

Abstract
The c-Jun N-terminal kinase (JNK) is an important mediator of physiological and pathophysiological processes in the central nervous system. Importantly, JNK not only is involved in neuronal cell death, but also plays a significant role in neuronal differentiation and regeneration. For example, nerve growth factor induces JNK-dependent neuronal differentiation in several model systems. The mechanism by which JNK mediates neuronal differentiation is not well understood. Here, we employed a proteomic strategy to better characterize the function of JNK during neuronal differentiation. We used SILAC-based quantitative proteomics to identify proteins that interact with JNK in PC12 cells in a nerve growth factor–dependent manner. Intriguingly, we found that JNK interacted with neuronal transport granule proteins such as Sfpq and Nono upon NGF treatment. We validated the specificity of these interactions by showing that they were disrupted by a specific peptide inhibitor that blocks the interaction of JNK with its substrates. Immunoprecipitation and Western blotting experiments confirmed the interaction of JNK1 with Sfpq/Nono and demonstrated that it was RNA dependent. Confocal microscopy indicated that JNK1 associated with neuronal granule proteins in the cytosol of PC12 cells, primary cortical neurons, and P19 neuronal cells. Finally, siRNA experiments confirmed that Sfpq was necessary for neurite outgrowth in PC12 cells and that it most likely acted in the same pathway as JNK. In summary, our data indicate that the interaction of JNK1 with transport granule proteins in the cytosol of differentiating neurons plays an important role during neuronal development.
The members of the c-Jun N-terminal kinase (JNK)1 family are important mediators of a broad range of biological processes in the brain (1, 2). JNK belongs to the family of mitogen-activated protein kinases and is encoded by three different genes: jnk1, jnk2, and jnk3. Alternative splicing combined with alternative exon usage of the three genes leads to at least 10 different JNK isoforms (3). JNK is induced by various stimuli such as cytokines, ligands of Toll-like receptors, or growth factors (4).
JNK is well known to induce neuronal cell death. However, it has become clear within the past decade that JNK plays an important role in neuronal regeneration, migration, and differentiation (5–8). JNK controls dendritic microtubule assembly and disassembly in sympathetic neurons (2, 9) and is responsible for dendritic elongation during brain development in mice (10). Furthermore, several cytoskeleton-regulating proteins such as doublecortin, superior cervical ganglion-10 protein, microtubule-associated protein 1B, microtubule-associated protein 2, and MARCKS-like protein 1 have been identified as JNK substrates (11–16). Collectively, these observations have established that JNKs not only are involved in neuronal death, but also play an important role during differentiation and regeneration. Although stress-induced pro-apoptotic JNK signaling is well characterized, the mechanisms involved in JNK-dependent neuronal differentiation remain enigmatic.
Quantitative mass-spectrometry-based proteomics has emerged as a powerful technology for investigating mammalian signaling pathways (17, 18). Specifically, stable isotope labeling by amino acids in cell culture (SILAC) has been shown to be a powerful approach for studying dynamic changes in protein–protein interactions during cell signaling. We reasoned that an unbiased proteomic analysis of JNK interaction partners during neuronal differentiation could provide novel insights into the mechanisms involved. For that purpose, we used PC12 cells as a well-characterized classical model for neuronal differentiation (19). Stimulation of PC12 cells with nerve growth factor (NGF) induces a major shift in phenotype, from proliferating tumor cells to non-dividing neurons showing characteristics of sympathetic neurons such as the growth of long neurites and electrical excitability (19, 20). In PC12 cells, NGF binds to neurotrophic tyrosine kinase receptor 1, low-affinity neurotrophin receptor, and fibroblast growth factor receptors (21, 22). This leads to a coordinated activation of downstream signaling cascades such as the Ras/Raf/Erk1–2, PI3K, and JNK pathways, resulting in increased expression of genes that are involved in neuronal differentiation (23, 24). Furthermore, G-proteins including Ras, Rap, and Cdc42 have been shown to link receptor activity to downstream kinase activation (25–27).
Here, we used quantitative interaction proteomics to analyze dynamic changes in JNK interaction partners during NGF-induced differentiation of PC12 cells. Our results show that JNK dynamically interacts with G-proteins, cytoskeletal proteins, and RNA binding proteins with distinct kinetic patterns. Intriguingly, several of the identified RNA binding proteins are known components of transport granules involved in mRNA localization and localized translation in neurons. Western blotting and co-localization experiments using confocal microscopy and proximity ligation assays validated the NGF-induced association of JNK1 with two RNA binding proteins, Sfpq and Nono. Furthermore, Sfpq knockdown decreased NGF-induced neurite outgrowth in PC12 cells, supporting the hypothesis that the interaction of JNK and Sfpq may contribute to neuronal differentiation.
EXPERIMENTAL PROCEDURES
Cell Culture
The rat pheochromocytoma cell line PC12 (ATCC, Manassas, VA) was cultured essentially as described before (19). In brief, PC12 cells were grown at 37 °C in 5% CO2 in either 10-cm or 15-cm cell culture dishes coated with rat tail type 1 collagen (Sigma-Aldrich, St. Louis, MO) at ∼30% confluency in RPMI 1640 medium (PAA Laboratories GmbH, Pasching, Austria) containing 10% horse serum (Invitrogen), 5% FCS (Sigma-Aldrich), 4 mm glutamine (PAA Laboratories GmbH), 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). For SILAC labeling, media contained 10% dialyzed horse serum (produced in house with an 8000 to 10,000 molecular weight cutoff), 5% dialyzed FCS (Sigma-Aldrich), and one of the following Lys–Arg combinations: 48 μg/ml Lys-C6H14N2O2 (Lys-0) and 28 μg/ml Arg-C6H14N4O2 (Arg-0) (Sigma-Aldrich), 48 μg/ml Lys-C6H10D4N2O2 (Lys-4) and 28 μg/ml Arg-13C6H14N4O2 (Arg-6), or 48 μg/ml Lys-13C6H1415N2O2 (Lys-8) and 28 μg/ml Arg-13C6H1415N4O2 (Arg-10) (Sigma Isotec, Miamisburg, OH). For cell differentiation, PC12 cells were starved for 24 h in media containing 1% horse serum or 1% dialyzed horse serum and were treated with 100 ng/ml NGF (Sigma-Aldrich) for the indicated length of time. For JNK inhibition experiments, PC12 cells were pretreated for 1 h with 10 μm SP600125 (Calbiochem, San Diego, CA), 10 μm d-JNKI-1, or 10 μm control peptide d-TAT (Enzo Life Sciences, Lörrach, Germany).
Primary cortical neurons were prepared from E18.5 mouse embryos as described elsewhere (28). Briefly, cortices were dissected in Hanks solution under a stereomicroscope and dissociated enzymatically in neurobasal medium (Invitrogen) containing 0.1% trypsin (Sigma-Aldrich) and 0.001% DNaseI (Roche) at 37 °C for 15 min. Trypsin was inactivated by 10% FBS in neurobasal media at 37 °C for 5 min, and cells were dissociated by trituration with a sterile pipette tip. The resulting suspension was centrifuged at 1000 × g for 3 min, and the supernatant was discarded. Next, neurons were resuspended and plated in neurobasal media containing B27 supplement (Invitrogen) and 100 μg/ml penicillin/streptomycin. Neurons were cultured for 4, 6, 8, 12, 24, 36, and 48 h.
Cell suspensions of the murine teratocarcinoma cell line P19 (ATCC) were cultured for 4 days in DMEM containing 5% FBS, 1 mm retinoic acid (Sigma), and 100 μg/ml penicillin/streptomycin. Subsequently, aggregates of P19 cells were dissociated enzymatically in DMEM (Invitrogen) containing 0.25% trypsin and 0.001% DNaseI at 37 °C for 10 min. Trypsin was inactivated by 10% FBS in DMEM at 37 °C for 5 min, and cells were dissociated by trituration with a sterile pipette tip. Afterward, cells were resuspended and plated in DMEM containing 5% FBS and 100 μg/ml penicillin/streptomycin for 2 days. 48 h after plating, cells were treated for an additional 24 h with 10 μm ARAC (Sigma) to eliminate proliferating glial-like cells. Last, P19-differentiated neurons were enzymatically dissociated and plated in DMEM containing 5% FBS and 100 μg/ml penicillin/streptomycin. Neurons were cultured for 4, 6, 8, 12, 24, 36, and 48 h.
Quantification of Neurite Outgrowth
Neurites of PC12 cells were measured essentially as described elsewhere (29, 30). PC12 cells were grown in six-well plates, and five pictures per well were randomly taken 24 h after treatment using a camera (Canon, Krefeld, Germany) linked to an inverted microscope with phase contrast illumination (Olympus, Hamburg, Germany). Each visual field contained on average 40 living cells. The number of neurites per visual field was measured using Adobe Photoshop (Adobe Systems, San Jose, CA). This number was divided by the total number of cells per visual field and expressed either as the percentage of untreated cells or as the percent inhibition of NGF-treated cells within the corresponding experiment.
Immunoblot Analysis
PC12 cells were lysed for 30 min on ice in modified radio-immunoprecipitation buffer (50 mm Tris HCl, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 0.25% Na-deoxycholate, 1 mm EDTA) containing 0.1% SDS and the following inhibitors: 1× protease inhibitor mixture (Roche), 1 mm sodium orthovanadate, 20 mm sodium pyrophosphate, and 1× phosphatase inhibitor mixture 1 (Sigma-Aldrich). The preparation of nuclear and cytosolic extracts was performed as described elsewhere (31). In brief, PC12 cells were incubated in a hypotonic buffer containing 10 mm HEPES (pH 7.9), 10 mm KCl, 0.1 mm EDTA, 0.1 mm EGTA, 0.025% Nonidet P-40 to extract cytosolic proteins. Afterward, nuclear proteins were extracted with a hypertonic buffer containing 20 mm HEPES (pH 7.9), 400 mm NaCl, 1 mm EDTA, 1 mm EGTA. The protein concentration was determined using the Coomassie Plus Protein Assay Kit (Pierce, Rockford, IL). Equal amounts of protein were boiled in LDS sample buffer (Invitrogen) under reducing conditions and separated on a 4–12% NuPAGE gradient gel (Invitrogen) according to the manufacturer's instructions. After blotting onto a PVDF membrane, nonspecific binding sites were blocked for 1 h with 5% non-fat dry milk (Carl Roth, Karlsruhe, Germany) in Tris-buffered saline containing 0.1% Tween 20. The membrane was incubated overnight using antibodies against phospho-JNK Thr183/Tyr185, phospho-c-Jun Ser73, c-Jun (Cell Signaling Technologies, Beverly, MA), pan-JNK, Sfpq, Nono (Santa Cruz Biotechnology, Santa Cruz, CA), JNK1 (BD Biosciences, Franklin Lakes, NJ), and JNK2 (Novus Biologicals, Littleton, CO). Horseradish-peroxidase-conjugated sheep anti-mouse antibody and donkey anti-rabbit (GE Healthcare, Buckinghamshire, UK) were used as secondary antibodies. To verify that equal amounts of protein were loaded, membranes were incubated at 37 °C for 20 min in stripping buffer (62.5 mm Tris (pH 6.7), 2% SDS, 100 mm β-mercaptoethanol) and reprobed with an antibody against β-actin (Sigma-Aldrich). Blots were incubated with enhanced chemiluminescence substrate (PerkinElmer, Waltham, MA), and band intensities were measured and quantified using Scion Image 4.0 (Scion Corporation, Frederick, MD).
Immunoprecipitation
For immunoprecipitation followed by LC-MS/MS analysis, PC12 cells were lysed for 10 min on ice in 50 mm Tris HCl (pH 7.4), 150 mm NaCl, 1% Triton X-100, 0.25% sodium deoxycholate, 1 mm EDTA, and all the protease and phosphatase inhibitors as described above. The protein concentration was determined using the Coomassie Plus Protein Assay Kit (Pierce). JNK1 (Biovision, Milpitas, CA) and JNK2 (Novus Biologicals) were incubated for 1 h at 4 °C with protein A Sepharose beads (Biovision) and then cross-linked for 1 h at room temperature in 200 mm sodium borate (pH 9.0), 25 mm dimethyl pimelimidate (Sigma-Aldrich). After three washes in 200 mm sodium borate, the antibody–bead complexes were blocked in 200 mm ethanolamine (pH 8.0) for 2 h at 4 °C. Equal amounts of protein extracts were incubated with bead-coupled antibody overnight at 4 °C. After two washes with lysis buffer and one with 5 mm Tris HCl (pH 7.4), proteins were eluted with 100 mm glycine (pH 3.0). For immunoprecipitation followed by immunoblot analysis, cells were lysed for 30 min on ice in 20 mm HEPES (pH 7.9), 400 mm NaCl, 0.025% Nonidet P-40, 1 mm EDTA, 1 mm EGTA, and all the protease and phosphatase inhibitors as described above. The protein concentration was determined using the Coomassie Plus Protein Assay Kit (Pierce). JNK1/3 or Nono antibody (both from Santa Cruz Biotechnology) was bound to protein A Sepharose beads (Biovision) in PBS for 3 h at 4 °C. Equal amounts of protein extracts were incubated with bead-coupled antibody in lysis buffer without Nonidet P-40 overnight at 4 °C. After three washes with lysis buffer without Nonidet P-40, samples were boiled in LDS sample buffer (Invitrogen) and further processed via immunoblot analysis as described above.
Sample Preparation for LC-MS/MS
Eluted proteins from JNK1/2 immunoprecipitation were precipitated in EtOH, 133 mm sodium acetate (pH 5.0), 0.06% GlycoBlue (Ambion Invitrogen Ltd., Paisley, UK) overnight at 4 °C. After centrifugation, the protein pellet was dried and dissolved in 10 mm HEPES (pH 8.0), 6 m urea, 2 m thiourea and reduced with 10 mm DTT in 50 mm ammonium bicarbonate. Proteins were alkylated in 50 mm ammonium bicarbonate, 55 mm iodacetamide, pre-digested with Lysyl endopeptidase (LysC) (Wako, Osaka, Japan), and subjected to trypsin digestion (Promega) overnight. Digestion was stopped by trifluoroacetic acid. Stop-and-go extraction (STAGE) tips containing C18 empore disks (3M, Minneapolis, MN) were used to purify and store peptide extracts (32).
LC-MS/MS
LC-MS/MS analysis was performed as described previously (33). Peptide mixtures were separated via reversed phase chromatography using the Eksigent NanoLC-1D Plus system (Eksigent, Dublin, CA) on in-house-manufactured 10-cm fritless silica microcolumns with an inner diameter of 75 μm. Columns were packed with ReproSil-Pur C18-AQ 3-μm resin (Dr. Maisch GmbH, Ammerbuch, Germany) (34). Separation was performed using a 10% to 60% acetonitrile gradient (155 min) with 0.5% acetic acid at a flow rate of 200 nl/min. Eluting peptides were directly ionized via electrospray ionization and transferred into the orifice of an LTQ-Orbitrap hybrid mass spectrometer (Thermo Fisher, Waltham, MA). Mass spectrometry was performed in the data-dependent mode with one full scan in the Orbitrap (m/z = 300–1700; r = 60,000; target value = 1 × 106). The five most intense ions with a charge state greater than 1 were selected (target value = 5000; monoisotopic precursor selection enabled) and fragmented in the LTQ using collision-induced dissociation (35% normalized collision energy, wideband activation enabled). The dynamic exclusion for selected precursor ions was 60 s.
MS Data Processing
The MaxQuant software package (version 1.3.0.5) was used to identify and quantify proteins (35). SILAC triplets or duplets were quantified using the following settings: heavy-label Lys-8, Arg-10 and Lys-4, Arg-6; a maximum of three labeled amino acids per peptide; and top 10 MS/MS peaks per 100 Da. Carbamidomethylation of cysteine was selected as a fixed modification, and oxidation of methionine and acetylation of the protein N terminus were used as variable modifications. Trypsin was selected as the protease (full specificity) with a maximum of two missed cleavages. MS/MS spectra were searched using the Andromeda search engine (36) against a UniProt rat database (release 2012–06, 37,327 entries) with an additional 248 common contaminants. All protein sequences were also reversed to generate a target-decoy database. A first search with a precursor mass tolerance of 20 ppm was used, and the main search was performed with a mass tolerance of 6 ppm (37). A mass tolerance of 20 ppm was selected for fragment ions. A minimum of six amino acids per identified peptide and at least one peptide per protein group were required. The false discovery rate was set at 1% at both the peptide and the protein level. Protein ratios were calculated from the median of all normalized peptide ratios using only unique peptides or peptides assigned to the protein group with the greatest number of peptides (razor peptides). Only protein groups with at least three SILAC counts were considered for further analysis.
Immunofluorescence Analysis
PC12 cells were incubated on poly-l-lysine-coated glass coverslips, fixed with 4% paraformaldehyde, and blocked for 1 h in PBS containing 10% goat serum and 0.3% Triton-X 100. The cells were incubated at a dilution of 1:500 with antibodies against JNK1 (Novus Biologicals), Nono, and Sfpq (Santa Cruz Biotechnology) overnight at 4 °C. Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 568 goat anti-mouse IgG (Invitrogen) were used as secondary antibodies at a dilution of 1:1000. PC12 cells were counterstained with DAPI and mounted with ProLong Gold Antifade (Invitrogen). Nonspecific staining was assessed through incubation of cells in the absence of primary antibodies. Images were taken with a Leica TCS SP5 confocal microscope (Leica Microsystems, Wetzlar, Germany) using a ×63 objective and recorded by Leica LAS AF software. Images were processed using Adobe Photoshop (Adobe Systems). P19 neuronal cells and primary cortical neurons were fixed with 4% paraformaldehyde in 0.1 m sodium phosphate buffer (pH 7.4). Primary antibodies against JNK1, Sfpq (as described above), and doublecortin (1:2000; Santa Cruz) were used. Secondary antibodies conjugated to Cy2, Cy3, or Cy5 were used at a dilution of 1:500 (Jackson ImmunoResearch Laboratories, Suffolk, UK). Fluorescence was imaged with a ×63 objective on a Zeiss LSM 700 confocal microscope, and images were processed using Adobe Photoshop software. Primary cortical and P19 neuronal cultures were visualized with a ×40 objective, and co-localization signals of JNK1 and Sfpq were quantified on at least 10 randomly taken visual fields containing ∼15 to 20 cells each.
Proximity Ligation Assay
PC12 cells were incubated on poly-l-lysine-coated 18-well μ-Slides (ibidi, Martinsried, Germany) and then subjected to primary antibody incubation as described above. The interactions between Sfpq-Nono and JNK1-Sfpq were assessed using the Duolink proximity ligation assay (Olink, Uppsala, Sweden) according to the manufacturer's instructions. Images were taken with a Leica TCS SP5 confocal microscope (Leica Microsystems) using a ×63 objective. Fluorescence spots were counted using ImageJ software (National Institutes of Health). Negative controls using JNK1 antibody alone with both secondary antibodies or secondary antibodies only did not show any signal.
siRNA Transfection
Transfection was carried out using the Amaxa Cell Line Nucleofector system according to the manufacturer's instructions with PC12-cell-specific settings (Lonza, Cologne, Germany). In brief, 2 × 106 cells were transfected with 200 nmol Sfpq siRNA (Thermo Fisher) and seeded in a six-well plate. After 4 days, cells were starved for 24 h and then differentiated as described above. Sfpq knockdown efficiency was confirmed by immunoblot before NGF treatment.
Statistical Analysis
Band intensities of Western blots were evaluated densitometrically using Scion Image 4.0 (Scion Corporation) and are expressed as a percentage relative to unstimulated cells (“relative pixel density”). Comparisons between multiple groups were performed via one-way analysis of variance followed by a Bonferroni post hoc test, and comparisons between two groups were assessed via the standard Student's t test (Prism 4.0, GraphPad Software, San Diego, CA). All data are expressed as mean ± S.E. Differences were considered statistically significant at p < 0.05. Cluster analysis was performed using Perseus (MPI, Munich, Germany) and PANTHER molecular function terms in the DAVID bioinformatics database (DAVID Bioinformatics Resources, National Cancer Institute, Frederick, MD) (38).
RESULTS
Nerve Growth Factor Induces JNK-dependent Neurite Outgrowth in PC12 Cells
In order to study JNK interaction partners during neuronal differentiation via quantitative mass spectrometry, we decided to use the very well-characterized rat pheochromocytoma cell line PC12 (19). The hallmark of PC12 cells is that treatment with NGF induces a major shift in phenotype, from proliferating tumor cells to non-dividing neurons. For our proteomics experiments, PC12 cells were grown in SILAC media containing dialyzed horse and calf sera and lower concentrations of lysine and arginine than in standard RPMI media. 24 h before differentiation, cells were starved in SILAC media containing 1% dialyzed horse serum. To assess the role of JNK in NGF-induced neuronal differentiation, we stimulated PC12 cells with a single dose of NGF (100 ng/ml) in the presence or absence of the ATP-competitive JNK inhibitor SP600125 (Fig. 1A). Without the inhibitor, NGF-induced neurite outgrowth was clearly visible at both 6 and 24 h after stimulation. When NGF was not replenished, neurites were largely degenerated after 48 h. Pretreatment of cells with SP600125 decreased NGF-induced neurite outgrowth by ∼60% at 6 h and 24 h. This result is in line with previous studies showing that JNK is an important mediator for neuronal differentiation in PC12 cells (23, 26, 39, 40). To analyze JNK activity, we quantified phosphorylation levels via Western blotting (Fig. 1B). We found increased JNK activation beginning at 1 h after treatment, with a maximum at about 2 h. Total JNK levels did not change significantly. Western blot results also showed that both JNK isoforms (p54 and p46) were phosphorylated in response to NGF. In summary, these results confirm that JNK activity is required for neurite outgrowth and establish PC12 cells as a model for the study of NGF-induced JNK-dependent signaling via SILAC-based quantitative mass spectrometry.
Fig. 1.
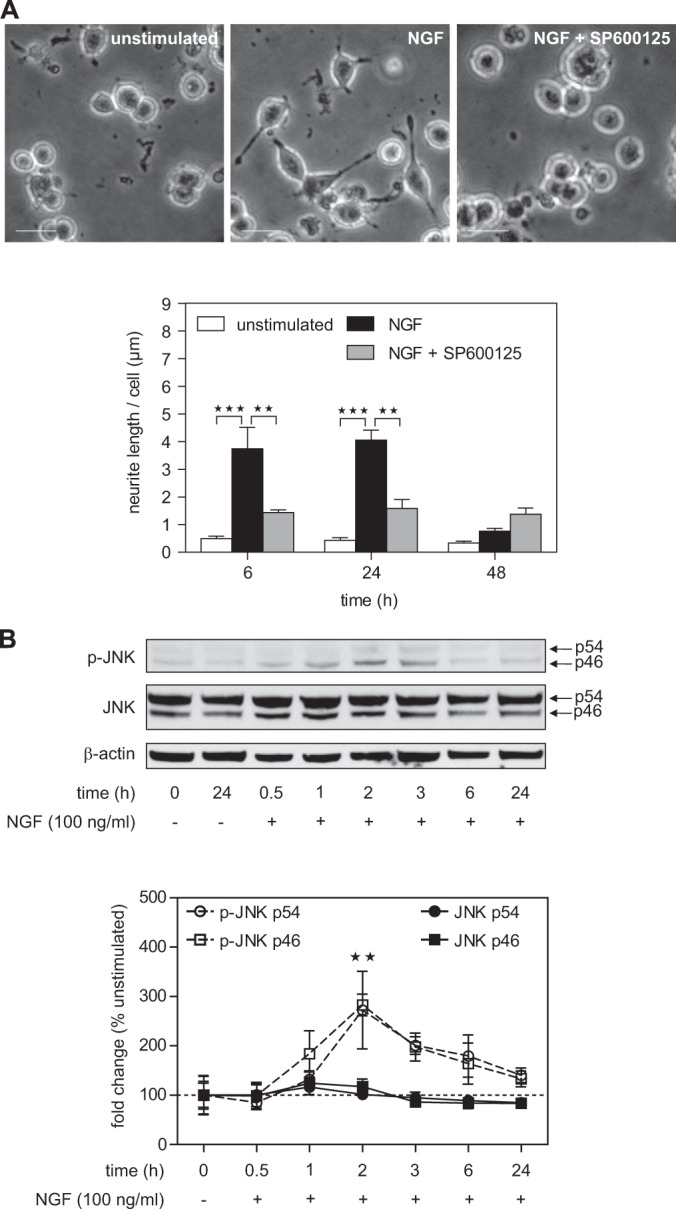
NGF treatment leads to JNK-dependent neurite formation in PC12 cells. A, treatment with NGF (100 ng/ml) induces a major shift in the phenotype of PC12 cells from proliferating tumor cells to non-dividing neurons showing growth of neurites at 6 h after stimulation. Pretreatment with the JNK inhibitor SP600125 (10 μm) for 1 h led to a significant decrease in neurite outgrowth. **p < 0.01, ***p < 0.001 versus unstimulated or NGF treated (n = 3 each). Scale bar: 20 μm. B, neurite outgrowth was accompanied by NGF-induced JNK phosphorylation (∼3-fold increase) peaking 2 h after NGF treatment. NGF treatment had no effect on total JNK levels. Arrows indicate the two different p54 and p46 JNK isoforms. **p < 0.01 versus unstimulated (n = 4 to 7).
JNK Interacts with Proteins of Different Molecular Functions during Neuronal Differentiation
Phosphoproteomics is a powerful approach for the study of cellular signal transduction (41). However, in the case of JNK, this strategy is complicated by several factors. First, the consensus JNK phosphorylation site is a typical proline-directed phosphorylation motif shared by several other kinases such as ERK1/2 and cyclin-dependent kinases. Second, it has been shown that JNK sites can also be phosphorylated by other kinases (6). These two factors make it difficult to discern JNK substrates from substrates of other kinases in a classical phosphoproteomic experiment. Finally, focusing exclusively on JNK substrates would not provide any information about upstream signaling events that lead to JNK activation. Because of these limitations, we pursued an alternative proteomic approach. We took advantage of the fact that JNK physically associates with effectors via a docking site, the so-called JNK binding domain (JBD). JBDs mediate the interaction of JNK with substrates, as well as with adaptor proteins and upstream activators (6). Therefore, studying dynamic changes in JNK interaction partners can provide insights into both upstream and downstream signaling events.
Our experimental strategy was to precipitate JNK from stimulated PC12 cells and characterize dynamic changes in the JNK interactome via SILAC-based quantitative shotgun proteomics (Fig. 2A). Metabolic labeling of PC12 cells with either “medium” (Lys-4, Arg-6) or “heavy” (Lys-8, Arg-10) SILAC media for six passages resulted in essentially complete labeling (data not shown). Labeled cells were then treated with NGF for different lengths of time. To cover the dynamic changes in JNK-associated proteins, we performed three parallel triple SILAC experiments with one time point as an internal reference. This procedure allows analysis of temporal changes of the interactome (42). Cells were lysed, and JNK and associated proteins were immunoprecipitated with bead-coupled JNK1- and JNK2-specific antibodies. Immunoprecipitates from the three corresponding SILAC states were combined, washed, and analyzed via high-resolution quantitative shotgun proteomics. Protein data from the three time-series measurements were combined into a single time course using the common 2-h time point as a reference (i.e. by calculating the ratio of ratios). To further increase the robustness of the data, we combined data from two independent biological replicates, and only proteins that were quantified with at least three SILAC ratio counts at each time point were considered. This analysis resulted in a list of 728 proteins. The list of identified proteins was expected to contain three different groups of proteins: (i) NGF-regulated JNK interaction partners, (ii) constitutive JNK interactors, and (iii) nonspecific contaminants. As our aim was to assess the role of JNK in NGF-induced neurite outgrowth, we focused our analysis on only the first group. To extract potential NGF-regulated interaction partners, we selected all proteins with a log2 fold change ≥ 0.7 and/or ≤ −0.7 at any time point (n = 201) (supplemental Table S1). We then used hierarchical clustering to classify these proteins according to their dynamic behavior. The abundance profiles of proteins fell into three major clusters (Fig. 2B).
Fig. 2.

Quantitative mass spectrometry timecourse analysis of potential JNK interacting proteins after NGF treatment. A, PC12 cells were grown in light, medium, or heavy SILAC media and were stimulated with NGF for different lengths of time. After JNK immunoprecipitation, samples were combined, digested with LysC and trypsin, and analyzed via LC-MS/MS. NGF-induced changes in JNK protein binding were calculated by using medium (2-h NGF-treated) cells as an internal reference. The illustration shows an example for a protein that transiently binds to JNK with the highest affinity at 6 h after NGF treatment. B, data from two independent biological experiments were combined and analyzed with MaxQuant. Proteins with a log2 fold change ≥ 0.7 and/or log2 fold change ≤ −0.7 at any time point given were extracted and z-scored. Proteins with similar JNK binding kinetics were clustered using Perseus software (Euclidean distance, average linkage, n = 201). Analysis of the molecular function of potential JNK interacting proteins using Protein Analysis through Evolutionary Relationships (PANTHER) revealed an enrichment for cytoskeletal protein (cluster 1, n = 25), nucleic acid binding (cluster 2, n = 73) and G-proteins (cluster 3, n = 104). The three most significant PANTHER terms per cluster are shown.
The first cluster contained proteins that showed increased abundance at both 0.5 to 1 h and 24 h of NGF stimulation with transiently decreased levels between 2 and 6 h. This cluster mainly contained actin-binding cytoskeletal proteins such as tropomodulin-2, tropomyosin α-4 chain, and several members of the myosin family involved in cytoskeleton reorganization and ATP-dependent movement along microfilaments. Furthermore, the motor protein kinesin light chain was found in this cluster, which is in line with previous studies demonstrating that JNK is involved in kinesin-dependent transport processes and microtubule dynamics in neurons (11, 13, 43).
The third cluster contained proteins that belonged to the families of large G-proteins and small GTPases including guanine nucleotide-binding protein G(o) subunit α, guanine nucleotide-binding protein G(i) subunit α-2, Rap1b, Rac1, RhoA, and Cdc42. These proteins showed increased levels 0.5 h to 2 h after NGF treatment followed by a steady decrease over time below control levels at 24 h. This finding suggests that NGF-dependent JNK activation might be induced via multiple G-proteins. Indeed, it has been shown that several G-proteins are involved in JNK activation during PC12 cell differentiation (25, 26) and that Rap1 is involved in NGF-induced PC12 cell differentiation (44, 45).
The second cluster contained proteins that showed delayed association with JNK starting 2 to 24 h after NGF stimulation. We were surprised to find that this cluster was strongly enriched in RNA binding proteins. For example, this subset contained proteins involved in mRNA splicing such as Fus and SF1 (46) and RNA helicases such as Ddx-1, -5, and -17. Intriguingly, two mRNA binding proteins in this cluster showed transient JNK binding at 2 to 3 h of stimulation. This kinetic behavior is similar to the activation profile of JNK (Fig. 1B). These two proteins, namely, non-POU domain-containing octamer-binding protein (Nono) and splicing factor proline- and glutamine-rich (Sfpq), are components of mRNA transport granules in dendrites (47). Additionally, we identified paraspeckle component 1 (Pspc1) in the cluster. Pspc1 is a paralogue of Nono and a known component of neuronal transport granule protein (47). Pspc1, Nono, and Sfpq are the three mammalian members of the Drosophila behavior and human splicing (DBHS) family involved in different aspects of RNA biology (48, 49). As components of paraspeckles and neuronal granules, they are found in the nucleus and in the cytosol, respectively. DBHS proteins have been linked to neuronal function. Sfpq mediates neuronal development in zebrafish, and Nono is required for normal vision and courtship behavior in fruit flies (50, 51).
d-JNKI-1 Specifically Blocks NGF-induced Neurite Outgrowth and Interaction of JNK with RNA-binding Proteins
Next, we asked whether NGF-mediated interactions are mediated by JBDs. To this end, we used the peptide inhibitor d-JNKI-1, which blocks interaction of JNK with its substrates (52). d-JNKI-1 consists of the protease-resistant d-retroinverso form of a 10-amino-acid HIV-Tat sequence, which allows for cellular uptake, and the 20-amino-acid sequence of the JBD of JNK interacting protein 1 (53–55). Thus, d-JNKI-1 specifically blocks interactions of JNK mediated by JBDs. d-JNKI-1 has been shown to be highly specific and has been used to block JNK activity in several neuronal disease model systems (56–58).
To see whether d-JNKI-1 blocks NGF-induced neurite outgrowth, we pretreated PC12 cells with d-JNKI-1 for 1 h. d-JNKI-1 dose-dependently decreased NGF-induced neurite outgrowth as assessed 24 h after stimulation (Fig. 3A). The inhibitory effect of d-JNKI-1 at a concentration of 10 μm was ∼60%, which is comparable to the inhibitory effect of the ATP competitive inhibitor SP600125 at the same concentration. Pretreatment with the control peptide d-TAT alone had no effect on NGF-induced neurite outgrowth, indicating that the inhibitory effect of d-JNKI-1 on neurite outgrowth is JNK specific. Additionally, we tested the inhibitory effect of d-JNKI-1 via phospho-specific Western blot analysis of the major JNK downstream target c-Jun. As expected, d-JNKI-1 (10 μm) significantly inhibited NGF-induced c-Jun phosphorylation at 3 h after NGF treatment (data not shown). These results corroborate our previous findings and suggest that JNK interaction with its substrates is necessary for NGF-induced neurite outgrowth.
Fig. 3.

The JNK-specific peptide inhibitor d-JNKI-1, which blocks the interaction of JNK with its substrates, decreased NGF-induced neurite outgrowth and NGF-induced interaction of JNK with cytoskeletal proteins and RNA transport granule proteins. A, PC12 cells were pretreated for 1 h with different concentrations of d-JNKI-1 or the control peptide d-TAT before NGF stimulation. Assessment of neurite elongation after 24 h revealed that d-JNKI-1 dose-dependently decreased neurite outgrowth by ∼60% at a concentration of 10 μm. The control peptide d-TAT had no effect on neurite outgrowth. **p < 0.01, ***p < 0.001 versus NGF + d-TAT (n = 3 each). Scale bar: 20 μm. B, PC12 cells were grown in light, medium, or heavy media and were stimulated with NGF for 3 h. Medium-labeled cells were pretreated for 1 h with d-JNKI-1. The heavy-labeled cells were pretreated with the control peptide d-TAT. After JNK immunoprecipitation (IP), samples were combined and analyzed via LC-MS/MS. C, 701 proteins were quantified with at least three ratio counts. PANTHER analysis showed an enrichment for cytoskeletal proteins (n = 65) and nucleic acid binding proteins (n = 94), which specifically co-immunoprecipitated with JNK after NGF treatment. A log2 fold change of 0.7 was applied as a cutoff. The three most significant PANTHER terms per cluster are shown. Highlighted proteins are discussed in the “Results” section.
To investigate the effect of d-JNKI-1 on the JNK interactome, we focused on 3-h NGF stimulation, as this time point coincides with high JNK activation. Light cells were left untreated, medium-heavy cells were pretreated for 1 h with d-JNKI-1 (10 μm), and heavy cells were pretreated with the control peptide d-TAT. Both medium-heavy and heavy cells were then stimulated with NGF for 3 h (Fig. 3B). JNK was precipitated from all three cell populations, and JNK-associated proteins were identified via mass spectrometry as described above. As a control we analyzed supernatants from the immunoprecipitations, and we did not observe global effects of NGF or d-JNKI-1 on the PC12 proteome (supplemental Fig. S1A). For JNK-associated proteins, we plotted ratios of NGF+d-TAT treated samples versus unstimulated samples (x-axis) and of NGF+d-TAT treated samples versus NGF+d-JNKI-1 treated samples (y-axis) against each other (Fig. 3C). The resulting plot shows which proteins interacted with JNK in an NGF- and/or d-JNKI-1-dependent manner.
We found that NGF-induced and d-JNKI-1-inhibited interactions were clearly correlated. Specifically, most of the NGF-induced interactions were inhibited by d-JNKI-1. Thus, NGF-induced interactions seem largely JBD dependent. Interestingly, interactors fell into two groups. The first group was strongly NGF dependent and moderately affected by the inhibitor (highlighted in green in Fig. 3C). This set of proteins was enriched in actin-binding cytoskeletal proteins such as drebrin, a protein that is involved in axonal growth (59). This subset also included filamin-B, an actin regulatory protein that has been described as a JNK scaffold protein that tethers Rac1 and a JNK-specific module (60). The second group of proteins was strongly inhibited by d-JNKI-1 and showed a moderate response to NGF (highlighted in red in Fig. 3C). This group contained the above-mentioned DBHS family proteins Sfpq, Nono, and Pspc1 and several other known components of RNA transport granules (Pur α and Pur β, several FMRPs, DDX1, DDX5, DDX6, and DDX17). To test whether other proteins in our dataset were also involved in RNA transport, we compared our data to a proteomic analysis of RNA transport granules (47). 26 out of 42 RNA transport granule proteins from a study by Kanai et al. were found in our dataset (supplemental Fig. S1B), and 18 out of these 26 were classified as JNK-specific interaction partners (log2 fold change ≥ 0.7). This is a highly significant enrichment (p = 2.81E-07, hypergeometric test) for mRNA transport granule proteins in the NGF- and d-JNKI-1-dependent JNK interactome. In summary, our experiments with the JNK-specific blocking peptide d-JNKI-1 confirm that JNK interacts with cytoskeletal proteins and RNA transport granule proteins in an NGF-dependent manner. The interactions can be specifically blocked with d-JNKI-1 and are thus apparently mediated by JBDs. JNK interacting proteins from this experiment are listed in supplemental Table S2.
JNK1 Interacts with the Nono–Sfpq Heterodimer in an RNA-dependent Way
To the best of our knowledge, the interaction of JNK with RNA transport granule proteins had not been described at the time of this study. We therefore performed follow-up experiments to validate and further characterize this observation. First, we sought to validate the mass spectrometry data via immunoprecipitation and Western blotting. We observed that JNK1 co-immunoprecipitated with Sfpq, Nono, and Pspc1 (Fig. 4A). Whereas NGF treatment increased the interaction, pretreatment of cells with d-JNKI-1 decreased the NGF-induced interaction to baseline levels. These results confirm that (i) JNK interacts with DBHS family proteins in PC12 cells, (ii) the interaction is increased by NGF stimulation, and (iii) the interaction is JBD dependent.
Fig. 4.
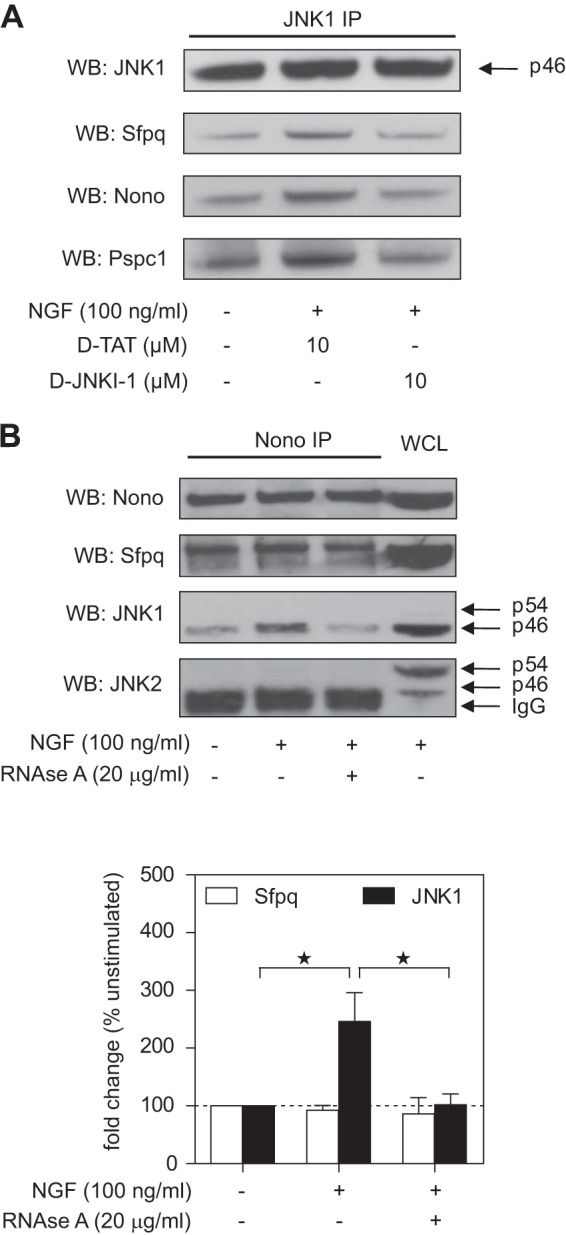
JNK1 interacts with the Nono–Sfpq heterodimer in a d-JNKI-1- and RNA-dependent way in NGF-treated PC12 cells. A, JNK1 was immunoprecipitated from whole cell lysates in unstimulated or NGF-treated cells and in the presence or absence of d-TAT or d-JNKI-1. Western blots against Nono, Sfpq, and Pspc1 showed that d-JNKI-1 decreased NGF-induced interaction of JNK1 with these proteins. A representative blot is shown. B, Nono was immunoprecipitated from whole cell lysates of unstimulated or NGF-treated cells and in the presence or absence of RNase A. Densitometric analysis of Western blots showed a ∼2.5-fold increase in JNK1–Nono interaction after 3 h of NGF treatment, which was inhibited by RNase A treatment (*p < 0.05 versus unstimulated (n = 4)). Interaction of Sfpq and Nono was not affected by this treatment. Reprobing with JNK2-specific antibody did not show any detectable signal in the more prominent JNK2 band p54. PC12 whole cell lysate (WCL) was used as a positive control.
Although co-immunoprecipitation of endogenous proteins is considered the gold-standard assay for protein–protein interactions, it is still affected by possible antibody cross-reactivities. To verify the specificity of the assay, we performed the inverse experiment and used an antibody against Nono for immunoprecipitation. In addition, we used RNase A treatment to test whether the interactions were RNA dependent. In these experiments we found that NGF treatment led to a ∼2.5-fold induction of JNK1 binding to Nono (Fig. 4B). RNase A treatment decreased the interaction, indicating that it was RNA dependent. The interaction seemed to be specific for JNK1, as JNK2 was not detected in the Nono pull-downs. Sfpq was also observed to co-precipitate with Nono, and this interaction was not affected by NGF or RNase A treatment. This result is in line with a previous study demonstrating that Nono and Sfpq form a stable, RNase-resistant, mRNA-binding heterodimer in neurons (47). Collectively, these results further confirm the interaction of JNK and mRNA transport granule proteins and indicate that JNK1, rather than JNK2, is the main interactor. Because we observed that the interaction was RNA dependent, we suggest that JNK1 associates with RNA-containing multi-protein complexes rather than with individual proteins.
NGF Treatment Induces JNK1 Interaction with Sfpq in the Cytosol
DBHS family proteins and JNK can be found in the nucleus and the cytosol. We therefore analyzed the subcellular distribution of Nono and Sfpq in PC12 cells via confocal microscopy. Representative images of three independent experiments showed that Nono and Sfpq were most abundant in the nucleus but were also found in the cytosol of PC12 cells (Fig. 5A, upper panel). This result was also confirmed by Western blot analyses of cytosolic and nuclear fractions from two independent experiments (Fig. 5B). NGF treatment for 3 h did not significantly alter the distribution of the total cellular Nono and Sfpq pools (Fig. 5B). To analyze the interaction of JNK1 and Sfpq, we stained stimulated and unstimulated PC12 cells with JNK1- and Sfpq-specific antibodies. Interestingly, increased co-localization of JNK1 and Sfpq was observed in outgrowing neurites of NGF-treated cells relative to unstimulated cells (Fig. 5A, lower panel). Evaluation of JNK1 stained cells alone revealed that JNK1 was mainly expressed in the cytosol of untreated and NGF-treated cells (Fig. 5A, lower panel, arrowheads). Consistent results were obtained in Western blots of cytosolic and nuclear extracts (Fig. 5B). These results are also supported by a previous study showing that JNK1 is mainly located in the cytosol in PC12 cells (61). Similar to JNK1, JNK2 was mainly detected in the cytosol, but it also showed considerable nuclear localization. NGF-induced phosphorylation of both JNK isoforms occurred mainly in the cytosol (Fig. 5B).
Fig. 5.

Co-localization and cellular distribution of Nono, Sfpq, and JNK1. A, confocal microscopy images of Nono (green), Sfpq (red), and nuclei (blue) co-stained PC12 cells show that Nono and Sfpq were expressed in the nucleus as well as in the cytosol. Co-localization mainly occurred in the nucleus, where the greater fraction of Nono and Sfpq is located (upper panel). The cellular distribution of JNK1 (green) showed that JNK1 was mainly localized in the cytosol (lower panel). NGF treatment for 3 h led to an increased co-localization signal of JNK1 (green) and Sfpq (red) in the cytosol. Scale bar: 6 μm. B, Western blot analysis of cytosolic and nuclear fractions revealed that NGF treatment for 3 h led to JNK phosphorylation mainly in the cytosol. Reprobing with JNK1- and JNK2-specific antibodies showed that JNKs were mainly localized in the cytosol, whereas the JNK2 p54 isoform was also found in the nucleus. Reprobing with Nono- and Sfpq-specific antibodies corroborated the cellular distribution of Nono and Sfpq noted in confocal microscopy analysis. Representative immunoblots are shown (n = 2).
Unbiased assessment of changes in protein co-localization via microscopy is challenging. Therefore, to confirm the increased co-localization of JNK1 and Sfpq, we employed the proximity ligation assay (PLA) (62). PLA uses the enzymatic ligation of connector oligonucleotides that are attached to different antibodies and rolling circle amplification to increase the signal derived from individual interactions. With this method one can detect the co-localization of endogenous proteins in an unbiased and quantitative manner. As a positive control, we tested the interaction of Nono and Sfpq. Evaluation of PLA signals in cells co-stained with DAPI revealed that the Nono–Sfpq heterodimer was located mainly in the nucleus and to a minor extent in the cytosol (Fig. 6A). Next, we used PLA to quantify the co-localization of JNK1 and Sfpq (Fig. 6B). Quantitative analysis of PLA signals revealed that NGF treatment led to a significant induction (∼5-fold) of JNK1–Sfpq interaction. In line with our previous findings, PLA signals of the JNK1–Sfpq interaction in NGF-treated cells were mainly observed in the cytosol, whereas JNK1–Sfpq interaction in unstimulated cells was found in the nucleus. Together, these results indicate that the Nono–Sfpq heterodimer is mainly located in the nucleus, but JNK1 interacts with Nono–Sfpq in an NGF-dependent manner mainly in the cytosol.
Fig. 6.
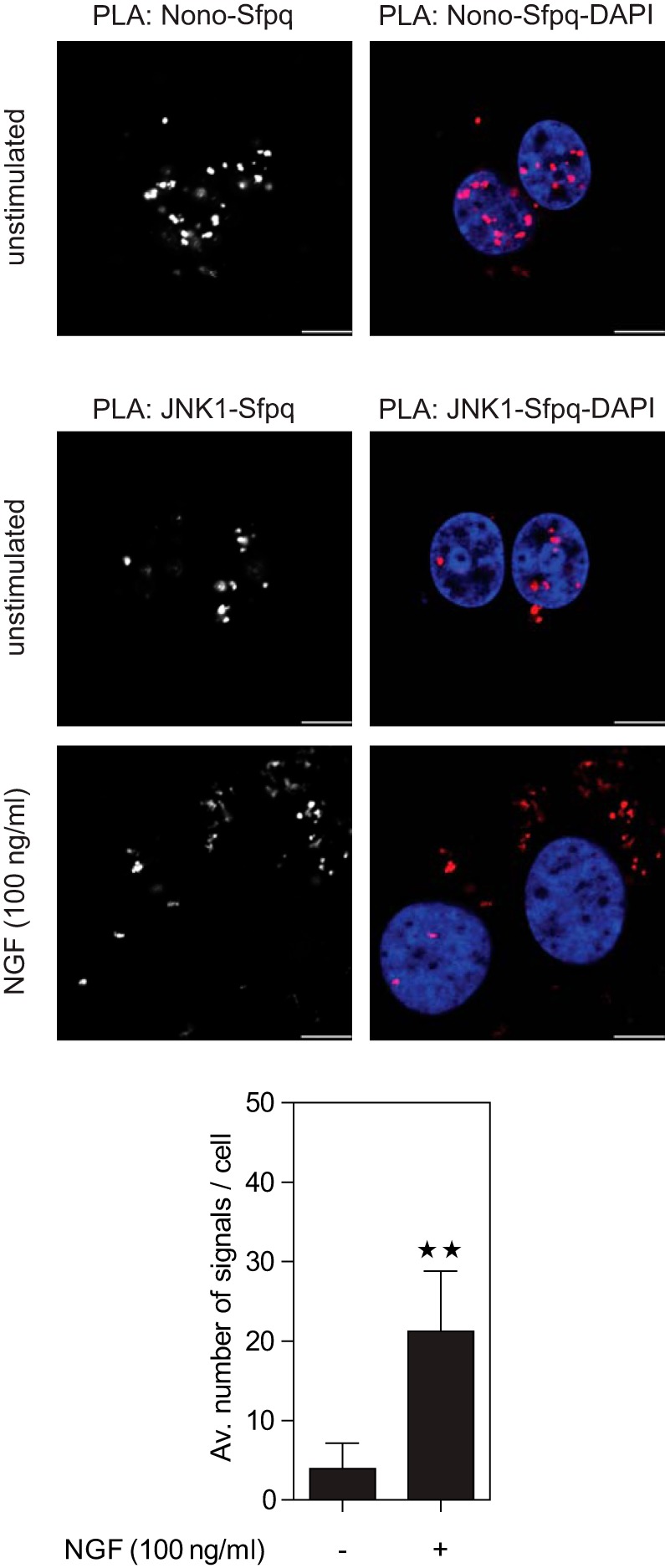
Co-localization and cellular distribution of Nono, Sfpq, and JNK1 assessed via proximity ligation assay (PLA). PC12 cells were incubated with Nono- and Sfpq-specific antibodies followed by PLA secondary antibodies (red) and were analyzed via confocal microscopy. Nuclear counterstaining with DAPI (blue) revealed that the Nono–Sfpq heterodimer was mainly located in the nucleus (upper panel). Incubation with JNK1- and Sfpq-specific antibodies followed by PLA staining showed a ∼5.4-fold increase in JNK1–Sfpq interaction after NGF treatment for 3 h (lower panel). **p < 0.01 versus unstimulated (n = 8 to 9). Nuclear counterstaining with DAPI showed that JNK1 and Sfpq mainly interacted in the cytosol of NGF-treated cells. Results were obtained from three independent experiments. Scale bar: 6 μm.
JNK1 and Sfpq Transiently Co-localize in Primary Cortical Neurons and P19 Neuronal Cells
To further study the association of JNK1 and Sfpq in the cytosol of differentiating neurons, we made use of two additional neuronal models, (i) mouse primary cortical neurons and (ii) neurons derived from the murine embryonic carcinoma cell line P19. Primary cortical neurons spontaneously form neurites after plating, continuing in vitro their differentiation process (28). P19 cells differentiate into neurons upon treatment with retinoic acid and produce numerous neurites similar to primary cortical neurons (63).
Confocal microscopy of primary cortical neurons and P19 neuronal cells revealed that JNK1 and Sfpq, like in PC12 cells, were present in the nucleus and in the cytosol, particularly during the first 12 h of differentiation, which coincides with the polarization and outgrowth of primary neurites in both neuronal models (Figs. 7A–7C and 7E–7G; arrowheads in insets 7A′–7C′ and 7E′–7G′). Confocal images showed that JNK1 was mainly localized in the cytosol (Figs. 7A″–7H″), whereas Sfpq localized in the cytosol only during the polarization and outgrowth of primary neurites (Figs. 7A‴–7H‴). At later stages, Sfpq was found exclusively in the nucleus (Figs. 7D, 7H, 7D‴, and 7H‴). Quantification of neurons stained for JNK1 and Sfpq revealed a maximal co-localization of both molecules in the cytosol of immature neurons during the first 12 h of neuronal differentiation, whereas no co-localization was observed after 24 h (Fig. 7I).
Fig. 7.

Co-localization and cellular distribution of Sfpq and JNK1 in primary cortical neurons and P19 retinoic-acid-treated neuronal cells. Confocal microscopy images of JNK1 (green), Sfpq (red), and the neuronal marker doublecortin (DCX; blue) co-stained in primary cortical (A–D) and P19-diferrentiated neurons (E–H). JNK1 and Sfpq co-localized in the cytosol during the first 12 h after the start of differentiation (see arrows), coinciding with the polarization and outgrowth of primary neurites (A–C, E–F; arrowheads in insets A′–C′ and E′–G′). The cellular distribution of JNK1 (green) during the time course showed that JNK1 was mainly localized in the cytosol (A″–H″). In contrast, Sfpq (red) was localized in the cytosol only during the polarization and outgrowth of primary neurites (A‴–H‴). At later stages, Sfpq was found exclusively in the nucleus (D, H, D‴, and H‴). Scale bar: 5 μm. I, quantification of neurons with co-localization of JNK1 and Sfpq in the cytosol of growing and elongating neurites (n = 150 to 200 neurons per stage). J, schematic representation of the interaction between JNK1 and Sfpq during neurite outgrowth.
Together, these results are consistent with the data obtained from PC12 cells. The fact that transient co-localization of JNK1 and Sfpq occurred in the same subcellular location and with similar kinetics in three distinct neuronal models strongly suggests that interaction of JNK with RNA transport granules is a generic feature during neuronal differentiation (Fig. 7J).
Sfpq Contributes to NGF-induced Neurite Outgrowth in PC12 Cells
To further investigate whether the JNK interaction partner Sfpq is involved in neuronal differentiation, we treated PC12 cells with Sfpq siRNA for 4 days. Western blot results for PC12 whole cell lysates showed that Sfpq was dose-dependently knocked down (Fig. 8A). Semi-quantitative analysis of band intensities revealed that knockdown efficiency was almost complete (97%) at a dose of 200 nmol siRNA. We then treated Sfpq knockdown cells with NGF (100 ng/ml) and measured neurite outgrowth 24 h after stimulation (Fig. 8B). Sfpq knockdown led to a significant decrease (∼50%) in NGF-induced neurite outgrowth, demonstrating that Sfpq is involved in the neuronal differentiation of PC12 cells. In parallel, we pretreated mock-transfected or Sfpq siRNA transfected cells with JNK inhibitor SP600125 (10 μm) for 1 h before NGF stimulation. SP600125 significantly decreased NGF-induced neurite outgrowth by ∼60% in mock-transfected cells. Thus, Sfpq knockdown and JNK inhibition have a similar effect on neurite outgrowth. Interestingly, treating cells with both SP600125 and Sfpq siRNA did not further increase the inhibition of neurite outgrowth. The lack of a cumulative effect is consistent with the hypothesis that JNK and Sfpq mediate NGF-induced neurite outgrowth via the same pathway and that interaction between JNK and RNA transport granule proteins is involved in neuronal differentiation.
Fig. 8.
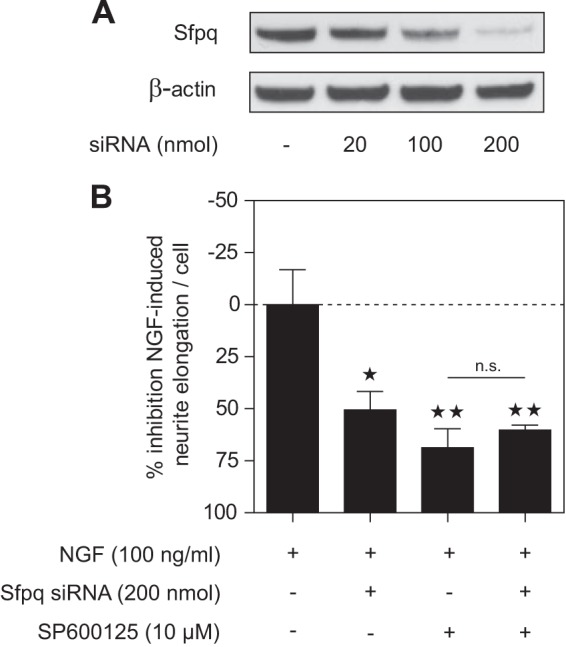
Knockdown of Sfpq led to decreased NGF-induced neurite outgrowth. A, PC12 cells were transfected with different amounts of Sfpq siRNA. Quantification of band intensities of PSF Western blots showed a knockdown efficiency after 4 days of 97% using 200 nmol of Sfpq siRNA. B, PC12 cells were mock-transfected or transfected with 200 nmol Sfpq siRNA for 4 days. NGF-induced neurite outgrowth in PSF knockdown cells was decreased by ∼50%, whereas 10 μm SP600125 inhibited NGF-induced neurite formation in mock-transfected cells by ∼69%. Pretreatment with SP600125 of Sfpq knockdown cells did not significantly further decrease NGF-induced neurite outgrowth. *p < 0.05, **p < 0.01 versus NGF treated; n.s. = non-significant (n = 3 each).
DISCUSSION
Signal transduction cascades are typically represented as graphs that begin with the activation of a cell surface receptor and end with the activation of a transcription factor. However, it is now clear that gene expression is also regulated at the posttranscriptional level. For example, microRNAs and RNA binding proteins interact with mRNAs and control their splicing, export, stability, localization, and translation (64, 65). In fact, translation efficiency appears to be the single best predictor of cellular protein abundance (66). Posttranscriptional regulation is particularly relevant in highly compartmentalized cells such as neurons, where specific functions occur in restricted subcellular domains (67). Although the link between signaling cascades and transcriptional control is quite well studied, the connections between cell signaling and posttranscriptional regulatory events are still poorly characterized.
It is known that JNKs play an important role in both neuronal cell death and neuronal differentiation (5–8). Whereas apoptosis induction appears to depend on nuclear JNK, cytosolic JNK activity is required for neurite outgrowth (5, 11). Here, we have shown that JNK dynamically associates with RNA binding proteins in an early phase during neuronal differentiation. First, we showed that JNK activity is required for NGF-induced neurite outgrowth, consistent with previous reports (23, 24). We then used quantitative interaction proteomics to identify proteins associating with JNK in an NGF-dependent manner. Surprisingly, we observed that several RNA-binding transport granule proteins including Nono, Sfpq, and Pspc1 temporally associated with JNK. We validated the specificity of these interactions by showing that they could be blocked by d-JNKI-1, a highly specific peptide inhibitor of JNK. Moreover, we used Western blotting to show that Nono, Sfpq, and Pspc1 co-precipitate with JNK1 but not JNK2 in an RNA-dependent manner. Using fractionation experiments, confocal microscopy, and proximity ligation assay, we found that this interaction mainly occurred in the cytosol of differentiating PC12 cells. Importantly, we were able to replicate our findings in P19-derived and primary neurons, strongly suggesting that the interaction is a generic feature of neuronal differentiation. The fact that the temporal interaction was observed during the early stages of neurite extension suggests that this interaction might play a role in the initiation of posttranscriptional gene regulation events.
Nono, Sfpq, and Pspc1 are the mammalian members of the DBHS family involved in many aspects of RNA biology (48, 49). Most research on these proteins so far has focused on their role in the nucleus, where they are components of subnuclear bodies termed paraspeckles. Although the function of paraspeckles is not well understood, they have been reported to retain mRNAs in the nucleus to allow their rapid release into the cytosol upon stress (68). In neuronal cells, all three proteins have been described as components of RNA transport granules (47). Sfpq has been shown to be involved in neuronal development in zebrafish (50), and we show here that knocking down Sfpq impaired neurite outgrowth. Nono is required for normal vision and courtship behavior in fruit flies (51). Thus, DBHS proteins have neuronal functions in vivo. We observed that JNK1 interacted with Sfpq in the cytosol of PC12 cells. Therefore, our data suggest that JNK1 mediates neuronal differentiation at least partially via interaction with DBHS family proteins in the cytosol. Interestingly, we found that the NGF-induced interaction of JNK1 with Nono/Sfpq was RNA-dependent. This suggests that JNK1 associates with entire transport granules in an NGF-dependent manner. In fact, the NGF-induced JNK interactome was strongly enriched in known components of RNA transport granules. It should therefore be stressed that we have no evidence that JNK1 is a direct binder of Sfpq and/or Nono. In fact, given that the interaction was RNA dependent, it appears more likely that JNK1 associates with higher order complexes containing these proteins and RNAs, possibly via its low-complexity region (see below). Both JNK and neuronal granule proteins are known to be involved in neuronal differentiation. However, to the best of our knowledge, a link between JNK signaling and neuronal granules has not yet been described.
RNA granules are particularly important in neurons for the targeting of mRNAs to distinct cellular compartments for localized translation (67, 69). How proteins and RNAs are targeted to and released from RNA granules is not yet entirely understood. Recently, it has been reported that proteins can assemble into RNA granules via low-complexity regions (70, 71). How this assembly and disassembly are regulated is largely unknown. One possible mechanism involves phosphorylation. For example, the kinase DYRK3 has been shown to interact with stress granules, presumably via a low-complexity region (72). DYRK3 inhibition stabilizes the granules, suggesting that this kinase can disassemble granules via the phosphorylation of specific target proteins. Similar to DYRK3, JNK1 has a predicted low-complexity region that is disordered in the crystal structure (73). It is tempting to speculate that JNK1 might be involved in regulating transport granules. For example, granule-associated JNK1 might phosphorylate substrate proteins in the granules to induce their release. As for DYRK3, these putative JNK1 substrates remain to be identified. In addition to RNA binding proteins, we found that JNK1 also interacted with proteins involved in dendritic and/or axonal transport via microfilaments and microtubules (kinesin light chain, filamin-B, and drebrin). It is therefore possible that JNK1 controls the transport of RNA granules. Consistently, JNK1 is known to control the phosphorylation of microtubule-associated proteins and to be required for the maintenance of neuronal microtubules (13).
In summary, our unbiased proteomic analysis of the dynamic JNK interactome reveals that JNK dynamically associates with RNA transport granule proteins during neuronal differentiation. Although the exact mechanisms involved in the interaction of JNK with neuronal granules remain to be investigated, blocking the interaction with the highly selective peptide d-JNKI-1 inhibits neurite outgrowth. Thus, interactions mediated via JBDs appear to be functionally important for neuronal differentiation. Furthermore, we show that Sfpq, a member of RNA transport granules, is involved in neuronal differentiation in PC12 cells. However, several important questions remain. First, it is not known which RNAs are located in the JNK-associated neuronal granules. Furthermore, it might be informative to see which proteins are phosphorylated by JNK upon NGF treatment, although the identification of direct kinase substrates in vivo is challenging. We also note that several well-described JNK interaction partners such as microtubule-associated protein, JNK interacting protein, and c-Jun were not identified in our dataset. It is not clear whether this is due to the specialized function of JNK in PC12 cells or to limitations of our experimental approach. Finally, JNK is not the only mediator of neuronal differentiation. How neurons integrate different signaling events in a spatio-temporal manner remains to be explored.
Supplementary Material
Acknowledgments
We thank Christophe Bonny for providing us with d-JNKI-1. We also thank Christian Sommer for excellent technical assistance.
Footnotes
Author contributions: M.D.S., C.B., and M.S. designed research; M.D.S., E.M., and L.H. performed research; M.D.S. analyzed data; M.D.S. and M.S. wrote the paper.
* This work was supported by the Helmholtz Association and the Swiss National Science Foundation.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- JNK
- c-Jun N-terminal kinase
- JBD
- c-Jun N-terminal kinase binding domain
- DBHS
- Drosophila behavior and human splicing
- Nono
- non-POU domain-containing octamer-binding protein
- NGF
- nerve growth factor
- PLA
- proximity ligation assay
- Pspc1
- paraspeckle component 1
- Sfpq
- splicing factor proline- and glutamine-rich
- SILAC
- stable isotope labeling by amino acids in cell culture.
REFERENCES
- 1. Haeusgen W., Boehm R., Zhao Y., Herdegen T., Waetzig V. (2009) Specific activities of individual c-Jun N-terminal kinases in the brain. Neuroscience 161, 951–959 [DOI] [PubMed] [Google Scholar]
- 2. Waetzig V., Zhao Y., Herdegen T. (2006) The bright side of JNKs—multitalented mediators in neuronal sprouting, brain development and nerve fiber regeneration. Prog. Neurobiol. 80, 84–97 [DOI] [PubMed] [Google Scholar]
- 3. Barr R. K., Bogoyevitch M. A. (2001) The c-Jun N-terminal protein kinase family of mitogen-activated protein kinases (JNK MAPKs). Int. J. Biochem. Cell Biol. 33, 1047–1063 [DOI] [PubMed] [Google Scholar]
- 4. Weston C. R., Davis R. J. (2007) The JNK signal transduction pathway. Curr. Opin. Cell Biol. 19, 142–149 [DOI] [PubMed] [Google Scholar]
- 5. Bjorkblom B., Vainio J. C., Hongisto V., Herdegen T., Courtney M. J., Coffey E. T. (2008) All JNKs can kill, but nuclear localization is critical for neuronal death. J. Biol. Chem. 283, 19704–19713 [DOI] [PubMed] [Google Scholar]
- 6. Bogoyevitch M. A., Kobe B. (2006) Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 70, 1061–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eminel S., Roemer L., Waetzig V., Herdegen T. (2007) c-Jun N-terminal kinases trigger both degeneration and neurite outgrowth in primary hippocampal and cortical neurons. J. Neurochem. 104, 957–969 [DOI] [PubMed] [Google Scholar]
- 8. Waetzig V., Herdegen T. (2003) A single c-Jun N-terminal kinase isoform (JNK3-p54) is an effector in both neuronal differentiation and cell death. J. Biol. Chem. 278, 567–572 [DOI] [PubMed] [Google Scholar]
- 9. Neidhart S., Antonsson B., Gillieron C., Vilbois F., Grenningloh G., Arkinstall S. (2001) c-Jun N-terminal kinase-3 (JNK3)/stress-activated protein kinase-beta (SAPKbeta) binds and phosphorylates the neuronal microtubule regulator SCG10. FEBS Lett. 508, 259–264 [DOI] [PubMed] [Google Scholar]
- 10. Tararuk T., Ostman N., Li W., Bjorkblom B., Padzik A., Zdrojewska J., Hongisto V., Herdegen T., Konopka W., Courtney M. J., Coffey E. T. (2006) JNK1 phosphorylation of SCG10 determines microtubule dynamics and axodendritic length. J. Cell Biol. 173, 265–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bjorkblom B., Ostman N., Hongisto V., Komarovski V., Filen J. J., Nyman T. A., Kallunki T., Courtney M. J., Coffey E. T. (2005) Constitutively active cytoplasmic c-Jun N-terminal kinase 1 is a dominant regulator of dendritic architecture: role of microtubule-associated protein 2 as an effector. J. Neurosci. 25, 6350–6361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bjorkblom B., Padzik A., Mohammad H., Westerlund N., Komulainen E., Hollos P., Parviainen L., Papageorgiou A. C., Iljin K., Kallioniemi O., Kallajoki M., Courtney M. J., Magard M., James P., Coffey E. T. (2012) c-Jun N-terminal kinase phosphorylation of MARCKSL1 determines actin stability and migration in neurons and in cancer cells. Mol. Cell. Biol. 32, 3513–3526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chang L., Jones Y., Ellisman M. H., Goldstein L. S., Karin M. (2003) JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Dev. Cell 4, 521–533 [DOI] [PubMed] [Google Scholar]
- 14. Gdalyahu A., Ghosh I., Levy T., Sapir T., Sapoznik S., Fishler Y., Azoulai D., Reiner O. (2004) DCX, a new mediator of the JNK pathway. EMBO J. 23, 823–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Podkowa M., Zhao X., Chow C. W., Coffey E. T., Davis R. J., Attisano L. (2010) Microtubule stabilization by bone morphogenetic protein receptor-mediated scaffolding of c-Jun N-terminal kinase promotes dendrite formation. Mol. Cell. Biol. 30, 2241–2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Westerlund N., Zdrojewska J., Padzik A., Komulainen E., Bjorkblom B., Rannikko E., Tararuk T., Garcia-Frigola C., Sandholm J., Nguyen L., Kallunki T., Courtney M. J., Coffey E. T. (2011) Phosphorylation of SCG10/stathmin-2 determines multipolar stage exit and neuronal migration rate. Nat. Neurosci. 14, 305–313 [DOI] [PubMed] [Google Scholar]
- 17. Francavilla C., Rigbolt K. T., Emdal K. B., Carraro G., Vernet E., Bekker-Jensen D. B., Streicher W., Wikstrom M., Sundstrom M., Bellusci S., Cavallaro U., Blagoev B., Olsen J. V. (2013) Functional proteomics defines the molecular switch underlying FGF receptor trafficking and cellular outputs. Mol. Cell 51, 707–722 [DOI] [PubMed] [Google Scholar]
- 18. Rigbolt K. T., Blagoev B. (2012) Quantitative phosphoproteomics to characterize signaling networks. Semin. Cell Dev. Biol. 23, 863–871 [DOI] [PubMed] [Google Scholar]
- 19. Greene L. A., Tischler A. S. (1976) Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. U.S.A. 73, 2424–2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Greene L. A., Aletta J. M., Rukenstein A., Green S. H. (1987) PC12 pheochromocytoma cells: culture, nerve growth factor treatment, and experimental exploitation. Methods Enzymol. 147, 207–216 [DOI] [PubMed] [Google Scholar]
- 21. Chevet E., Lemaitre G., Janjic N., Barritault D., Bikfalvi A., Katinka M. D. (1999) Fibroblast growth factor receptors participate in the control of mitogen-activated protein kinase activity during nerve growth factor-induced neuronal differentiation of PC12 cells. J. Biol. Chem. 274, 20901–20908 [DOI] [PubMed] [Google Scholar]
- 22. Kaplan D. R., Stephens R. M. (1994) Neurotrophin signal transduction by the Trk receptor. J. Neurobiol. 25, 1404–1417 [DOI] [PubMed] [Google Scholar]
- 23. Waetzig V., Herdegen T. (2003) The concerted signaling of ERK1/2 and JNKs is essential for PC12 cell neuritogenesis and converges at the level of target proteins. Mol. Cell. Neurosci. 24, 238–249 [DOI] [PubMed] [Google Scholar]
- 24. Leppa S., Saffrich R., Ansorge W., Bohmann D. (1998) Differential regulation of c-Jun by ERK and JNK during PC12 cell differentiation. EMBO J. 17, 4404–4413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Heasley L. E., Storey B., Fanger G. R., Butterfield L., Zamarripa J., Blumberg D., Maue R. A. (1996) GTPase-deficient G alpha 16 and G alpha q induce PC12 cell differentiation and persistent activation of cJun NH2-terminal kinases. Mol. Cell. Biol. 16, 648–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tso P. H., Morris C. J., Yung L. Y., Ip N. Y., Wong Y. H. (2009) Multiple Gi proteins participate in nerve growth factor-induced activation of c-Jun N-terminal kinases in PC12 cells. Neurochem. Res. 34, 1101–1112 [DOI] [PubMed] [Google Scholar]
- 27. York R. D., Yao H., Dillon T., Ellig C. L., Eckert S. P., McCleskey E. W., Stork P. J. (1998) Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature 392, 622–626 [DOI] [PubMed] [Google Scholar]
- 28. Hernandez-Miranda L. R., Cariboni A., Faux C., Ruhrberg C., Cho J. H., Cloutier J. F., Eickholt B. J., Parnavelas J. G., Andrews W. D. (2011) Robo1 regulates semaphorin signaling to guide the migration of cortical interneurons through the ventral forebrain. J. Neurosci. 31, 6174–6187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ishima T., Hashimoto K. (2012) Potentiation of nerve growth factor-induced neurite outgrowth in PC12 cells by ifenprodil: the role of sigma-1 and IP(3) receptors. PLoS One 7, e37989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nishimura T., Ishima T., Iyo M., Hashimoto K. (2008) Potentiation of nerve growth factor-induced neurite outgrowth by fluvoxamine: role of sigma-1 receptors, IP3 receptors and cellular signaling pathways. PLoS One 3, e2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lahiri D. K., Ge Y. (2000) Electrophoretic mobility shift assay for the detection of specific DNA-protein complex in nuclear extracts from the cultured cells and frozen autopsy human brain tissue. Brain Res. Brain Res. Protoc. 5, 257–265 [DOI] [PubMed] [Google Scholar]
- 32. Rappsilber J., Ishihama Y., Mann M. (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75, 663–670 [DOI] [PubMed] [Google Scholar]
- 33. Sury M. D., Chen J. X., Selbach M. (2010) The SILAC fly allows for accurate protein quantification in vivo. Mol. Cell. Proteomics 9, 2173–2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ishihama Y., Rappsilber J., Andersen J. S., Mann M. (2002) Microcolumns with self-assembled particle frits for proteomics. J. Chromatogr. A 979, 233–239 [DOI] [PubMed] [Google Scholar]
- 35. Cox J., Matic I., Hilger M., Nagaraj N., Selbach M., Olsen J. V., Mann M. (2009) A practical guide to the MaxQuant computational platform for SILAC-based quantitative proteomics. Nat. Protoc. 4, 698–705 [DOI] [PubMed] [Google Scholar]
- 36. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 37. Cox J., Michalski A., Mann M. (2011) Software lock mass by two-dimensional minimization of peptide mass errors. J. Am. Soc. Mass Spectrom. 22, 1373–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 39. Eriksson M., Taskinen M., Leppa S. (2007) Mitogen activated protein kinase-dependent activation of c-Jun and c-Fos is required for neuronal differentiation but not for growth and stress response in PC12 cells. J. Cell. Physiol. 210, 538–548 [DOI] [PubMed] [Google Scholar]
- 40. Kita Y., Kimura K. D., Kobayashi M., Ihara S., Kaibuchi K., Kuroda S., Ui M., Iba H., Konishi H., Kikkawa U., Nagata S., Fukui Y. (1998) Microinjection of activated phosphatidylinositol-3 kinase induces process outgrowth in rat PC12 cells through the Rac-JNK signal transduction pathway. J. Cell Sci. 111, 907–915 [DOI] [PubMed] [Google Scholar]
- 41. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 42. Blagoev B., Ong S. E., Kratchmarova I., Mann M. (2004) Temporal analysis of 2 phosphotyrosine-dependent signaling networks by quantitative proteomics. Nat. Biotechnol. 22, 1139–1145 [DOI] [PubMed] [Google Scholar]
- 43. Cavalli V., Kujala P., Klumperman J., Goldstein L. S. (2005) Sunday Driver links axonal transport to damage signaling. J. Cell Biol. 168, 775–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mochizuki N., Yamashita S., Kurokawa K., Ohba Y., Nagai T., Miyawaki A., Matsuda M. (2001) Spatio-temporal images of growth-factor-induced activation of Ras and Rap1. Nature 411, 1065–1068 [DOI] [PubMed] [Google Scholar]
- 45. Jeon C. Y., Kim H. J., Lee J. Y., Kim J. B., Kim S. C., Park J. B. (2010) p190RhoGAP and Rap-dependent RhoGAP (ARAP3) inactivate RhoA in response to nerve growth factor leading to neurite outgrowth from PC12 cells. Exp. Mol. Med. 42, 335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rappsilber J., Ryder U., Lamond A. I., Mann M. (2002) Large-scale proteomic analysis of the human spliceosome. Genome Res. 12, 1231–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kanai Y., Dohmae N., Hirokawa N. (2004) Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 43, 513–525 [DOI] [PubMed] [Google Scholar]
- 48. Fox A. H., Lamond A. I. (2010) Paraspeckles. Cold Spring Harb. Perspect. Biol. 2, a000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nakagawa S., Hirose T. (2012) Paraspeckle nuclear bodies—useful uselessness? Cell. Mol. Life Sci. 69, 3027–3036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lowery L. A., Rubin J., Sive H. (2007) Whitesnake/sfpq is required for cell survival and neuronal development in the zebrafish. Dev. Dyn. 236, 1347–1357 [DOI] [PubMed] [Google Scholar]
- 51. Kulkarni S. J., Steinlauf A. F., Hall J. C. (1988) The dissonance mutant of courtship song in Drosophila melanogaster: isolation, behavior and cytogenetics. Genetics 118, 267–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bogoyevitch M. A., Arthur P. G. (2008) Inhibitors of c-Jun N-terminal kinases: JuNK no more? Biochim. Biophys. Acta 1784, 76–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bonny C., Oberson A., Negri S., Sauser C., Schorderet D. F. (2001) Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes 50, 77–82 [DOI] [PubMed] [Google Scholar]
- 54. Brugidou J., Legrand C., Mery J., Rabie A. (1995) The retro-inverso form of a homeobox-derived short peptide is rapidly internalised by cultured neurones: a new basis for an efficient intracellular delivery system. Biochem. Biophys. Res. Commun. 214, 685–693 [DOI] [PubMed] [Google Scholar]
- 55. Schwarze S. R., Ho A., Vocero-Akbani A., Dowdy S. F. (1999) In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 285, 1569–1572 [DOI] [PubMed] [Google Scholar]
- 56. Borsello T., Clarke P. G., Hirt L., Vercelli A., Repici M., Schorderet D. F., Bogousslavsky J., Bonny C. (2003) A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat. Med. 9, 1180–1186 [DOI] [PubMed] [Google Scholar]
- 57. Sury M. D., Agarinis C., Widmer H. R., Leib S. L., Christen S. (2008) JNK is activated but does not mediate hippocampal neuronal apoptosis in experimental neonatal pneumococcal meningitis. Neurobiol. Dis. 32, 142–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang J., Van De Water T. R., Bonny C., de Ribaupierre F., Puel J. L., Zine A. (2003) A peptide inhibitor of c-Jun N-terminal kinase protects against both aminoglycoside and acoustic trauma-induced auditory hair cell death and hearing loss. J. Neurosci. 23, 8596–8607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mizui T., Kojima N., Yamazaki H., Katayama M., Hanamura K., Shirao T. (2009) Drebrin E is involved in the regulation of axonal growth through actin-myosin interactions. J. Neurochem. 109, 611–622 [DOI] [PubMed] [Google Scholar]
- 60. Jeon Y. J., Choi J. S., Lee J. Y., Yu K. R., Ka S. H., Cho Y., Choi E. J., Baek S. H., Seol J. H., Park D., Bang O. S., Chung C. H. (2008) Filamin B serves as a molecular scaffold for type I interferon-induced c-Jun NH2-terminal kinase signaling pathway. Mol. Biol. Cell 19, 5116–5130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Haeusgen W., Herdegen T., Waetzig V. (2010) Specific regulation of JNK signalling by the novel rat MKK7gamma1 isoform. Cell Signal. 22, 1761–1772 [DOI] [PubMed] [Google Scholar]
- 62. Soderberg O., Gullberg M., Jarvius M., Ridderstrale K., Leuchowius K. J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L. G., Landegren U. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 [DOI] [PubMed] [Google Scholar]
- 63. McBurney M. W., Reuhl K. R., Ally A. I., Nasipuri S., Bell J. C., Craig J. (1988) Differentiation and maturation of embryonal carcinoma-derived neurons in cell culture. J. Neurosci. 8, 1063–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Keene J. D. (2007) RNA regulons: coordination of post-transcriptional events. Nat. Rev. Genet. 8, 533–543 [DOI] [PubMed] [Google Scholar]
- 65. Sonenberg N., Hinnebusch A. G. (2009) Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schwanhausser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 67. Holt C. E., Schuman E. M. (2013) The central dogma decentralized: new perspectives on RNA function and local translation in neurons. Neuron 80, 648–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Prasanth K. V., Prasanth S. G., Xuan Z., Hearn S., Freier S. M., Bennett C. F., Zhang M. Q., Spector D. L. (2005) Regulating gene expression through RNA nuclear retention. Cell 123, 249–263 [DOI] [PubMed] [Google Scholar]
- 69. Kiebler M. A., Bassell G. J. (2006) Neuronal RNA granules: movers and makers. Neuron 51, 685–690 [DOI] [PubMed] [Google Scholar]
- 70. Han T. W., Kato M., Xie S., Wu L. C., Mirzaei H., Pei J., Chen M., Xie Y., Allen J., Xiao G., McKnight S. L. (2012) Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell 149, 768–779 [DOI] [PubMed] [Google Scholar]
- 71. Kato M., Han T. W., Xie S., Shi K., Du X., Wu L. C., Mirzaei H., Goldsmith E. J., Longgood J., Pei J., Grishin N. V., Frantz D. E., Schneider J. W., Chen S., Li L., Sawaya M. R., Eisenberg D., Tycko R., McKnight S. L. (2012) Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149, 753–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wippich F., Bodenmiller B., Trajkovska M. G., Wanka S., Aebersold R., Pelkmans L. (2013) Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell 152, 791–805 [DOI] [PubMed] [Google Scholar]
- 73. Heo Y. S., Kim S. K., Seo C. I., Kim Y. K., Sung B. J., Lee H. S., Lee J. I., Park S. Y., Kim J. H., Hwang K. Y., Hyun Y. L., Jeon Y. H., Ro S., Cho J. M., Lee T. G., Yang C. H. (2004) Structural basis for the selective inhibition of JNK1 by the scaffolding protein JIP1 and SP600125. EMBO J. 23, 2185–2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.