M.L., Greenberg R.A. Links between genome integrity and BRCA1 tumor suppression. Trends Biochem. Sci. 2012;37:418–424. doi: 10.1016/j.tibs.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Silver D.P., Livingston D.M. Mechanisms of BRCA1 tumor suppression. Cancer Discov. 2012;2:679–684. doi: 10.1158/2159-8290.CD-12-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elledge S.J., Amon A. The BRCA1 suppressor hypothesis: an explanation for the tissue-specific tumor development in BRCA1 patients. Cancer Cell. 2002;1:129–132. doi: 10.1016/s1535-6108(02)00041-7. [DOI] [PubMed] [Google Scholar]
- 57.Elia A.E., Elledge S.J. BRCA1 as tumor suppressor: lord without its RING. Breast Cancer Res. 2012;14:306. doi: 10.1186/bcr3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Masuda K., Abdelmohsen K., Kim M.M., Srikantan S., Lee E.K., Tominaga K., Selimyan R., Martindale J.L., Yang X., Lehrmann E., et al. Global dissociation of HuR-mRNA complexes promotes cell survival after ionizing radiation. EMBO J. 2011;30:1040–1053. doi: 10.1038/emboj.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lal S., Burkhart R.A., Beeharry N., Bhattacharjee V., Londin E.R., Cozzitorto J.A., Romeo C., Jimbo M., Norris Z.A., Yeo C.J., et al. HuR posttranscriptionally regulates WEE1: implications for the DNA damage response in pancreatic cancer cells. Cancer Res. 2014;74:1128–1140. doi: 10.1158/0008-5472.CAN-13-1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.