

Abstract
Background:
Aspirin-exacerbated respiratory disease (AERD) is explained in part by overexpression of 5-lipoxygenase and leukotriene C4 synthase (LTC4S), resulting in constitutive overproduction of cysteinyl leukotrienes (CysLTs) and driving the surge in CysLT production that occurs with aspirin ingestion. Similarly, AERD is characterized by the overexpression of CysLT receptors. Increased levels of both interleukin (IL)-4 and interferon (IFN)-γ are present in the tissue of AERD subjects. Previous studies demonstrated that IL-4 is primarily responsible for the up-regulation of LTC4S by mast cells.
Methods:
Literature review.
Results:
Our previous studies demonstrated that IFN-γ, but not IL-4, drives this process in eosinophils. These published studies also extend to both IL-4 and IFN-γ the ability to up-regulate CysLT receptors. Prostaglandin E2 (PGE2) acts to prevent CysLT secretion by inhibiting mast cell and eosinophil activation. PGE2 concentrations are reduced in AERD, and our published studies confirm that this reflects diminished expression of cyclooxygenase (COX)-2. A process again that is driven by IL-4. Thus, IL-4 and IFN-γ together play an important pathogenic role in generating the phenotype of AERD. Finally, induction of LTC4S and CysLT1 receptors by IL-4 reflects in part the IL-4-mediated activation of signal transducer and activator of transcription 6 (STAT6). Our previous studies demonstrated that aspirin blocks trafficking of STAT6 into the nucleus and thereby prevents IL-4-mediated induction of these transcripts, thereby suggesting a modality by which aspirin desensitization could provide therapeutic benefit for AERD patients.
Conclusion:
This review will examine the evidence supporting this model.
Keywords: Leukotriene, cyclooxygenase, prostaglandin, aspirin-exacerbated respiratory disease
Aspirin-exacerbated respiratory disease (AERD) or Samter's triad was originally defined by the presence of nasal polyps, aspirin sensitivity, and asthma.1 It is now recognized that this disorder is characterized by hypersensitivity not only to aspirin but also to other nonselective cyclooxygenase (COX) inhibitors.2–4 Asthma is not always present and thus the preferred terminology AERD. Other characteristics include the less common association with atopy,3,5 hypereosinophilia, and a tendency to develop de novo in adulthood.3,5–7 Aspirin hypersensitivity is found in as many as 10%–20% of adult asthmatics and up to 30% of asthmatics with nasal polyposis.5,8 When asthma is present in this disorder, it often becomes severe and is associated with aggressive airway remodeling.9 Similarly, the sinusitis present in this disorder is often severe and associated with complete or near complete sinus opacification.7
A central feature of AERD is its association with profound overproduction and overresponsiveness to cysteinyl leukotrienes (CysLTs)10,11 occurring concomitantly with a profound underproduction and underresponsiveness to prostaglandins.12–14 These CysLTs have important proinflammatory and profibrotic effects that contribute to the asthma severity and to the extensive hyperplastic sinusitis and nasal polyposis.7,15,16 And, conversely, the down-regulation of prostaglandin pathways reduces the constraints that would normally act to attenuate these proinflammatory pathways.17 This review will focus on the dysregulation of these respective pro- and antiinflammatory pathways and the cytokine mechanisms that underlie this dysregulation and, finally, will discuss implications of aspirin desensitization as a therapeutic intervention that acts by altering these pathways.
CYSTEINYL LEUKOTRIENE OVERPRODUCTION AND OVERRESPONSIVENESS IN AERD
AERD is characterized by the constitutive overproduction of CysLTs and a massive, potentially life-threatening, further surge in CysLT production in response to aspirin and other nonselective COX inhibitors that block COX-1.18 This includes not only nonselective nonsteroidal antiinflammatory drugs (NSAIDs) but also other inhibitors of COX-1, including alcoholic products.19,20 Overproduction of CysLTs in AERD reflects the increased expression of its primary synthesis enzymes 5-lipoxygenase and especially leukotriene C4 synthase (LTC4S). Up-regulation of these enzymes is observed in the lungs, sinuses, and nasal polyps of AERD subjects, localized in large part to the infiltrating eosinophils, and resident mast cells.12,15,21,22
AERD subjects also demonstrate an increased sensitivity to CysLTs,23 reflecting in part their up-regulation of CysLT 1 receptors.24 The two originally characterized CysLT receptors were distinguished by their differing potency for the CysLTs: CysLT1 receptors primarily respond to LTD4, whereas CysLT2 receptors respond equally to LTD4 and LTC4. However, this pattern could not explain a body of literature demonstrating the capacity of LTE4, and not either of these other CysLT receptors, to drive smooth muscle contraction and proinflammatory influences on both airway explants25 and, via inhalation challenges, on the airway itself of AERD subjects.23,26,27 The relative insensitivity of either CysLT1 or CysLT2 receptors to LTE4, in contrast to the sensitivity of AERD subjects to this lipid mediator, led to the exploration for and ultimate identification of additional CysLT receptors that selectively respond to LTE4.28–30 CysLT type 1 receptors are prominently expressed on airway smooth muscle,31 and these receptors do mediate much of the CysLT-induced bronchospasm associated with aspirin challenges or desensitizations,32–35 as evinced by the ability of leukotriene receptor antagonists to attenuate much of the bronchospasm that occurs with these procedures. Thus, AERD is characterized by enhanced sensitivity to leukotrienes, reflecting in part the overexpression of CysLT1 receptors. The enhanced responsiveness of these subjects to LTE4 is intriguing, although as of now, the expression and function of putative LTE4 receptors in this disorder remains unstudied.
PROSTAGLANDIN E2 (PGE2) AND PGE2 RECEPTOR DYSREGULATION IN AERD
PGE2 displays both pro- and antiinflammatory functions reflecting its ability to interact with four distinct receptors (EP1–EP4), each having various activating or inhibitory functions. However, it is the role of PGE2 acting through antiinflammatory EP2 receptors to block eosinophil and mast cell degranulation that is central to the pathogenesis of AERD. AERD patients constitutively display low levels of PGE212,36 attenuating the antiinflammatory constraints provided by this lipid. The further reduction of tissue PGE2 concentrations by aspirin and other NSAIDs through COX-1 inhibition precipitates the activation of eosinophils and mast cells in AERD, as demonstrated by the ability of an infusion of PGE2 to protect against these reactions.37,38 This sensitivity of AERD patients to low tissue PGE2 concentrations is amplified by their reduced expression of the antiinflammatory EP2 receptor.14
Several studies have investigated the mechanism behind the reduced levels of PGE2 in AERD and, perhaps not surprisingly, have correlated this with a decrease in the responsible upstream metabolic enzymes. The production of PGE2 from arachidonic acid involves the sequential synthesis of PGG2/PGH2 by the two COX enzymes (COX-1 and COX-2) followed by the synthesis of PGE2 by the microsomal PGE2 synthases (mPGES-1 and mPGES-2) and cytosolic PGE2 synthase. It is mPGES-1 that is most relevant to PGE2 production in inflammatory disorders such as AERD, because it is the enzyme primarily functionally coupled to COX-2.39 COX-2 mRNA and protein expression are markedly diminished in AERD.12,13,40 Our studies have confirmed this diminished expression of COX-2.41 We found no significant change in COX-1 and a trend toward diminished mPGES-1 expression.
Diminished COX-2 expression and the reduced capacity to synthesize PGE2 contributes to the severity of inflammation observed in AERD and accentuates the sensitivity of these individuals to the inhibition of PGE2 synthesis associated with aspirin and other NSAIDs. With this relative absence of COX-2, AERD subjects become dependent upon COX-1 for the PGE2 that is necessary to restrain mast cell and eosinophil activation. Most AERD patients tolerate selective COX-2 inhibitors, supporting this concept regarding the unique importance of COX-1-derived PGE2. In summary, AERD represents a state characterized by constitutive overexpression and overresponsiveness to CysLTs occurring concomitantly with diminished expression and underresponsiveness to PGE2. This dysfunctional state reflects the cytokine milieu of AERD (Fig. 1).
Figure 1.
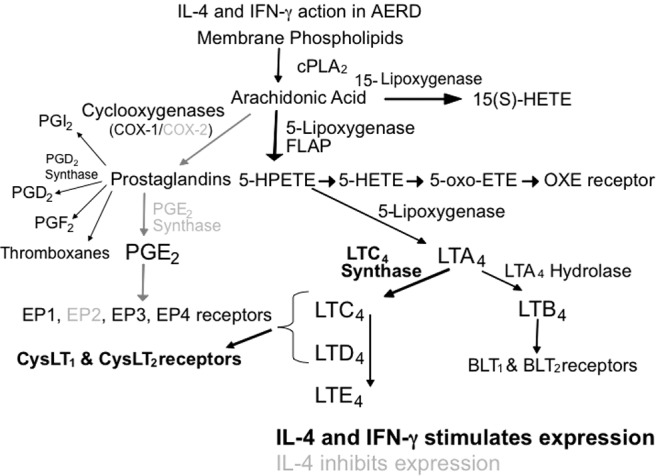
Summary of IL-4/IFN-γ activity on the leukotriene and prostaglandin synthesis pathways. Activation of gene synthesis by IL-4/IFN-γ is shown in boldface while inhibition of gene synthesis by IL-4 is shown in grey.
CYTOKINE EXPRESSION IN AERD
Numerous studies have addressed the cytokine milieu of eosinophilic sinusitis and, to a lesser extent, AERD. Not surprisingly, most of these studies have demonstrated a prominent T helper (Th)2-like profile, as would be expected in any eosinophilic process.42–48 However, there are numerous observations that suggest that in contrast to asthmatic or eosinophilic sinusitis patients who tolerate aspirin, AERD seems much more to be a mixed Th2- and Th1-like milieu with prominent expression of interferon (IFN)-γ. This was first suggested in studies demonstrating enhanced IFN-γ expression in a group labeled “nonallergic” sinusitis patients. One characteristic of this group was a higher prevalence of AERD in comparison with the other sinusitis groups.49 Other investigators have also described high IFN-γ levels in “chronic sinusitis” although, again, did not specify the extent to which this was driven by inclusion of AERD subjects.50 More recently, a group of adult-onset severe asthmatics with hypereosinophilia and absence of allergy, a cohort in whom AERD subjects were known to be particularly overexpressed, were distinctly characterized by their expression of a high IFN-γ gene signature (Sally Wenzel, AAAAI National Meeting, San Diego, CA; March 2014). The only other study that evaluated IFN-γ expression in AERD demonstrated enhanced levels of IFN-γ in circulating CD8+ cells.51
The concept that AERD might reflect a prominent Th1-like component was confirmed in our recently published studies, in which nasal polyp tissue derived from AERD subjects was contrasted from those obtained from aspirin tolerant and control subjects by their overexpression of IFN-γ mRNA transcripts (Fig. 2) and protein.52 Surprisingly, we subsequently also demonstrated that eosinophils themselves were the most important source for this cytokine52 consistent with recognition that IFN-γ can be expressed by eosinophils in substantial amounts.53–55 Eosinophils secrete numerous cytokines and chemokines,54 and unlike lymphocytes, eosinophils store these cytokines preformed within granules that can be instantly released with activation.55,56 The robust expression of interleukin (IL)-4 in AERD did not extent to IL-5 (Fig. 2), likely representing the physiology of eosinophils, specifically that IL-4 but not IL-5 is a prominent eosinophil-derived cytokine. Given the capacity of IFN-γ to block IgE class switch recombination, it is intriguing to speculate that this coexpression of IFN-γ contributes to the frequent absence of allergy in AERD previously noted,3,5 despite the observed expression of IL-4. The recognition of AERD as a combined Th2-/Th1-like disease prompted us to query the role of this mixed cytokine milieu in driving the AERD phenotype.
Figure 2.
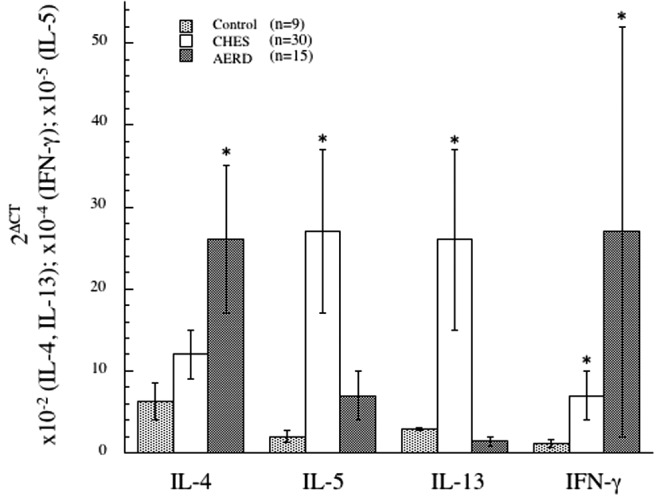
Th1/Th2 cytokine signature in control, CHES, and AERD sinus tissue by qPCR. Tissue samples were homogenized after surgical removal and RNA isolated. Transcript levels for IL-4, IL-5, IL-13, and IFN-γ were quantified using PCR with SYBR-green detection. Data (mean ± SEM) reflect relative expression of each gene in comparison with β-actin (2ΔCT). Control samples (n = 9) are depicted in black bars, CHES (n = 30) in gray bars and AERD (n = 15) in white bars. *p < 0.05 as compared with control. Reprinted from Steinke et al.,52 with permission from Elsevier.
HYPEREOSINOPHILIA IN AERD
AERD sinonasal and lung tissue are characterized by dramatic (∼10-fold) up-regulation in the number of infiltrating eosinophils.6,22 An IFN-γ-induced transcription factor (interferon consensus sequence binding protein) can drive the differentiation of eosinophils,57 leading us to speculate on its role in contributing to this eosinophilia. Consistent with this, we were struck by the capacity of IFN-γ, acting in synergy with IL-5, to promote both the survival and differentiation of mature bilobed, CC chemokine receptor 3- and sialic acid-binding immunoglobulin-like lectin 8-expressing eosinophils from CD34+ hematopoietic progenitors,52 confirming earlier studies58 regarding the influence of this cytokine on eosinophil-mediated inflammation. However, for the remainder of this review, we will primarily focus on influences of cytokines on CysLT and PG pathways.
CYTOKINE DYSREGULATION OF LTC4S AND CysLT RECEPTORS
Mast cells typically express modest levels of LTC4S and its up-regulation can be mediated by IL-4 (but not by IL-5 or IL-13).59 Interestingly, a role for IFN-γ in up-regulating LTC4S expression in umbilical cord-derived mast cell progenitors was also observed (J. Boyce, Harvard Medical School, 2014, personal communications). However, as noted,22 studies investigating the source of CysLTs in AERD have suggested that eosinophils are likely the more important cell type overexpressing LTC4S. In our studies, we were unable to demonstrate an ability of numerous innate or adaptive cytokines, including IL-3, IL-4, IL-5, granulocyte-macrophage colony-stimulating factor, IL-1, tumor necrosis factor-α, or even IFN-γ to modulate LTC4S expression in circulating eosinophils, presumably reflecting their terminal differentiation state. And our studies also failed to demonstrate an influence of IL-4 on LTC4S expression by eosinophils differentiated from progenitors in the presence of IL-3 and IL-5. In contrast, we demonstrated a significant increase in LTC4S expression by de novo IFN-γ-differentiated eosinophils.52 Most importantly, this translated into increased capacity of these newly differentiated eosinophils to secrete CysLTs upon activation. Thus, IFN-γ at present uniquely carries the capacity to drive this disease-defining characteristic of AERD.
As with LTC4S expression, the expression of the CysLT receptors is regulated by cytokines, including, most prominently, by IL-4 and IFN-γ. IL-4 increases expression of CysLT1 on mast cells60,61 and monocytes.62 In our studies, IL-4 also increased the expression of CysLT1 on T and B lymphocytes and eosinophils.63 Interestingly, our studies also demonstrated robust up-regulation of CysLT1 receptor in response to IFN-γ on T cells and eosinophils.63 This was an effect that, at the time of publication, we did not appreciate in regard to its likely relevance to AERD. More recently, we have also extended this capacity of IFN-γ to up-regulate CysLT1 receptors to eosinophils newly differentiated from CD34+ progenitors.52
CYTOKINE DYSREGULATION OF PGE2 SYNTHESIS AND EP2 RECEPTORS
We investigated the molecular mechanism underlying inhibition of PGE2 synthesis pathways in AERD. For these studies we focused on influences of IL-4, reflecting again, its prominent expression in AERD, its previously described influences on the prostaglandin metabolic pathways,64,65 and its involvement in the other facets of arachidonate dysregulation previously discussed. IFN-γ is unlikely to be involved in this aspect of the AERD phenotype, given its established role in up-regulating inducible COX (COX-2). Our studies were performed on nasal polyp-derived fibroblasts and mononuclear phagocytic cells. Monocytes were used both as representative inflammatory cells, but also because PGE2 is their dominant prostaglandin product. Significant inhibition of COX-2 and mPGES-1 (but not COX-1) mRNA and protein expression was observed in response to IL-4.41 This appears to be a generalized effect of IL-4 insofar as similar inhibition was also observed in fibroblasts. Inhibition of COX-2 and mPGES-1 synergize to result in dramatically less stimulated PGE2 secretion by monocytes.41 Thus, in addition to up-regulating CysLT pathways, IL-4 contributes to the AERD phenotype by inhibiting their PG pathways.
However, it is necessary to remark that more than just loss of the tempering influences of PGE2 underlies these reactions, otherwise all asthmatics and, indeed, even healthy subjects would react to aspirin/NSAID ingestion with activation of their mast cells and eosinophils. We therefore questioned the capacity of aspirin (and other NSAIDs) to directly drive the activation of eosinophils and mast cells.
DIRECT ACTIVATION OF EOSINOPHILS AND MAST CELLS BY ASPIRIN
How aspirin triggers these non-IgE-mediated reactions has been an enigma. We evaluated the capacity of aspirin and other NSAIDs to directly activate eosinophils and mast cells. For these studies, we used the water-soluble aspirin-like compound lysine aspirin (LysASA). Both eosinophils and mast cells displayed Ca+2 fluxes after stimulation with LysASA,66 and similar results were observed with eosinophil eosinophil-derived neurotoxin secretion. Similar results were obtained with ketorolac but not sodium salicylates. To our surprise, when eosinophils from control, aspirin tolerant, and aspirin intolerant subjects were compared, no differences were observed. We suspect the explanation as to why hypersensitivity reactions to aspirin/NSAID are not observed in these control cohorts reflects the relative implication of COX inhibition. As previously discussed, PGE2 acts through the antiinflammatory EP2 receptor to prevent the acute reactions to aspirin/NSAIDs.17,37 However, with the diminished expression of and responsiveness to PGE2 in AERD,12,14,67 these subjects exist on the precipice of cellular activation, such that even a modest decrease in PGE2 can have the observed catastrophic effects. The baseline expression and persistence of, by comparison, much higher levels of PGE2 protects these non-AERD subjects from cellular activation. This model is consistent with recent observations that in a murine model of AERD, knockout of the rate-limiting enzyme responsible for PGE2 synthesis, specifically mPGES-1, renders these mice susceptible to anaphylactoid reactions after exposure to aspirin.68
A further explanation for lack of reactivity in the control cohorts is that a large component of the anaphylactoid response to aspirin is driven by CysLTs (as shown as previously mentioned by the ability of LT modifiers to greatly attenuate their severity32,33). Interestingly, we never observed LysASA or ketorolac-mediated CysLT secretion from circulating eosinophils, even when obtained from AERD donors. AERD sinonasal and lung tissue is characterized by high numbers of eosinophilic hematopoietic progenitor (CD34+IL-5Rα+) cells69,70 that will mature in the presence of IFN-γ and, as previously discussed, acquire the ability to produce CysLTs. This would suggest that the robust CysLT production would be limited to airway, and not circulating, eosinophils. We therefore investigated whether eosinophils differentiated from progenitor cells in the presence of IFN-γ would recapitulate the sensitivity to aspirin displayed by tissue eosinophils in vivo in AERD. Consistent with their increased LTC4S expression, CysLT secretion was subsequently detected upon LysASA activation (Fig. 3).66
Figure 3.
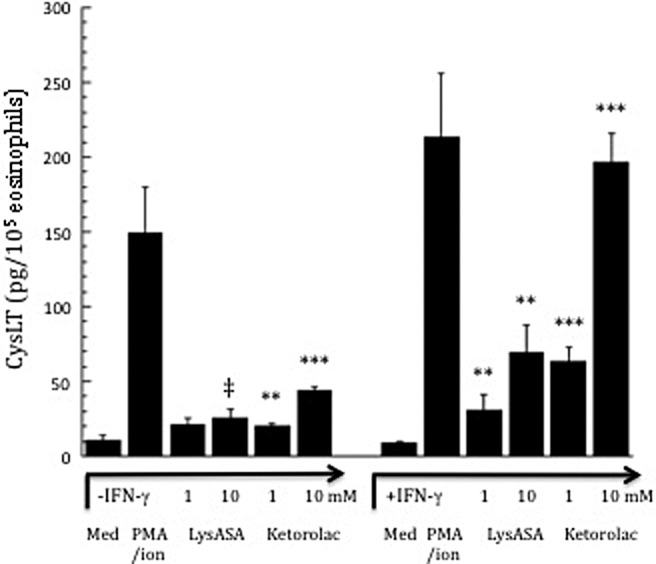
Activation of eosinophils differentiated from hematopoietic precursors in the additional presence of IFN-γ. CD34+-enriched hematopoietic progenitor cells were cultured for three days with SCF, TPO, Flt3L, IL-3, and IL-5, after which they were cultured for three additional weeks with just IL-3 and IL-5 with or without the additional presence of IFN-γ. Eosinophils matured from CD34+ progenitors were activated with or without calcium ionophore/PMA, LysASA (1mM and 10mM) or ketorolac (1mM and 10mM) for 30 minutes. Supernatants were collected and CysLT levels quantified and measured in pg/mL. Data are presented as the mean ± SEM of a minimum of six separate studies. ‡p = 0.05, **p < 0.03, ***p < 0.002 as compared with unstimulated cells. Reprinted with permission of the Journal of Immunology. Source: Reference 66.
ASPIRIN DESENSITIZATION IN AERD, TARGETING OF IL-4
Aspirin desensitization is an effective treatment for AERD and has been associated with the diminished need for nasal endoscopic surgery, improved sense of smell, fewer bouts of acute sinusitis, reduced need for oral corticosteroids, and less severe asthma.2,71,72 The molecular mechanisms driving the beneficial effects of aspirin have not been determined, but we believe it is due in part to the ability of this compound to inhibit the biologic activities of IL-4 (and perhaps, by extension, IFN-γ). Consistent with this concept are the observations that successful aspirin desensitization is associated with reversal of many of the IL-4-modulated features of AERD discussed above, including the ability of desensitization to down regulate CysLT synthesis and responsiveness pathways.23,24,73 There are many mechanisms by which aspirin may induce these effects. However, we focused on recognition that engagement of the IL-4 receptor by IL-4 induces activation of signal transducer and activator of transcription (STAT)6. A STAT6 site in the CysLT1 receptor promoter is central to its IL-4-mediated transcription, and similarly, a putative STAT site has been identified in the LTC4S gene.74 Aspirin inhibits the activation of STAT6,75 suggesting to us that aspirin may produce its clinical utility in AERD through direct inhibition of the IL-4-activated STAT6 pathway. Our studies investigated the inhibition by aspirin and other NSAIDs of the STAT6-mediated regulation of the CysLT1 receptor and LTC4S genes.41 In a dose-dependent fashion, aspirin inhibited transcription of IL-4-induced CysLT1 receptors. Subsequently, via electrophoretic mobility shift, oligomer competition, and supershift assays, we confirmed the presence of STAT6-binding sites within the CysLT1R and LTC4S promoters. Both by electrophoretic mobility shift and Western hybridization assays, our data demonstrated the absence of phosphoSTAT6 protein within the nuclei of aspirin-treated cells (Fig. 4). These results were extended to other NSAIDs, including ketorolac, but not sodium salicylate. The mechanism by which aspirin blocks pSTAT6 nuclear expression is not known but has been suggested to involve nuclear trafficking and recycling of transcription factors.76 These data suggest that aspirin desensitization may provide effective therapy for AERD, in part through mitigation of STAT6 activation, leading to down-regulation of the leukotriene pathways. That similar mechanisms could be involved in blocking IFN-γ-activated pathways (e.g., STAT1 trafficking) would be an intriguing area for future research.
Figure 4.
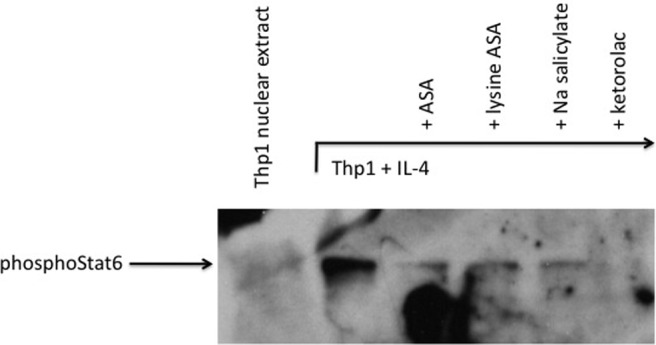
Western hybridization of nuclear extracts. Nuclear extract proteins were separated on a SDS polyacrylamide gel and transferred to a nitrocellulose membrane. Presence of phosphoStat6 was determined via probing with an anti-phosphoStat6 antibody and chemiluminescent detection. Reprinted from Steinke et al.,41 with permission from Elsevier.
SUMMARY
Toward a Generalized Model for the Induction of the AERD Phenotype
Although the exact mechanisms driving AERD are not fully understood, part of the explanation is the marked overexpression of the 5-lipoxygenase and LTC4S genes, resulting in constitutive overproduction of CysLTs, and the decrease in PGE2 expression that would normally act to constrain mast cell and eosinophil activation. We present a series of studies that strongly suggest that this AERD phenotype is derived, in large part, from the increased expression of both IL-4 and IFN-γ and that, as such, this disease reflects a mixed Th1/Th2 process. The increased expression of proinflammatory mediators and loss of protective PGE2 leads to uncontrolled release of mediators, and especially CysLTs, when eosinophils and mast cells are directly trigged by aspirin in AERD subjects. This increased understanding of the cellular reactions provides an opportunity to develop new therapeutic approaches aimed at dampening the severe impacts of this disease.
Footnotes
Presented at the North American Rhinology & Allergy Conference, January 23, 2014, Puerto Rico
This work was supported by National Institutes of Health RO1 AI47737 and PO1 AI50989
The authors have no conflicts of interest to declare pertaining to this article
REFERENCES
- 1. Samter M, Beers RF., Jr Intolerance to aspirin. Clinical studies and consideration of its pathogenesis. Ann Intern Med 68:975–983, 1968. [DOI] [PubMed] [Google Scholar]
- 2. Szczeklik A, Stevenson DD. Aspirin-induced asthma: advances in pathogenesis and management. J Allergy Clin Immunol 104:5–13, 1999. [DOI] [PubMed] [Google Scholar]
- 3. Szczeklik A, Nizankowska E. Clinical features and diagnosis of aspirin induced asthma. Thorax 55:S42–SS44, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berges-Gimeno MP, Simon RA, Stevenson DD. The natural history and clinical characteristics of aspirin-exacerbated respiratory disease. Ann Allergy Asthma Immunol 89:474–478, 2002. [DOI] [PubMed] [Google Scholar]
- 5. Vally H, Taylor ML, Thompson PJ. The prevalence of aspirin intolerant asthma (AIA) in Australian asthmatic patients. Thorax 57:569–574, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Payne SC, Early SB, Huyett P, et al. Evidence for distinct histologic profile of nasal polyps with and without eosinophilia. Laryngoscope 121:2262–2267, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mascia K, Borish L, Patrie J, et al. Chronic hyperplastic eosinophilic sinusitis as a predictor of aspirin-exacerbated respiratory disease. Ann Allergy Asthma Immunol 94:652–657, 2005. [DOI] [PubMed] [Google Scholar]
- 8. Szczeklik A, Sanak M. Molecular mechanisms in aspirin-induced asthma. ACI International 12/ 4:171–176, 2000. [Google Scholar]
- 9. Mascia K, Haselkorn T, Deniz YM, et al. Aspirin sensitivity and severity of asthma: evidence for irreversible airway obstruction in patients with severe or difficult-to-treat asthma. J Allergy Clin Immunol 116:970–975, 2005. [DOI] [PubMed] [Google Scholar]
- 10. Antczak A, Montuschi P, Kharitonov S, et al. Increased exhaled cysteinyl-leukotrienes and 8-isoprostane in aspirin-induced asthma. Am J Respir Crit Care Med 166:301–306, 2002. [DOI] [PubMed] [Google Scholar]
- 11. Daffern PJ, Muilenburg D, Hugli TE, Stevenson DD. Association of urinary leukotriene E4 excretion during aspirin challenges with severity of respiratory responses. J Allergy Clin Immunol 104:559–564, 1999. [DOI] [PubMed] [Google Scholar]
- 12. Perez-Novo CA, Watelet JB, Claeys C, et al. Prostaglandin, leukotiene, and lipoxin balance in chronic rhinosinusitis with and without nasal polyposis. J Allergy Clin Immunol 2005; 115:1189–1196, 2005. [DOI] [PubMed] [Google Scholar]
- 13. Picado C, Fernandez-Morata JC, Juan M, et al. Cyclooxygenase-2 mRNA is downexpressed in nasal polyps from aspirin-sensitive asthmatics. Am J Respir Crit Care Med 160:291–296, 1999. [DOI] [PubMed] [Google Scholar]
- 14. Ying S, Meng Q, Scadding G, et al. Aspirin-sensitive rhinosinusitis is associated with reduced E-prostanoid 2 receptor expression on nasal mucosal inflammatory cells. J Allergy Clin Immunol 117:312–318, 2006. [DOI] [PubMed] [Google Scholar]
- 15. Steinke JW, Bradley D, Arango P, et al. Cysteinyl leukotriene expression in chronic hyperplastic sinusitis-nasal polyposis: importance to eosinophilia and asthma. J Allergy Clin Immunol 111:342–349, 2003. [DOI] [PubMed] [Google Scholar]
- 16. Beller TC, Friend DS, Maekawa A, et al. Cysteinyl leukotriene 1 receptor controls the severity of chronic pulmonary inflammation and fibrosis. Proc Natl Acad Sci USA 101:3047–3052, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Steinke JW. Editorial: Yin-Yang of EP receptor expression. J Leuk Biol 92:1129–1131, 2012. [DOI] [PubMed] [Google Scholar]
- 18. Christie PE, Tagari P, Ford-Hutchinson AW, et al. Urinary leukotriene E4 concentrations increase after aspirin challenge in aspirin-sensitive asthmatic subjects. Am Rev Respir Dis 143:1025–1029, 1991. [DOI] [PubMed] [Google Scholar]
- 19. Cardet JC, White AA, Barrett NA, et al. Alcohol-induced respiratory symptoms are common in patients with aspirin exacerbated respiratory disease. J Allergy Clin Immunol. In practice 2:208–213, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Payne SC. Re: Alcohol-induced respiratory symptoms are common in patients with aspirin exacerbated respiratory disease. J Allergy Clin Immunol Pract 2:644, 2014. [DOI] [PubMed] [Google Scholar]
- 21. Sampson AP, Cowburn AS, Sladek K. Profound overexpression of leukotriene C4 synthase in bronchial biopsies from aspirin-intolerant asthmatic patients. Int Archives Allergy Immunol 113:355–357, 1997. [DOI] [PubMed] [Google Scholar]
- 22. Cowburn AS, Sladek K, Soja J, et al. Overexpression of leukotriene C4 synthase in bronchial biopsies from patients with aspirin-intolerant asthma. J Clin Invest 101:834–846, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Arm JP, O'Hickey SP, Spur BW, Lee TH. Airway responsiveness to histamine and leukotriene E4 in subjects with aspirin-induced asthma. Am Rev Respir Dis 140:148–153, 1989. [DOI] [PubMed] [Google Scholar]
- 24. Sousa AR, Parikh A, Scadding G, et al. Leukotriene-receptor expression on nasal mucosal inflammatory cells in aspirin-sensitive rhinosinusitis. N Engl J Med 347:1493–1499, 2002. [DOI] [PubMed] [Google Scholar]
- 25. Lee TH, Austen KF, Corey EJ, Drazen JM. Leukotriene E4-induced airway hyperresponsiveness of guinea pig tracheal smooth muscle to histamine and evidence for three separate sulfidopeptide leukotriene receptors. Proc Natl Acad Sci USA 81:4922–4925, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Christie PE, Schmitz-Schumann M, Spur BW, Lee TH. Airway responsiveness to leukotriene C4 (LTC4), leukotriene E4 (LTE4) and histamine in aspirin-sensitive asthmatic subjects. Eur Respir J 6:1468–1473, 1993. [PubMed] [Google Scholar]
- 27. Laitinen LA, Laitinen A, Haahtela T, et al. Leukotriene E4 and granulocytic infiltration into asthmatic airways. Lancet 341:989–990, 1993. [DOI] [PubMed] [Google Scholar]
- 28. Nonaka Y, Hiramoto T, Fujita N. Identification of endogenous surrogate ligands for human P2Y12 receptors by in silico and in vitro methods. Biochem Biophys Res Commun 337:281–288, 2005. [DOI] [PubMed] [Google Scholar]
- 29. Paruchuri S, Tashimo H, Feng C, et al. Leukotriene E4-induced pulmonary inflammation is mediated by the P2Y12 receptor. J Exp Med 206:2543–2555, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kanaoka Y, Maekawa A, Austen KF. Identification of GPR99 protein as a potential third cysteinyl leukotriene receptor with a preference for leukotriene E4 ligand. J Biol Chem 288:10967–10972, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lynch KR, O'Neill GP, Liu Q, et al. Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature 399:789–793, 1999. [DOI] [PubMed] [Google Scholar]
- 32. Christie PE, Smith CM, Lee TH. The potent and selective sulfidopeptide leukotriene antagonists, SK&F 104353, inhibits aspirin-induced asthma. Am Rev Resir Dis 144:957–958, 1991. [DOI] [PubMed] [Google Scholar]
- 33. Dahlén B. Treatment of aspirin-intolerant asthma with antileukotrienes. Am J Respir Crit Care Med 161:S137–S141, 2000. [DOI] [PubMed] [Google Scholar]
- 34. Walch L, Norel X, Bäck M, et al. Pharmacological evidence for a novel cysteinyl-leukotriene receptor subtype in human pulmonary artery smooth muscle. Br J Pharmacol 137:1339–1345, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. White A, Bigby T, Stevenson D. Intranasal ketorolac challenge for the diagnosis of aspirin-exacerbated respiratory disease. Ann Allergy Asthma Immunol 97:190–195, 2006. [DOI] [PubMed] [Google Scholar]
- 36. Schmid M, Göde U, Schäfer D, Wigand ME. Arachidonic acid metabolism in nasal tissue and peripheral blood cells in aspirin intolerant asthmatics. Acta Otolaryngol 119:277–280, 1999. [DOI] [PubMed] [Google Scholar]
- 37. Sestini P, Armetti L, Gambaro G, et al. Inhaled PGE2 prevents aspirin-induced bronchoconstriction and urinary LTE4 excretion in aspirin-sensitive asthma. Am J Respir Crit Care Med 153:572–575, 1996. [DOI] [PubMed] [Google Scholar]
- 38. Feng C, Beller EM, Bagga S, Boyce JA. Human mast cells express multiple EP receptors for prostaglandin E2 that differentially modulate activation responses. Blood 107:3243–3250, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Murakami M, Nakashima K, Kamei D, et al. Cellular prostaglandin E2 production by membrane-bound prostaglandin E synthase-2 via both cyclooxygenases-1 and -2. J Biol Chem 278:37937–37947, 2003. [DOI] [PubMed] [Google Scholar]
- 40. Gosepath J, Brieger J, Mann WJ. New immunohistologic findings on the differential role of cyclooxygenase 1 and cyclooxygenase 2 in nasal polyposis. Am J Rhinol 19:111–116, 2005. [PubMed] [Google Scholar]
- 41. Steinke JW, Culp JA, Kropf E, Borish L. Modulation by aspirin of nuclear phospho-signal transducer and activator of transcription 6 expression: possible role in therapeutic benefit associated with aspirin desensitization. J Allergy Clin Immunol 124:724–730 e4, 2009. [DOI] [PubMed] [Google Scholar]
- 42. Bachert C, Wagenmann M, Hauser U, Rudack C. IL-5 synthesis is upregulated in human nasal polyp tissue. J Allergy Clin Immunol 99:837–842, 1997. [DOI] [PubMed] [Google Scholar]
- 43. Bachert C, Gevaert P, van Cauwenberge P. Nasal polyposis- A new concept on the formation of polyps. ACI International 11:130–135, 1999. [Google Scholar]
- 44. Minshall EM, Cameron L, Lavigne F, et al. Eotaxin mRNA and protein expression in chronic sinusitis and allergen-induced nasal responses in seasonal allergic rhinitis. Amer J Resp Cell Mol Biol 17:683–690, 1997. [DOI] [PubMed] [Google Scholar]
- 45. Hamilos DL, Leung DY, Huston DP, et al. GM-CSF, IL-5, and RANTES immunoreactivity and mRNA expression in chronic hyperplastic sinusitis with nasal polyposis (NP). Clin Exp Allergy 28:1145–1152, 1998. [DOI] [PubMed] [Google Scholar]
- 46. Kamil A, Ghaffar O, Lavigne F, et al. Comparison of inflammatory cell profile and Th2 cytokine expression in the ethmoid sinuses, maxillary sinuses, and turbinates of atopic subjects with chronic sinusitis. Otolaryngol Head Neck Surg 18:804–809, 1998. [DOI] [PubMed] [Google Scholar]
- 47. Riechelmann H, Deutschle T, Rozsasi A, et al. Nasal biomarker profiles in acute and chronic rhinosinusitis. Clin Exp Allergy 35:1186–1191, 2005. [DOI] [PubMed] [Google Scholar]
- 48. Van Bruaene N, Perez-Novo CA, Basinski TM, et al. T-cell regulation in chronic paranasal sinus disease. J Allergy Clin Immunol 121:1435–1441.e1–e3, 2008. [DOI] [PubMed] [Google Scholar]
- 49. Hamilos DL, Leung DY, Wood R, et al. Evidence for distinct cytokine expression in allergic versus nonallergic chronic sinusitis. J Allergy Clin Immunol 96:537–544, 1995. [DOI] [PubMed] [Google Scholar]
- 50. Van Zele T, Claeys S, Gevaert P, et al. Differentiation of chronic sinus diseases by measurement of inflammatory mediators. Allergy 61:1280–1289, 2006. [DOI] [PubMed] [Google Scholar]
- 51. Shome GP, Tarbox J, Shearer M, Kennedy R. Cytokine expression in peripheral blood lymphocytes before and after aspirin desensitization in aspirin-exacerbated respiratory disease. Allergy Asthma Proc 28:706–710, 2007. [DOI] [PubMed] [Google Scholar]
- 52. Steinke JW, Liu L, Huyett P, et al. Prominent role of interferon-γ in aspirin-exacerbated respiratory disease. J Allergy Clin Immunol 132:856–865.e3, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kanda A, Driss V, Hornez N, et al. Eosinophil-derived IFN-γ induces airway hyperresponsiveness and lung inflammation in the absence of lymphocytes. J Allergy Clin Immunol 124:573–582, 2009. [DOI] [PubMed] [Google Scholar]
- 54. Akuthota P, Xenakis JJ, Weller PF. Eosinophils: offenders or general bystanders in allergic airway disease and pulmonary immunity? J Innate Immun 3:113–119, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Spencer LA, Szela CT, Perez SA, et al. Human eosinophils constitutively express multiple Th1, Th2, and immunoregulatory cytokines that are secreted rapidly and differentially. J Leukoc Biol 85:117–123, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Spencer LA, Melo RC, Perez SA, et al. Cytokine receptor-mediated trafficking of preformed IL-4 in eosinophils identifies an innate immune mechanism of cytokine secretion. Proc Natl Acad Sci USA 103:3333–3338, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Milanovic M, Terszowski G, Struck D, et al. IFN consensus sequence binding protein (Icsbp) is critical for eosinophil development. J Immunol 181:5045–5053, 2008. [DOI] [PubMed] [Google Scholar]
- 58. de Bruin AM, Buitenhuis M, van der Sluijs KF, et al. Eosinophil differentiation in the bone marrow is inhibited by T cell-derived IFN-γ. Blood 116:2559–2569, 2010. [DOI] [PubMed] [Google Scholar]
- 59. Hsieh FH, Lam BK, Penrose JF, et al. T helper cell type 2 cytokines coordinately regulate immunoglobulin E-dependent cysteinyl leukotriene production by human cord blood-derived mast cells: profound induction of leukotriene C(4) synthase expression by interleukin 4. J Exp Med 193:123–133, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mellor EA, Austen KF, Boyce JA. Cysteinyl leukotrienes and uridine diphosphate induce cytokine generation by human mast cells through an interleukin 4-regulated pathway that is inhibited by leukotriene receptor antagonists. J Exp Med 195:583–592, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mellor EA, Frank N, Soler D, et al. Expression of the type 2 receptor for cysteinyl leukotrienes (CysLT2R) by human mast cells: functional distinction from CysLT1R. Proc Natl Acad Sci USA 100:11589–11593, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Thivierge M, Stanková J, Rola-Pleszczynski M. IL-13 and IL-4 up-regulate cysteinyl leukotriene 1 receptor expression in human monocytes and macrophages. J Immunol 167:2855–2860, 2001. [DOI] [PubMed] [Google Scholar]
- 63. Early SB, Barekzi E, Negri J, et al. Concordant modulation of cysteinyl leukotriene receptor expression by IL-4 and IFN-γ on peripheral immune cells. Am J Respir Cell Mol Biol 36:715–720, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yano T, Hopkins HA, Hempel SL, et al. Interleukin-4 inhibits lipopolysaccharide-induced expression of prostaglandin H synthase-2 in human alveolar macrophages. J Cell Physiol 165:77–82, 1995. [DOI] [PubMed] [Google Scholar]
- 65. Dworski R, Sheller JR. Differential sensitivities of human blood monocytes and alveolar macrophages to the inhibition of prostaglandin endoperoxide synthase-2 by interleukin-4. Prostaglandins 53:237–251, 1997. [DOI] [PubMed] [Google Scholar]
- 66. Steinke JW, Negri J, Liu L, et al. Aspirin activation of eosinophils and mast cells: implications in the pathogenesis of aspirin-exacerbated respiratory disease. J Immunol 193:41–47, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kowalski ML, Pawliczak R, Wozniak J, et al. Differential metabolism of arachidonic acid in nasal polyp epithelial cells cultured from aspirin-sensitive and aspirin-tolerant patients. Am J Respir Crit Care Med 161:391–398, 2000. [DOI] [PubMed] [Google Scholar]
- 68. Liu T, Laidlaw TM, Katz HR, Boyce JA. Prostaglandin E2 deficiency causes a phenotype of aspirin sensitivity that depends on platelets and cysteinyl leukotrienes. Proc Natl Acad Sci USA 110:16987–16992, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kim YK, Uno M, Hamilos DL, et al. Immunolocalization of CD34 in nasal polyposis. Effect of topical corticosteroids. Am J Respir Cell Mol Biol 20:388–397, 1999. [DOI] [PubMed] [Google Scholar]
- 70. Denburg JA. Haemopoietic mechanisms in nasal polyposis and asthma. Thorax 55:S24–S25, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sweet JM, Stevenson DD, Simon RA, Mathison DA. Long-term effects of aspirin desensitization–treatment for aspirin-sensitive rhinosinusitis-asthma. J Allergy Clin Immunol 85:59–65, 1990. [DOI] [PubMed] [Google Scholar]
- 72. Stevenson DD, Simon RA. Selection of patients for aspirin desensitization treatment. J Allergy Clin Immunol 118:801–804, 2006. [DOI] [PubMed] [Google Scholar]
- 73. Juergens UR, Christiansen SC, Stevenson DD, Zuraw BL. Inhibition of monocyte leukotriene B4 production after aspirin desensitization. J Allergy Clin Immunol 96:148–156, 1995. [DOI] [PubMed] [Google Scholar]
- 74. Woszczek G, Pawliczak R, Qi HY, et al. Functional characterization of human cysteinyl leukotriene 1 receptor gene structure. J Immunol 175:5152–5159, 2005. [DOI] [PubMed] [Google Scholar]
- 75. Perez-G M, Melo M, Keegan AD, Zamorano J. Aspirin and salicylates inhibit the IL-4- and IL-13-induced activation of STAT6. J Immunol 168:1428–1434, 2002. [DOI] [PubMed] [Google Scholar]
- 76. Tegeder I, Pfeilschifter J, Geisslinger G. Cyclooxygenase-independent actions of cyclooxygenase inhibitors. FASEB J 15:2057–2072, 2001. [DOI] [PubMed] [Google Scholar]