

Abstract
Background
Breast cancer brain metastases (BCBM) are challenging complications that respond poorly to systemic therapy. The role of the blood–tumor barrier in limiting BCBM drug delivery and efficacy has been debated. Herein, we determined tissue and serum levels of capecitabine, its prodrug metabolites, and lapatinib in women with BCBM resected via medically indicated craniotomy.
Methods
Study patients with BCBM requiring surgical resection received either single-dose capecitabine (1250 mg/m2) 2–3 h before surgery or 2–5 doses of lapatinib (1250 mg) daily, the last dose 2–3 h before surgery. Serum samples were collected serially on the day of surgery. Drug concentrations were determined in serum and BCBM using liquid chromatography tandem mass spectrometry.
Results
Twelve patients were enrolled: 8 for capecitabine and 4 for lapatinib. Measurable drug levels of capecitabine and metabolites, 5′-deoxy-5-fluorocytidine, 5′-deoxy-5-fluorouridine, and 5-fluorouracil, were detected in all BCBM. The ratio of BCBM to serum was higher for 5-fluorouracil than for capecitabine. As for lapatinib, the median BCBM concentrations ranged from 1.0 to 6.5 µM. A high variability (0.19–9.8) was noted for lapatinib BCBM-to-serum ratio.
Conclusions
This is the first study to demonstrate that capecitabine and lapatinib penetrate to a significant though variable degree in human BCBM. Drug delivery to BCBM is variable and in many cases appears partially limiting. Elucidating mechanisms that limit drug concentration and innovative approaches to overcome limited drug uptake will be important to improve clinical efficacy of these agents in the central nervous system.
Trial registration ID: NCT00795678.
Keywords: blood–tumor barrier, brain metastases, breast cancer, capecitabine, lapatinib
Breast cancer brain metastases (BCBM) are a devastating complication and portend a poor prognosis.1 The role of systemic therapy is limited and secondary for BCBM. The normal blood–brain barrier excludes entry of drugs, and the degree to which it remains patent with a brain metastasis (BM), as the blood–tumor barrier (BTB), is debated.2 Contrast agents show uptake into BCBM, but chemotherapy has been generally ineffective; presumably the BTB prevents attainment of active drug levels.
Very limited data exist on drug uptake in human BCBM.1 Evidence for CNS drug penetration is usually based on preclinical xenograft models or evaluation of cerebrospinal fluid in human clinical samples.3–6 In experimental BCBM models, most lesions exhibit drug uptake greater than that of the normal brain, but brain lesion uptake may be heterogeneous and insufficient to mount an apoptotic response.6
Capecitabine and lapatinib have demonstrated efficacy in clinical trials among patients with BCBM.7,8 Capecitabine is an oral prodrug of 5-flourouracil (5-FU), an antimetabolite, and undergoes 3-step enzymatic conversion to 5-FU (Supplementary Fig. S1). Lapatinib is an oral small-molecule tyrosine kinase inhibitor (TKI) that reversibly targets epidermal growth factor receptor and human epidermal growth factor receptor 2 (HER2). The uptake of lapatinib in BCBM has been demonstrated in a preclinical model.5
Clinical evidence for BM uptake of drugs is scarce, and it is unclear whether preclinical models reflect human BM pharmacokinetics (PK). Prospective evaluation of BM drug uptake in humans is challenging but valuable for future drug development. In this study, we examined the uptake of capecitabine, its metabolites, and lapatinib in BCBM resected from patients undergoing medically indicated craniotomy.
Materials and Methods
Patients
The study was conducted at the Memorial Sloan-Kettering Cancer Center and the Cleveland Clinic Foundation with approval from their respective local institutional review boards and the United States Army Medical Research and Materiel Command's Office of Research Protections, Human Research Protection Office. Written informed consent was obtained from all the patients prior to enrollment in accordance with the local institutional review board policies. Patients with BCBM with a clinical indication for surgical resection providing informed consent enrolled in this clinical trial. Eligibility criteria included: age ≥18 years, histologically or cytologically documented breast cancer with known or suspected parenchymal BM for which surgical resection or biopsy was clinically indicated, KPS ≥50, preserved bone marrow and liver function with no significant abnormalities in baseline blood tests including complete blood count and chemistry panel, off cytotoxic chemotherapy ≥3 weeks (6 wk for nitrosoureas and mitomycin-C), no bevacizumab in the past 60 days, no cranial radiation ≥4 weeks, no pregnancy with use of active contraception if of childbearing potential, and no other serious medical or cardiac condition, with no limit to the previous number or types of chemotherapy or types of radiotherapy.
Clinical Data Collection
Data collected included primary tumor characteristics, clinical information regarding BM lesions, prior therapy, and concurrent medication use. As an exploratory variable, lesions were described by the study investigators as “noncystic” if they were solid enhancing lesions on imaging and otherwise as “cystic.”
Drug Administration and Sample Collection
Patients with HER2− metastatic breast cancer (MBC) received a single preoperative oral dose of capecitabine (1250 mg/m2) 2–3 h before surgery. Those with HER2+ MBC received oral lapatinib (1250 mg) daily for 2–5 days as clinical circumstances permitted, with the last dose 2–3 h before surgery. One HER2+ patient received capecitabine rather than lapatinib. On the day of surgery, serum was collected serially at the following time points—serum 1: immediately before drug administration; serum 2: within 1 h after drug administration; serum 3: at the start of surgery; serum 4: at tumor identification; serum 5: immediately after resection; and serum 6: 1 h after the conclusion of surgery. The time points were selected to provide sufficiently detectable serum levels based on the known half-lives of both agents. For those with multiple BCBM lesions, tissue was sampled from only a single lesion due to clinical feasibility. Samples were immediately snap frozen, shipped on dry ice to the analysis site, and stored at −80°C until analysis.
Drug Analysis
Serum and BCBM samples were analyzed in duplicate for lapatinib or capecitabine and its prodrug metabolites: 5′-deoxy-5-fluorocytidine (5′-DFCR), 5′-deoxy-5-fluorouridine (5′-DFUR), and 5-FU, using liquid chromatography tandem mass spectrometry (LC-MS/MS) with stable isotope internal standards. Serum was thawed on ice. From each sample, duplicate 20-μL aliquots were collected, which were vortex mixed with buffer and internal standards. BCBM were thawed slightly on ice prior to collection of ∼10–40 mg specimens (average ∼20 mg) from various locations within the tumor. BCBM specimens were homogenized with buffer by microsonication together with internal standards. Calibration curves were created using rat brain for brain standards and either the first serum draw sample for capecitabine or human serum for lapatinib.
For capecitabine, samples were processed and analyzed using methods adapted from Salvador et al.9 Briefly, after vortex mixing and centrifugation, the supernatant was processed through preconditioned ATOLL XWP-60/3 SPE cartridges (Interchim). The eluent from the cartridges was evaporated under N2 and reconstituted with water for injection onto the LC-MS/MS. For lapatinib, samples were protein precipitated with 3 parts acetonitrile and centrifuged.10 An aliquot of the supernatant was diluted with 4 parts aqueous mobile phase and directly injected onto the LC-MS/MS. Processing/sample transfer was minimized to avoid lapatinib surface binding.
The chromatographic system consisted of a Shimadzu Nexera ultra high performance liquid chromatograph and an ABSciex QTRAP 5500 MS/MS with electrospray interface operating in negative mode for capecitabine and in positive mode for lapatinib. For capecitabine, compounds were separated on an Atlantis T3 column (100 × 2.1 mm, 3 µm; Waters) held at 40°C. Mobile phase was 10 mM ammonium acetate (pH 4) and acetonitrile using gradient elution from 100% to 15% aqueous over 15 min, followed by 4 min reequilibration. For lapatinib, compounds were separated on an ACE3 C18-AR column (100 × 2.1 mm) (MAC-MOD) using 20 mM ammonium formate (pH 3.75) and acetonitrile. Gradient elution from 35% to 5% aqueous over 7 min was followed by 3 min reequilibration. Internal standards (d11-capecitabine for capecitabine, 13C15N2–5-FU for 5-FU, 13C15N2 -DFCR for 5′-DFCR and 5′-DFUR, and 13C15N2-lapatinib for lapatinib) were used to correct for possible compound-specific extraction and ionization-suppression effects. Conditions and mass transitions for analytes were optimized using the autotune feature of the software. Instrument control and data acquisition were performed using Analyst 1.5.2 software, and quantitation was accomplished with MultiQuant 2.1 software.
Lapatinib, due to its marked lipophilicity (cLogP >5), was expected to bind highly (>95%) to tissue proteins and lipids.11 Such binding would invalidate comparison of BCBM total lapatinib concentration with free extracellular-fluid half-maximal inhibitory concentration (IC50). Therefore, the unbound fraction (fu) of lapatinib was determined from the in vitro equilibrium distribution of lapatinib between BCBM tissue slices (20 µm) and buffered saline at 37°C.12 The unbound fraction was then used along with the measured BCBM total lapatinib concentration to calculate the unbound (free) concentration of lapatinib in the tumor, [lapatinib]u = fu×[lapatinb]total, for comparison with the extracellular lapatinib IC50. It has been shown that 5-FU exhibits minimal tissue binding (fu ∼1.00).13 Therefore, the BCBM total 5-FU concentration was directly compared with extracellular 5-FU values of IC50 without correction for free fraction. Tissue blood volume was determined colorimetrically in BCBM samples from the absorbance of hemoglobin.14 The enzymatic conversion rate of capecitabine to 5′-DFCR in BCBM homogenates was determined using the method of Miwa et al.15
Statistical Analysis
Given the absence of previous data on drug uptake in human BCBM and likely heterogeneity among lesion characteristics and individuals, no formal sample size calculation was conducted. Descriptive statistics were used to summarize and compare drug concentrations in serum, plasma, and tumor tissue. Drug levels within individual BCBM were presented as averages and ranges of determinations. Drug levels among patients' BCBM were presented as medians with quartiles and ranges. Integrated area-under-the-curve (AUC) serum drug concentration was calculated, ending at the time of BCBM identification. Statistics were calculated using STATA version 12 and GraphPad Prism version 6.
Results
Patient Characteristics
A total of 12 patients consented and were enrolled in the study from October 2008 to February 2013. Patient and tumor characteristics are summarized in Table 1. Of the 12 patients, 8 received capecitabine and 4 received lapatinib. No patient received both drugs. All had surgical pathologic confirmation of the diagnosis of MBC from the craniotomy. One patient had received stereotactic radiosurgery to the sampled BM ∼1.5 years prior to the craniotomy.
Table 1.
Patient characteristics
| Patient | Study Drug | No. of Preop Doses | Age, y | Concurrent AED (levetiracetam) | Receptor Status ER/PR/HER2 | No. of Lesionsa | BCBM Location | Largest BCBM Size, mm | Cystic | Concurrent Steroid (dexamethasone) |
|---|---|---|---|---|---|---|---|---|---|---|
| C1 | Capecitabine | 1 | 51 | None | unknown | >20 | Cerebellum | 39 × 27 | Yes | Yes |
| C2b | Capecitabine | 1 | 71 | None | +/−/− | 1 | Cerebellum | 20 × 15 | No | Yes |
| C3 | Capecitabine | 1 | 53 | Yes | +/+/unknown | 2 | Frontal | 28 × 24 | No | No |
| C4 | Capecitabine | 1 | 52 | None | −/−/− | 1 | Temporal | 16 × 17 | No | No |
| C5 | Capecitabine | 1 | 36 | None | −/−/− | 1 | Parietal | 40 × 35 | Yes | Yes |
| C6 | Capecitabine | 1 | 46 | None | +/+/+ | 1 | Cerebellum | 26 × 24 | No | Yes |
| C7 | Capecitabine | 1 | 41 | None | −/−/− | 1 | Parietal, temporal | 25 × 22 | Unknown | Yes |
| C8 | Capecitabine | 1 | 48 | None | +/+/− | 2 | Parietal, temporal | 42 × 41 | Yes | No |
| L1 | Lapatinib | 5 | 58 | None | +/−/+ | >20 | Cerebellum | 25 × 14 | No | Yes |
| L2 | Lapatinib | 3 | 60 | None | −/−/+ | 1 | Frontal | 37 × 34 | Yes | No |
| L3 | Lapatinib | 3 | 63 | Yes | −/−/+ | 1 | Occipital | 32 × 31 | No | Yes |
| L4 | Lapatinib | 2 | 59 | None | +/+/+ | 2 | Frontal, occipital | 37 × 25 | Yes | No |
Abbreviations: AED, antiepileptic drug; ER, estrogen receptor; PR, progesterone receptor.
aOnly one lesion was sampled and analyzed for the study.
bPatient had stereotactic radiosurgery 1.5 y prior to study ; no other patients had earlier treatment.
Capecitabine Distribution
The time courses of serum capecitabine and metabolite concentrations are presented for one representative patient (#C7) in Fig. 1. Capecitabine, 5′-DFCR, and 5′-DFUR concentrations rose quickly in serum to peak values within 0.6–2.7 h of dosing. Serum 5-FU concentration remained low throughout the experiment. Table 2 summarizes serum maximum concentration (Cmax) and AUC values. Median Cmax values were 9.16 µM for capecitabine, 19.0 µM for 5′-DFCR, 26.1 µM for 5′-DFUR, and 1.53 µM for 5-FU.
Fig. 1.
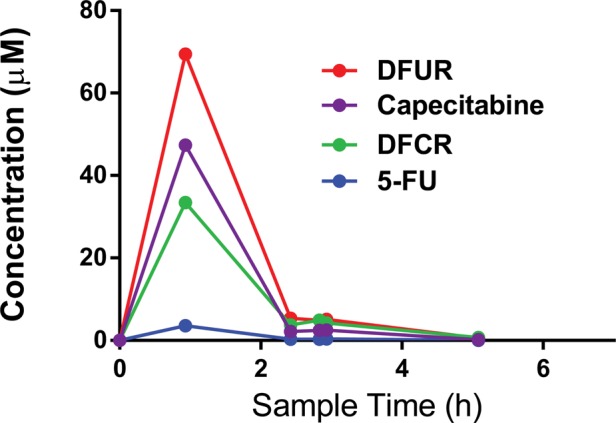
Time course of serum capecitabine and metabolite concentrations. Example time course of capecitabine and metabolite serum concentrations (patient C7). Six serum draws are plotted against time (h). BCBM was identified at the 4th serum draw
Table 2.
Capecitabine and metabolite medians (range) of average concentrations
| Compound | BCBM, µM | Serum 4, µM | Serum Cmax, µM | Serum AUC | BCBM/Serum 4 |
|---|---|---|---|---|---|
| Capecitabine | 0.81 (0.12–1.97) | 3.17 (0.63–5.57) | 9.16 (0.63–47.38) | 12.40 (1.05–75.34) | 0.28 (0.031–0.81) |
| 5′-DFCR | 1.52 (0.19–3.86) | 4.78 (0.57–9.67) | 18.98 (0.57–33.68) | 24.73 (0.74–77.0) | 0.36 (0.11–0.67) |
| 5′-DFUR | 0.27 (0.03–3.19) | 5.15 (0.49–15.96) | 26.09 (0.49–69.94) | 42.03 (0.67–170.9) | 0.06 (0.010–0.58) |
| 5-FU | 1.81 (0.10–4.57) | 0.38 (0.02–0.79) | 1.53 (0.53–8.06) | 2.25 (0.67–17.8) | 5.64 (1.67–12.9) |
For each patient, serum 4 is taken at the time the breast cancer BM is identified. Serum AUCs are calculated until the time of BCBM identification.
BCBM were procured at 2.5–5 h after preoperative capecitabine administration. Measurable drug levels were obtained in every BCBM sample, with median BCBM concentrations equaling 0.81 μM for capecitabine, 1.5 μM for 5′-DFCR, 0.27 μM for 5′-DFUR, and 1.8 μM for 5-FU. However, values varied markedly among patients. Figure 2 presents BCBM concentrations of capecitabine and its 3 prodrug metabolites for each of the 8 patients. Among the patients, BCBM values varied 15- to 150-fold. One patient (#C3) exhibited a very low serum AUC, suggesting poor gastrointestinal absorption. Even after removal of patient C3 with low systemic exposure, BCBM concentrations still varied by ∼5-fold for 5-FU and >10-fold for 5′-DFCR and capecitabine. Marked variability was also noted for capecitabine and metabolite concentrations within individual BCBM specimens. Supplementary Fig. S2 presents selected examples of intralesional variability in BCBM concentrations from 2 patients (#C6 and #C7). For patient C6, 5-FU concentrations varied ∼10-fold among individual regions of the BCBM (Supplementary Fig. S2A). In contrast, in patient C7, 5-FU distribution was more uniform (±20%). Thus, significant heterogeneity was also observed among patients.
Fig. 2.
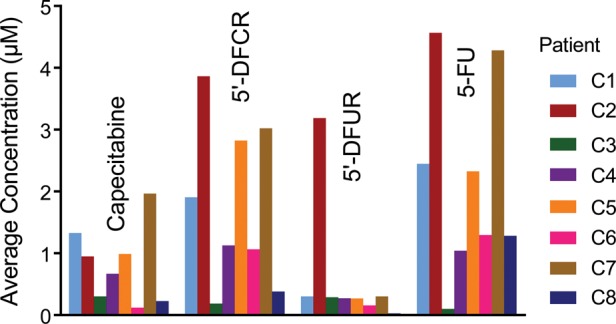
Interpatient variability in capecitabine and metabolite concentrations. Mean concentrations of capecitabine, 5′-DFCR, 5′-DFUR, and 5-FU in all patients. Each patient is color coded.
To assess the extent to which residual blood influenced measured BCBM concentration, BM concentration was expressed as a tumor-to-serum ratio, which was then compared with the measured BM blood volume. BCBM blood content averaged 0.01–0.02 mL/g for most specimens in the study, with the highest samples approaching 0.05 mL/g. Median BCBM-to-serum ratio at the time of tumor sampling (serum 4) equaled 0.28, 0.36, 0.06, and 5.64 for capecitabine, 5′-DFCR, 5′-DFUR, and 5-FU, respectively (Table 2). Therefore, vascular blood contributed on average <10% to measured BCBM concentration for capecitabine, 5′-DFCR, and 5-FU, but ∼20% for 5′-DFUR.
The higher level of 5-FU in BCBM compared with other metabolites (Fig. 2) and the large BCBM-to-serum ratio (Supplementary Fig. S3B) suggested that BCBM 5-FU was formed locally from precursors, as opposed to 5-FU uptake from the circulation. Consistent with this, the BCBM-to-serum ratio for 5-FU was 10- to 100-fold greater than that for the other metabolites (Table 2). The ∼5-fold variation in BCBM 5-FU concentration was not reduced by normalization to circulating 5-FU prodrug species concentrations, as variation in the ratio of BCBM 5-FU to serum (5′-DFCR + 5′-DFUR) AUC (Supplementary Fig. S3C) actually exceeded that of BCBM 5-FU concentration alone (Supplementary Fig. S3A). Brain metastasis exhibited the capability of converting capecitabine to 5′-DFCR, with a measured rate of 1.16 ± 0.17 nmol/h/mg tumor protein in vitro at 37°C in BCBM homogenate. This rate, converted to undiluted BCBM tissue, gave a rate coefficient of 0.016 ± 0.002 h−1. Thus, 5-FU formation in BM was from capecitabine, as well as from 5′-DFCR and 5′-DFUR. Further, no associations were noted between BCBM 5-FU concentration and tumor properties, such as BCBM size, corticosteroid use, and/or cystic structure.
Lapatinib
Four patients received lapatinib daily for 2–5 days prior to craniotomy. Table 3 summarizes lapatinib concentrations from serum and BCBM samples. All patients had comparable average serum lapatinib concentrations at the time of tumor resection, ranging 2.7-fold from 2.4 to 6.5 µM. Within each patient, serum lapatinib levels generally varied by <2-fold over the ∼6-h time course of the procedure (Fig. 3A).
Table 3.
Lapatinib concentrations at time of BCBM resection and BCBM-to-serum ratioa
| Patient | No. Preop Doses | BCBM Avg Concentration (range), µMa | Serum 4 Concentration,b µM | BCBM/Serum 4 |
|---|---|---|---|---|
| L1 | 5 | 63.6 (43.9–77.2) | 6.5 | 9.8 |
| L2 | 3 | 14.6 (11.7–22.2) | 2.4 | 6.0 |
| L3 | 3 | 18.6 (11.3–31.0) | 3.5 | 5.3 |
| L4 | 2 | 1.0 (0.7–1.5) | 5.3 | 0.19 |
aBCBM average concentrations were calculated from multiple samples of a single collected lesion.
bSerum 4 is taken at the time the BCBM is identified.
Fig. 3.
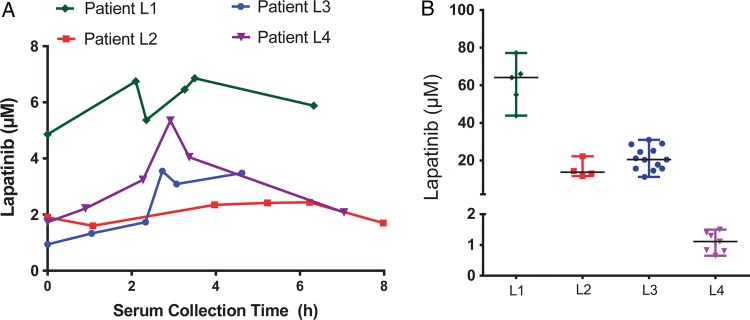
Serum, intratumor, and interpatient BCBM lapatinib concentration variability. (A) Time course of serum lapatinib concentration for all 4 patients. (B) Lapatinib concentrations in all patient BCBM, showing range, median, and quartile of concentration.
BCBM concentrations differed greatly, ranging from 1.0 µM to 63 µM (Fig. 3B). Despite a very small sample size, there was a strong correlation between the number of preoperative doses of lapatinib and BM drug concentration (r = 0.99, P < .01). The ratio of BCBM to serum also exhibited marked variation, ranging ∼50-fold, demonstrating that heterogeneity among tumors could not be explained simply by serum exposure level. Intratumor sampling was conducted in all 4 cases. In one case 13 specimens were analyzed from a single BCBM sample exhibiting a 3-fold range of values. Vascular correction did not significantly impact drug tumor-to-serum ratio, as demonstrated by the large BCBM-to-serum ratios. Correction of measured total BCBM concentration to unbound or “free” lapatinib concentrations lowered values by 3 orders of magnitude, to 64, 15, 19, and 1 nM.
Discussion
This is the first study to demonstrate uptake of capecitabine, its metabolites, and lapatinib in human BCBM. Although capecitabine and lapatinib have been investigated for BCBM in clinical trials, no tissue-based evidence of drug penetration in BCBM has been reported for humans. Our primary objective was to provide evidence for drug uptake in BCBM and offer a feasible template to inform drug development for BCBM in the future.
The serum capecitabine PK after a single preoperative dose of capecitabine were generally comparable to previous studies in breast cancer patients with Cmax values of 7.5–11 µM and 1.7–2.4 µM for capecitabine and 5-FU, respectively.16 In BCBM tissue, capecitabine and its metabolites were all detected at measurable levels. Considerable variation was noted both among patients and within individual lesions. This large variation in drug distribution in human BCBM is consistent with prior preclinical models using autoradiography and in vivo tissue measurement.6
Given a paucity of data on capecitabine PK in human tissue and the fact that a single preoperative dose was given (due to feasibility), we are unable to benchmark whether the observed concentrations were at clinically relevant levels. However, our data have similarities to the published study of capecitabine PK from colorectal cancer patients who were treated with the drug for 5–7 days preoperatively. For example, in most cases, the BCBM 5-FU concentration was higher than that of the other capecitabine prodrug metabolites, and the 5-FU BCBM-to-serum ratio was much greater than similar such ratios for capecitabine, 5′-DFCR, and 5′-DFUR. In a colorectal tumor study, similar preferential conversion to 5-FU was noted in tumor tissue and thought to be due to differential expression of the conversion enzyme thymidine phosphorylase.17 Thus, the significance of enzymatic conversion in BCBM may play a role in capecitabine PK and needs further evaluation.
Capecitabine has been associated with CNS clinical response in case series/reports.18 In a prospective phase II clinical trial, the addition of capecitabine in BCBM patients who had progressed on lapatinib therapy yielded an objective CNS response of 20% (95% CI: 3.0–33.7).19 Further investigation into mechanisms that affect capecitabine uptake and conversion in BCBM may aid in improving capecitabine efficacy for BCBM. Individual roles likely exist for plasma exposure, BTB permeability, active efflux transport, enzymatic conversion, and basic tumor cell sensitivity to 5-FU concentration.
As for lapatinib, after 2–5 consecutive daily 1250-mg oral doses, serum Cmax varied by less than ∼3-fold among patients and was comparable to previously published serum levels from phase I studies.20,21 Although the tissue levels were likely not at steady state based on lapatinib PK, as evidenced by the trend for increased BCBM levels with increased doses, BCBM concentrations were comparable to the previously reported preclinical IC50 of 0.1–4 µM in breast cancer cell lines.22 However, the calculated “free” lapatinib concentrations to account for lipophilicity of the drug were lower. This is a hypothesis-generating finding that motivates further clinical study of dose and length of exposure for optimal BM drug uptake and efficacy. In fact, nonstandard dosing and scheduling of TKI is currently being explored to improve CNS penetration and antitumor activity for CNS disease.
A preclinical model demonstrated that a higher drug concentration in brain may be achieved with a higher dose of lapatinib administered systemically. In the BCBM murine study by Polli and colleagues,23 parenteral administrations of lapatinib at 1 mg/kg and 10 mg/kg were compared. There was an ∼8-fold increase in the BM lapatinib concentrations when a higher dose of lapatinib was administered. The ability to safely administer high-dose lapatinib has been demonstrated by use of a pulsatile dosing schedule in a phase I study.24 In a retrospective study, pulsatile high-dose erlotinib was associated with CNS responses in lung cancer patients who developed CNS metastases while on standard-dose TKI.25,26 Pulsatile high-dose erlotinib is currently being studied prospectively for primary brain tumor patients (NCT01257594).
Despite its variability, in general, the ratio of BM to serum concentrations noted in our study was markedly higher than expected based on previously published murine studies with BM-to-plasma ratios ranging from 0.09 to 0.26.10,23 Polli et al23 reported that lapatinib is a substrate of the efflux transporters (eg, P-glycoprotein [Pgp], breast cancer resistance protein [BCRP]) located in the blood–brain barrier and that mice deficient in Pgp-BCRP had a markedly higher brain-to-plasma ratio for lapatinib.23 The nonvascular corrected lapatinib concentrations in our study were also considerably higher than those observed in the study of recurrent glioblastoma patients who were given 750 mg of oral lapatinib daily for 7 days preoperatively.27 The median concentration reported among the study patients was 497 nM (range: 70–3826 nM). However, given the differences in tumor type and lapatinib doses administered, it is difficult to compare the result to ours.
Multiple factors, including plasma exposure, BTB permeability, and active efflux transport, likely contribute to BCBM lapatinib AUC and in vivo antitumor efficacy. In fact, addressing the role of drug transporters may be one way to enhance drug delivery. For example, in preclinical studies, coadministration of efflux transporter inhibitors (eg, elacridar) has been shown to improve the brain-to-plasma ratio of small-molecule drugs.28,29 Although lapatinib uptake was much higher than expected, it is still unclear whether the concentrations noted in our study are sufficient to produce significant clinical responses in patients. Given the modest response rate seen for lapatinib as a single agent in BCBM patients, further evaluation to improve its antitumor activity for BCBM is warranted now that we have demonstrated its ability to cross the BTB.
Given that BCBM patients do not routinely undergo surgical resection, unlike those with primary brain tumors, and systemic therapy is commonly withheld prior to a major surgical intervention, evaluating drug concentration in BCBM tissue can be a challenge. We were unable to evaluate pharmacodynamic endpoints due to lack of pretreatment (control) tissue. Furthermore, presurgical administration of drugs long enough to achieve therapeutically relevant or steady-state levels was impractical. Biopsy of BCBM prior to surgical resection to obtain control tissue and delaying of the medically necessary surgical procedure to ensure adequate steady-state levels of chemotherapeutic drugs preoperatively were limited by real-life constraints and safety concerns. Nevertheless, we were able to achieve our primary goal of providing evidence for drug penetration through the BTB in human BM tissue. This is one of the largest studies evaluating drug concentration in BCBM reported to date.30–32 We found differences in drug uptake between our human BCBM and the murine data, especially for lapatinib. Evaluation of clinical evidence for drug uptake is important for future drug development for BM, and our study demonstrates the feasibility of such endeavors.
We were able to demonstrate BCBM uptake of capecitabine and lapatinib in mostly nonirradiated tissue, thus providing further support for use of systemic therapy in BCBM. This may be especially relevant where these systemic agents are evaluated in management of BCBM prior to radiation therapy to possibly avoid or delay whole-brain radiotherapy, as suggested by a recent phase II trial of capecitabine and lapatinib combination therapy in patients with BCBM.8 Thus, lapatinib and capecitabine are promising systemic therapeutic agents for BCBM, but their administration likely requires further optimization for CNS penetration. High-dose pulsatile lapatinib is safe to administer24 and may offer an approach that could maximize CNS efficacy.23 Motivated by this work and our experience described herein, we plan to evaluate high-dose pulsatile lapatinib with or without capecitabine in breast cancer patients with HER2+ CNS metastases. In addition, further evaluation of mechanisms related to drug uptake and conversion/metabolism may shed light on reasons for the observed variability and inform strategies to improve BM uptake. These studies are ongoing in preclinical models and await translation into the clinical setting.
Supplementary Material
Funding
This work was supported by the Department of Defense Center of Excellence Breast Cancer Research Program Award (W81XWH-06-0033) and the Judah Gubbay Memorial Fund.
Supplementary Material
Acknowledgments
The authors thank all participating patients and clinical and research teams at the Memorial Sloan-Kettering Cancer Center and the Cleveland Clinic Foundation.
Conflict of interest statement. A.D.S. declares an association with Genentech/Roche.
References
- 1.Lin NU, Bellon JR, Winer EP. CNS metastases in breast cancer. J Clin Oncol. 2004;22(17):3608–3617. doi: 10.1200/JCO.2004.01.175. [DOI] [PubMed] [Google Scholar]
- 2.Deeken JF, Loscher W. The blood–brain barrier and cancer: transporters, treatment, and Trojan horses. Clin Cancer Res. 2007;13(6):1663–1674. doi: 10.1158/1078-0432.CCR-06-2854. [DOI] [PubMed] [Google Scholar]
- 3.Pestalozzi BC, Brignoli S. Trastuzumab in CSF. J Clin Oncol. 2000;18(11):2349–2351. doi: 10.1200/JCO.2000.18.11.2349. [DOI] [PubMed] [Google Scholar]
- 4.Stemmler HJ, Schmitt M, Willems A, et al. Ratio of trastuzumab levels in serum and cerebrospinal fluid is altered in HER2-positive breast cancer patients with brain metastases and impairment of blood–brain barrier. Anticancer Drugs. 2007;18(1):23–28. doi: 10.1097/01.cad.0000236313.50833.ee. [DOI] [PubMed] [Google Scholar]
- 5.Taskar KS, Rudraraju V, Mittapalli RK, et al. Lapatinib distribution in HER2 overexpressing experimental brain metastases of breast cancer. Pharm Res. 2012;29(3):770–781. doi: 10.1007/s11095-011-0601-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lockman PR, Mittapalli RK, Taskar KS, et al. Heterogeneous blood–tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin Cancer Res. 2010;16(23):5664–5678. doi: 10.1158/1078-0432.CCR-10-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin NU, Carey LA, Liu MC, et al. Phase II trial of lapatinib for brain metastases in patients with human epidermal growth factor receptor 2–positive breast cancer. J Clin Oncol. 2008;26(12):1993–1999. doi: 10.1200/JCO.2007.12.3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bachelot T, Romieu G, Campone M, et al. Lapatinib plus capecitabine in patients with previously untreated brain metastases from HER2-positive metastatic breast cancer (LANDSCAPE): a single-group phase 2 study. Lancet Oncol. 2013;14(1):64–71. doi: 10.1016/S1470-2045(12)70432-1. [DOI] [PubMed] [Google Scholar]
- 9.Salvador A, Milllerioux L, Renou A, et al. Simultaneous LC-MS-MS analysis of capecitabine and its metabolites (5′-deoxy-5-fluorocytidine, 5′-deoxy-5-fluorouridine, 5-fluorouracil) after off-line SPE from human plasma. Chromatographia. 2006;63(11–12):609–615. [Google Scholar]
- 10.Polli JW, Olson KL, Chism JP, et al. An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino }methyl)-2-furyl]-4-quinazolinamine; GW572016) Drug Metab Dispos. 2009;37(2):439–442. doi: 10.1124/dmd.108.024646. [DOI] [PubMed] [Google Scholar]
- 11.Wan H, Rehngren M, Giordanetto F, et al. High-throughput screening of drug-brain tissue binding and in silico prediction for assessment of central nervous system drug delivery. J Med Chem. 2007;50(19):4606–4615. doi: 10.1021/jm070375w. [DOI] [PubMed] [Google Scholar]
- 12.Uchida Y, Ohtsuki S, Kamiie J, et al. Blood–brain barrier (BBB) pharmacoproteomics: reconstruction of in vivo brain distribution of 11 P-glycoprotein substrates based on the BBB transporter protein concentration, in vitro intrinsic transport activity, and unbound fraction in plasma and brain in mice. J Pharmacol Exp Ther. 2011;339(2):579–588. doi: 10.1124/jpet.111.184200. [DOI] [PubMed] [Google Scholar]
- 13.Tsukamoto Y, Kato Y, Ura M, et al. A physiologically based pharmacokinetic analysis of capecitabine, a triple prodrug of 5-FU, in humans: the mechanism for tumor-selective accumulation of 5-FU. Pharm Res. 2001;18(8):1190–1202. doi: 10.1023/a:1010939329562. [DOI] [PubMed] [Google Scholar]
- 14.Cohn W. The determination of hemoglobin in tissue extracts or other turbid solutions. J Biol Chem. 1943;1(148):219–223. [Google Scholar]
- 15.Miwa M, Ura M, Nishida M, et al. Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur J Cancer. 1998;34(8):1274–1281. doi: 10.1016/s0959-8049(98)00058-6. [DOI] [PubMed] [Google Scholar]
- 16.Reigner B, Blesch K, Weidekamm E. Clinical pharmacokinetics of capecitabine. Clin Pharmacokinet. 2001;40(2):85–104. doi: 10.2165/00003088-200140020-00002. [DOI] [PubMed] [Google Scholar]
- 17.Schuller J, Cassidy J, Dumont E, et al. Preferential activation of capecitabine in tumor following oral administration to colorectal cancer patients. Cancer Chemother Pharmacol. 2000;45(4):291–297. doi: 10.1007/s002800050043. [DOI] [PubMed] [Google Scholar]
- 18.Ekenel M, Hormigo AM, Peak S, et al. Capecitabine therapy of central nervous system metastases from breast cancer. J Neurooncol. 2007;85(2):223–227. doi: 10.1007/s11060-007-9409-0. [DOI] [PubMed] [Google Scholar]
- 19.Lin NU, Dieras V, Paul D, et al. Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clin Cancer Res. 2009;15(4):1452–1459. doi: 10.1158/1078-0432.CCR-08-1080. [DOI] [PubMed] [Google Scholar]
- 20.Burris HA, Hurwitz HI, Dees EC, et al. Phase I safety, pharmacokinetics, and clinical activity study of lapatinib (GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas. J Clin Oncol. 2005;23(23):5305–5313. doi: 10.1200/JCO.2005.16.584. [DOI] [PubMed] [Google Scholar]
- 21.Burris HA, Taylor CW, Jones SF, et al. A phase I and pharmacokinetic study of oral lapatinib administered once or twice daily in patients with solid malignancies. Clin Cancer Res. 2009;15(21):6702–6708. doi: 10.1158/1078-0432.CCR-09-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rusnak DW, Lackey K, Affleck K, et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther. 2001;1(2):85–94. [PubMed] [Google Scholar]
- 23.Polli JW, Humphreys JE, Harmon KA, et al. The role of efflux and uptake transporters in [N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino}methyl)-2-furyl]-4-quinazolinamine (GW572016, lapatinib) disposition and drug interactions. Drug Metab Dispos. 2008;36(4):695–701. doi: 10.1124/dmd.107.018374. [DOI] [PubMed] [Google Scholar]
- 24.Chien AJ, Auerback G, Rugo HS, et al. A phase I dose-escalation study of 5-day intermittent oral lapatinib therapy with biomarker anlaysis in patients with HER-2-overexpressing breast cancer. J Clin Onc. 2014;32(14):1472–1479. doi: 10.1200/JCO.2013.52.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grommes C, Oxnard GR, Kris MG, et al. ‘Pulsatile’ high-dose weekly erlotinib for CNS metastases from EGFR mutant non-small cell lung cancer. Neuro Oncol. 2011;13(12):1364–1369. doi: 10.1093/neuonc/nor121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.EGFR inhibition using high dose administration of erlotinib weekly for recurrent malignant gliomas with EGFR variant III mutation NCT01257594. Bethesda, MD, USA: National Library of Medicine; Available at http://clinicaltrials.gov . Accessed February 1, 2014. [Google Scholar]
- 27.Vivanco I, Robins HI, Rohle D, et al. Differential sensitivity of glioma- versus lung cancer–specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov. 2012;2(5):458–471. doi: 10.1158/2159-8290.CD-11-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Durmus S, Sparidans RW, Wagenaar E, et al. Oral availability and brain penetration of the B-RAFV600E inhibitor vemurafenib can be enhanced by the P-GLYCOprotein (ABCB1) and breast cancer resistance protein (ABCG2) inhibitor elacridar. Mol Pharm. 2012;9(11):3236–3245. doi: 10.1021/mp3003144. [DOI] [PubMed] [Google Scholar]
- 29.Tang SC, Lagas JS, Lankheet NA, et al. Brain accumulation of sunitinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by oral elacridar and sunitinib coadministration. Int J Cancer. 2012;130(1):223–233. doi: 10.1002/ijc.26000. [DOI] [PubMed] [Google Scholar]
- 30.Stewart DJ, Mikhael NZ, Nair RC, et al. Platinum concentrations in human autopsy tumor samples. Am J Clin Oncol. 1988;11(2):152–158. doi: 10.1097/00000421-198804000-00013. [DOI] [PubMed] [Google Scholar]
- 31.Lien EA, Wester K, Lonning PE, et al. Distribution of tamoxifen and metabolites into brain tissue and brain metastases in breast cancer patients. Br J Cancer. 1991;63(4):641–645. doi: 10.1038/bjc.1991.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boogerd W, Tjahja IS, van de Sandt MM, et al. Penetration of idarubicin into malignant brain tumor tissue. J Neurooncol. 1999;44(1):65–69. doi: 10.1023/a:1006335517191. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.