

Abstract
The objective of this study was to investigate whether a single defect in skin barrier function simulated by filaggrin silencing could induce Th2-predominant inflammation. Filaggrin gene expression was silenced in cultured normal human epidermal keratinocytes (NHEKs) using small hairpin RNA (shRNA, GTTGGCTCAAGCATATTATTT). The efficacy of silencing was confirmed by polymerase chain reaction (PCR) and Western blotting. Filaggrin-silenced cells (LV group), shRNA control cells (NC group), and noninfected cells (Blank group) were evaluated. The expression of cornified cell envelope-related proteins, including cytokeratin (CK)-5, -10, -14, loricrin, involucrin, and transglutaminase (TGM)-1, was detected by Western blotting. Interleukins (IL)-2, IL-4, IL-5, IL-12p70, IL-13, and interferon-gamma (IFN-γ) were detected by enzyme-linked immunosorbent assay (ELISA). After filaggrin was successfully silenced by shRNA, the expressions of CK-5, -10, -14, involucrin, and TGM-1 in NHEKs were significantly downregulated compared to the Blank and NC groups (P<0.05 or P<0.01); only loricrin expression was markedly upregulated (P<0.01). Filaggrin silencing also resulted in significant increases of IL-2, IL-4, IL-5, and IL-13 (P<0.05 or P<0.01), and significant decreases of IL-12p70 and IFN-γ (P<0.01) compared with cells in the Blank and NC groups. Filaggrin silencing impaired normal skin barrier function mainly by targeting the cornified cell envelope. The immune response after filaggrin silencing was characterized by Th2 cells, mainly because of the inhibition of IFN-γ expression. Lack of filaggrin may directly impair skin barrier function and then further induce the immune response.
Keywords: Atopic dermatitis, T helper 2 cytokines, Filaggrin silencing, Pathogenesis
Introduction
Atopic dermatitis (AD) is an inflammatory, immunologically mediated skin disease characterized by intensely pruritic and subacute or chronic eczematous plaques (1). AD usually occurs in infants and children, and its prevalence of over 20% in both developed and developing countries is increasing (2). It has been reported that an interaction between keratinocytes and immune cells resulting in the activation of keratinocytes by T cells through soluble factors is essential in the development of AD (3). Immunoglobulin E (IgE) hyperproduction and the expression of T-helper 2 (Th2) cytokines have also been reported to occur in the AD model established in NC/Nga mice (1). Moreover, serum levels of cytokines produced by Th2 cells including CCL17, CCL12 and CCL27 are significantly correlated with the severity of AD in infants (4). Therefore, a Th2 cell bias can be regarded as a characteristic immune response in atopic dermatitis.
Filaggrin is a filament-associated protein derived via dephosphorylation from profilaggrin, which is the major component of keratohyalin granules within epidermal granular cells. The primary function of filaggrin is modulation of epidermal homeostasis via interaction with keratin filaments (5). Filaggrin is important for the retention of water in the stratum corneum (SC), which serves as the external barrier of the human body. Filaggrin mutations can lead to reduced SC natural moisturizing factor (NMF) components, increased pH in the SC, and consequent dysfunction of the skin barrier (6). The reduction or complete loss of filaggrin expression can lead to abnormal skin barrier function, which permits enhanced percutaneous transfer of allergens, and finally induces AD. Research by Morar et al. further confirmed the importance of filaggrin mutations and the role of the skin barrier in AD pathogenesis (7). In Chinese AD patients, more than 18 filaggrin mutations have been reported, of which 3321 delA and K4671X are two of the most common (8). A recent study found that the frequency distribution of f3321 delA was significantly correlated with a number of clinical presentations of AD (9). Chinese AD patients with filaggrin mutations were more susceptible to bacterial skin infection than those without filaggrin mutations (10).
Although most studies have found that immune dysregulation resulting in Th2-predominant inflammation and an intrinsic defect in skin barrier function are closely associated with the pathogenesis of AD, the precise mechanism is still unknown (11). There is intense debate as to whether the observed changes in skin barrier function are the cause or the consequence of the inflammation that accompanies AD (12). We conducted the current study to investigate whether the single defect in skin barrier function simulated by filaggrin gene silencing in normal human epidermal keratinocytes (NHEKs) could induce Th2-predominant inflammation. We first silenced the filaggrin gene in NHEKs, detected the expression of proteins related to skin barrier function, and then compared the generation of typical inflammatory cytokines with that seen in normal cells. The results of our study will provide new insights into the precise pathogenesis of AD.
Material and Methods
Cell culture
NHEKs were obtained from Invitrogen (USA) and cultured in EpiLife medium (Invitrogen) with 5% CO2 at 37°C on 6-cm dishes. The EpiLife medium consisted of 10% fetal calf serum (FCS, Gibco, USA), 1.5 mM L-glutamine, 100 IU/mL penicillin, and 100 µg/mL streptomycin (Gibco). The culture medium was replaced twice weekly.
Silencing by lentivirus infection
We chose a small hairpin RNA (shRNA) with the sequence GTTGGCTCAAGCATATTATTT (position: nt-274), which had the best silencing effect based on the results of a preliminary experiment. The cDNA of the selected shRNA was inserted into the lentivirus gene transfer vector, and the double-stranded shRNA oligo was cloned into pGLV-H1-GFP (GenePharma Co., China) with the help of BamHI and EcoRI. The construct was verified by real-time polymerase chain reaction (PCR) and Western blotting. The infected cells comprised the LV group, the NC group included cells infected with a control shRNA unrelated to the filaggrin sequence, and cells without infection treatment served as a control or Blank group.
The construct system solution was diluted with 4 volumes of 10% fetal calf serum (FBS), supplemented with Polybrene (Shanghai Genechem Co., Ltd., China) and adjusted to a final concentration of 5 µg/mL to make the lentivirus working solution. When the NHEK cultures reached 90% confluence, the cells were digested using 1 mL Trypsin-EDTA solution, and a single cell suspension was prepared. Cells were transferred to 6-well plates (10×105 cells/well) and incubated with 5% CO2 at 37°C for 24 h. Then, 1 mL lentivirus working solution was added to each experimental well and incubated for 24 h before adding EpiLife medium. After incubation in EpiLife medium for a further 24 h, the cells were observed by fluorescence microscopy (Olympus America, USA).
Real-time PCR
To measure the silencing efficiency of shRNA at the mRNA level, quantitative RT-PCR was performed using an Mx3000 real-time PCR Instrument (Stratagen, USA) according to the manufacturer's instructions. The primers used were filaggrin forward, 5′-CACAAGATTCTGCGTATCACTCAGG-3′ and reverse, 5′-GCCTTTCAGTGCCCTCAGATTG-3′; and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) forward, 5′-CATGAGAAGTATGACAACAGCCT-3′ and reverse, 5′-AGTCCTTCCACGATACCAAAGT-3′. GAPDH primers were used as an internal control. Amplification was performed for 40 cycles under the following conditions: denaturation at 95°C for 3 min, annealing at 95°C for 30 s, and extension at 62°C for 40 s. PCR products were separated by electrophoresis on 2% agarose gels and visualized by staining with ethidium bromide.
Western blot analysis
Cells in the LV, NC, and Blank groups were collected, washed twice in ice-cold PBS, and then lyzed and homogenized using lysis buffer for 30 min on ice. The resulting samples were diluted 1:1 with 2× sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer, followed by thermal denaturation at 100°C for 5 min. After cooling, the supernatants were centrifuged at 10,000 g for 10 min at 4°C to remove the insoluble precipitate. The samples were separated on 10% SDS-polyacrylamide gels, followed by 2 h blocking with blocking buffer. After that, the membranes were incubated overnight at 4°C with primary antibodies, and then with secondary antibodies for 2 h at room temperature, followed by enhancement with a chemiluminescence reagent (Pierce, USA) in the dark. The level of protein expression was quantified by the Gel-Pro Analyzer software (Media Cybernetics, USA). GAPDH was used as the internal reference. The following polyclonal primary antibodies were obtained from GeneTex (Irvine, USA): rabbit anti-human cytokeratin 5 (CK-5, Cat #GTX113219), cytokeratin 10 (CK-10, Cat #GTX108883), cytokeratin 14 (CK-14, Cat #GTX104124), loricrin (Cat #GTX116013), and involucrin (Cat #GTX116012); rabbit anti-human transglutaminase-1 (TGM-1) polyclonal antibody (Cat #12912-3-AP) was from Proteintech (USA).
ELISA assays
After infection or treatment, cells were collected from each study group and washed twice with ice-cold PBS. The cells were lyzed and homogenized in lysis buffer for 30 min on ice, and then centrifuged at 10,000 g at 4°C for 10 min to remove the insoluble precipitate. The supernatants were assayed for IL-2, IL-4, IL-5, IL-12p70, IL-13, and IFN-γ using commercial ELISA kits (ExCell Biology Inc., China), following the manufacturer's protocols. The ELISA plates were read with an ELX-800 plate reader (Bio-Tek Instruments Inc., USA) at 450 nm.
Statistical analysis
Data are reported as means±SD, and statistical analysis was performed using the SPSS software (version 13.0; SPSS, USA). Differences among the 3 experimental groups were analyzed using one-way analysis of variance (ANOVA) with the least significant difference (LSD) post hoc test. P<0.05 was considered to be statistically significant.
Results
Filaggrin silencing by lentivirus encoding a filaggrin shRNA
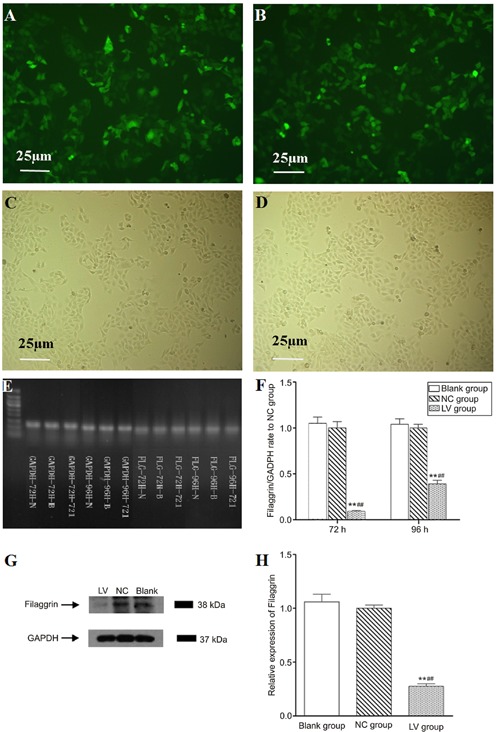
NHEKs exposed to green fluorescent protein (GFP) and the lentivirus encoding filaggrin shRNA or the control shRNA were observed and photographed with an inverted fluorescence microscope (Eclipse TE300; Nikon Corp., Japan) at 72 and 96 h after infection. The GFP sequence was encoded in the lentivirus transduction vector under the control of a separate promoter. GFP was expressed in lentivirus-infected cells as the marker for cell sorting and to indicate that the cells were successfully infected with the shRNA for filaggrin (Figure 1A and B). The PCR results showed that the knockdown efficiencies were 91 and 61%, respectively, at 72- and 96-h after infection (Figure 1E and F). Protein expression was virtually eliminated in NHEKs after filaggrin shRNA transduction, indicating efficient targeting of filaggrin mRNA. The lentivirus carrying unrelated shRNA did not influence the expression of filaggrin (Figure 1G and H). Because filaggrin silencing by lentivirus encoding shRNA at 72 h was found to be successful and efficient, cells undergoing 72 h transfection were used for the subsequent experiments.
Figure 1. Filaggrin silencing in normal human epidermal keratinocytes (NHEKs). Cell sorting was carried out by selecting cells expressing the green fluorescent protein (GFP) marker. NHEKs after 72 and 96 h infection were analyzed by PCR and Western blotting analysis, respectively. A and B, Photographs of NHEKs in the LV (filaggrin-silenced cells) and NC (shRNA control cells) groups taken after cell sorting under a fluorescent microscope (100×). C and D, Photographs of NHEKs in the LV and NC groups taken after cell sorting under a light microscope (100×). E and F, PCR results of filaggrin targeted mRNA expression rate to NC group and GADPH primer as the internal reference. Infection for 72 h could effectively reduce the expression of filaggrin targeted mRNA (>75%, P<0.01) whereas for 96 h the silencing efficacy was not efficient, by 61% (<75%). G and H, Western blotting results of NHEKs after 72-h infection and GADPH as the internal reference. After infection with lentivirus encoding shRNA of filaggrin, the expression of filaggrin protein was significantly inhibited by the shRNA. **P<0.01 vs Blank group (noninfected cells); ##P<0.01 vs NC group (least significant difference test).
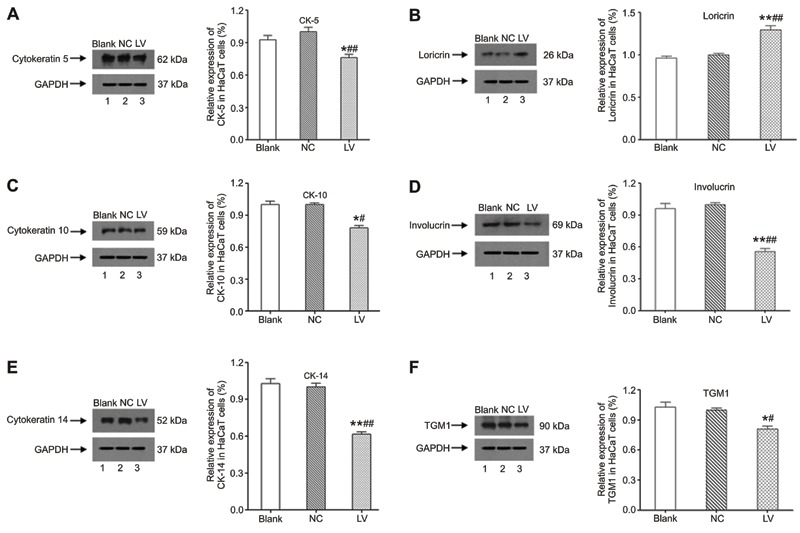
Influence of filaggrin silencing on epidermal protein expression
After shRNA silencing, the expression of CK-5, -10, -14, involucrin, and TGM-1 in NHEKs was significantly downregulated (Figure 2A,C-F, P<0.05 or P<0.01) compared with cells in the Blank and NC groups. As shown in Figure 2B, only loricrin expression was markedly upregulated in the LV group compared to the Blank and NC groups (P<0.01).
Figure 2. Western blotting detection for cytokeratin 5, 10, 14, and loricrin, involucrin, and transglutaminase-1 (TGM1) expression in normal human epidermal keratinocytes (NHEKs) after shRNA silencing. LV group: filaggrin-silenced cells; NC group: shRNA control cells; Blank group: noninfected cells. *P<0.05, **P<0.01 vs Blank group; #P<0.05, ##P<0.01 vs NC group (least significant difference test).
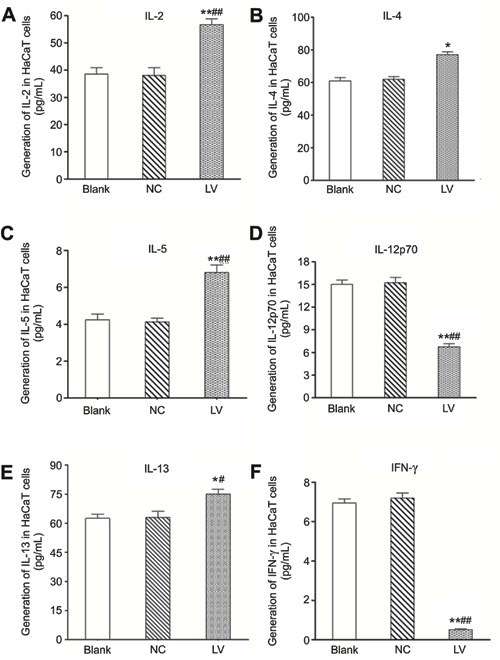
Influence of filaggrin silencing on cytokines determined by ELISA
Filaggrin silencing resulted in significant increases in the generation of IL-2, IL-4, IL-5, and IL-13 (P<0.05 or P<0.01, Figure 3A, B, C and E) and significant decreases in IL-12p70 and IFN-γ (P<0.01, Figure 3D and F) compared to the Blank and NC groups.
Figure 3. Influence of filaggrin silencing on cytokine generation in normal human epidermal keratinocytes (NHEKs). LV group: filaggrin-silenced cells; NC group: shRNA control cells; Blank group: noninfected cells. *P<0.05, **P<0.01 vs Blank group; #P<0.05, ##P<0.01 vs NC group (least significant difference test).
Discussion
AD is a chronic, intensely painful disease with frequent relapses, which affects a large number of patients, especially children. It is generally accepted that AD might be caused by immune dysregulation characterized by Th2-predominant inflammation and/or an intrinsic defect in skin barrier function (11). However, the precise pathogenesis of AD is still controversial, especially the relationship between skin barrier dysfunction and inflammatory changes. In this study, we carried out shRNA silencing of filaggrin to simulate in vivo loss-of-function mutations of filaggrin, and found that it was followed by abnormal expression of proteins related to skin barrier function. We, thus, confirmed that filaggrin silencing caused the expected skin barrier dysfunction, and also observed significant alterations in the generation of inflammatory cytokines in response to silencing.
Filaggrin is a protein in the SC that plays a vital role in the regulation of epidermal homeostasis, and loss-of-function mutations in the filaggrin gene can disturb normal skin barrier function (13). In the SC, filaggrin monomers become incorporated into the lipid envelope, which is responsible for the skin barrier function. Loricrin and involucrin are proteins important for skin barrier formation and integrity. Involucrin contributes to the formation of a cell envelope that protects corneocytes in the skin by binding loricrin (14,15). TGM-1 is an enzyme needed for the formation of the cornified cell envelope because it facilitates the crosslinking of proteins (16). Moreover, CK-5, -10, and -14 are all well known as essential proteins of the cellular skeleton and are of great importance in skin barrier function by participating in the proliferation and differentiation of epithelial cells. In our study, shRNA silencing succeeded in significantly inhibiting the expression of filaggrin protein and mRNA. Filaggrin silencing also resulted in decreased expression of CK-5, -10, -14, involucrin, and TGM-1, but overexpression of loricrin compared to cells in the NC and Blank groups. Obviously, normal skin barrier function was disturbed by the direct silencing of filaggrin, as evidenced by the significant alterations to the cornified cell envelope-related proteins. Of interest is the observation that the interruption of filaggrin expression not only attenuated involucrin expression but also resulted in the upregulation of loricrin. Mantel et al. (17) reported that silencing of aldo-keto reductase family 1, member C3 (AKR1C3) resulted in decreased expression of K10 but upregulation of loricrin, which is consistent with our results. Maybe there is an interaction between K10 and loricrin. However, it is clear that the successful silencing of filaggrin impaired normal skin barrier function mainly by targeting the formation of the cornified cell envelope.
The defective skin barrier in NHEKs would allow increased allergen and pathogen penetration through the skin, which would then induce an inflammatory response in the lesion. More than 80% of AD patients have increased levels of total IgE, which is closely associated with Th2 cells in the inflamed area (18). It is widely accepted that AD is a kind of cutaneous inflammation reaction characterized by a Th2 cell-predominant phenotype initially with the additional acquisition of a Th1 cell phenotype during the chronic eczematous phase (19). IL-4, IL-5, and IL-13 are the cytokines typically secreted by Th2 cells, whereas IL-2 and IFN-γ are secreted by Th1 cells. In addition, IL-12p70 is the determinant factor for the differentiation of TH0 cells into Th1 or Th2 cells. IL-17 and IL-22 are cytokines secreted by activated T cells. Recent studies indicate that IL-17- and IL-22-producing T cells are involved in the progression of AD. Moreover, IL-31 is also derived from Th2 cells (20,21). The occurrence of IL-31-producing T cells was observed to be increased in AD lesions compared to that in autologous blood and donor skin (22).
In the present study, levels of IL-4, IL-5, and IL-13 produced by Th2 cells and IL-2 produced by Th1 cells were significantly elevated by filaggrin silencing and levels of IFN-γ from Th1 cells and IL-12p70 were significantly downregulated. Therefore, the activated immune response associated with defective skin barrier function in filaggrin-silenced NHEKs was characterized by a Th2 cell bias. This bias may have been caused by decreased expression of IFN-γ, which can inhibit the differentiation of Th1 cells but facilitate the differentiation of Th2 cells (23). Further, the elevated IL-2 production in NHEKs after filaggrin silencing may reflect the partial activation of Th1 cells. It is known that the pathogenesis and development of AD involve a complex interplay of genetic, pharmacological, environmental, and psychological factors (12,24). However in vitro experiments lack environmental and psychological factors and systematic feedback. Therefore, the immune response activated by filaggrin silencing can also have a Th2-cell bias mainly because of the low expression of IFN-γ after silencing, and the immune response can occur mainly in response to the defect in skin barrier function.
As stated above, the Th2-cell bias is an immune response involving a great number of Th2 cell-producing cytokines and plays a critical role in the development of AD. Also, recent studies have shown that the frequency and spectrum of filaggrin mutations detected in AD patients varies in different populations (25-27). So far, the precise relationship between Th2-cell dysfunction and filaggrin mutation has not been clearly defined, but the importance of filaggrin mutation in the pathogenesis of AD has been supported by a great number of studies. In order to provide novel insight into this relationship, future in vivo and in vitro studies will need to focus on the discrepancy in immune response between AD patients with and without filaggrin mutations.
In this study, the silencing of filaggrin impaired normal skin barrier function mainly by targeting formation of the cornified cell envelope. The immune response activated by filaggrin silencing was also characterized by a Th2-cell bias, mainly because of decreased expression of IFN-γ after silencing. Filaggrin silencing may directly impair skin barrier function, with the consequent induction of a Th2-biased immune response.
Acknowledgments
Research supported by the National Natural Science Foundation of China (NSFC; #81101183, #81170771, #81272588) and China Postdoctoral Science Foundation (CPSF; #2014M550370).
Footnotes
First published online.
References
- 1.Takakura M, Takeshita F, Aihara M, Xin KQ, Ichino M, Okuda K, et al. Hyperproduction of IFN-gamma by CpG oligodeoxynucleotide-induced exacerbation of atopic dermatitis-like skin lesion in some NC/Nga mice. J Invest Dermatol. 2005;125:1156–1162. doi: 10.1111/j.0022-202X.2005.23928.x. [DOI] [PubMed] [Google Scholar]
- 2.Flohr C, Mann J. New insights into the epidemiology of childhood atopic dermatitis. Allergy. 2014;69:3–16. doi: 10.1111/all.12270. [DOI] [PubMed] [Google Scholar]
- 3.van den Bogaard EH, Tjabringa GS, Joosten I, Vonk-Bergers M, van Rijssen E, Tijssen HJ, et al. Crosstalk between keratinocytes and T cells in a 3D microenvironment: a model to study inflammatory skin diseases. J Invest Dermatol. 2014;134:719–727. doi: 10.1038/jid.2013.417. [DOI] [PubMed] [Google Scholar]
- 4.Nakazato J, Kishida M, Kuroiwa R, Fujiwara J, Shimoda M, Shinomiya N. Serum levels of Th2 chemokines, CCL17, CCL22, and CCL27, were the important markers of severity in infantile atopic dermatitis. Pediatr Allergy Immunol. 2008;19:605–613. doi: 10.1111/j.1399-3038.2007.00692.x. [DOI] [PubMed] [Google Scholar]
- 5.Presland RB, Bassuk JA, Kimball JR, Dale BA. Characterization of two distinct calcium-binding sites in the amino-terminus of human profilaggrin. J Invest Dermatol. 1995;104:218–223. doi: 10.1111/1523-1747.ep12612770. [DOI] [PubMed] [Google Scholar]
- 6.Kezic S, O'Regan GM, Lutter R, Jakasa I, Koster ES, Saunders S, et al. Filaggrin loss-of-function mutations are associated with enhanced expression of IL-1 cytokines in the stratum corneum of patients with atopic dermatitis and in a murine model of filaggrin deficiency. J Allergy Clin Immunol. 2012;129:1031–1039. doi: 10.1016/j.jaci.2011.12.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morar N, Cookson WO, Harper JI, Moffatt MF. Filaggrin mutations in children with severe atopic dermatitis. J Invest Dermatol. 2007;127:1667–1672. doi: 10.1038/sj.jid.5700739. [DOI] [PubMed] [Google Scholar]
- 8.Zhang H, Guo Y, Wang W, Shi M, Chen X, Yao Z. Mutations in the filaggrin gene in Han Chinese patients with atopic dermatitis. Allergy. 2011;66:420–427. doi: 10.1111/j.1398-9995.2010.02493.x. [DOI] [PubMed] [Google Scholar]
- 9.Meng L, Wang L, Tang H, Tang X, Jiang X, Zhao J, et al. Filaggrin gene mutation c.3321delA is associated with various clinical features of atopic dermatitis in the Chinese Han population. PLoS One. 2014;9: doi: 10.1371/journal.pone.0098235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai SC, Chen H, Koh WP, Common JE, van Bever HP, McLean WH, et al. Filaggrin mutations are associated with recurrent skin infection in Singaporean Chinese patients with atopic dermatitis. Br J Dermatol. 2012;166:200–203. doi: 10.1111/j.1365-2133.2011.10541.x. [DOI] [PubMed] [Google Scholar]
- 11.Abramo F, Campora L, Albanese F, della Valle MF, Cristino L, Petrosino S, et al. Increased levels of palmitoylethanolamide and other bioactive lipid mediators and enhanced local mast cell proliferation in canine atopic dermatitis. BMC Vet Res. 2014;10:21. doi: 10.1186/1746-6148-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olivry T. Is the skin barrier abnormal in dogs with atopic dermatitis? Vet Immunol Immunopathol. 2011;144:11–16. doi: 10.1016/j.vetimm.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 13.Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38:441–446. doi: 10.1038/ng1767. [DOI] [PubMed] [Google Scholar]
- 14.Kim BE, Leung DY, Boguniewicz M, Howell MD. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin Immunol. 2008;126:332–337. doi: 10.1016/j.clim.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eckert RL, Green H. Structure and evolution of the human involucrin gene. Cell. 1986;46:583–589. doi: 10.1016/0092-8674(86)90884-6. [DOI] [PubMed] [Google Scholar]
- 16.Radner FP, Fischer J. The important role of epidermal triacylglycerol metabolism for maintenance of the skin permeability barrier function. Biochim Biophys Acta. 2014;1841:409–415. doi: 10.1016/j.bbalip.2013.07.013. [DOI] [PubMed] [Google Scholar]
- 17.Mantel A, Carpenter-Mendini AB, Vanbuskirk JB, De Benedetto A, Beck LA, Pentland AP. Aldo-keto reductase 1C3 is expressed in differentiated human epidermis, affects keratinocyte differentiation, and is upregulated in atopic dermatitis. J Invest Dermatol. 2012;132:1103–1110. doi: 10.1038/jid.2011.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fukiwake N, Furusyo N, Kubo N, Takeoka H, Toyoda K, Morita K, et al. Incidence of atopic dermatitis in nursery school children - a follow-up study from 2001 to 2004, Kyushu University Ishigaki Atopic Dermatitis Study (KIDS) Eur J Dermatol. 2006;16:416–419. [PubMed] [Google Scholar]
- 19.Novak N, Bieber T. The role of dendritic cell subtypes in the pathophysiology of atopic dermatitis. J Am Acad Dermatol. 2005;53:S171–S176. doi: 10.1016/j.jaad.2005.04.060. [DOI] [PubMed] [Google Scholar]
- 20.Souwer Y, Szegedi K, Kapsenberg ML, de Jong EC. IL-17 and IL-22 in atopic allergic disease. Curr Opin Immunol. 2010;22:821–826. doi: 10.1016/j.coi.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 21.Nograles KE, Zaba LC, Shemer A, Fuentes-Duculan J, Cardinale I, Kikuchi T, et al. IL-22-producing “T22” T cells account for upregulated IL-22 in atopic dermatitis despite reduced IL-17-producing TH17 T cells. J Allergy Clin Immunol. 2009;123:1244–1252. doi: 10.1016/j.jaci.2009.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szegedi K, Kremer AE, Kezic S, Teunissen MB, Bos JD, Luiten RM, et al. Increased frequencies of IL-31-producing T cells are found in chronic atopic dermatitis skin. Exp Dermatol. 2012;21:431–436. doi: 10.1111/j.1600-0625.2012.01487.x. [DOI] [PubMed] [Google Scholar]
- 23.Goriely S, Goldman M. Interleukin-12 family members and the balance between rejection and tolerance. Curr Opin Organ Transplant. 2008;13:4–9. doi: 10.1097/MOT.0b013e3282f406c4. [DOI] [PubMed] [Google Scholar]
- 24.Grewe M, Bruijnzeel-Koomen CA, Schopf E, Thepen T, Langeveld-Wildschut AG, Ruzicka T, et al. A role for Th1 and Th2 cells in the immunopathogenesis of atopic dermatitis. Immunol Today. 1998;19:359–361. doi: 10.1016/S0167-5699(98)01285-7. [DOI] [PubMed] [Google Scholar]
- 25.Chen H, Common JE, Haines RL, Balakrishnan A, Brown SJ, Goh CS, et al. Wide spectrum of filaggrin-null mutations in atopic dermatitis highlights differences between Singaporean Chinese and European populations. Br J Dermatol. 2011;165:106–114. doi: 10.1111/j.1365-2133.2011.10331.x. [DOI] [PubMed] [Google Scholar]
- 26.Thawer-Esmail F, Jakasa I, Todd G, Wen Y, Brown SJ, Kroboth K, et al. South African amaXhosa patients with atopic dermatitis have decreased levels of filaggrin breakdown products but no loss-of-function mutations in filaggrin. J Allergy Clin Immunol. 2014;133:280–282. doi: 10.1016/j.jaci.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winge MC, Bilcha KD, Lieden A, Shibeshi D, Sandilands A, Wahlgren CF, et al. Novel filaggrin mutation but no other loss-of-function variants found in Ethiopian patients with atopic dermatitis. Br J Dermatol. 2011;165:1074–1080. doi: 10.1111/j.1365-2133.2011.10475.x. [DOI] [PubMed] [Google Scholar]