


Abstract
Altering zinc bioavailability to bacterial pathogens is a key component of host innate immunity. Thus, the ability to sense and adapt to the alterations in zinc concentrations is critical for bacterial survival and pathogenesis. To understand the adaptive responses of group A Streptococcus (GAS) to zinc limitation and its regulation by AdcR, we characterized gene regulation by AdcR. AdcR regulates the expression of 70 genes involved in zinc acquisition and virulence. Zinc-bound AdcR interacts with operator sequences in the negatively regulated promoters and mediates differential regulation of target genes in response to zinc deficiency. Genes involved in zinc mobilization and conservation are derepressed during mild zinc deficiency, whereas the energy-dependent zinc importers are upregulated during severe zinc deficiency. Further, we demonstrated that transcription activation by AdcR occurs by direct binding to the promoter. However, the repression and activation by AdcR is mediated by its interactions with two distinct operator sequences. Finally, mutational analysis of the metal ligands of AdcR caused impaired DNA binding and attenuated virulence, indicating that zinc sensing by AdcR is critical for GAS pathogenesis. Together, we demonstrate that AdcR regulates GAS adaptive responses to zinc limitation and identify molecular components required for GAS survival during zinc deficiency.
INTRODUCTION
Zinc is an essential nutrient for bacterial growth and pathogenesis (1). Zinc is a critical cofactor for protein folding and catalytic activity of various enzymes (2). However, when in excess, zinc can also be toxic as it competes for non-cognate metal binding sites in metalloproteins and renders them dysfunctional (3). The host can exploit this paradoxical nutritional requirement of bacteria to impede their growth by altering the amount of zinc available to the invading pathogens (4,5). Depending on the host microenvironment and the pathogen, different molecular strategies are employed to either sequester zinc from the microbial population or poison the pathogen with toxic concentrations of zinc (4,6–10). Infiltrating neutrophils at the bacterial colonization surface produce large amounts of the secreted protein calprotectin. Extracellular calprotectin sequesters zinc and manganese from the infection site and deprives the pathogen of these critical cofactors (4,5,7,11). Conversely, macrophages inundate phagocytosed pathogens (e.g. pneumococci and Mycobacterium tuberculosis) with bactericidal zinc levels as part of their protective immune response (8,12,13). Bacterial countermeasures against these host defense mechanisms include tight coordination of the expression of zinc uptake and efflux systems in response to varying zinc concentrations (3,14). Bacterial adaptive responses to zinc starvation include the upregulation of zinc import systems, zinc sparing responses and mobilization of intracellular zinc storage (15). In most bacteria, these counterstrategies to host-mediated zinc limitation are controlled by the metal-sensing transcription regulator, zinc uptake regulator (Zur). However, in streptococci, similar responses are controlled by the structurally dissimilar adhesin-competence regulator, AdcR (3,16).
Group A Streptococcus (GAS, Streptococcus pyogenes) is an exclusive human pathogen and a causative agent for a variety of diseases that range from mild pharyngitis to complicated necrotizing fasciitis (17). Given the abundance of neutrophils at the GAS colonization interface (18), it is likely that GAS faces zinc limitation within the host. Consistent with this, genes encoding zinc uptake systems, zinc storage systems and zinc sparing responses are significantly upregulated during subcutaneous GAS infection (19). A recent study also demonstrated that internalization of GAS by neutrophils triggers a rapid influx of zinc into the cytoplasm, and a gene encoding a zinc efflux pump, czcD, is critical for GAS survival against neutrophil-mediated zinc cytotoxicity (10). The significance of zinc homeostasis systems to GAS pathogenesis is further underscored by the finding that the metal transporters, both uptake and efflux systems, are critical for GAS virulence and inactivation of these genes results in significantly attenuated strains (10,20). Together, these data indicate that GAS encounters host-mediated zinc defense mechanisms during infection and the components of the zinc homeostasis machinery contribute to GAS survival and virulence.
Adaptive responses to zinc limitation in streptococci are controlled by AdcR (21). AdcR regulators form a subclass of multiple antibiotic resistance family regulators (MarR) that use metal ions as co-repressors (21). AdcR regulators sense the alterations in intracellular zinc concentrations and negatively regulate the expression of genes involved in metal uptake, metal storage and metal-sparing responses (21,22). Although adcR has been identified in most streptococci, AdcR from S. pneumoniae is the best studied. The pneumococcal adc regulon is comprised of ∼15 genes, most of which are implicated in zinc uptake and storage (21). Genes regulated by AdcR in S. pneumoniae include an ATP-binding cassette (ABC)-family high affinity transporter (adcABC), an extracellular zinc-binding protein (adcAII) and putative zinc storage proteins, known as polyhistidine triad proteins (phtA, phtD, phtE and phpA) (21). Both AdcA and AdcAII are critical for zinc uptake, albeit by different mechanisms, and inactivation of either gene significantly attenuates pneumococcal virulence (23). Similar studies in GAS show that AdcAII, also known as Lsp, plays a critical role in zinc procurement, as deletion of adcAII results in defective growth in zinc-limited conditions and attenuates GAS virulence (20). The Pht proteins are implicated in streptococcal zinc homeostasis as zinc scavengers, in immune evasion by preventing complement deposition and in host cell attachment by unknown mechanisms (24,25). Collectively, these data indicate that AdcR is the master regulator of adaptive responses to zinc limitation and AdcR-dependent gene regulation plays a critical role in pneumococcal pathogenesis.
AdcR is a homodimeric DNA-binding protein that has two functional domains: a DNA-binding domain with winged helix-turn-helix (wHTH) motif and a dimerization domain (22). The zinc binding sites are located in the interdomain region of AdcR, and metal occupancy produces conformation changes that enable DNA binding (22). Thus, when sufficient zinc is available, the metallated form of AdcR binds operator sequences (adc motif) in AdcR-regulated promoters (21) resulting in repression of gene expression. Conversely, apo-AdcR dissociates from the promoter sequences resulting in derepression of the target genes. Structurally, AdcR from S. pneumoniae consists of a central domain with the wHTH motif and a dimerization domain with the N- and C-terminal helices forming the dimer interface (22). The crystal structure has two zinc molecules bound to AdcR within the interdomain region (22). Although the regulatory role of the primary site has been identified, the role of second site remains unknown. Much of what is known regarding AdcR structure is based on pneumococci, but the role of AdcR in zinc metalloregulation, the breadth of the GAS adc regulon and the contribution of AdcR-dependent gene regulation to GAS pathogenesis remain largely unknown.
To elucidate the molecular and mechanistic basis of zinc metalloregulation by AdcR in GAS, we performed genetic, biochemical and animal infection studies to characterize AdcR from a GAS serotype M3 strain. Transcriptome analyses using an isogenic ΔadcR mutant determined that AdcR influences the expression of a much larger regulon in GAS compared to that of pneumococci and identified the genes involved in the GAS adaptive response to zinc limitation. We also demonstrated that AdcR negatively regulates gene expression by directly binding promoter sequences containing the adc motif. Interestingly, our data revealed a novel role for AdcR as a transcriptional activator responsible for upregulation of the operon encoding the genes involved in GAS capsule biosynthesis. Finally, using a mouse model of infection, we demonstrated that the metal sensing and gene regulation functions of AdcR are critical for GAS pathogenesis in vivo.
MATERIALS AND METHODS
Ethics statement
Mouse experiments were performed according to protocols approved by the Methodist Hospital Research Institute Institutional Animal Care and Use Committee. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of laboratory Animals, eighth edition. The protocol was approved by the Institutional Animal Care and Use Committee of TMHRI (OLAW assurance no A4555–01; USDA assurance no 740R-0192). No surgery was performed. All efforts were made to minimize animal suffering.
Bacterial strains, plasmids and growth conditions
Bacterial strains and plasmids used in this study are listed in Supplementary Table S1. Strain MGAS10870 is a fully sequenced invasive serotype M3 isolate that has wild-type (WT) sequence for all major regulatory genes (26). GAS was grown routinely on Trypticase Soy agar containing 5% sheep blood (bovine serum albumin; Becton Dickinson) or in Todd–Hewitt broth containing 0.2% (w/v) yeast extract (THY; DIFCO). When required, ampicillin or chloramphenicol was added to a final concentration of 80 and 8 μg/ml, respectively. All GAS growth experiments were done in triplicate on three separate occasions. Overnight cultures were inoculated into fresh medium to achieve an initial OD600 of < 0.1. Growth was monitored by measuring optical density at A600. The Escherichia coli strain used for protein overexpression was grown in Luria–Bertani broth (LB broth; Fisher Scientific) and when appropriate ampicillin was added to a final concentration of 80 μg/ml.
Construction of adcR-inactivated mutant strain
A three-step process was used to generate a polymerase chain reaction (PCR) fragment in which 414 bases of the adcR open reading frame (ORF) were removed. Primer pair adcR-For and adcRKO-Rev were used to generate a 1060-bp fragment including the first 15 bases of the adcR ORF. Likewise, primer pair adcR-Rev and adcRKO-For were used to generate a 1151-bp fragment containing the last 15 bases of the adcR ORF. The two PCR products were subsequently used to generate a 2171-bp product containing an in-frame deletion of adcR using primer pair adcR-For and adcR-Rev. The final PCR product was subsequently cloned into the temperature-sensitive E. coli—Gram-positive shuttle vector pJL1055 (27,28) and used for allelic replacement as previously described (29,30). Inactivation of the gene was confirmed by DNA sequencing. Primers used for the construction of strain 10870ΔadcR are listed in Supplementary Table S2.
Construction of adcR trans-complementation plasmid
To complement isogenic mutant strain ΔadcR, the coding sequence of the full-length adcR gene together with the putative adcR promoter region were cloned into the E. coli-GAS shuttle vector pDC123 (31). Using the primers listed in Supplementary Table S2, the respective fragments were amplified by PCR from GAS genomic DNA, digested with BglII and NdeI and ligated into digested vector to generate the complementation plasmid, pDC-adcR. The inserts were verified by DNA sequencing, and pDC-adcR was electroporated into the strain MGAS10870-ΔadcR.
RNA sequencing (RNA-seq)
GAS strains were grown to late exponential phase of growth (A600 ∼ 1.0) in THY broth and cells were were harvested by centrifugation. RNA isolation and purification were performed using an RNeasy (Qiagen) mini kit according to the manufacturer's protocol and treated with DNase using the TURBO DNA-free kit (Ambion). RNA was analyzed for quality and concentration with an Agilent 2100 Bioanalyzer. The ribosomal RNA was then removed using a Ribo-Zero treatment kit (Epicenter) according to manufacturer's protocol and further purified using the Min-Elute RNA purification kit (Qiagen). The ribosomally depleted RNA was then used to synthesize adaptor-tagged cDNA libraries using the ScriptSeq V2 RNA-seq library preparation kit (Epicenter). cDNA libraries were then run on a MiSeq using the MiSeq v2 reagent kit (Illumina). Approximately 2 million reads were obtained per sample and the reads were mapped to the MGAS315 genome using the CLC-Genomics WorkBench, version 5 (CLC Bio). For RNA-seq analysis, the total number of reads per gene between the replicates was normalized by RPKM [(reads/kilobase of gene)/(million reads aligning to the genome)]. Using the RPKM values, pair-wise comparisons were carried out between the two strains to identify the differentially expressed genes. Genes with 2-fold difference and P < 0.05 after applying Bonferroni's correction considered to be statistically significant.
Transcript analysis by quantitative reverse transcriptase-PCR (qRT-PCR)
GAS strains were grown to late exponential phase (A600 ∼ 1.0) and incubated with two volumes of RNAprotect (Qiagen) for 10 min at room temperature. Bacteria were harvested by centrifugation and the cell pellets were snap frozen with liquid nitrogen. RNA isolation and purification were performed using an RNeasy kit (Qiagen). Purified RNA was analyzed for quality and concentration with an Agilent 2100 Bioanalyzer. cDNA was synthesized from the purified RNA using Superscript III (Invitrogen) and qRT-PCR was performed with an ABI 7500 Fast System (Applied Biosystems). Comparison of transcript levels was done using ΔCT method of analysis using tufA as the endogenous control gene (32). The Taqman primers and probes used are listed in Supplementary Table S2.
Electrophoretic mobility shift assay
Oligonucleotides containing the putative adc motif from the adcRCB promoter (top strand 5′-GTTATAATTAACTGGTTAATGAAAGA) were annealed by heating equimolar mixture of top and bottom strand oligonucleotides at 95°C for 5 min followed by slow cooling to room temperature. Binding reactions were carried out in 20 μl volume of binding buffer (20 mM Tris pH 7.5, 0.8 M NaCl, 12% glycerol and 100 μM Zinc sulfate) containing 0.5 μM of oligoduplex, and increasing concentrations of AdcR. After 15-min incubation at 37°C, the reaction mixtures were resolved on a 10% native polyacrylamide gel supplemented with 5% glycerol (native polyacrylamide gel electrophoresis (PAGE)) for 90 min at 100 V at 4°C in Tris Borate buffer with 5% glycerol. The gels were stained with ethidium bromide and analyzed on a BioRad Gel Electrophoresis Systems.
Protein overexpression and purification
The adcR gene of strain MGAS10870 was cloned into plasmid pET-21b and protein was overexpressed in E. coli strain BL21 (DE3) after induction with 1 mM IPTG at 30°C for 5 h. Cell pellets were suspended in 50 ml of buffer A (20 mM Tris HCl pH 8.0, 100 mM NaCl and 1 mM Tris 2-carboxyethyl phosphine hydrochloride (TCEP)) supplemented with DNaseI to a final concentration of 5 μg/ml. Cells were lysed by a cell disruptor (Constant Systems) and cell debris was removed by centrifugation at 15 000 revolutions per minute for 30 min. AdcR was purified by affinity chromatography using a Ni-NTA agarose column. Purified AdcR was subjected to two rounds of overnight dialysis against buffer containing 10 mM ethylenediaminetetraacetic acid (EDTA) and one more round of dialysis against buffer containing 100 mM EDTA to remove any trace metals bound to the protein. All subsequent steps were carried out with buffer treated with chelex beads (Sigma). The concentrated AdcR was further purified by size exclusion chromatography with a Superdex 200G column and stored in buffer B (20 mM Tris HCl pH 8.0, 200 mM NaCl, 1 mM TCEP and 1mM EDTA). The protein was purified to >95% homogeneity and concentrated to a final concentration of ∼65 mg/ml. To reconstitute the AdcR:Zn form of AdcR, the apo AdcR was dialyzed overnight against chelexed buffer A containing 50 μM ZnSO4.
Modeling studies of AdcR
The structure of AdcR was modeled using the I-TASSER structure prediction server (http://zhanglab.ccmb.med.umich.edu/I-TASSER). The top template identified by I-TASSER to model AdcR was AdcR from S. pneumoniae (22). The algorithm for structure prediction by I-TASSER involves template identification by threading alignments, assembly by iterative simulations and refinement of the resulting model (33,34). The accuracy of the predicted model is validated by two parameters: the confidence score (c-score) and the template modeling score (TM-score) (33,34). The predicted AdcR model has a high c-score of 0.41, indicating the accuracy of the prediction, and a very reliable TM-score of 0.77, pointing to the topological match of the model to the template. The resulting model covers all AdcR residues. The AdcR dimer was generated by superimposition of individual subunits of AdcR to the pneumococcal AdcR dimer using COOT (35).
Site-directed mutagenesis of recombinant AdcR
Plasmid pET21b-adcR or pDC-adcR containing the WT adcR-coding region was used as template for the site-directed mutagenesis. Quick change site-directed mutagenesis kit (Strategene) was used to introduce single amino acid substitutions within adcR coding region and substitutions were confirmed by DNA sequencing.
Mouse infection studies
Virulence of the isogenic mutant GAS strains was tested using an intraperitoneal mouse model of infection. Twenty female 3 to 4-week-old CD1 mice (Harlan Laboratories) were used for each GAS strain. Animals were inoculated intraperitoneally with 1 × 107 colony-forming units (CFUs) and survival was monitored daily. Data were graphically displayed as a Kaplan–Meier survival curve and analyzed using the log-rank test.
RESULTS AND DISCUSSION
GAS adaptive responses to zinc limitation
To elucidate the molecular components of GAS adaptive responses to zinc limitation, we compared the transcription profile of serotype M3 strain MGAS10870 grown in zinc-depleted and zinc-replete conditions by RNA-seq. We used chemically defined medium (CDM) with no zinc supplementation to mimic zinc limitation. Since the GAS growth medium THY contains 10–15 μM zinc and the level of AdcR-mediated repression of target gene expression is unaffected within this range (Supplementary Figure S1A) (36), we used the CDM supplemented with 15 μM zinc to mimic the zinc replete condition. GAS grown under either of these conditions did not show any differences in growth phenotype (Supplementary Figure S1B). Briefly, cells were grown to late exponential phase (A600 ∼ 1.0) and RNA isolation and cDNA library preparation was performed using commercial protocols. For the purpose of statistical analysis, three biological replicates were used for each growth condition. Transcript levels of GAS genes with ≥2-fold changes with statistical significance (P < 0.05) are summarized in Supplementary Table S3. As expected, GAS senses zinc bioavailablity and upregulates (≥3-fold) the expression of genes involved in zinc uptake (adcA,adcAII and adcBC), zinc sparing responses (rpsN) and zinc storage systems (phtD and phtY) (Supplementary Table S3). Additional genes that are differentially regulated in response to zinc limitation include the operon encoding dipeptide permeases (dppA-E) and manganese uptake systems (mtsABC) (Supplementary Table S3). Since most of these genes are implicated in zinc homeostasis in streptococci, and the promoter sequences of adcA,adcAII,adcRBC,rpsN,phtD and phtY contain putative AdcR-binding sites in their promoter sequences, it is likely that the GAS adaptive responses to zinc limitation are predominantly regulated by AdcR.
AdcR regulates zinc metabolism and virulence in GAS
To investigate the global regulatory influence of AdcR in GAS and define the broader regulon controlled by AdcR, we constructed an in-frame adcR deletion in parental serotype M3 strain MGAS10870 (ΔadcR). Since adcR is the first gene of the adcRCB operon, disruption of adcR in the isogenic ΔadcR mutant strain could cause polar effect and alter expression of the downstream genes. To rule out this possibility, we measured the transcript levels of adcC, the second gene in the adcRCB operon, by qRT-PCR assay. Consistent with its role as a repressor, inactivation of adcR gene resulted in the upregulation of adcC gene compared to the parental serotype, indicating the absence of polar effect in ΔadcR (Supplementary Figure S2).
Comparative genome-wide transcription profiling of parental strain MGAS10870 and ΔadcR strains was performed by RNA-seq. AdcR exerts significant influence on GAS gene expression: a total of 70 genes were significantly regulated (∼4% of the 1951 predicted genes in the genome), with 31 genes repressed and 39 genes upregulated by AdcR (Supplementary Tables S4 and S5). Out of the 31 negatively regulated genes, the promoters of 15 genes contain AdcR operator sequences, suggesting that these genes are under direct regulation by AdcR. As observed in the pneumococcal transcriptome studies, the negatively regulated genes include the ABC zinc uptake system (adcBC), two outer membrane zinc-binding proteins (adcA and adcAII) and polyhistidine triad proteins (phtD and phtY) (Supplementary Table S4). In S. pneumoniae, the extracellular zinc-binding protein (AdcA) is co-transcribed as part of adcRABC operon. In GAS, however, the adcA gene is not in the adcRBC operon and encoded elsewhere on the GAS genome. Although the physiological significance of this uncoupling of adcA from the adcRCB operon in GAS is not clear, it results in a higher level of induction of adcA gene in GAS (15-fold in GAS compared to 8-fold in pneumococci). Genes encoding the two Pht proteins, phtD gene (SpyM3_1724), which is co-transcribed as a bicistronic transcript with adcAII, and the phtY gene (SpyM3_1035) were also highly regulated (Supplementary Table S4). The pneumococcal genome encodes for 4 Pht proteins and the expression of all 4 pht genes is controlled by AdcR. Interestingly, the GAS genome also contains four copies of predicted pht genes (SpyM3_1724, SpyM3_1035, SpyM3_0072 and SpyM3_1504), but only two of them are under the control of AdcR. This was further corroborated by the promoter analysis, which indicated that the promoter sequences of both spyM3_0072 and spyM3_1504 lack AdcR operator sequences, and are therefore not likely targets of AdcR-mediated regulation.
Although the adc regulon in GAS shares similarities with its pneumococcal counterpart, the regulatory circuit controlled by AdcR in GAS is much larger and contains several additional components. Importantly, the transcript levels of rpsN (spyM3_1615), the gene encoding the zinc-free paralog for 30S ribosomal protein S14, was increased in the ΔadcR strain (Supplementary Table S4). Most bacteria encode two sets of ribosomal proteins: zinc-containing and their zinc-free paralogs (37). During zinc starvation, the AdcR-mediated repression of zinc-free paralogs is relieved and results in the upregulation of non-zinc containing paralogs (37–39). The zinc-free paralogs replace their zinc-bound counterparts from the ribosome and this process of zinc recycling constitutes the zinc sparing or zinc mobilization responses (37–39). Further, the genes encoding alcohol dehydrogenases (adh) (spyM3_0036 and spyM3_0037) were significantly upregulated in the ΔadcR strain (Supplementary Table S4). This is contrary to the observations made in pneumococci in which both the genes were downregulated in the ΔadcR strain (21,40). The GAS genome has three genes encoding adh isozymes (spyM3_0772, spyM3_0036 and spyM3_0037), among which only the spyM3_0772 is annotated as zinc-containing ADH. The adh2 gene (spyM3_0036) encodes a putative iron-containing ADH. Although adh1 is a zinc-dependent ADH, the catalytic activity of ADH1 requires fewer zinc atoms per subunit compared to the zinc-containing ADH (spyM3_0772). Given that ADHs are among the most abundant zinc-containing proteins in the cell and require ∼8 zinc atoms per molecule as structural and functional cofactors (41–43), it is plausible that substitution of zinc-dependent ADH with zinc-independent isozymes will likely reduce the bacterial zinc requirement and release the zinc for essential activities during zinc limitation. Similar zinc conservation mechanisms have been identified as a component of yeast adaptive responses to zinc limitation (43,44). Thus, we speculate that the regulation of rpsN and adh genes by AdcR constitute the zinc sparing responses in GAS.
Transcriptome analysis of GAS grown in zinc-limited conditions indicated that GAS upregulates the five gene operon (dppABCDE) encoding the dipeptide permeases in response to decreased zinc levels (Supplementary Table S4). Interestingly, similar observations were made in the RNA-seq studies of ΔadcR mutant strain, suggesting that AdcR regulates dppABCDE operon in response to changing zinc availability (Supplementary Tables S3 and S4). Given that the promoter sequence of the dpp operon contains the characteristic adc motif immediately upstream of the −35 promoter element, it is likely that AdcR directly controls the expression of dipeptide permeases. Increased transcript levels were observed for 16 additional genes in ΔadcR strain (Supplementary Table S4), but since their promoter sequences lack consensus or near-consensus AdcR binding sites, these genes may be indirectly regulated by AdcR.
Contrary to the known role of AdcR as a transcription repressor, a significant number of genes were downregulated in ΔadcR strain. Genes with decreased transcript included several major GAS virulence factors, genes involved in metal transport and ribosomal subunits (Supplementary Table S5). AdcR-regulated virulence genes include well-characterized virulence factors, such as the hasABC operon encoding capsule biosynthesis enzymes (45), genes encoding cell envelope protease (prtS) (46,47), C5a peptidase (scpA) (48), streptokinase A (ska) (49) and exotoxin A (speA3) (50), suggesting a role for AdcR and its gene regulation in GAS pathogenesis (Supplementary Table S5). Together, these results demonstrate that AdcR is the master regulator of GAS adaptive responses to zinc limitation and identify the major molecular determinants that contribute to GAS survival during low zinc availability.
Mechanism of negative gene regulation by AdcR
The promoter sequences of the core adcRBC regulon possess the putative AdcR binding site (adc motif) upstream of their respective transcription start sites (TSSs). Thus, we used gel mobility shift assays to determine whether AdcR directly binds to the adc motif sequences in a zinc-dependent manner. First, we assessed the metal-requirement of AdcR for its DNA binding by assaying the DNA binding by apo and zinc-metallated AdcR. When apo AdcR was incubated with oligoduplexes containing the putative adc motif from the adcRCB promoter, AdcR failed to bind the cognate DNA. However, when the DNA binding was measured in the presence of zinc, AdcR specifically bound the adc motif (Figure 1A and B). To ensure that the observed interaction was sequence specific, we performed the same experiment with oligoduplexes containing the binding sites for peroxide sensing regulator (per box) from GAS (51). AdcR did not bind to the per box, even in the presence of zinc (Figure 1C). Based on these results, we conclude that AdcR interacts directly and specifically with adc motif sequences and these interactions require the presence of zinc.
Figure 1.
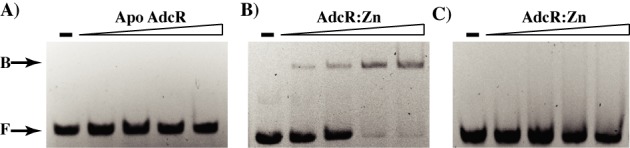
Zinc-dependent DNA binding by AdcR. Analysis of AdcR-adc motif interactions in the absence (A) and presence (B) of 100 μM zinc by gel mobility shift assay. Oligoduplexes containing the sequences corresponding to either the putative adc motif from the adcRCB promoter were incubated with increasing concentrations (250, 500 and 750 ng) of either purified apo-AdcR (A) or zinc-bound AdcR (B). (C) Assessment of sequence-specificity of AdcR-DNA interactions. Zinc-bound AdcR was incubated with non-cognate DNA containing the per box sequence. The reaction mixtures were resolved on a 10% native PAGE and visualized by ethidium bromide staining. The positions of free probe (F) and protein-bound probe (B) are labeled and indicated by arrows.
Although it is evident that AdcR mediates negative regulation by directly binding to the target promoters, the mechanism of transcription repression remains unclear. To elucidate the likely mechanism of repression by AdcR, we analyzed the promoter sequences for the location of various promoter elements and their proximity to the adc motifs. Using the program, BPROM (52), we identified the putative TSS and the location of RNA polymerase (RNAP) recognition elements, the −10 and −35 hexamers. Subsequently, we mapped the AdcR binding sites to determine the relative proximity of the adc motifs to the identified promoter elements. Interestingly, the adc motifs in the analyzed promoters are situated either immediately upstream or downstream of or overlapping the RNAP recognition elements in the promoter. These observations suggest that AdcR binding to the operator sequences in the negatively regulated promoters might sterically hinder the RNAP-promoter interactions, thereby negatively regulates the transcription from these promoters.
Differential gene regulation by AdcR
Transcriptome data revealed that AdcR-regulated genes exhibit varied levels of regulation in response to zinc deficiency. Genes encoding adcA, adcAII, phtD, phtY and rpsN were upregulated 15-fold or higher, whereas only a ≤4.5-fold induction was observed for the adcRCB and dppABCDE operons, and adh and sdn genes (Supplementary Tables S4 and S5). To understand the basis for the differential induction of target genes by AdcR, we analyzed the promoters of the target genes. With the exception of phtY, the promoters of all the highly regulated genes contain two adc motifs, while the promoters in the second group with more moderate repression had only one AdcR-binding site (Figure 2A and B). These observations led us to hypothesize that AdcR might bind to the target promoters in different oligomeric states and mediate differential gene regulation. To test this hypothesis, we carried out gel mobility shift assays with oligoduplexes containing either single adc motif from adcRCB operon or two sites from the adcAII-phtD operon. Consistent with our hypothesis, addition of zinc metallated AdcR to adcRCB operator sequences generated single shifted species, which likely corresponds to an AdcR dimer binding to the adc motif (Figure 2C). However, when the similar experiment was performed with two-site operator sequences, it produced two shifted bands at lower AdcR concentrations, and the highest concentration of AdcR yielded a single super-shifted band (Figure 2D). Collectively, these data indicate that AdcR binds to target promoters with differing number of adc motifs in different stoichiometry. Although the results from the binding experiments revealed the differences in the sizes of macromolecular complexes containing AdcR bound to either single or two adc motifs, the precise stoichiometry between AdcR and its DNA interacting partners remains to be elucidated. Our next line of investigation will focus on deducing the oligomeric state of AdcR bound to differing number of adc motifs. Possible approaches to elucidate the molar ratio between AdcR and the varying number of adc motifs include mass estimation of protein-DNA complexes by size exclusion chromatography and calculation of molar ratio of the interacting partners by fluorescent polarization (FP) assays. The elution profiles of individual components of the macromolecular complexes as well as the different AdcR-DNA complexes, as assessed by size exclusion chromatography, can be used to deduce the molecular weight and the corresponding oligomeric states of AdcR-DNA complexes. Alternatively, FP assays in which binding experiments are carried out with DNA concentrations in molar excess of the Kd of protein-DNA interactions. Subsequently, the stoichometry of protein to DNA can be derived by measuring the protein concentration at which the transition from specific (high-affinity) to non-specific (low-affinity) interactions occur. Similar methods have been used to compute the oligomeric states of MepR from S. aureus bound to different target operator sites (53).
Figure 2.
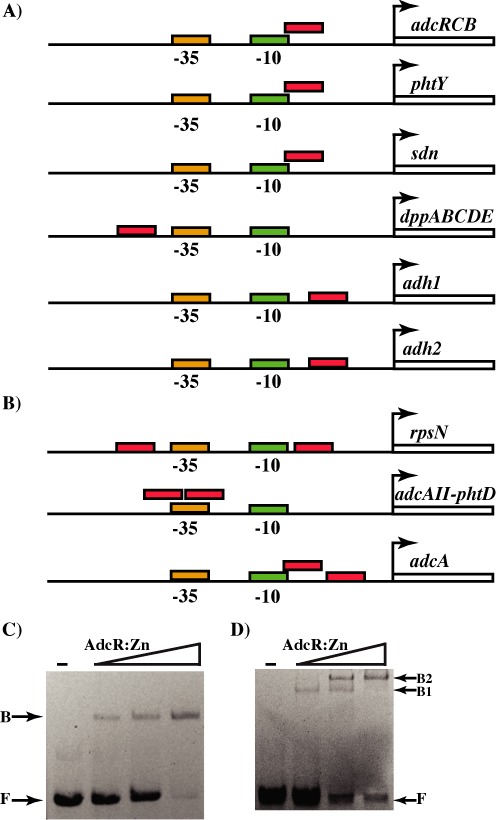
Organization of regulatory elements in target promoters negatively regulated by AdcR. The positions of predicted -10 and -35 hexamers, and the TSS (bent arrow) in the AdcR-regulated promoters containing one (A) and two (B) AdcR-binding sites are shown. The locations of adc motifs are indicated in red. Interactions between the metallated form of AdcR and operator sequences containing one (C) and two (D) adc motifs as assessed by gel mobility shift assay. Increasing concentrations of AdcR (250, 500 and 750 ng) were incubated with the respective oligoduplexes and reactions mixtures were resolved on a 10% native PAGE. The positions of free (F) and AdcR-bound (B) forms of probe are indicated.
Although the correlation between the number of adc motifs and level of induction is evident, the underlying mechanism remains unknown. Possibililites include tighter repression of genes with two adc motifs by the multimeric AdcR and/or differential sensitivity of AdcR-DNA complexes to reductions in zinc availability. Thus, we investigated AdcR-mediated gene regulation in GAS undergoing zinc withdrawal by measuring the transcript levels of target genes. To mimic zinc withdrawal, GAS was grown in zinc-rich conditions to late exponential growth phase and subsequently treated with the zinc-chelator N,N,N′,N′-tetrakis-(2-pyridylmethyl) ethane-1,2-diamine (TPEN). Samples were collected over 5 min in one-minute intervals and the transcript level of target genes was analyzed by qRT-PCR. We tested the transcript level of adcC and phtD as representative genes for the AdcR-regulated promoters with one or two adc motifs, respectively. Since AdcR exists predominantly in a zinc-metallated state during growth in zinc-rich conditions, transcript levels of AdcR-regulated genes in cells not treated with TPEN were considered repressed. Accordingly, gene expression was repressed by AdcR in untreated cells and repression was relieved with the addition of TPEN (Figure 3A and B). Interestingly, the mode of regulation by AdcR appears to differ for the two tested genes. During the repressed state, the relative abundance of phtD transcript level was significantly lower compared to that of adcC, indicating that phtD was strongly repressed by AdcR. Additionally, the zinc withdrawal had distinct effects on the derepression of adcC and phtD. As shown in Figure 3, exposure to TPEN caused an earlier induction of phtD expression, as a >6-fold increase in phtD transcript levels occurs as early as 2 min after addition of TPEN and continues to increase in a gradual manner over the tested time points. In contrast, the derepression of adcC was less sensitive to zinc withdrawal, and most of the induction occurs in one step, 3 min after TPEN addition. Together, these data indicate that AdcR exerts tighter repression of genes with two AdcR binding sites during zinc sufficiency and causes early onset of derepression during mild zinc deficiency. Conversely, genes with single adc motif might be less tightly repressed by AdcR under zinc-rich conditions while the repression is relieved only during severe zinc deficiency.
Figure 3.
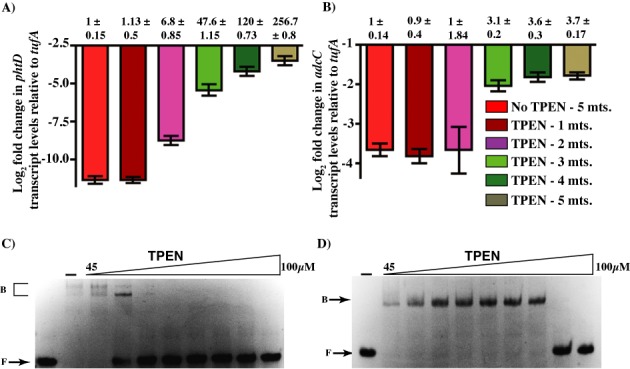
Differential gene regulation by AdcR. Strain MGAS10870 was grown to late exponential phase (A600 ∼ 1.0) in zinc-rich growth medium and treated with 50 μM TPEN. Samples were collected at one-minute intervals for 5 min. Transcript levels of phtD (A) and adcC (B) were measured by qRT-PCR. Three biological replicates were used. Data graphed are mean ± standard deviation. Average values for untreated samples were used as reference and the fold changes in the transcript levels in the treated samples relative to untreated sample were shown above. The sensitivity of AdcR bound to two- (C) and one-site (D) operator sequences to zinc chelation was assessed by gel mobility shift assay. The metallated form of AdcR bound to DNA was challenged by increasing concentrations of TPEN (45, 55, 60, 65, 70, 75, 80 and 100 μM) and the reaction mixtures were resolved on a 10% native PAGE. Representative image from three independent experiments is shown. The positions of free (F) and AdcR-bound (B) forms of probe are indicated.
To better understand the differential induction of the two classes of target promoters by AdcR, we subsequently tested the effect of TPEN on the DNA-binding ability of AdcR by gel mobility shift assay. Consistent with the qRT-PCR results, AdcR-DNA complexes on the two-site operator sequence displayed higher sensitivity to zinc withdrawal and readily dissociated at lower TPEN concentration (60–65 μM) (Figure 3C). However, AdcR bound to a single adc motif was more resistant to zinc chelation by TPEN, and dissociation occurred at 80–100 μM (Figure 3D). Together, these data indicate that the differential induction of target genes by AdcR are due to the combined effects of varying levels of repression during zinc sufficiency and responsiveness to alterations in zinc availability.
Differential gene regulation by zinc-sensing regulators has been observed in bacteria and yeast (43,44,54). Zur from Streptomyces coelicolor (ZurSc) displays varying affinities for the operator sequences in different promoters, and mediates graded gene regulation in concert with varying zinc concentration (54). High-affinity interactions occur between ZurSc and a subset of target promoters at low zinc concentration, which leads to tighter repression and delayed derepression (54). Alternatively, higher zinc concentration is required for the association of ZurSc with lower affinity promoters. Consequently, gene expression from these promoters is more sensitive to zinc withdrawal and results in early onset of derepression (54). Despite the similarities in the mode of gene regulation between ZurSc and AdcR, fundamental differences exist in the underlying mechanism of differential gene regulation. ZurSc has two zinc-binding sites and occupancy of one or two sites modulate its DNA-binding activity with high- or low-affinity promoters, respectively (54). Similarly, the crystal structure of AdcR from S. pneumoniae has two zinc-binding sites per subunit (22). However, the second site of pneumococcal AdcR is dispensable for DNA binding, high-affinity metal binding and gene regulation by AdcR (21). Thus, the physiological relevance of zinc binding at the second site to gene regulation by AdcR is insignificant. Additionally, our unpublished crystal structure of zinc-bound AdcR from GAS has only one zinc per subunit and this site corresponds to the primary zinc-sensing site identified in pneumococcal AdcR. Although the structure of AdcR from GAS does not have zinc at site 2, the metal ligands of site 2 are conserved. Thus, contrary to ZurSc, the differential gene regulation by AdcR is primarily controlled by the number of AdcR-binding sites in the target promoters. Interestingly, the promoters that are tightly repressed by AdcR during zinc sufficiency are derepressed more readily than the less-stringently regulated promoters. This paradoxical regulatory mechanism might be explained by a simplified model in which AdcR mediates two layers of adaptive responses to the alterations in zinc availability. Growth in zinc-replete conditions leads to varying levels of gene repression by AdcR, tight repression of the target promoters with two sites by multimeric AdcR and weaker repression of genes with single site by AdcR dimer. Importantly, tighter repression of the highly regulated genes requires the occupancy of both operator sites by AdcR. However, when the bacteria encounters slight decrease in zinc concentration, disruption of the higher order AdcR-DNA complex occurs first, which leads to partial derepression of the highly regulated genes. As the zinc concentration drops further, it promotes further dissociation of AdcR from the target promoters and causes additional derepression. This mechanism allows GAS to constantly monitor alterations in zinc availability and mediate a multilayered adaptive response. The first layer of response to mild zinc deficiency includes the upregulation of pht and adcAII genes to acquire extracellular zinc and mobilize internal zinc storage by energy-independent mechanisms, and induction of rpsN gene to conserve zinc utilization by switching to zinc-independent mechanisms. The second layer of response to severe zinc deficiency involves the upregulation of energy-dependent, highly efficient zinc uptake systems to obtain sparsely available zinc from the environment. Given that pathogenic bacteria encounter zinc-limiting conditions in the host, the proposed model provides a glimpse of how GAS might sense and respond to various zinc-limiting conditions in the host.
AdcR activates hasABC expression by directly binding to hasABC promoter
Transcriptome analysis in GAS identified several genes that were downregulated in the ΔadcR mutant strain, suggesting a novel role for AdcR as an activator. The genes that are upregulated by AdcR include well-characterized virulence factors and virulence regulators. Among these genes, two of the highly regulated genes are the hasABC operon encoding the enzymes involved in capsule biosynthesis, and prtS that codes for cell envelope-bound membrane protease (Supplementary Table S5).
Although the role of AdcR as a transcription repressor is well characterized, its function as an activator is not documented. To validate these findings from our transcription profiling, we investigated the positive gene regulation by AdcR in detail by measuring hasA and prtS transcript levels using qRT-PCR. The parental strain MGAS10870 trans-complemented with empty vector pDC123 was used as WT, and the isogenic adcR mutant trans-complemented with empty vector pDC123 was used as a negative control. To trans-complement the isogenic mutant ΔadcR strain, we constructed plasmid pDC-adcR containing the coding region of adcR and its putative promoter region. The transcriptional competence of pDC-adcR was verified by comparing adcR transcript levels in the WT (10870:pDC) and trans-complemented (ΔadcR:pDC-adcR) strains (Supplementary Figure S3). To determine the AdcR-dependent upregulation of target genes, three biological replicates of the WT strain, isogenic ΔadcR mutant strain and the complemented strain were grown in zinc-replete CDM to late exponential growth phase (A600 ∼ 1.0) and transcript levels were assessed. Consistent with our observations in RNA-seq studies, a 10-fold reduction in hasA transcript levels were observed in the ΔadcR mutant strain compared to WT (Figure 4A), the transcription of which is restored to WT levels in the adcR-complemented strain (Figure 4A). However, overexpression of adcR from the high copy number trans-complementation plasmid, pDC-adcR, (Supplementary Figure S3) caused an increase in the transcript levels of hasA in the complemented strain compared to the parental strain (10870:pDC), indicating that AdcR is responsible for the transcription upregulation of the hasABC operon (Figure 4A). The transcript level analysis of prtS exhibited statistically significant AdcR-dependent upregulation, but the level of induction is reduced in WT and the trans-complemented strains compared to the fold changes observed in RNA-seq studies (Figure 4B). Together, these data indicate that AdcR is involved in the transcription activation of prtS and hasABC operon.
Figure 4.
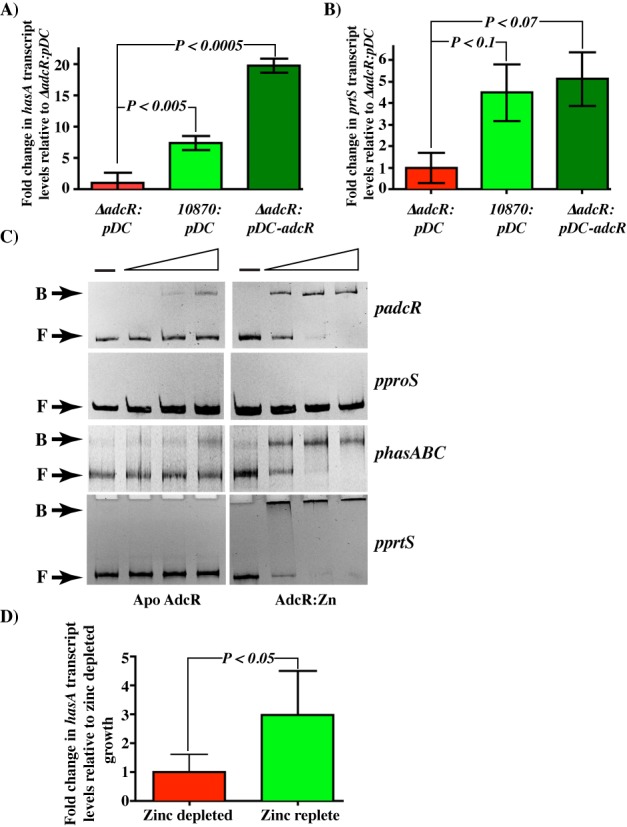
AdcR directly upregulates hasABC and prtS expression. Taqman qRT-PCR analysis of hasA (A) and prtS (B) transcript levels in indicated strains relative to ΔadcR trans-complemented with empty vector, pDC123 (ΔadcR:pDC). Data graphed are mean ± standard deviation for three biological replicates. (C) AdcR-DNA interactions as assessed by gel mobility shift assay. DNA fragments of similar size were amplified from the promoters of adcRCB (padcR), non-specific control proS (pproS), hasABC (phasABC) and prtS (pprtS) genes. Increasing concentrations of apo- (left panels) and zinc-bound AdcR (right panels) (50, 100 and 200 ng) were incubated with the indicated DNA fragments and reaction mixtures were analyzed on a 10% native PAGE. The positions of free (F) and AdcR-bound (B) probe are indicated. (D) Transcript level analysis of hasA gene measured by qRT PCR. Fold changes in GAS grown in zinc-rich media relative to cells grown under zinc-depleted conditions were shown.
Since transcription activation can occur by direct or indirect mechanisms, we next assessed whether AdcR activates transcription of hasA and prtS by direct binding to the respective promoters using gel mobility shift assays. Using genomic DNA of parental strain MGAS10870 as template, we amplified a 400-bp DNA fragment upstream of the hasA or the prtS translation start codon comprising the entire promoter region and tested for AdcR binding. Recombinant AdcR was purified to near homogeneity under metal-free conditions and used for DNA-binding experiments in the absence and presence of zinc. We used the promoter region of adcRCB operon that has the adc motif as positive control and an intergenic region of a constitutively expressed gene (proS) of similar length as negative control. Results from these experiments demonstrated that AdcR bound to the hasA promoter sequence in a zinc-dependent manner (Figure 4C). Although AdcR bound to the prtS promoter, the shifted species was too large to be resolved (Figure 4C). Attempts to vary the DNA length, the protein or DNA concentration, or the porosity of the gel matrix failed to allow resolution. Regardless, the data demonstrate that AdcR binds to the prtS promoter sequences in a sequence-specific manner. Together, these results indicate that the interactions between AdcR and the two activated promoters, hasABC and prtS, are specific and zinc-dependent, and AdcR has a direct role in the activation of the hasABC operon.
To determine whether the transcription activation of hasABC by AdcR requires zinc, we compared the transcript levels of hasA in GAS grown in zinc-depleted and zinc-replete conditions. Consistent with the role of AdcR as a zinc-dependent regulator, a zinc-dependent upregulation of hasA expression was observed. GAS grown under zinc-rich conditions exhibited a 3-fold increase in the hasA transcript levels compared to cells grown under zinc deficiency (Figure 4D) (P < 0.05 by t-test). These results indicate that hasABC expression is induced during zinc-rich growth conditions and downregulated during zinc limitation. Since metal binding by AdcR is required for adc motif binding and repression, we investigated the influence of zinc on AdcR interactions with hasABC promoter. When tested for the DNA-binding ability to hasABC promoter by gel mobility shift assay, the apo AdcR displayed weak to no binding to its cognate DNA sequences, whereas the zinc-bound AdcR binding to hasABC promoter was comparable to its interactions with adcRCB operator elements (Figure 4C). Collectively, these data demonstrate that AdcR activates hasABC expression in a zinc-dependent fashion by directly binding to the operator sequences in the promoter.
Analysis of the hasA promoter region for AdcR binding site
Since our analysis of the hasA promoter failed to identify adc motif-like sequences, it is possible that AdcR might bind to distinct operator sequences to activate transcription. To identify the minimal region within the hasA promoter required for AdcR binding, we screened different fragments of the hasA promoter for AdcR binding. Fragments with truncations up to 281-bp upstream of the translation start site retained AdcR binding comparable to that of the full-length promoter-AdcR interactions (Figure 5A and B). However, truncations that removed an additional 80 and 189 bp resulted in complete loss of AdcR binding, suggesting that the putative AdcR binding site is located within a 80-bp region between −281 and −201 bp upstream of the hasA start codon (Figure 5A and B). Given the lack of recognizable adc motifs in this 80-bp segment, we examined the region for the presence of cis-acting motifs. Our analysis identified two inverted repeats: a 16-bp inverted repeat from −228 to −213 bp, and a second 27-bp inverted repeat from –223 to –199 bp that overlaps with the first (Figure 5C). Interestingly, the identified repeats share no significant similarity with the canonical adc motif present in the negatively regulated promoters. Collectively, these data demonstrate that AdcR binds to DNA sequences that are distinct from the adc motif to mediate activation of hasABC expression.
Figure 5.
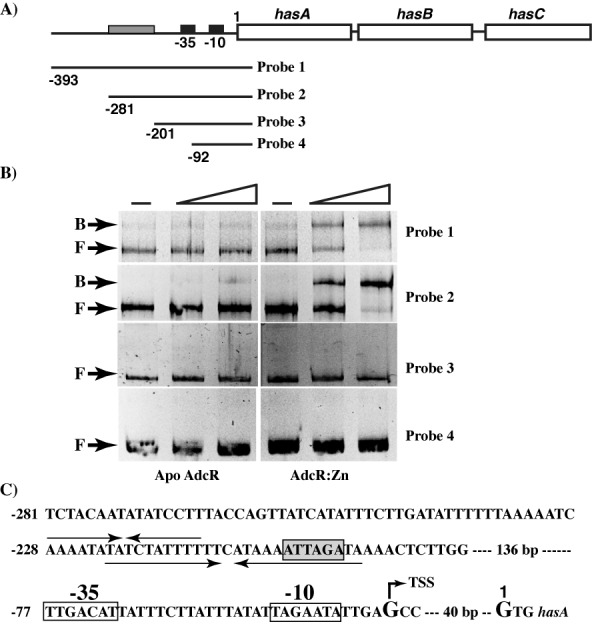
Analysis of the hasABC promoter region. The significance of hasABC promoter sequences for AdcR binding was assessed by gel mobility shift assay. (A) Genetic organization of hasABC operon on the chromosome of MGAS10870. The −10 and −35 promoter elements are indicated with black boxes, whereas the AdcR binding region, as identified in this study, is indicated in gray. Truncated promoter fragments (probes 1–4) tested for AdcR binding are shown and the numbers to the left denote the 5′ end of the deletions relative to the first nucleotide of the hasA start codon. (B) Increasing concentrations (50 and 100 ng) of purified apo- (left) or zinc-bound AdcR (right) were incubated with the indicated DNA fragments and reaction mixtures were resolved on a 10% native PAGE. The positions of free (F) and AdcR-bound (B) probe are indicated. (C) Architecture of the hasABC promoter. The locations of −10 hexamer, −35 hexamer and TSS, as identified by Ashbaugh et al. (56) are marked and labeled. The positions of the short and long inverted repeats within the hasABC promoter are indicated by arrows above and below the sequence, respectively. The putative CovR-binding site with the characteristic 5′ATTARA 3′ sequence located within the inverted repeat is boxed and shaded in gray.
To identify the AdcR binding motif within hasABC promoter, we investigated the interactions between AdcR and the two candidate inverted repeat elements, IR1 and IR2, by gel mobility shift assay. AdcR failed to bind oligoduplexes containing the longer 27-bp inverted repeat (IR1) even in the presence of zinc (Figure 6A and B). On the other hand, AdcR exhibited sequence-specific interactions with oligoduplexes containing the shorter 16-bp inverted repeat (IR2) (Figure 6A and D). However, these interactions require zinc metallation of AdcR as the metal-free apo AdcR failed to bind DNA (Figure 6C). To further demonstrate that the interactions between AdcR and IR2 are sequence specific, we introduced three substitutions within the inverted repeat, IR2, and tested the oligoduplexes containing the mutant promoter for AdcR binding by gel shift assay. Consistent with the specific nature of these interactions, substitutions at 3 out of total 14 bases within IR2 motif drastically reduced AdcR binding (Figure 6E). Together, these data indicate that AdcR binds to a novel non-canonical DNA-binding motif within hasABC promoter and mediates upregulation of gene expression.
Figure 6.
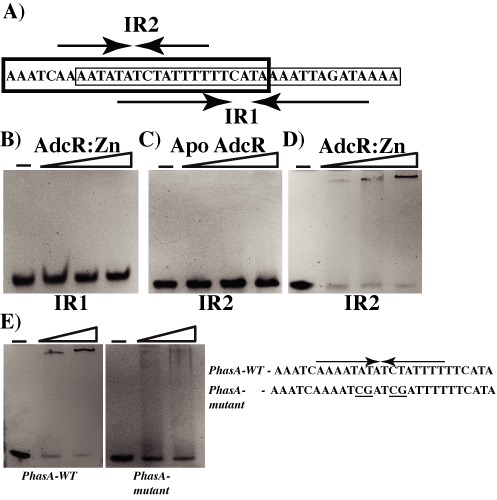
Identification of AdcR binding site in the hasABC promoter. (A) The nucleotide sequence of the two inverted repeats, IR1 (thin lined box) and IR2 (bold box), that has the putative AdcR binding site within the hasABC promoter is shown and labeled. Oligoduplexes containing either IR1 or IR2 were tested for AdcR binding by gel mobility shift assay. Increasing concentrations (50, 75 and 100 ng) of purified zinc-bound AdcR (B and D), and apo AdcR (C) were incubated with the indicated oligoduplexes and reaction mixtures were resolved on a 10% native PAGE. (E) The ability of zinc-bound AdcR (50 and 100 ng) to bind WT IR2 (left) and mutant IR2 (right) was assessed by gel mobility shift assay. The nucleotide sequences of the WT (PhasA-WT) and mutant (PhasA-mutant) IR2 are shown and the mutagenized bases are underlined. The positions of free (F) and AdcR-bound (B) probe are indicated.
Although our studies demonstrate that AdcR upregulates hasABC operon expression by direct binding, the molecular mechanism remains unclear. However, the location of AdcR binding sites in the hasA promoter and zinc dependency of AdcR for hasA upregulation offers clues into the likely mechanism of transcription activation by AdcR. One of the well-documented mechanisms of transcription activation by bacterial regulators involves the binding of the regulator to the sequences immediately upstream or downstream of −10 and −35 promoter elements to recruit/stabilize the binding of RNAP and subsequently activate transcription (55). Typically, the activator-dependent promoters lack recognizable −10 or −35 motifs, which necessitates an activator to facilitate RNAP binding to an otherwise incompetent promoter (55). However, the hasA promoter contains strong −10 and −35 hexamers (Figure 5C), which has been previously demonstrated to be sufficient for basal transcription (56). In addition, the AdcR-binding site in the hasA promoter identified in this study is located >150 bp upstream of the −10 and −35 motifs (Figure 5C). Thus, it is unlikely that AdcR upregulates hasABC expression by recruitment of RNAP to the promoter.
Another intriguing possibility is that AdcR binding site in the hasA promoter is located within or overlapping a hasABC repressor site and AdcR-mediated activation of hasABC occurs by the steric occlusion of repressor binding by bound AdcR. A similar mechanism of activation has been reported in the upregulation of the norB gene by Fur in N. gonorrohoeae. The norB promoter has operator elements that are shared by both Fur and a repressor protein, ArsR. Fur binding to the norB promoter precludes the ArsR from accessing its binding site and results in Fur-dependent activation of norB (57). The expression of hasABC operon in GAS is negatively regulated by global regulator CovR, and the hasA promoter has multiple binding sites for CovR that span the entire promoter region (58). Thus, it is plausible that AdcR might upregulate hasABC operon by prevention of CovR binding to the hasA promoter. Typically, CovR binds to a conserved hexameric element with a nucleotide sequence of ATTARA to mediate repression of target genes (58). To investigate this possibility, we analyzed the hasA promoter sequences for CovR binding sites in the proximity of the putative AdcR binding element. Interestingly, we have identified a CovR binding site with a sequence of ATTAGA that is located only 8-bp away from the putative AdcR binding site (Figure 5C). Although these observations lend support to our hypothesis that AdcR precludes CovR from binding to the hasA promoter and promote transcription activation by relieving CovR-mediated repression, experimental evidence is needed to validate these findings. Thus, our future work will focus on elucidating the mechanisms by which AdcR binding to the hasA promoter influences CovR binding and activates transcription.
Metal sensing and gene regulation by AdcR
To better understand the molecular mechanism of metal binding and gene regulation by AdcR, we carried out modeling studies using the structure of AdcR from S. pneumoniae (22). As in other MarR family regulators, AdcR is a triangular-shaped molecule with its DNA-binding domain located at the base and the amino and carboxy-terminal helices of each subunit form the dimerization domain (53,59). Although pneumococcal AdcR has two zinc-binding sites per subunit, occupation of only one of the two sites is critical for zinc sensing (22). Thus, the critical site is designated as the primary site, while the role of the second site remains unclear. The primary site is made of side chains from residues E24 and H42 from the N-terminal domain, and H108 and H112 from the C-terminal domain (Supplementary Figure S4A and B) (22). The co-ordination of zinc by the amino acids from the two functional domains promote a closed conformation in AdcR that is compatible for DNA binding and repression (22). Alignment of the amino acid sequence of AdcR from GAS and pneumococci revealed that the metal ligands of the primary metal sensing site is highly conserved and ideally positioned in the model of GAS AdcR to interact with zinc.
To test the hypothesis that the metal ligands of AdcR are critical for regulatory metal binding, DNA binding and gene regulation, we chose one metal ligand from each domain, H42 from the N-terminal domain and H108 from the C-terminal domain, for further analysis (Supplementary Figure S4A and B). We introduced single alanine substitutions at each of these two metal-binding histidines, purified the recombinant proteins and tested the mutant proteins for DNA binding using gel shift assays (Figure 7A). Even in the presence of 100 μM zinc, neither mutant protein bound the oligoduplexes containing the adc motif (Figure 7A). These results demonstrate that the metal ligands of AdcR, H42 and H108, are required for DNA binding and indicate these residues are critical components for zinc sensing of GAS AdcR.
Figure 7.
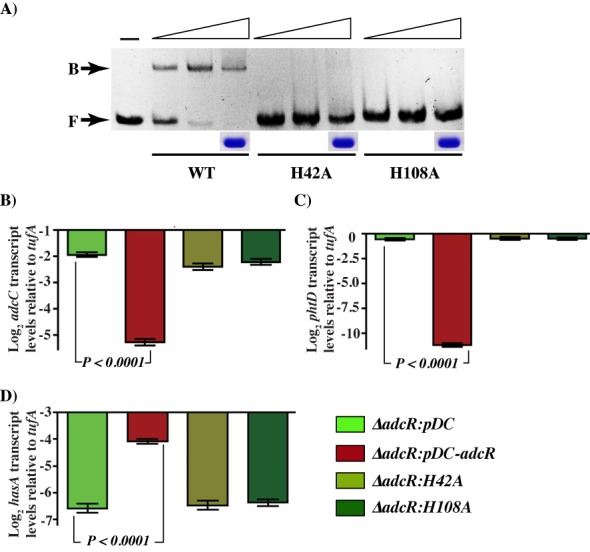
Metal binding ligands of AdcR are critical for AdcR-DNA interactions. (A) DNA binding activites of WT and mutant derivatives of AdcR, as assessed by gel mobility shift assay. Increasing concentrations of purified WT or AdcR mutant proteins were incubated with oligoduplexes containing the adc motif and resolved on native PAGE. The positions of unbound (F) and AdcR-bound (B) probes are indicated. Coomassie-stained bands corresponding to purified WT and mutant derivatives of AdcR are shown. Taqman qRT-PCR analysis of adcC (B), phtD (C) and hasA (D) transcript levels in indicated strains relative to constitutively expressed endogenous control, tufA. Data graphed are mean ± standard deviation for three biological replicates.
To investigate whether the alanine substitutions at the metal ligands of AdcR affect the gene regulation, we measured the transcript levels of AdcR-regulated genes by qRT-PCR. The trans-complementation plasmid, pDC-adcR, was used to introduce single alanine substitutions at 2 (H42A and H108A) out of the total 4 metal-binding ligands of AdcR by quick-change site-directed mutagenesis. When grown in standard medium, the parental serotype (10870:pDC), ΔadcR mutant strain (ΔadcR:pDC) and mutant strain trans-complemented with either the WT adcR (ΔadcR:pDC-adcR) or one of the mutant strains exhibited similar growth characteristics, indicating that the single amino acid substitutions introduced in AdcR do not affect GAS growth (data not shown). The isogenic ΔadcR mutant strain trans-complemented with the WT adcR (ΔadcR-pDC-adcR) was used as the WT control. Consistent with the RNA-seq data, AdcR-mediated repression of adcC and phtD was relieved in the mutant ΔadcR strain as the transcript levels of both genes were elevated (Figure 7B and C) and the defective phenotype was rescued in the WT control (Figure 7B and C). However, the ΔadcR strain trans-complemented with either H42A or H108A mutant adcR failed to reverse the defective phenotype of ΔadcR mutant and transcript levels of adcC and phtD genes in these mutant strains were comparable to that of ΔadcR strain (Figure 7B and C). Interestingly, similar phenotype was observed for AdcR-dependent upregulation of hasA expression. Consistent with the requirement for AdcR for transcription activation of hasA, the upregulation was absent in the mutant ΔadcR strain (Figure 7D). Although the upregulation was restored in the WT control, both mutant strains exhibited hasA transcript levels similar to ΔadcR mutant strain (Figure 7D). Together, these results indicate that metal binding at the primary metal-sensing site of AdcR is critical for positive and negative gene regulation by AdcR.
Contribution of metal sensing and gene regulation by AdcR to GAS virulence
To test the hypothesis that zinc-binding ligands of AdcR are critical for in vivo metal sensing, gene regulation and GAS virulence, we carried out animal infection studies using an intraperitoneal mouse model of infection. Mice were infected intraperitoneally and monitored for near-mortality for 9 days post-infection. Consistent with our hypothesis, the ΔadcR mutant strain (ΔadcR:pDC) was significantly less virulent compared to the WT strain (10870:pDC). Although the attenuated virulence phenotype of the isogenic ΔadcR mutant strain was restored to WT virulence phenotype in the trans-complemented strain (ΔadcR:pDC-adcR), the strains with alanine substitutions at the metal ligands failed to complement the virulence phenotype of ΔadcR mutant strain (Figure 8). The virulence phenotype of the ΔadcR mutant strain (ΔadcR:pDC) is similar to the mutant strains (P = n.s.) (Figure 8). Together, these data indicate that the metal sensing function of AdcR is critical for GAS pathogenesis and impaired zinc sensing by AdcR significantly attenuates GAS virulence, likely due to the dysregulation of adc regulon and zinc homeostasis.
Figure 8.
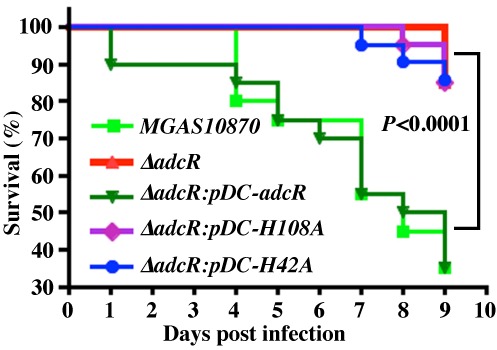
Zinc sensing and gene regulation by AdcR is critical for GAS virulence. Fifteen CD-1 mice were infected with indicated strains intraperitoneally and near-mortality was recorded. The Kaplan–Meier survival curve with P-values derived by log rank test is shown.
In conclusion, data presented in this study revealed the molecular strategies used by GAS to survive under zinc-limiting growth conditions. Our results demonstrate that AdcR senses the zinc availability and responds by regulating the expression of genes involved in zinc homeostasis, metabolism and virulence. Importantly, the regulatory mechanism of AdcR involves both repression and activation. Interestingly, the regulon controlled by AdcR goes beyond the genes involved in metal homeostasis and include several virulence genes that are known to participate in GAS disease production. However, further studies are required to understand the contribution of upregulation of AdcR-regulated virulence factors to GAS survival during zinc limitation.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Kathryn Stockbauer, PhD, for editorial assistance.
Footnotes
These authors contributed equally to the paper as first authors.
FUNDING
National Institutes of Health [1R21AI103708–01 to M.K.]. Escuela de Medicina y Ciencias de la Salud, Tecnológico de Monterrey y Consejo Nacional de Ciencia y Tecnología [CONACyT, 421460/263885 to M.S.]. Fondren Foundation [to J.M.M.]. Funding for open access charge: National Institutes of Health [1R21AI103708-01].
Conflict of interest statement. None declared.
REFERENCES
- 1.Andreini C., Banci L., Bertini I., Rosato A. Zinc through the three domains of life. J. Prot. Res. 2006;5:3173–3178. doi: 10.1021/pr0603699. [DOI] [PubMed] [Google Scholar]
- 2.Coleman J.E. Zinc enzymes. Curr. Opin. Chem. Biol. 1998;2:222–234. doi: 10.1016/s1367-5931(98)80064-1. [DOI] [PubMed] [Google Scholar]
- 3.Blencowe D.K., Morby A.P. Zn(II) metabolism in prokaryotes. FEMS Microbiol. Rev. 2003;27:291–311. doi: 10.1016/S0168-6445(03)00041-X. [DOI] [PubMed] [Google Scholar]
- 4.Kehl-Fie T.E., Chitayat S., Hood M.I., Damo S., Restrepo N., Garcia C., Munro K.A., Chazin W.J., Skaar E.P. Nutrient metal sequestration by calprotectin inhibits bacterial superoxide defense, enhancing neutrophil killing of Staphylococcus aureus. Cell Host Microbe. 2011;10:158–164. doi: 10.1016/j.chom.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kehl-Fie T.E., Skaar E.P. Nutritional immunity beyond iron: a role for manganese and zinc. Curr. Opin. Chem. Biol. 2010;14:218–224. doi: 10.1016/j.cbpa.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Corbin B.D., Seeley E.H., Raab A., Feldmann J., Miller M.R., Torres V.J., Anderson K.L., Dattilo B.M., Dunman P.M., Gerads R., et al. Metal chelation and inhibition of bacterial growth in tissue abscesses. Science. 2008;319:962–965. doi: 10.1126/science.1152449. [DOI] [PubMed] [Google Scholar]
- 7.Kuo C.-F., Luo Y.-H., Lin H.-Y., Huang K.-J., Wu J.-J., Lei H.-Y., Lin M.T., Chuang W.-J., Liu C.-C., Jin Y.-T., et al. Histopathologic changes in kidney and liver correlate with streptococcal pyrogenic exotoxin B production in the mouse model of group A streptococcal infection. Microb. Pathog. 2004;36:273–285. doi: 10.1016/j.micpath.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 8.McDevitt C.A., Ogunniyi A.D., Valkov E., Lawrence M.C., Kobe B., McEwan A.G., Paton J.C. A molecular mechanism for bacterial susceptibility to zinc. PLoS Pathog. 2011;7:e1002357. doi: 10.1371/journal.ppat.1002357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hood M.I., Skaar E.P. Nutritional immunity: transition metals at the pathogen-host interface. Nat. Rev. Microbiol. 2012;10:525–537. doi: 10.1038/nrmicro2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ong C.-l.Y., Gillen C.M., Barnett T.C., Walker M.J., McEwan A.G. An antimicrobial role for zinc in innate immune defense against group A Streptococcus. J. Infect. Dis. 2014;209:1500–1508. doi: 10.1093/infdis/jiu053. [DOI] [PubMed] [Google Scholar]
- 11.Hood M.I., Mortensen B.L., Moore J.L., Zhang Y., Kehl-Fie T.E., Sugitani N., Chazin W.J., Caprioli R.M., Skaar E.P. Identification of an Acinetobacter baumannii zinc acquisition system that facilitates resistance to calprotectin-mediated zinc sequestration. PLoS Pathog. 2012;8:e1003068. doi: 10.1371/journal.ppat.1003068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Botella H., Stadthagen G., Lugo-Villarino G., de Chastellier C., Neyrolles O. Metallobiology of host-pathogen interactions: an intoxicating new insight. Trends Microbiol. 2012;20:106–112. doi: 10.1016/j.tim.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Botella H., Peyron P., Levillain F., Poincloux R., Poquet Y., Brandli I., Wang C., Tailleux L., Tilleul S., Charrière G.M., et al. Mycobacterial P1-Type ATPases mediate resistance to zinc poisoning in human macrophages. Cell Host Microbe. 2011;10:248–259. doi: 10.1016/j.chom.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klaus H. Bacterial zinc uptake and regulators. Curr. Opin. Microbiol. 2005;8:196–202. doi: 10.1016/j.mib.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Gaballa A., Wang T., Ye R.W., Helmann J.D. Functional analysis of the bacillus subtilis zur regulon. J. Bacteriol. 2002;184:6508–6514. doi: 10.1128/JB.184.23.6508-6514.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore C.M., Helmann J.D. Metal ion homeostasis in Bacillus subtilis. Curr. Opin. Microbiol. 2005;8:188–195. doi: 10.1016/j.mib.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 17.Olsen R.J., Shelburne S.A., Musser J.M. Molecular mechanisms underlying group A streptococcal pathogenesis. Cell Microbiol. 2009;11:1–12. doi: 10.1111/j.1462-5822.2008.01225.x. [DOI] [PubMed] [Google Scholar]
- 18.Lukomski S., Montgomery C.A., Rurangirwa J., Geske R.S., Barrish J.P., Adams G.J., Musser J.M. Extracellular cysteine protease produced by Streptococcus pyogenes participates in the pathogenesis of invasive skin infection and dissemination in mice. Infect. Immun. 1999;67:1779–1788. doi: 10.1128/iai.67.4.1779-1788.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brenot A., Weston B.F., Caparon M.G. A PerR-regulated metal transporter (PmtA) is an interface between oxidative stress and metal homeostasis in Streptococcus pyogenes. Mol. Microbiol. 2007;63:1185–1196. doi: 10.1111/j.1365-2958.2006.05577.x. [DOI] [PubMed] [Google Scholar]
- 20.Weston B.F., Brenot A., Caparon M.G. The metal homeostasis protein, Lsp, of Streptococcus pyogenes is necessary for acquisition of zinc and virulence. Infect. Immun. 2009;77:2840–2848. doi: 10.1128/IAI.01299-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reyes-Caballero H., Guerra A.J., Jacobsen F.E., Kazmierczak K.M., Cowart D., Koppolu U.M.K., Scott R.A., Winkler M.E., Giedroc D.P. The metalloregulatory zinc site in Streptococcus pneumoniae AdcR, a zinc-activated MarR family repressor. J. Mol. Biol. 2010;403:197–216. doi: 10.1016/j.jmb.2010.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guerra A.J., Dann C.E., Giedroc D.P. Crystal structure of the zinc-dependent MarR family transcriptional regulator AdcR in the Zn(II)-bound state. J. Am. Chem. Soc. 2011;133:19614–19617. doi: 10.1021/ja2080532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Plumptre C.D., Eijkelkamp B.A., Morey J.R., Behr F., Couñago R.M., Ogunniyi A.D., Kobe B., O'Mara M.L., Paton J.C., McDevitt C.A. AdcA and AdcAII employ distinct zinc acquisition mechanisms and contribute additively to zinc homeostasis in Streptococcus pneumoniae. Mol. Microbiol. 2014;91:834–851. doi: 10.1111/mmi.12504. [DOI] [PubMed] [Google Scholar]
- 24.Ogunniyi A.D., Grabowicz M., Mahdi L.K., Cook J., Gordon D.L., Sadlon T.A., Paton J.C. Pneumococcal histidine triad proteins are regulated by the Zn2+-dependent repressor AdcR and inhibit complement deposition through the recruitment of complement factor H. FASEB J. 2009;23:731–738. doi: 10.1096/fj.08-119537. [DOI] [PubMed] [Google Scholar]
- 25.Plumptre C.D., Ogunniyi A.D., Paton J.C. Polyhistidine triad proteins of pathogenic streptococci. Trends Microbiol. 2012;20:485–493. doi: 10.1016/j.tim.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 26.Beres S.B., Carroll R.K., Shea P.R., Sitkiewicz I., Martinez-Gutierrez J.C., Low D.E., McGeer A., Willey B.M., Green K., Tyrrell G.J. Molecular complexity of successive bacterial epidemics deconvoluted by comparative pathogenomics. Proc. Natl. Acad. Sci. U.S.A. 2010;107:4371–4376. doi: 10.1073/pnas.0911295107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gryllos I., Grifantini R., Colaprico A., Cary M.E., Hakansson A., Carey D.W., Suarez-Chavez M., Kalish L.A., Mitchell P.D., White G.L., et al. PerR confers phagocytic killing resistance and allows pharyngeal colonization by group A Streptococcus. PLoS Pathog. 2008;4:e1000145. doi: 10.1371/journal.ppat.1000145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J., Kasper D.L., Ausubel F.M., Rosner B., Michel J.L. Inactivation of the alpha C protein antigen gene, bca, by a novel shuttle/suicide vector results in attenuation of virulence and immunity in group B Streptococcus. Proc. Natl. Acad. Sci. U.S.A. 1997;94:13251–13256. doi: 10.1073/pnas.94.24.13251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flores A.R., Olsen R.J., Wunsche A., Kumaraswami M., Shelburne S.A., Carroll R.K., Musser J.M. Natural variation in the promoter of the gene encoding the Mga regulator alters host-pathogen interactions in group A Streptococcus carrier strains. Infect. Immun. 2013;81:4128–4138. doi: 10.1128/IAI.00405-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carroll R.K., Shelburne S.A., III, Olsen R.J., Suber B., Sahasrabhojane P., Kumaraswami M., Beres S.B., Shea P.R., Flores A.R., Musser J.M. Naturally occurring single amino acid replacements in a regulatory protein alter streptococcal gene expression and virulence in mice. J. Clin. Invest. 2011;121:1956–1968. doi: 10.1172/JCI45169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chaffin D.O., Rubens C.E. Blue/white screening of recombinant plasmids in Gram-positive bacteria by interruption of alkaline phosphatase gene (phoZ) expression. Gene. 1998;219:91–99. doi: 10.1016/s0378-1119(98)00396-5. [DOI] [PubMed] [Google Scholar]
- 32.Virtaneva K., Porcella S.F., Graham M.R., Ireland R.M., Johnson C.A., Ricklefs S.M., Babar I., Parkins L.D., Romero R.A., Corn G.J., et al. Longitudinal analysis of the group A Streptococcus transcriptome in experimental pharyngitis in cynomolgus macaques. Proc. Natl. Acad. Sci. U.S.A. 2005;102:9014–9019. doi: 10.1073/pnas.0503671102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roy A., Kucukural A., Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protocols. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeLano W.L. The PyMol Molecular Graphics System. Palo Alto, CA, USA: DeLano Scientific; 2002. [Google Scholar]
- 36.Grifantini R., Toukoki C., Colaprico A., Gryllos I. Peroxide stimulon and role of PerR in group A Streptococcus. J. Bacteriol. 2011;193:6539–6551. doi: 10.1128/JB.05924-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nanamiya H., Kawamura F. Towards an elucidation of the roles of the ribosome during different growth phases in Bacillus subtilis. Biosci. Biotechnol. Biochem. 2010;74:451–461. doi: 10.1271/bbb.90859. [DOI] [PubMed] [Google Scholar]
- 38.Gabriel S.E., Helmann J.D. Contributions of zur-controlled ribosomal proteins to growth under zinc starvation conditions. J. Bacteriol. 2009;191:6116–6122. doi: 10.1128/JB.00802-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Natori Y., Nanamiya H., Akanuma G., Kosono S., Kudo T., Ochi K., Kawamura F. A fail-safe system for the ribosome under zinc-limiting conditions in Bacillus subtilis. Mol. Microbiol. 2007;63:294–307. doi: 10.1111/j.1365-2958.2006.05513.x. [DOI] [PubMed] [Google Scholar]
- 40.Shafeeq S., Kloosterman T.G., Kuipers O.P. Transcriptional response of Streptococcus pneumoniae to Zn2+ limitation and the repressor/activator function of AdcR. Metallomics. 2011;3:609–618. doi: 10.1039/c1mt00030f. [DOI] [PubMed] [Google Scholar]
- 41.Brandt E.G., Hellgren M., Brinck T., Bergman T., Edholm O. Molecular dynamics study of zinc binding to cysteines in a peptide mimic of the alcohol dehydrogenase structural zinc site. Phys. Chem. Chem. Phys. 2009;11:975–983. doi: 10.1039/b815482a. [DOI] [PubMed] [Google Scholar]
- 42.Leskovac V., Trivić S., Peričin D. The three zinc-containing alcohol dehydrogenases from baker's yeast, Saccharomyces cerevisiae. FEMS Yeast Res. 2002;2:481–494. doi: 10.1111/j.1567-1364.2002.tb00116.x. [DOI] [PubMed] [Google Scholar]
- 43.Eide D.J. Homeostatic and adaptive responses to zinc deficiency in Saccharomyces cerevisiae. J. Biol. Chem. 2009;284:18565–18569. doi: 10.1074/jbc.R900014200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu C.-Y., Bird A.J., Chung L.M., Newton M.A., Winge D.R., Eide D.J. Differential control of Zap1-regulated genes in response to zinc deficiency in Saccharomyces cerevisiae. BMC Genom. 2008;9:370. doi: 10.1186/1471-2164-9-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wessels M.R., Moses A.E., Goldberg J.B., DiCesare T.J. Hyaluronic acid capsule is a virulence factor for mucoid group A streptococci. Proc. Natl. Acad. Sci. U.S.A. 1991;88:8317–8321. doi: 10.1073/pnas.88.19.8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaur S.J., Nerlich A., Bergmann S., Rohde M., Fulde M., Zähner D., Hanski E., Zinkernagel A., Nizet V., Chhatwal G.S., et al. The CXC chemokine-degrading protease SpyCep of Streptococcus pyogenes promotes its uptake into endothelial cells. J. Biol. Chem. 2010;285:27798–27805. doi: 10.1074/jbc.M109.098053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurupati P., Turner C.E., Tziona I., Lawrenson R.A., Alam F.M., Nohadani M., Stamp G.W., Zinkernagel A.S., Nizet V., Edwards R.J., et al. Chemokine-cleaving Streptococcus pyogenes protease SpyCEP is necessary and sufficient for bacterial dissemination within soft tissues and the respiratory tract. Mol. Microbiol. 2010;76:1387–1397. doi: 10.1111/j.1365-2958.2010.07065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ji Y., McLandsborough L., Kondagunta A., Cleary P.P. C5a peptidase alters clearance and trafficking of group A streptococci by infected mice. Infect. Immun. 1996;64:503–510. doi: 10.1128/iai.64.2.503-510.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun H., Xu Y., Sitkiewicz I., Ma Y., Wang X., Yestrepsky B.D., Huang Y., Lapadatescu M.C., Larsen M.J., Larsen S.D. Inhibitor of streptokinase gene expression improves survival after group A streptococcus infection in mice. Proc. Natl. Acad. Sci. U.S.A. 2012;109:3469–3474. doi: 10.1073/pnas.1201031109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kline J.B., Collins C.M. Analysis of the superantigenic activity of mutant and allelic forms of streptococcal pyrogenic exotoxin A. Infect. Immun. 1996;64:861–869. doi: 10.1128/iai.64.3.861-869.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fuangthong M., Helmann J.D. Recognition of DNA by three ferric uptake regulator (Fur) homologs in Bacillus subtilis. J. Bacteriol. 2003;185:6348–6357. doi: 10.1128/JB.185.21.6348-6357.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Solovyev V.V., Shahmuradov I.A., Salamov A.A. Identification of promoter regions and regulatory sites. Methods Mol. Biol. 2010;674:57–83. doi: 10.1007/978-1-60761-854-6_5. [DOI] [PubMed] [Google Scholar]
- 53.Kumaraswami M., Schuman J.T., Seo S.M., Kaatz G.W., Brennan R.G. Structural and biochemical characterization of MepR, a multidrug binding transcription regulator of the Staphylococcus aureus multidrug efflux pump MepA. Nucleic Acids Res. 2009;37:1211–1224. doi: 10.1093/nar/gkn1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shin J.-H., Jung H.J., An Y.J., Cho Y.-B., Cha S.-S., Roe J.-H. Graded expression of zinc-responsive genes through two regulatory zinc-binding sites in Zur. Proc. Natl. Acad. Sci. U.S.A. 2012;108:5045–5050. doi: 10.1073/pnas.1017744108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Busby S., Ebright R.H. Transcription activation by catabolite activator protein (CAP) J. Mol. Biol. 1999;293:199–213. doi: 10.1006/jmbi.1999.3161. [DOI] [PubMed] [Google Scholar]
- 56.Ashbaugh C.D., Albertí S., Wessels M.R. Molecular analysis of the capsule gene region of group A Streptococcus: the hasAB genes are sufficient for capsule expression. J. Bacteriol. 1998;180:4955–4959. doi: 10.1128/jb.180.18.4955-4959.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Isabella V., Wright L.F., Barth K., Spence J.M., Grogan S., Genco C.A., Clark V.L. cis- and trans-acting elements involved in regulation of norB (norZ), the gene encoding nitric oxide reductase in Neisseria gonorrhoeae. Microbiology. 2008;154:226–239. doi: 10.1099/mic.0.2007/010470-0. [DOI] [PubMed] [Google Scholar]
- 58.Federle M.J., Scott J.R. Identification of binding sites for the group A streptococcal global regulator CovR. Mol. Microbiol. 2002;43:1161–1172. doi: 10.1046/j.1365-2958.2002.02810.x. [DOI] [PubMed] [Google Scholar]
- 59.Wilkinson S.P., Grove A. Ligand-responsive transcriptional regulation by members of the MarR family of winged helix proteins. Curr. Issues Mol. Biol. 2006;8:51–62. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.