

Abstract
Cytosolic carboxypeptidases (CCPs) constitute a new subfamily of M14 metallocarboxypeptidases associated to axonal regeneration and neuronal degeneration, among others. CCPs are deglutamylating enzymes, able to catalyze the shortening of polyglutamate side-chains and the gene-encoded C termini of tubulin, telokin, and myosin light chain kinase. The functions of these enzymes are not entirely understood, in part because of the lack of information about C-terminal protein processing in the cell and its functional implications. By means of C-terminal COFRADIC, a positional proteomics approach, we searched for cellular substrates targets of CCP1, the most relevant member of this family. We here identified seven new putative CCP1 protein substrates, including ribosomal proteins, translation factors, and high mobility group proteins. Furthermore, we showed for the first time that CCP1 processes both glutamates as well as C-terminal aspartates. The implication of these C termini in molecular interactions furthermore suggests that CCP1-mediated shortening of acidic protein tails might regulate protein–protein and protein–DNA interactions.
Cytosolic carboxypeptidases (CCPs)1 form a recently described subfamily of M14 metallocarboxypeptidases (MCPs) associated with neurodegeneration (1–4). Human and mouse genomes encode for six CCP members (CCP1–6), all related to Nna1 (nervous system nuclear protein induced by axotomy), the first reported and best characterized member of the CCP subfamily. Nna1, afterward renamed CCP1, was identified when screening for genes up-regulated during axonal regeneration and found to be homologous to members of the MCP family (5). This finding suggested a role of CCPs in axonal regeneration, which was further substantiated by studies in C. elegans showing that CCP6 is required for axon regrowth after axotomy (6). Loss-of-function mutations in the Ccp1/Nna1 gene lead to an ataxic phenotype in Purkinje cell degeneration (pcd) mice (7), characterized by a late-onset degeneration of cerebellar Purkinje cells (3–5 weeks of age) (8). Next to displaying reproductive abnormalities, these mice also exhibit a selective degeneration of other cell types in the nervous system; that is, thalamic neurons, retinal photoreceptors, and olfactory bulb mitral cells (9–11). These findings put CCP forward as an unexpected molecular link between neuronal degeneration and regeneration. Although the molecular pathways through which the loss of CCP1 function leads to neuronal degeneration in pcd mice are not completely understood, different mechanisms have been proposed such as endoplasmic reticulum stress (12), dysfunctional mitochondria (13), dysregulation of microtubule stability by abnormal levels of microtubule associated proteins (MAPs) (14), increased levels of tubulin polyglutamylation (15), and a progressive transcriptional silencing caused by the accumulation of DNA lesions (16–18).
More recently, a primary function for CCPs in tubulin processing was shown. Four members of the CCP subfamily, CCPs 1, 4, 5, and 6, specifically remove the gene-encoded C-terminal glutamate residue of detyrosinated tubulin (15). Moreover, CCPs 1, 4, and 6 shorten post-translationally added polyglutamate side-chains on tubulin (15). CCP5 on the other hand preferentially removes the branching point of a polyglutamylation event (15, 19, 20). It was suggested that abnormally high polyglutamylation levels, caused by the lack of functional CCP1, are responsible for the neuronal degeneration in pcd mice (15). A recent study further supports the link between CCPs and microtubules as the taxonomic distribution of CCPs suggests their primary function to be associated with cilia and basal bodies (2). Likewise, the C. elegans CCP1 ortholog, CCPP-1, was described to regulate the structural integrity of microtubules in sensory cilia and transport along them (21).
CCP1 was also found to C-terminally process telokin and myosin light chain kinase 1 (MLCK1) (15), regulators of myosin function. As such, it was postulated that CCP1 might cleave additional substrates and identifying these might help to elucidate its function and understand some of the anomalies observed in pcd mice. For instance, a nuclear localization for CCP1 was described, but its possible function in nuclear processes remains unclear (5). In this respect, Baltanas et al. observed extensive chromatin reorganization in pcd mice, leading to the accumulation of unrepaired DNA and ultimately to Purkinje cells death (16).
Here, we performed a proteome-wide screen for CCP1 substrates in HEK293T cells using C-terminal COFRADIC, a recently developed C-terminomics approach (22). Overall, we identified seven new putative CCP1 substrates, all of them harboring acidic amino acids in their gene-encoded C terminus. Validation of two of these substrates confirmed the processing events identified by C-terminomics and proved that these are the result of the direct action of CCP1 on these proteins. Analysis of the role of these acidic tails in proteins like the high mobility group proteins B (HMGBs), led us to speculate that CCP1, by shortening these tails, might regulate protein–protein and protein–DNA interactions.
EXPERIMENTAL PROCEDURES
Cell Culture
HEK293T cells (American Type Culture Collection (ATCC), Manassas, VA) were cultured in Dulbecco's Modified Eagle's Medium (DMEM) with 4.5 g/L glucose and GlutaMAX-I supplemented with 10% fetal calf serum (Invitrogen, Carlsbad, CA) at 37 °C, 10% CO2 and 95% humidity. HEK293F cells (Invitrogen) were grown in FreeStyle 293 expression medium (Invitrogen) in flasks on a rotary shaker (120 rpm) at 37 °C, 8% CO2, and 70% humidity.
Molecular Cloning and Transfection
The full-length cDNA sequence of human CCP1 (Uniprot Q9UPW5–1) was cloned into the pOPINFS vector (23). Both a Strep-tag II (24) and a hemagglutinin (HA) epitope were introduced at the C terminus of CCP1. The CCP1 E270Q (using the numbering system of bovine CPA, E1102Q in human CCP1) mutant was generated by QuikChange site-directed mutagenesis according to the manufacturer's protocol (Stratagene, La Jolla, CA). The full-length cDNA sequences of TRAF-type zinc finger domain-containing protein (TRAD1, Uniprot O14545) and human high mobility group protein B3 (HMGB3, Uniprot O15347) were cloned into the pTriEx-6 vector (EMD Millipore, Billerica, MA) and as a result both a Strep-tag and a HA-tag were introduced on their N termini. Full-length coding sequences of human high mobility group protein B1 (HMGB1, Uniprot P09429) and B2 (HMGB2, Uniprot P26583) were subcloned into the MBU-I-8856 vector (kindly provided by Prof. Sven Eyckerman, VIB-Ghent University, Belgium) and this SRα promoter driven eukaryotic expression vector introduces a Myc-tag on the N termini of these proteins. Supplemental Fig. S1 illustrates the constructs generated for this study.
DNA transfections were carried out using linear 25-kDa polyethylenimine (PEI) (PolySciences, Warrington, PA). Briefly, DNA was mixed with PEI in a ratio of 1:3 and incubated in the presence of serum-free medium for 15 min at room temperature. At 40–50% confluency, HEK293T cells were exposed to the transfection complex at 1.4 μg DNA per ml of culture for 60 h. HEK293F cells, at 106 cells per ml, were exposed to the transfection complex at 1 μg DNA per ml of culture for 60 h.
Expression and Purification of Recombinant Proteins
CCP1, HMGB3, and TRAD1 were expressed in mammalian cells and affinity purified with a Strep-Tactin affinity column (IBA GmbH, Göttingen, Germany). Briefly, HEK293F cells were transfected as described above with the constructs encoding the Strep-tagged version of these proteins. After 60 h, cells were washed and resuspended at 2 × 107 cells per ml of binding buffer (100 mm Tris-HCl, pH 8.0, 150 mm NaCl supplemented, with EDTA-free protease inhibitor mixture Set III (EMD Millipore)). Cells were then subjected to three rounds of freeze-thaw lysis and cell lysates were cleared by centrifugation at 16,000 × g for 5 min at 4 °C. The cleared lysate was loaded on a Strep-Tactin column equilibrated with binding buffer. Following a washing step with seven column volumes of binding buffer, Strep-tagged proteins were eluted and fractionated using elution buffer (binding buffer supplemented with 2.5 mm desthiobiotin). The purest eluted fractions as analyzed by SDS-PAGE were pooled.
In Vitro Substrate Validation Assay
Affinity purified CCP1 was incubated at 5 ng/μl for 2 h or 16 h at 37 °C with affinity purified TRAD1 or HMGB3 at 25 ng/μl in CCP1 buffer (80 mm PIPES, pH 6.8, 1 mm MgCl2, complemented with EDTA-free protease inhibitor mixture Set III (EMD Millipore)). In the control experiment, 10 mm o-phenanthroline, a metalloprotease inhibitor, was added to the reaction (15, 25).
Protein analysis of intact and processed HMGB3 was performed by nano LC-MS on a Waters nano-Acquity HPLC (Waters Corporation, Milford, MA) in-line coupled to an ESI-quadrupole (Q)-time-of-flight (TOF) (Waters Corporation) mass spectrometer. Briefly, 10 μl of HMGB3 solution at 10 ng of protein/μl was first loaded on a trapping column (made in-house, 100 μm internal diameter (I.D.) × 20 mm length, 5 μm Reprosil-Pur Basic-C4-HD beads, Dr. Maisch, Ammerbuch-Entringen, Germany) at a flow rate of 10 μl/min with 0.1% TFA in 2% acetonitrile. After 4 min, the trapping column was placed in-line with the analytical column (made in-house, 75 μm I.D. × 170 mm length, 3 μm Reprosil-Pur Basic-C4-HD beads, Dr. Maisch). The proteins were eluted with a linear gradient of 1.7% of acetonitrile increase per min at a flow rate of 300 nL/min. MS spectra were calibrated using the continuous lock mass correction with Leu-enkephalin, obtaining an accuracy of 500 ppm. The MaxEnt 1 deconvolution algorithm of the MassLynx software (Waters Corporation) was used to transform the multiple-charged ions into a MaxEnt spectrum on a real-mass scale.
Peptide mass fingerprinting of purified recombinant HMGB3 was performed by in-gel tryptic digestion followed by MALDI-TOF-MS analysis using an UltrafleXtreme MALDI-TOF mass spectrometer (Bruker Daltonics, Bremen, Germany) as previously described (26). The Flexanalysis software (version 3.3, Bruker Daltonics) was used to create peak lists and the Biotools software package (version 3.2, Bruker Daltonics) was used for interpretation of the MS spectra (Matrix Science, London, UK) and for the identification of tryptic maps. Sequence Editor 3.2 (Biotools 3.2, Bruker Daltonics) was used to compare the fingerprint of the experimental tryptic peptides map with the tryptic peptides maps of an in silico trypsin-digest, in search for unmatched peaks hinting to post-translational modifications (PTMs). The following criteria were used for the in silico enzymatic digestion: up to three missed tryptic cleavages and only single peptide charges were allowed, 150 ppm was set as precursor mass tolerance and acetylation of the protein N terminus, oxidation of methionines, and carbamidomethylation of cysteine residues were set as variable modifications.
Immunoblot
For immunoblotting, transfected cells were resuspended in phosphate saline buffer, supplemented with 0.1% or 1% Nonidet P-40, and Complete Protease Inhibitor Mixture (Roche Diagnostics GmbH, Mannheim, Germany). The suspension was subjected to three rounds of freeze-thaw lysis and cell lysates were cleared by centrifugation at 16,000 × g for 7 min at 4 °C. Cleared extracts were mixed with 4× SDS-PAGE loading buffer and boiled for 10 min at 95 °C. The pellet after centrifugation was boiled in 1× SDS-PAGE to generate the insoluble fraction samples. For the whole cell lysate samples, 4× SDS-PAGE loading buffer was added to the cell lysate without previous centrifugation. Equal amounts of each lysate or fraction were analyzed by SDS-PAGE and the separated proteins were electroblotted onto PVDF membranes (EMD Millipore), which were subsequently blocked with 5% skimmed milk for 2 h. The PVDF membranes were then incubated with primary antibody for 16 h at 4 °C. The following primary antibodies were used: anti-Δ2-tubulin (1:1000; EMD Millipore), anti-polyE (anti-polyglutamylation; 1:2000; gift of M.A. Gorovsky), anti-HA (HA-7; 1:3000; Sigma), anti-Strep-tag II (StrepMAB-Classic; 1:1000; IBA GmbH), anti-tyrosinated tubulin (TUB-1A2; 1:5000; Sigma), anti-β-actin (1:100,000; Sigma), anti-HMGB2 (ab67282; 1:500; Abcam, Cambridge, UK), anti-GAPDH (ab9484; 1:3000; Abcam), anti-histone H3 (1:2000; Cell Signaling, Danvers, MA), and anti-PARP (1:4000; BD Pharmingen, San Diego, CA). After washing with 0.05% Tween 20 in PBS (PBS-T), the primary antibodies were detected with HRP-labeled goat donkey anti-rabbit (1:5000; GE Healthcare, Piscataway, NJ) or goat anti-mouse (1:5000; Bio-Rad, Munich, Germany). After three washes in PBS-T, immunoreacted bands were visualized with Immobilon Western Chemiluminiscent HRP substrate (EMD Millipore).
For immunoblots visualized using an Odyssey infrared imager (LI-COR Biosciences, Lincoln, NE) a similar protocol was performed, except that membranes were blocked with Odyssey blocking buffer (LI-COR Biosciences) and the secondary antibodies used were goat anti-mouse-IRDye 680 and goat anti-rabbit-IRDye 800CW (LI-COR Biosciences).
Isolation of N-terminal and C-terminal Peptides
HEK293T cells were washed in PBS and resuspended at 2 × 107 cells per ml in 50 mm sodium phosphate buffer, pH 8.0, 100 mm NaCl, and 0.5 mm EDTA supplemented with Complete Protease Inhibitor Mixture (Roche Diagnostics GmbH). Cells were subjected to three rounds of freeze-thaw lysis and cleared by centrifugation at 16,000 × g for 5 min at 4 °C. Protein concentrations were determined with Bio-Rad's Protein Assay according to the manufacturer's instructions, and 1 ml of cleared cell lysate containing 2.5 mg of proteins was processed as follows. Guanidinium hydrochloride was added to the cell lysates to a final concentration of 4 m to denature proteins. Primary amines were acetylated for 2 h at 30 °C using sulfo-N-hydroxysuccinimide D3-acetate to a final concentration of 10 mm. O-acetylation was reversed by adding hydroxylamine to a final concentration of 40 mm and incubation for 10 min at 30 °C. The acetylation reagent was further quenched by the addition of 20 mm glycine for 10 min at 30 °C. Excess reagents were removed by buffer exchange to 1.5 ml of 10 mm NH4HCO3, pH 7.9, using 1 ml Illustra NAPTM-10 columns (GE Healthcare). After heating the proteins for 5 min at 95 °C followed by placing them on ice for 5 min, N-acetylated proteins were digested overnight at 37 °C with sequencing-grade, modified trypsin (Promega, Madison, WI), at an enzyme/substrate ratio of 1/100 (w/w). Subsequent steps of the C-terminal COFRADIC protocol were performed as described previously (22). As a result of the RP-HPLC separation, fractionation, and pooling, a total of 96 samples were generated. Samples were dried and redissolved in 20 μl of 2 mm TCEP and 2% acetonitrile for subsequent LC-MS/MS analysis.
LC-MS/MS Analysis
LC-MS/MS analysis of 2 μl of the sample mixtures was performed using an Ultimate 3000 RSLC nano LC system (Dionex, Amsterdam, The Netherlands) in-line connected to a LTQ Orbitrap Velos (Thermo Fisher, Bremen, Germany). LC-MS/MS analysis and generation of MS/MS peak lists were performed as previously described (27). Mascot generic files (mgf) were created using the Mascot Distiller software (version 2.2.1.0, Matrix Science). The generated MS/MS peak lists were then searched with Mascot using the Mascot Daemon interface (version 2.3, Matrix Science). These searches were performed in the Swiss-Prot database with taxonomy set to human (2011_05 UniProtKB/Swiss-Prot database containing 20,286 human entries). The following search parameters were set: spectra were searched with semi-ArgC/P enzyme settings, allowing no missed cleavages. Mass tolerance on the precursor ion was set to 10 ppm and on fragment ions to 0.5 Da. In addition, Mascot's C13 setting was set to 1. The peptide charge was set to 1+, 2+, or 3+, and instrument setting was put on ESI-TRAP. For the identification of butyrylated peptides, variable modifications were set to pyroglutamate formation of N-terminal glutamine and fixed modifications included D3-acetylation at lysines, methionine oxidation to methionine-sulfoxide, and butyrylation (12C4 or 13C4) of peptide N termini. Additionally, we also searched for peptides containing D3-acetylation at lysines and methionine oxidation as fixed modifications, and pyroglutamate formation of N-terminal glutamine and acetylation and D3-acetylation of N termini as variable modifications. Only peptides that were ranked one and scored above the threshold score, set at 95% confidence, were withheld. This confidence level was chosen because we have generally found lower Mascot scores for butyrylated C-terminal peptides (22). The estimated false discovery rate by searching decoy databases (a reversed version of the 2011_05 human UniProtKB/Swiss-Prot database made by the DBToolkit algorithm (28)) was found to be 3% at the spectrum level (29). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://www.proteomexchange.org) via the PRIDE partner repository (30, 31) with the data set identifier PXD000834 and DOI 10.6019/PXD000834.
Peptide quantification (12C4- versus 13C4-butyrylated) was carried out using the Mascot Distiller Quantitation toolbox (version 2.2.1). The quantification method details were as follows: constrain search, yes; protein ratio type, average; report detail, yes; minimum peptides, 1; protocol, precursor; allow mass time match, yes; allow elution shift, no; all charge states, yes; and fixed modifications, mass values. Ratios for the proteins were calculated by comparing the extracted ion chromatogram peak areas of all matched light peptides with those of the heavy peptides. To identify significantly altered C termini, robust statistics (32) was applied to the base-2 logarithm values of the calculated ratios of all the identified (12C4-butyrylated/13C4-butyrylated) peptide doublets being set as TRUE. Then, the R software package was used for statistical computing to calculate the probability distributions of log2 transformed peptide ratios. We considered neo-C-terminal peptides to those processed C termini (i.e. the latter peptides lacking up to 20 amino acids from database annotated C terminus) that were present at much higher levels (according to the statistical analysis and using a stringent p value threshold of 0.0001) in the proteome of HEK293T cells overexpressing CCP1. Ratios being set as FALSE were all verified by individual inspection.
Analysis of the Occurrence of Swiss-Prot Protein Entries with Acidic C termini
An in silico search for putative human CCP1 substrates was performed using ScanProsite (33) and using the pattern [ED](n)>, where n represents the number of consecutive acidic residues at a protein's C terminus which was allowed to vary from 1 to 32. The pattern search was performed in the Homo sapiens taxon of the 2014_03 UniProtKB/Swiss-Prot database, using the default settings (except that no splice variants were allowed).
To estimate the probability of finding proteins with acidic C-terminal tails, we assumed a random model of sequences in which the different amino acids occur independently in protein sequences (34). As a consequence, the probability (p) of finding a motif n1n2n3 … ni is
where nj ∈ Z and Z is the set of naturally occurring amino acids, Z = (A,R,N,D,C,Q,E,G,H,I,L,K,M,F,P,S,T,W,Y,V); p(nj) is the natural occurrence of the amino acid nj in the proteome, and these numbers were obtained from the average occurrences calculated by the iceLogo program for the human Swiss-Prot database (35). When applied to the C terminus, j = 1 corresponds to the C-terminal position, j = 2 to the penultimate position, j = 3 to the antepenultimate position, and so on. For the search of acidic motifs, we are not interested in a unique amino acid, but rather in the presence of Glu or Asp. Thus, we are interested in the probability of finding any of these two amino acids, i.e. p(E or D). In consequence, based on (Eq. 1) the probability of finding, in the whole proteome, proteins with C termini of i or more consecutive acidic residues was calculated as follows
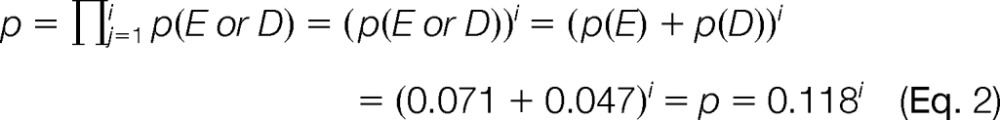 |
RESULTS
Study Design and Sample Preparation
It has previously been shown that the carboxypeptidases CCPs 1, 4, and 6 not only trim post-translationally added polyglutamate side chains of tubulin, but also act on the gene-encoded C-terminal glutamate stretches of two important regulators of myosin function; MLCK1 and telokin (15). It was then suggested that additional proteins carrying multiple glutamate residues at their C termini might also be CCP substrates. In this context, we performed an unbiased search for natural substrates of CCP1 using the C-terminal COmbined FRActional DIagonal Chromatography (C-terminal COFRADIC) positional proteomics approach (22). C-terminal COFRADIC is a quantitative approach, which enriches for C-terminal peptides, thereby pointing to peptides reporting C-terminal protein processing events (i.e. neo-C termini) and thus facilitating the discovery of natural substrates of carboxypeptidases (22).
We chose HEK293T cells for this study, since it was previously shown that this cell line constitute a simple and appropriate cellular system to study CCP1 function (36). We transiently transfected full-length CCP1 in HEK293T cells and analyzed its effect on different tubulin post-translational modifications (PTMs). After 72 h of transfection, Western blot analyses showed that overexpression of CCP1 generated increased levels of Δ2-tubulin (Fig. 1A and 1B), in line with previous reports (15, 36). Of note is that for overexpressed CCP1, besides the main 150-kDa band, CCP1 fragments of ∼120–130 kDa and 80–90 kDa can be observed (Fig. 1A). Similar patterns were reported for native and recombinant CCP1, both for the mouse (37) and human (2) orthologs.
Fig. 1.
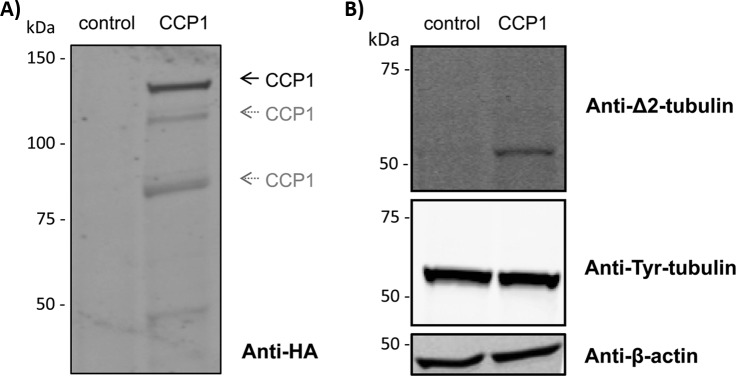
CCP1 overexpression increases the levels of Δ2-tubulin. A, CCP1 transfection in HEK293T cells. Protein extracts from control cells or cells expressing HA-tagged CCP1 were prepared. Samples (30 μg of protein extract) were analyzed by Western blot using an anti-HA antibody. Besides the main 150-kDa band, additional bands of ∼120–130 kDa and 80–90 kDa can be observed, similar to the patterns previously reported in literature (2, 37). B, Detection of tubulin PTMs. Extracts from control cells or CCP1 expressing cells were analyzed by Western blot using anti-Δ2-tubulin and anti-Tyr-tubulin antibodies. The Western blot probing anti-β-actin served as loading control.
C-terminal COFRADIC Analysis to Identify CCP1 Substrates
Knowing that expressed CCP1 is active in HEK293T cells, we proceeded with C-terminal COFRADIC (22) to identify CCP1 cleaved substrates on a proteome-wide level. Extracts from control HEK293T cells (i.e. mock-transfected) and HEK293T cells overexpressing CCP1 were processed in parallel, as described (22). In C-terminal COFRADIC, both N-terminal and C-terminal peptides are enriched by strong cation exchange (SCX) from internal peptides present in whole-proteome trypsin digests. Subsequent postmetabolic and chemical stable isotope-labeling of free amines with isotopic variants of N-hydroxysuccinimide (NHS)-butyrate (i.e. NHS ester of 12C4-butyrate for the control sample and 13C4-butyrate for the sample with CCP1 overexpression) allows for segregation of N-terminal and C-terminal peptides, and for MS-based quantification enabling the relative quantification of isolated C-terminal peptides per proteome.
Following LC-MS/MS analysis and database searching, we expected different readouts for the protein C termini identified and quantified. The majority of protein C termini were expected not be processed by CCP1 (Fig. 2A), and, as a consequence, their corresponding C-terminal peptides should be present at similar concentrations in both proteomes (Fig. 2C). Similarly, proteins C-terminally processed by a different carboxypeptidase (or other protease) were expected to give such a readout. CCP1 substrates however, should have been processed to higher extents in the proteome in which CCP1 is overexpressed (Fig. 2B). As a result, the corresponding intact C-terminal peptide (or database-annotated protein C-terminal peptide) should be found at higher levels in the control sample (Fig. 2D). Additionally, the generated neo-C-terminal peptide (C-terminal peptide generated by proteolytic cleavage of CCP1) is expected to be present at much higher levels in the proteome where CCP1 is overexpressed (Fig. 2E).
Fig. 2.

Identification of C-terminal proteolytic processing events in CCP1-transfected cells by means of C-terminal COFRADIC analysis. A, Representation of a protein that is not processed by CCP1. Scissors indicate trypsin cleavage sites. N-terminal tryptic peptides are colored in red. C-terminal tryptic peptides are colored in green/blue. Internal tryptic peptides are colored in yellow. B, Representation of a protein that is C-terminally processed by CCP1. C, Mass spectrum representation of a C-terminal peptide that is not processed in both proteomes (Peptide A). This mass spectrum would equally represent a C-terminal peptide showing basal degradation in both proteomes by a protease different from CCP1. D, Mass spectrum representation of the intact C-terminal peptide of a protein that is processed by CCP1 (Peptide B). E, Mass spectrum representation of the neo-C-terminal peptide of a protein that is processed by CCP1 (Peptide C). F, Identification of α-tubulin 1A/1B as CCP1 substrate. Mass spectrum of the triply charged neo-C terminus of α-tubulin 1A/1B, But13C4-EDMAALEKDYEEVGVDSVEGEGEEEG (residues 423–448; But13C4, α-amino group modified by 13C4-butyric acid).
In total, we identified 1259 unique database-annotated N termini, 886 unique database-annotated protein C termini, and 75 processed protein C termini (i.e. only considering peptides lacking up to 20 amino acids from database annotated C terminus) (supplemental Table S1). Most of the latter were found at similar levels in both proteomes, and thus point to proteins processed by proteases other than CCP1 or to artifacts (i.e. C-terminal ragging) generated during sample preparation. Interestingly, we identified nine processed C termini that were present at much higher levels (over sevenfold up, p < 0.0001) in the proteome of HEK293T cells overexpressing CCP1 (Table I). Annotated MS/MS spectra of the neo-C termini identified in Table I are provided in supplemental Fig. S2. We focused our analysis on these neo-C termini, because they directly point to putative CCP1 substrates. In particular, two of these neo-C termini corresponded to processed α-tubulin C termini, a protein known to be processed by CCP1 (Fig. 1B and references (15, 36, 37)). C-terminal COFRADIC identified the processed C terminus of α-tubulin 1A/1B (EDMAALEKDYEEVGVDSVEGEGEEEG; residues 423–448) as almost uniquely present in the proteome of HEK293T cells overexpressing CCP1 (Fig. 2F). Moreover, we also identified a neo-C terminus (EDMAALEKDYEEVGADSADGEDEG; residues 423–446) of a different α-tubulin isotype (α-tubulin 1C) as a singleton in CCP1 overexpression sample. Both peptides correspond to the C terminus of Δ3-tubulin, a tubulin form that lacks the last three residues of α-tubulin. Coincident with previous reports (36), our data confirm the ability of CCP1 to generate Δ2-tubulin (Fig. 1B) or Δ3-tubulin (Table I) by removing one or two Glu residues from the C terminus of detyrosinated α-tubulin; considering that another enzyme, different from CCP1, is responsible for releasing C-terminal Tyr from α-tubulin (15, 38).
Table I. List of proteins processed by CCP1 at their gene-encoded C-terminus.
| Protein | Accession number | Assay | Identified neo-C-terminus | Cleaved amino acids | L/H ratio | Subcellular Localization | Function |
|---|---|---|---|---|---|---|---|
| α-tubulin 1A/1B | Q71U36/P68363 | C-terminal COFRADIC | EDMAALEKDYEEVGVDSVEGEGEEEG | EE(Y)a | N/A | Cytoplasm | Microtubules |
| α-tubulin 1C | Q9BQE3 | C-terminal COFRADIC | EDMAALEKDYEEVGADSADGEDEG | EE(Y)a | N/A | Cytoplasm | Microtubules |
| Eukaryotic translation initiation factor 4H | Q15056 | C-terminal COFRADIC | EEVVQKEQ | E | N/A | Cytoplasm | Translation initiation |
| Stathmin | P16949 | C-terminal COFRADIC | KNKESKDPADETEA | D | 0.029 | Cytoplasm | Microtubules regulation |
| 40S ribosomal protein S9 | P46781 | C-terminal COFRADIC | KNAKKGQGGAGAGDDE | EED | N/A | Cytoplasm | Translation initiation |
| C-terminal COFRADIC | KNAKKGQGGAGAGDD | EEED | N/A | ||||
| TRAF-type zinc finger domain-containing protein | O14545 | C-terminal COFRADIC | TAKAKPSKQQGAGDA | EEEEEE | 0.14 | Cytoplasm | Regulation TLR4 and RLH pathways |
| High mobility group protein B3 | O15347 | C-terminal COFRADIC | KKVEEEDE | EEEEEEEEEEEEEDE | 0.083 | Nucleus/Cytoplasm | Transcription regulation, chromatin |
| C-terminal COFRADIC | KKVEEED | EEEEEEEEEEEEEEDE | N/A | ||||
| High mobility group protein B2 | P26583 | Western blot | EDEEEEEEEEDEDEEEEDEDEEb | Nucleus/Cytoplasm | Transcription regulation, chromatin | ||
| High mobility group protein B1 | P09429 | Western blot | EEEEDEEDEEDEEEEEDEEDEDEEEDDDDEb | Nucleus/Cytoplasm | Transcription regulation, chromatin | ||
| Myosin light chain kinase 1/Telokinc | Q6PDN3 | Western blot | EEEEEEd | Cytoplasm | Regulator of myosin function |
N/A, not applicable for the impossibility to calculate an m/z ratio due to the absence of light peptide ions (i.e. peptide not identified in the control sample).
a In the case of α-tubulin, we considered that CCP1 uses as substrate the pool of detyrosinated tubulin naturally present in the cell (15, 38).
b The C-terminus of these proteins is displayed, although no CCP1 cleavage sites have been identified.
c This substrate was not identified in our screen, but the orthologous mouse substrate was previously reported by Rogowski et al. (15).
d Based on western blots showing an increase in Δ2-tubulin.
Besides α-tubulin, we identified seven neo-C termini belonging to five new putative CCP1 substrates (Table I). Interestingly, in all cases the amino acids released by CCP1 are acidic amino acids, a substrate specificity that partially agrees with previous reports. Two of these peptides indicate CCP1-mediated removal of a single amino acid from the protein's C terminus: release of glutamate from the C terminus in case of the eukaryotic translation initiation factor 4H (eIF4H), and of aspartate in the case of stathmin. Previous reports demonstrated the action of CCP1 on C-terminal glutamates (15, 36, 37), and it was suggested that CCP1 does not recognize aspartate (15, 37). Our observed trimming of the C terminus of stathmin indicates that CCP1 is indeed able to release C-terminal aspartates. Moreover, two peptides pointed to C-terminal processing of the 40S ribosomal protein S9 (RPS9). Here, CCP1 removed respectively three and four amino acids from the C terminus of RSP9 (i.e. the C-terminal Asp and two or three subsequent Glu residues were removed, Table I). Given that M14 MCPs release only one amino acid at a time, it is expected that CCP1 sequentially releases these amino acids (37). The TRAF-type zinc finger domain-containing protein (TRAD1) was also identified as a putative CCP1 substrate. TRAD1 contains a C-terminal stretch of six glutamates and CCP1 seems to be capable of releasing all of them. Remarkably, we also identified two neo-C termini of the high mobility group protein B3 (HMGB3), which shows that up to 15 or 16 amino acids are removed by CCP1.
Interestingly, when identified and except for α-tubulin 1C, the levels of corresponding intact C termini of these putative CCP1 substrates are not significantly altered (supplemental Table S2). This might indicate that the extent of processing of these putative CCP1 substrates in cells is generally low. The intact C terminus of α-tubulin 1C with significantly altered expression, however, does not match the expected readout for a substrate (Fig. 2C), but rather seems to be at fivefold higher concentration in the sample with CCP1 overexpression. Western blot analysis using an antibody recognizing different tubulin isotypes however showed equal levels of total Tyr-tubulin in both samples (Fig. 1B).
Validation of Putative CCP1 Substrates
From the five new putative CCP1 substrates identified, two were selected for further validation; TRAD1 and HMGB3. TRAD1 contains a C-terminal stretch of six glutamates residues and CCP1 is able to sequentially remove all of them (Fig. 3A). We therefore used the polyE antibody that was designed to recognize a polyglutamate side-chain of at least three consecutive glutamate residues in tubulin (39), but additionally recognizes gene-encoded C termini of three or more glutamates (15). N-terminal Strep- and HA-tagged TRAD1 was overexpressed in HEK293F cells together with CCP1 or an inactive form of CCP1 (CCP1-E270Q). The latter contains a mutation of a key catalytic residue of M14 metallocarboxypeptidases (E270Q using the numbering system of bovine carboxypeptidase A, E1102Q in human CCP1) that polarizes the metal-bound water molecule responsible for attack of the scissile bond (37, 40). Because of the low sensitivity of the polyE antibody, we enriched TRAD1 by means of its N-terminal Strep-tag. Subsequent Western blot analysis of TRAD1 with antibodies directed to its N-terminal HA-tag (Fig. 3B) shows that TRAD1 is enriched from both cell lines with similar efficiency. Note that CCP1 is copurified because it also holds a Strep-tag at its C terminus (Fig. 3B). Interestingly, the polyE antibody detects TRAD1's C terminus when co-expressed with inactive CCP1, but not when co-expressed with active CCP1 (Fig. 3B). In addition, we directly incubated purified recombinant TRAD1 with purified recombinant CCP1 and found that TRAD1 was no longer recognized by the polyE antibody (Fig. 3C). However, in the presence of o-phenanthroline (32), a specific inhibitor of metalloproteases, the polyE antibody was still capable of recognizing affinity purified TRAD1, indicative of CCP1 dependent removal of the acidic tail of TRAD1. Hence, our data validate TRAD1 as a novel and direct CCP1 substrate.
Fig. 3.

TRAD1 and HMGB3 are direct CCP1 substrates. A, CCP1 is capable of removing all acidic C-terminal amino acids of TRAD1. The epitope recognized by the PolyE antibody is indicated. B, The C terminus of TRAD1 is processed by active CCP1. Strep/HA- tagged TRAD1 was cotransfected in HEK293F cells either with active CCP1 or inactive (E270Q) CCP1. TRAD1 was enriched from protein extracts using Strep-Tactin columns. Equal amounts of protein eluate were analyzed by Western blot using an anti-Strep antibody. CCP1 is also enriched by means of its C-terminal Strep-tag. The polyE antibody shows the integrity of TRAD1 C terminus when co-expressed with inactive CCP1, but not when co-expressed with the active enzyme. C, In vitro validation of TRAD1 as a direct CCP1 substrate. Purified recombinant TRAD1 was incubated with CCP1 for 2 h at 37 °C, in the presence or absence of the MCP inhibitor o-phenanthroline (o-Phen), and analyzed by Western blot using the polyE antibody. D, HMGB3 is composed of two central DNA binding domains (HMG-boxes) and a long acidic C-terminal tail. CCP1 is able to sequentially release up to 16 acidic amino acids from its C terminus. The size of the HMG boxes and the acidic tail are merely illustrative, and does not represent their real sizes. E, HMGB3 is processed by CCP1. Strep-tagged HMGB3 was cotransfected with active or inactive (E270Q) CCP1. Equal amounts of each cell extract were analyzed by Western blot using a HA-tag antibody, to demonstrate equal expression levels of both CCP1 variants. Western blotting using an anti-Strep antibody shows the appearance of a degraded form of HMGB3 when co-expressed with active CCP1. F, In vitro validation of HMGB3 as a direct CCP1 substrate. Purified recombinant HMGB3 was incubated with CCP1 ON at 37 °C, in the presence or absence of 10 mm o-Phen, and analyzed by SDS-PAGE.
As for HMGB3, CCP1 seems to remove quite a long stretch of acidic amino acids from its C terminus (Fig. 3D). Accordingly, we expected to observe a significant change in the MW of the HMGB3 precursor versus its processed counterpart(s) by means of SDS-PAGE. As such, N-terminally Strep-tagged HMGB3 was co-expressed in HEK293T cells with either active or inactive CCP1. Western blot analysis shows that in addition to the intact HMGB3 band, a proteolytic fragment of HMGB3 is found when HMGB3 is co-expressed with active CCP1 (Fig. 3E). The appearance of a HMGB3 fragment, with different electrophoretic mobility, is consistent with the CCP1-mediated processing identified by C-terminal COFRADIC. By assaying recombinantly produced and purified proteins, we could also show direct processing of HMGB3 by CCP1, but only in the absence of o-phenanthroline (Fig. 3F). Finally, we used protein mass spectrometry to measure the masses of the purified HMGB3 protein(-fragments). Q-TOF analyses determined the molecular weight of intact HMGB3 purified from HEK293F cells to be 25,300 ± 12.65 Da (supplemental Fig. S3A), which indicates that this protein lacks its initiator methionine, is N-terminally acetylated and lacks a C-terminal glutamate (predicted MW 25303.3 Da). Peptide mass fingerprint analysis confirms the presence of these PTMs (supplemental Fig. S3B and supplemental Tables S3 and S4). The mass of the HMGB3 fragment generated by CCP1 is 23,377 ± 11.67 Da (supplemental Fig. S3C), which corresponds to the release of 15 extra C-terminal residues of HMGB3 by CCP1 (predicted MW 23380.6 Da). Hence, the in vitro cleavage product fully correlates with one of the HMGB3 neo-C termini identified by C-terminal COFRADIC, and all of these results validate HMGB3 as a direct CCP1 substrate.
Prediction of Additional CCP1 Substrates
Our findings show that CCP1's substrate specificity is restricted to Glu and Asp residues. We exploited this characteristic to perform an in silico scan for other potential CCP1 targets using the ScanProsite tool (33). Our search (parameters indicated in the Experimental Procedures section) returned a list of 16, 36, and 111 human proteins containing respectively 7, 5, and 3 or more consecutive acidic amino acids at their C terminus. Table II shows the protein hits with five or more consecutive acidic C-terminal residues. Supplemental Table S5 displays hits containing C termini with three or more acidic residues. The number of proteins in Table II is 76 times higher than expected by chance alone; among the 20,257 human proteins in the 2014_03 UniProtKB/Swiss-Prot database less than one protein (or 1 out of 43,159 proteins) is expected to have five or more consecutive acidic C-terminal residues (considering that the natural frequencies of glutamate and aspartate for human proteins in Swiss-Prot are 7.1% and 4.7%, respectively). More so, finding proteins with 10 or more consecutive C-terminal acidic residues is highly unlikely (1 in 1.86 × 109 proteins); however, nine human proteins have acidic stretches of at least 10 residues.
Table II. List of human proteins containing a stretch of at least 5 consecutive acidic residues in their gene-encoded C-terminus.
| Number | Name | UniProt | C-terminus |
|---|---|---|---|
| 1 | High mobility group protein B1 | P09429 | SKKKKEEEEDEEDEEDEEEEEDEEDEDEEEDDDDE |
| 2 | Putative high mobility group protein B1-like 1 | B2RPK0 | SKKKKEEEEDEEDEEDEEEEDEEDEEDDDDE |
| 3 | High mobility group protein B2 | P26583 | KKNEPEDEEEEEEEEDEDEEEEDEDEE |
| 4 | High mobility group protein B3a | O15347 | ARKKVEEEDEEEEEEEEEEEEEEDE |
| 5 | Homeobox protein Hox-A7 | P31268 | AADKADEEDDDEEEEDEEE |
| 6 | Cytosolic purine 5′-nucleotidase | P49902 | ITHCHDEDDDEEEEEEEE |
| 7 | Translocation protein SEC63 homolog | Q9UGP8 | FEDSFEEEEEEEEDDD |
| 8 | mTERF domain-containing protein 2 | Q7Z6M4 | EAEDNDEDEDDDEEE |
| 9 | 60S ribosomal protein L22 | P35268 | FQINQDEEEEEDED |
| 10 | Calsequestrin-1 | P31415 | GEINTEDDDDDDDD |
| 11 | Ubiquitin thioesterase ZRANB1 | Q9UGI0 | SLSDGEEDEDDEDE |
| 12 | Calcium-dependent secretion activator 1 | Q9ULU8 | SMKDSDEEDEEDD |
| 13 | Putative high mobility group protein B3-like | P0C6E5 | AQKKVEEEDEDEE |
| 14 | RNA polymerase-associated protein LEO1 | Q8WVC0 | KYVISDEEEEDDD |
| 15 | LIM domain and actin-binding protein 1 | Q9UHB6 | RNRYYDEDEDEE |
| 16 | Single-pass membrane protein with aspartate-rich tail 1, mitochondrial | Q9H4I9 | DIFVPEDDDDDD |
| 17 | Calsequestrin-2 | O14958 | DNDDSDDDDDE |
| 18 | Cell cycle control protein 50B | Q3MIR4 | RYQDQDDDDEE |
| 19 | Dipeptidyl aminopeptidase-like protein 6 | P42658 | TVTAKEDEEED |
| 20 | Homeobox protein Hox-B7 | P09629 | DRAEAEEEEEE |
| 21 | Metaxin-1 | Q13505 | LGMAEEDEEE |
| 22 | 40S ribosomal protein S9a | P46781 | GAGAGDDEEED |
| 23 | Protein timeless homolog | Q9UNS1 | KRYQIEDDEDD |
| 24 | TRAF-type zinc finger domain-containing protein 1a | O14545 | GAGDAEEEEEE |
| 25 | Acidic leucine-rich nuclear phosphoprotein 32 family member E | Q9BTT0 | AEDDGEEEDD |
| 26 | Interferon alpha-inducible protein 6 | P09912 | KYLDSEEDEE |
| 27 | Myosin light chain kinase, smooth musclea | Q15746 | GEGEGEEEEE |
| 28 | Choline-phosphate cytidylyltransferase A | P49585 | AYDISEDEED |
| 39 | Protein SSX1 | Q16384 | EISDPEEDDE |
| 30 | Protein SSX2 | Q16385 | EISDPEEDDE |
| 31 | Protein SSX3 | Q99909 | EISDPEEDDE |
| 32 | Protein SSX4 | O60224 | EISDPEEDDE |
| 33 | Protein SSX7 | Q7RTT5 | EISDPEEDDE |
| 34 | Protein SSX8 | Q7RTT4 | EIRDPEEDDE |
| 35 | Protein SSX9 | Q7RTT3 | EISDPEEDDE |
| 36 | Transmembrane protein 190 | Q8WZ59 | EETEGEEEED |
a These proteins have already been identified as CCP1 substrates.
Our ScanProsite analysis pointed to different high mobility group proteins as potential CCP1 targets. In particular, the high mobility group protein B1 (HMGB1) and B2 (HMGB2) contain an unusually high number of acidic residues at their C termini (30 and 22 residues respectively). To confirm that in line with HMGB3, these could be CCP1 substrates, N-terminally Myc-tagged versions of both proteins were co-expressed in HEK293T cells with either active or inactive CCP1. Western blot analyses show that CCP1 indeed processes the C terminus of HMGB2 (Fig. 4B). A second cleavage product of HMGB2 with a lower molecular weight can also be observed, although at lower levels (Fig. 4B). CCP1 is also capable of processing the C terminus of HMGB1 (Fig. 4A), but to a lower extent when compared with HMGB3 or HMGB2. In this case, processing leads to several protein fragments.
Fig. 4.

HMGB2 and HMGB1 are processed by CCP1. CCP1 is able to cleave the C terminus of A, HMGB1 and B, HMGB2. N-terminally Myc-tagged HMGB1 or HMGB2 were cotransfected with active or inactive CCP1. Equal amounts of each cell extract were analyzed by Western blot using a HA-tag antibody, to show equal expression levels of both CCP1 variants. Western blotting using an anti-Myc antibody shows the appearance of C-terminal processed forms of the HMGB proteins when co-expressed with active CCP1.
CCP1 partially localizes in the nucleus, as was shown by Rodriguez de la Vega et al. by confocal microscopy analysis in HeLa cells, revealing a granular distribution for CCP1 both in the nucleus and cytoplasm (2). Furthermore, recently, a nuclear export signal (NES) in the N-terminal region of CCP1 was identified and characterized, showing that CCP1 is involved in a nuclear-to-cytoplasmic relocalization mediated by CRM1-dependent nuclear export (41). Because the function of CCP1 in nuclear processes remains unclear, the identification of possible nuclear substrates is of interest. HMGB1 and HMGB2 localize in the nucleus, but shuttle continuously to the cytoplasm by active transport mechanisms, with the equilibrium shifted toward nuclear accumulation (42–45). To exclude the possibility that cleavage of HMGB proteins occurs as a result of an aberrant localization of HMGB proteins (i.e. generated by their overexpression), we used an anti-HMGB2 antibody to analyze endogenous HMGB2 in HEK293T cell overexpressing CCP1 or CCP1-E270Q. Western blot analysis showed the appearance of C-terminal processed forms of endogenous HMGB2 in cells overexpressing active CCP1, absent when inactive CCP1 mutant was overexpressed (supplemental Fig. S4, lanes 1–2). Thus, we confirm the ability of CCP1 to process the acidic C-terminal tail of HMGB2, in this case by analyzing the endogenous HMGB2 protein. Given that HMGB2 partially associates with chromatin, we fractionated whole cell protein lysates into soluble fractions (containing cytosolic and nuclear soluble proteins) and chromatin- bound insoluble fractions, and found that most of the processed HMGB2 is present in the chromatin(-associated) fraction (supplemental Fig. S4, lanes 3–6). This could indicate that chromatin-bound HMGB2 is more susceptible to CCP1 cleavage or, alternatively, that processed HMGB2 display a higher affinity for chromatin (44, 46). Further research is however, needed to clarify this observation and to elucidate the functional implications of these and other proteolytic processing events observed in this work. Studies with the pcd knock-out might offer further validation of the putative substrates here identified.
DISCUSSION
Identification of carboxypeptidase substrates in complex proteomes has been challenging until the recent development of C-terminal positional proteomics techniques (22, 47–50). Previously, C-TAILS was used to investigate constitutive C-terminal proteolysis in E. coli (47), oxazolone chemistry enabled the study of natural neo-C termini in Thermoanaerobacter tengcongensis (49) and C-terminal COFRADIC was used to screen for substrates of human carboxypeptidase A4, when added to human PC3 cell lysates (22). In this work, we present the first proteomic study aimed at identifying protein substrates of carboxypeptidases in a cellular context. Here, C-terminal COFRADIC analyses resulted in the identification of nine processed C termini directly generated by human CCP1 in HEK293T cells. The putative natural substrates here identified together with the previously reported substrate MLCK1/telokin (Table I), support a role for CCP1 in the proteolytic processing of gene-encoded acidic C termini.
Notably, among the putative substrates identified by C-terminal COFRADIC, we found α-tubulin, a known CCP substrate that has already been extensively studied (15, 36, 37). In addition, the observed preference for acidic residues is in agreement with previous reports (15, 20, 36, 37). All putative CCP1 substrates here identified are located either in the cytoplasm or the nucleus, which is consistent with the subcellular localization of CCP1 (2, 3, 5). We further validated TRAD1 and HMGB3 as CCP1 substrates by cotransfecting cells with TRAD1 or HMGB3 and with CCP1. This allowed us to verify that in a cellular context, C-terminal trimming of both proteins occur in the presence of active CCP1. In addition, in vitro experiments making use of purified substrates confirmed that the identified neo-C termini are the result of the direct action of CCP1 on these proteins.
It was previously hypothesized that CCPs would generally recognize and process proteins with gene-encoded C termini ending with glutamate stretches (15). The here identified CCP1 substrates show for the first time that CCP1 processes both glutamate as well as aspartate C-terminal residues. We used an iceLogo representation (35) to visualize the derived substrate specificity for CCP1, which as expected is shown to be restricted to acidic residues, with a marked preference for glutamates over aspartates (Fig. 5). The iceLogo displays, in addition to the requirement of an extended substrate specificity profile, a critical role of nonprimed binding subsites in substrate selection. This result agrees with a recent report describing that biotin-3E is a better CCP1 substrate than biotin-2E, and therefore shows the preference of this enzyme for longer acidic tails (37). Previous reports stated that CCP1 was not capable of releasing C-terminal aspartate residues (15, 37). Given our data, we hypothesize that CCP1 does cleave off aspartates but less efficient than glutamates. Indeed, CCP1 cleaves aspartates from RPS9 and HMGB3, but their identified final products also suggest that CCP1 slows down or stalls its proteolytic activity when it encounters an aspartate residue in P1 or P1′ (Table I). Also, the higher proportion of aspartate residues residing at the C terminus of HMGB1, when compared with HMGB2 or HMGB3, might explain why the extent of cleavage is lower for this protein (Figs. 3E and 4). In vitro assays showed processing of the HMGB3 tail (Fig. 3F) and Biot-EEE, but failed to detect CCP1 cleavage of Biot-EED peptide in a colorimetric assay (data not shown) (37). Cleavage of Asp might be favored in the context of long acidic tails like those found in HMGB proteins, where nonprime positions are occupied by favoring acidic residues.
Fig. 5.
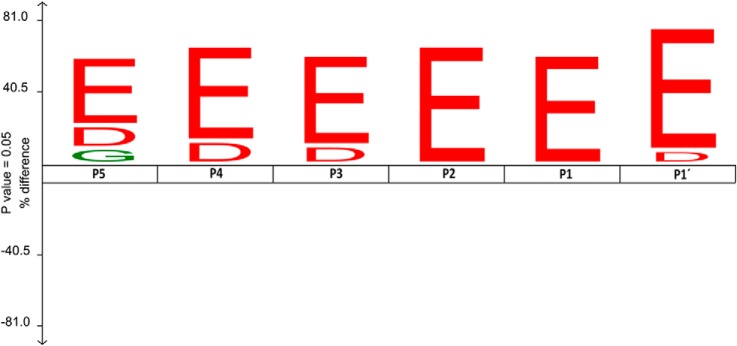
Substrate specificity profile derived from putative CCP1 substrates. IceLogo (35) was used to show the enriched residues present at the different identified CCP1 putative substrate positions as compared with the reference set (i.e. C termini of human proteins in the Swiss-Prot database). For the iceLogo preparation we took into consideration that MCPs sequentially release amino acids. As a result, when a few amino acids are released to get to the final identified product, intermediate products of digestion act as new substrate and are further digested by CCP1. Consequently, intermediate products of digestion were considered as different individual substrates when preparing the iceLogo and, as a result, 35 CCP1 substrates were considered. Only those substrates identified by proteomics were considered here. In the representation the substrate residues are depicted according to Schechter & Berger nomenclature. The frequency of the amino acid occurrence at each position in the sequence set was compared with the occurrence in the reference set. Only statistically significant residues with a p value ≤ 0.05 are plotted. Amino acids height shows the degree of difference in the positional amino acid frequencies in the experimental set as compared with the reference set. Residues that are statistically over- or underrepresented in the experimental set are shown in the upper or lower part of the iceLogo respectively.
To further support a role of CCP1 in gene-encoded C-terminal processing, we searched for reports describing the natural occurrence of the C-terminal modifications shown in Table I. In the case of α-tubulin, a pool of Δ2-tubulin is known to be naturally present in the cell (51–53). Richter-Cook et al. described the purification of rabbit eIF4H from rabbit reticulocyte lysates and identified a form of the protein with a more basic pI and postulated that this protein form might result from proteolysis of its C-terminal glutamate (54). Rusconi et al. analyzed by MALDI-TOF-MS the C terminus of chicken telokin and found forms of the protein from which one to six C-terminal glutamate residues had been removed (55). Furthermore, we searched for evidence of CCP1 being the responsible carboxypeptidase in vivo for some of these C-terminal processing events. Berezniuk et al. performed CCP1 knock-down experiments in HEK293T cells and observed that Δ2-tubulin levels were significantly decreased (36). Comparative Western blot analysis of wild type and pcd mouse show that MLCK1 and telokin have intact C termini in the absence of CCP1 (i.e. in pcd mouse), but processed C termini in wild type mice (15, 36).
HMGB proteins can provide clues to the functional implications of CCP1-mediated C-terminal processing, given that comprehensive research is done on how the lack of their acidic C-terminal tails affects their function. Most of these studies were performed with HMGB1 or HMGB2, and have not yet been performed on more recently discovered mammalian HMGB proteins such as HMGB3. HMGBs are nonhistone proteins that are involved in various nuclear chromatin-associated processes such as transcription, replication, recombination, DNA repair, and genomic stability (44, 56, 57). Mammalian HMGBs are composed of two central DNA binding domains (HMG-boxes A and B) and a negatively charged C-terminal region, which is highly conserved among different species (44). The acidic tail of HMGB1 and HMGB2 negatively affects binding to both linear and supercoiled DNA (44, 58) mediated by intra-molecular interactions of the acidic tail with the HMGB boxes and thereby shielding these domains from other interactions. However, these acidic tails seem to modulate the biological functions of HMGBs in many other ways. For instance, they are involved in the nucleocytoplasmic transport of HMGB1 and HMGB2, and affect their localization (59, 60). In addition, the C-terminal tail of HMGB1 is crucial for transcription stimulation (61, 62), essentially by its interaction with histones H1 and H3 (61, 63). Furthermore, the HMGB1 C-terminal tail was reported to play a major role in DNA repair, chromatin remodeling, and DNA replication (64–66). Marintcheva et al. built a model to explain the role for the acidic tail of the ssDNA-binding protein of bacteriophage T7, which can be extended to other proteins such as ribosomal proteins, translation factors and HMG proteins (67). According to this model, the acidic tail of these proteins would regulate the affinity to their binding partners (DNA or other proteins) by binding and thereby shedding their basic binding clefts. In this context, CCP1, by shortening the length of these acidic tails would regulate (the avidity of) protein–protein and DNA–protein interactions. In fact, this is in line with the proposed role for PTMs affecting the C terminus of tubulin; modulation of motor proteins, and MAPs binding to the C-terminal region of tubulin (52). Similarly, Rusconi et al. proposed that the observed heterogeneity of telokin's C terminus might regulate its interaction with other proteins (55).
The functions of the here identified putative CCP1 substrates are quite diverse (Table I), including microtubule-related proteins and proteins involved in transcription and chromatin remodeling. The latter could help to understand, for instance, the progressive transcriptional silencing or the large scale reorganization of chromatin in pcd mice. However, further work is needed to fully validate these proteins as in vivo CCP1 substrates. Studies in the pcd knockout mouse might provide the ultimate validation of these substrates. Future research should additionally assess the possible implications of C-terminal processing of these proteins on their function. Finally, further findings from C-terminomics studies will aid in the understanding of post-translational modifications that affect protein C termini and their role in the regulation of biological processes.
Supplementary Material
Acknowledgments
We thank the Cell Culture Facility of the Institute for Biotechnology and Biomedicine (IBB-UAB) for the technical assistance and the PRIDE Team for technical assistance with the submission of the mass spectrometry data to the repository. S.T is a Postdoctoral Fellow of the Belgian Federal Science (BELSPO). P.V.D. is a Postdoctoral Fellow of the Research Foundation - Flanders (FWO-Vlaanderen).
Footnotes
Author contributions: S.T., O.T., F.A., K.G., P.V., and J.L. designed research; S.T., O.T., H.D., P.V., and J.L. performed research; S.T., P.V., and J.L. analyzed data; S.T., F.A., K.G., P.V., and J.L. wrote the paper.
* F.X.A. and J.L. acknowledges support of the Spanish Ministry of Economy and Competitiveness (MINECO), grant BIO2013-44973-R, and the European Commission (project FP7-Health-2009-241919-LIVIMODE). K.G. has been supported by the PRIME-XS project, grant agreement number 262067, funded by the European Union 7th Framework Program.
 This article contains supplemental Figs. S1 to S4 and Tables S1 to S5.
This article contains supplemental Figs. S1 to S4 and Tables S1 to S5.
1 The abbreviations used are:
- CCPs
- Cytosolic carboxypeptidases
- COFRADIC
- COmbined FRActional DIagonal Chromatography
- eIF4H
- Eukaryotic translation initiation factor 4H
- HMGB
- High mobility group protein B
- I.D.
- Internal diameter
- MAPs
- Microtubule associated proteins
- MCPs
- Metallocarboxypeptidases
- MLCK1
- Myosin light chain kinase 1
- Nna1
- Nervous system nuclear protein induced by axotomy
- pcd
- Purkinje cell degeneration
- PEI
- Polyethylenimine
- RPS9
- 40S ribosomal protein S9
- TRAD1
- TRAF-type zinc finger domain-containing protein.
REFERENCES
- 1. Rodriguez de la Vega M., Sevilla R. G., Hermoso A., Lorenzo J., Tanco S., Diez A., Fricker L. D., Bautista J. M., Aviles F. X. (2007) Nna1-like proteins are active metallocarboxypeptidases of a new and diverse M14 subfamily. FASEB J. 21, 851–865 [DOI] [PubMed] [Google Scholar]
- 2. Rodriguez de la Vega M., Lorenzo J., Tort O., Aviles F. X., Bautista J. M. (2012) Functional segregation and emerging role of cilia-related cytosolic carboxypeptidases (CCPs). FASEB J. 27, 424–431 [DOI] [PubMed] [Google Scholar]
- 3. Kalinina E., Biswas R., Berezniuk I., Hermoso A., Aviles F. X., Fricker L. D. (2007) A novel subfamily of mouse cytosolic carboxypeptidases. FASEB J. 21, 836–850 [DOI] [PubMed] [Google Scholar]
- 4. Wang T., Morgan J. I. (2007) The Purkinje cell degeneration (pcd) mouse: an unexpected molecular link between neuronal degeneration and regeneration. Brain Res. 1140, 26–40 [DOI] [PubMed] [Google Scholar]
- 5. Harris A., Morgan J. I., Pecot M., Soumare A., Osborne A., Soares H. D. (2000) Regenerating motor neurons express Nna1, a novel ATP/GTP-binding protein related to zinc carboxypeptidases. Mol. Cell. Neurosci. 16, 578–596 [DOI] [PubMed] [Google Scholar]
- 6. Ghosh-Roy A., Goncharov A., Jin Y., Chisholm A. D. (2012) Kinesin-13 and tubulin posttranslational modifications regulate microtubule growth in axon regeneration. Dev. Cell 23, 716–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fernandez-Gonzalez A., La Spada A. R., Treadaway J., Higdon J. C., Harris B. S., Sidman R. L., Morgan J. I., Zuo J. (2002) Purkinje cell degeneration (pcd) phenotypes caused by mutations in the axotomy-induced gene, Nna1. Science 295, 1904–1906 [DOI] [PubMed] [Google Scholar]
- 8. Mullen R. J., Eicher E. M., Sidman R. L. (1976) Purkinje cell degeneration, a new neurological mutation in the mouse. Proc. Natl. Acad. Sci. U.S.A. 73, 208–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blanks J. C., Mullen R. J., LaVail M. M. (1982) Retinal degeneration in the pcd cerebellar mutant mouse. II. Electron microscopic analysis. J. Comp. Neurol. 212, 231–246 [DOI] [PubMed] [Google Scholar]
- 10. Greer C. A., Shepherd G. M. (1982) Mitral cell degeneration and sensory function in the neurological mutant mouse Purkinje cell degeneration (PCD). Brain Res. 235, 156–161 [DOI] [PubMed] [Google Scholar]
- 11. O'Gorman S., Sidman R. L. (1985) Degeneration of thalamic neurons in “Purkinje cell degeneration” mutant mice. I. Distribution of neuron loss. J. Comp. Neurol. 234, 277–297 [DOI] [PubMed] [Google Scholar]
- 12. Kyuhou S., Kato N., Gemba H. (2006) Emergence of endoplasmic reticulum stress and activated microglia in Purkinje cell degeneration mice. Neurosci. Lett. 396, 91–96 [DOI] [PubMed] [Google Scholar]
- 13. Chakrabarti L., Eng J., Ivanov N., Garden G. A., La Spada A. R. (2009) Autophagy activation and enhanced mitophagy characterize the Purkinje cells of pcd mice prior to neuronal death. Mol. Brain 2, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li J., Gu X., Ma Y., Calicchio M. L., Kong D., Teng Y. D., Yu L., Crain A. M., Vartanian T. K., Pasqualini R., Arap W., Libermann T. A., Snyder E. Y., Sidman R. L. (2010) Nna1 mediates Purkinje cell dendritic development via lysyl oxidase propeptide and NF-kappaB signaling. Neuron 68, 45–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rogowski K., van Dijk J., Magiera M. M., Bosc C., Deloulme J. C., Bosson A., Peris L., Gold N. D., Lacroix B., Grau M. B., Bec N., Larroque C., Desagher S., Holzer M., Andrieux A., Moutin M. J., Janke C. (2010) A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell 143, 564–578 [DOI] [PubMed] [Google Scholar]
- 16. Baltanas F. C., Casafont I., Lafarga V., Weruaga E., Alonso J. R., Berciano M. T., Lafarga M. (2011) Purkinje cell degeneration in pcd mice reveals large scale chromatin reorganization and gene silencing linked to defective DNA repair. J. Biol. Chem. 286, 28287–28302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Baltanas F. C., Casafont I., Weruaga E., Alonso J. R., Berciano M. T., Lafarga M. (2011) Nucleolar disruption and cajal body disassembly are nuclear hallmarks of DNA damage-induced neurodegeneration in purkinje cells. Brain Pathol. 21, 374–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Valero J., Berciano M. T., Weruaga E., Lafarga M., Alonso J. R. (2006) Preneurodegeneration of mitral cells in the pcd mutant mouse is associated with DNA damage, transcriptional repression, and reorganization of nuclear speckles and Cajal bodies. Mol. Cell. Neurosci. 33, 283–295 [DOI] [PubMed] [Google Scholar]
- 19. Kimura Y., Kurabe N., Ikegami K., Tsutsumi K., Konishi Y., Kaplan O. I., Kunitomo H., Iino Y., Blacque O. E., Setou M. (2010) Identification of tubulin deglutamylase among Caenorhabditis elegans and mammalian cytosolic carboxypeptidases (CCPs). J. Biol. Chem. 285, 22936–22941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berezniuk I., Lyons P. J., Sironi J. J., Xiao H., Setou M., Angeletti R. H., Ikegami K., Fricker L. D. (2013) Cytosolic carboxypeptidase 5 removes alpha- and gamma-linked glutamates from tubulin. J. Biol. Chem. 288, 30445–30453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. O'Hagan R., Piasecki B. P., Silva M., Phirke P., Nguyen K. C., Hall D. H., Swoboda P., Barr M. M. (2011) The tubulin deglutamylase CCPP-1 regulates the function and stability of sensory cilia in C. elegans. Curr. Biol. 21, 1685–1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Van Damme P., Staes A., Bronsoms S., Helsens K., Colaert N., Timmerman E., Aviles F. X., Vandekerckhove J., Gevaert K. (2010) Complementary positional proteomics for screening substrates of endo- and exoproteases. Nat. Methods 7, 512–515 [DOI] [PubMed] [Google Scholar]
- 23. Berrow N. S., Alderton D., Sainsbury S., Nettleship J., Assenberg R., Rahman N., Stuart D. I., Owens R. J. (2007) A versatile ligation-independent cloning method suitable for high-throughput expression screening applications. Nucleic Acids Res. 35, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schmidt T. G., Skerra A. (2007) The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2, 1528–1535 [DOI] [PubMed] [Google Scholar]
- 25. Felber J. P., Coombs T. L., Vallee B. L. (1962) The mechanism of inhibition of carboxypeptidase A by 1,10-phenanthroline. Biochemistry 1, 231–238 [DOI] [PubMed] [Google Scholar]
- 26. Torres M. J., Trejo S. A., Obregon W. D., Aviles F. X., Lopez L. M., Natalucci C. L. (2012) Characterization of the proteolytic system present in Vasconcellea quercifolia latex. Planta 236, 1471–1484 [DOI] [PubMed] [Google Scholar]
- 27. Tanco S., Lorenzo J., Garcia-Pardo J., Degroeve S., Martens L., Aviles F. X., Gevaert K., Van Damme P. (2013) Proteome-derived peptide libraries to study the substrate specificity profiles of carboxypeptidases. Mol. Cell. Proteomics 12, 2096–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martens L., Vandekerckhove J., Gevaert K. (2005) DBToolkit: processing protein databases for peptide-centric proteomics. Bioinformatics 21, 3584–3585 [DOI] [PubMed] [Google Scholar]
- 29. Kall L., Storey J. D., MacCoss M. J., Noble W. S. (2008) Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J. Proteome Res. 7, 29–34 [DOI] [PubMed] [Google Scholar]
- 30. Vizcaino J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Perez-Riverol Y., Reisinger F., Rios D., Wang R., Hermjakob H. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang R., Fabregat A., Rios D., Ovelleiro D., Foster J. M., Cote R. G., Griss J., Csordas A., Perez-Riverol Y., Reisinger F., Hermjakob H., Martens L., Vizcaino J. A. (2012) PRIDE Inspector: a tool to visualize and validate MS proteomics data. Nat. Biotechnol. 30, 135–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huber P. J., Ronchetti E. M. (2009) Robust Statistics, John Wiley & Sons, Inc. [Google Scholar]
- 33. de Castro E., Sigrist C. J., Gattiker A., Bulliard V., Langendijk-Genevaux P. S., Gasteiger E., Bairoch A., Hulo N. (2006) ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 34, W362–W365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Karlin S., Altschul S. F. (1990) Methods for assessing the statistical significance of molecular sequence features by using general scoring schemes. Proc. Natl. Acad. Sci. U.S.A. 87, 2264–2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Colaert N., Helsens K., Martens L., Vandekerckhove J., Gevaert K. (2009) Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 6, 786–787 [DOI] [PubMed] [Google Scholar]
- 36. Berezniuk I., Vu H. T., Lyons P. J., Sironi J. J., Xiao H., Burd B., Setou M., Angeletti R. H., Ikegami K., Fricker L. D. (2012) Cytosolic carboxypeptidase 1 is involved in processing alpha- and beta-tubulin. J. Biol. Chem. 287, 6503–6517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu H. Y., Wang T., Li L., Correia K., Morgan J. I. (2012) A structural and functional analysis of Nna1 in Purkinje cell degeneration (pcd) mice. FASEB J. 26, 4468–4480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sahab Z. J., Hall M. D., Me Sung Y., Dakshanamurthy S., Ji Y., Kumar D., Byers S. W. (2011) Tumor suppressor RARRES1 interacts with cytoplasmic carboxypeptidase AGBL2 to regulate the alpha-tubulin tyrosination cycle. Cancer Res. 71, 1219–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shang Y., Li B., Gorovsky M. A. (2002) Tetrahymena thermophila contains a conventional gamma-tubulin that is differentially required for the maintenance of different microtubule-organizing centers. J. Cell Biol. 158, 1195–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Arolas J. L., Vendrell J., Aviles F. X., Fricker L. D. (2007) Metallocarboxypeptidases: emerging drug targets in biomedicine. Curr. Pharm. Des. 13, 349–366 [DOI] [PubMed] [Google Scholar]
- 41. Thakar K., Karaca S., Port S. A., Urlaub H., Kehlenbach R. H. (2013) Identification of CRM1-dependent nuclear export cargos using quantitative mass spectrometry. Mol. Cell. Proteomics 12, 664–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mosevitsky M. I., Novitskaya V. A., Iogannsen M. G., Zabezhinsky M. A. (1989) Tissue specificity of nucleo-cytoplasmic distribution of HMG1 and HMG2 proteins and their probable functions. Eur. J. Biochem. 185, 303–310 [DOI] [PubMed] [Google Scholar]
- 43. Kuehl L., Salmond B., Tran L. (1984) Concentrations of high-mobility-group proteins in the nucleus and cytoplasm of several rat tissues. J. Cell Biol. 99, 648–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stros M. (2010) HMGB proteins: interactions with DNA and chromatin. Biochim. Biophys. Acta 1799, 101–113 [DOI] [PubMed] [Google Scholar]
- 45. Bustin M., Neihart N. K. (1979) Antibodies against chromosomal HMG proteins stain the cytoplasm of mammalian cells. Cell 16, 181–189 [DOI] [PubMed] [Google Scholar]
- 46. Lee K. B., Thomas J. O. (2000) The effect of the acidic tail on the DNA-binding properties of the HMG1,2 class of proteins: insights from tail switching and tail removal. J. Mol. Biol. 304, 135–149 [DOI] [PubMed] [Google Scholar]
- 47. Schilling O., Barre O., Huesgen P. F., Overall C. M. (2010) Proteome-wide analysis of protein carboxy termini: C terminomics. Nat. Methods 7, 508–511 [DOI] [PubMed] [Google Scholar]
- 48. Xu G., Shin S. B., Jaffrey S. R. (2011) Chemoenzymatic labeling of protein C-termini for positive selection of C-terminal peptides. ACS Chem. Biol. 6, 1015–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu M., Zhang L., Yao J., Yang P., Lu H. (2013) Approach for identification and quantification of C-terminal peptides: incorporation of isotopic arginine labeling based on oxazolone chemistry. Anal. Chem. 85, 10745–10753 [DOI] [PubMed] [Google Scholar]
- 50. Sonomura K., Kuyama H., Matsuo E., Tsunasawa S., Nishimura O. (2009) The specific isolation of C-terminal peptides of proteins through a transamination reaction and its advantage for introducing functional groups into the peptide. Rapid Commun. Mass Spectrom. 23, 611–618 [DOI] [PubMed] [Google Scholar]
- 51. Paturle-Lafanechere L., Manier M., Trigault N., Pirollet F., Mazarguil H., Job D. (1994) Accumulation of delta 2-tubulin, a major tubulin variant that cannot be tyrosinated, in neuronal tissues and in stable microtubule assemblies. J. Cell Sci. 107, 1529–1543 [DOI] [PubMed] [Google Scholar]
- 52. Janke C., Bulinski J. C. (2011) Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat. Rev. Mol. Cell Biol. 12, 773–786 [DOI] [PubMed] [Google Scholar]
- 53. Sahab Z. J., Kirilyuk A., Zhang L., Khamis Z. I., Pompach P., Sung Y., Byers S. W. (2012) Analysis of tubulin alpha-1A/1B C-terminal tail post-translational poly-glutamylation reveals novel modification sites. J. Proteome Res. 11, 1913–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Richter-Cook N. J., Dever T. E., Hensold J. O., Merrick W. C. (1998) Purification and characterization of a new eukaryotic protein translation factor. Eukaryotic initiation factor 4H. J. Biol. Chem. 273, 7579–7587 [DOI] [PubMed] [Google Scholar]
- 55. Rusconi F., Potier M. C., Le Caer J. P., Schmitter J. M., Rossier J. (1997) Characterization of the chicken telokin heterogeneity by time-of-flight mass spectrometry. Biochemistry 36, 11021–11026 [DOI] [PubMed] [Google Scholar]
- 56. Lange S. S., Vasquez K. M. (2009) HMGB1: the jack-of-all-trades protein is a master DNA repair mechanic. Mol. Carcinog. 48, 571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Travers A. A. (2003) Priming the nucleosome: a role for HMGB proteins? EMBO Rep. 4, 131–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sheflin L. G., Fucile N. W., Spaulding S. W. (1993) The specific interactions of HMG 1 and 2 with negatively supercoiled DNA are modulated by their acidic C-terminal domains and involve cysteine residues in their HMG 1/2 boxes. Biochemistry 32, 3238–3248 [DOI] [PubMed] [Google Scholar]
- 59. Ditsworth D., Zong W. X., Thompson C. B. (2007) Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. J. Biol. Chem. 282, 17845–17854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shirakawa H., Tanigawa T., Sugiyama S., Kobayashi M., Terashima T., Yoshida K., Arai T., Yoshida M. (1997) Nuclear accumulation of HMG2 protein is mediated by basic regions interspaced with a long DNA-binding sequence, and retention within the nucleus requires the acidic carboxyl terminus. Biochemistry 36, 5992–5999 [DOI] [PubMed] [Google Scholar]
- 61. Ueda T., Chou H., Kawase T., Shirakawa H., Yoshida M. (2004) Acidic C-tail of HMGB1 is required for its target binding to nucleosome linker DNA and transcription stimulation. Biochemistry 43, 9901–9908 [DOI] [PubMed] [Google Scholar]
- 62. Aizawa S., Nishino H., Saito K., Kimura K., Shirakawa H., Yoshida M. (1994) Stimulation of transcription in cultured cells by high mobility group protein 1: essential role of the acidic carboxyl-terminal region. Biochemistry 33, 14690–14695 [DOI] [PubMed] [Google Scholar]
- 63. Cato L., Stott K., Watson M., Thomas J. O. (2008) The interaction of HMGB1 and linker histones occurs through their acidic and basic tails. J. Mol. Biol. 384, 1262–1272 [DOI] [PubMed] [Google Scholar]
- 64. Bonaldi T., Langst G., Strohner R., Becker P. B., Bianchi M. E. (2002) The DNA chaperone HMGB1 facilitates ACF/CHRAC-dependent nucleosome sliding. EMBO J. 21, 6865–6873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mitkova E., Ugrinova I., Pashev I. G., Pasheva E. A. (2005) The inhibitory effect of HMGB-1 protein on the repair of cisplatin-damaged DNA is accomplished through the acidic domain. Biochemistry 44, 5893–5898 [DOI] [PubMed] [Google Scholar]
- 66. Topalova D., Ugrinova I., Pashev I. G., Pasheva E. A. (2008) HMGB1 protein inhibits DNA replication in vitro: a role of the acetylation and the acidic tail. Int. J. Biochem. Cell Biol. 40, 1536–1542 [DOI] [PubMed] [Google Scholar]
- 67. Marintcheva B., Marintchev A., Wagner G., Richardson C. C. (2008) Acidic C-terminal tail of the ssDNA-binding protein of bacteriophage T7 and ssDNA compete for the same binding surface. Proc. Natl. Acad. Sci. U.S.A. 105, 1855–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.