

Abstract
Background
Chemoradiation, followed by adjuvant temozolomide, is the standard treatment for newly diagnosed glioblastoma. Adding other active agents may enhance treatment efficacy.
Methods
The primary objective of this factorial phase II study was to determine if one of 3 potential chemotherapy agents added to dose-dense temozolomide (ddTMZ) improves progression-free survival (PFS) for patients with newly diagnosed glioblastoma. A prior phase I trial established the safety of combining ddTMZ with isotretinoin, celecoxib, and/or thalidomide. Adults with good performance status and no evidence of progression post chemoradiation were randomized into 8 arms: ddTMZ alone (7 days on/7 days off) or doublet, triplet, and quadruplet combinations with isotretinoin, celecoxib, and thalidomide.
Results
The study enrolled 155 participants with a median age of 53 years (range, 18-84 y). None of the agents demonstrated improved PFS when compared with arms not containing that specific agent. There was no difference in PFS for triplet compared with doublet regimens, although a trend for improved overall survival (OS) was seen (20.1 vs 17.0 months, P = .15). Compared with ddTMZ, the ddTMZ + isotretinoin doublet had worse PFS (10.5 vs 6.5 months, P = .043) and OS (21.2 vs 11.7 months, P = .037). Trends were also seen for worse outcomes with isotretinoin-containing regimens, but there was no impact with celecoxib or thalidomide combinations. Treatment was well tolerated with expected high rates of lymphopenia.
Conclusions
The results do not establish a benefit for these combinations but indicate that adding isotretinoin to ddTMZ may be detrimental. This study demonstrated the feasibility and utility of the factorial design in efficiently testing drug combinations in newly diagnosed glioblastoma.
Clinicaltrials.gov identifier
Keywords: factorial design, glioblastoma, isotretinoin, temozolomide, celecoxib, thalidomide
Despite optimal therapy with radiation and concurrent and adjuvant temozolomide (TMZ),1 survival is poor for most patients with glioblastoma (GBM), and effective second-line therapies are scarce.2,3 Even in the context of clinical trials, 3-year survival is only 16%, and 5-year survival is 10%.4 There is an urgent need for more effective first-line therapies. The implementation of novel clinical trial designs is an important step for increasing efficiency when testing new treatments.
Molecular studies have uncovered many potential targets for treatment of malignant gliomas.5 However, despite promising preclinical testing, the results of trials evaluating single-agent targeted therapies have been generally disappointing and have shown very low response rates and no improvement in progression-free survival (PFS) or overall survival (OS).6,7 Combinatorial strategies targeting more than one critical pathway have been developed with the goal of overcoming resistance to single agents.8–10 However, the increasing number of promising treatments exponentially increases the potential combinations to be tested. Novel clinical trial designs are needed to test combinations of targeted agents more efficiently.
One of the novel designs that can address this challenge is the factorial design,11 which can be planned as a randomized, multiple arm study involving different combinations of the agents to be tested. For instance, a 2 × 2 × 2 factorial design can accommodate 3 investigational agents to be tested with a standard cytotoxic agent. The cytotoxic agent can be tested alone, combined with each of the 3 agents (doublets), added to each of 3 pairs of agents (triplets), and, finally, given together with all 3 (quadruplet combination), for a total of 8 treatment arms.
There has been interest in examining the utility of combining TMZ with various cytostatic agents that have non-overlapping toxicity profiles and overall better tolerance than cytotoxic drugs. Several combinations of TMZ and cytostatics have been evaluated in recurrent malignant gliomas with promising preliminary results. Reported 6-month PFS rates were 24% for TMZ plus thalidomide, a putative antiangiogenic agent, and 35% for TMZ plus isotretinoin, a differentiation- and growth-inhibitory agent.12–16 COX-2 enzyme is upregulated in high-grade gliomas, and high expression in tumor cells is a strong predictor of poor survival.17 Preclinical studies have shown that celecoxib exerts both cytostatic and potentially cytotoxic effects in vitro,18 and a study using a C6 rat glioma orthotopic model suggested synergy between TMZ and celecoxib.19 At the time our trial was designed, dose-dense temozolomide (ddTMZ) at 150 mg/m2/day, week-on/week-off, was felt to be a promising schema due to rapid, marked, and sustained inactivation of MGMT and preliminary evidence of activity in recurrent malignant gliomas.20,21
The results of the phase I component of the study evaluating the different combinations of ddTMZ with isotretinoin, celecoxib, and thalidomide have been previously published.22 Overall, all combinations were well tolerated. Herein we present the results of the first phase II factorial randomized trial in participants with newly diagnosed GBM. This study was designed to evaluate efficacy measured by PFS and gauge the feasibility of the factorial design.
Patients and Methods
Patients
Eligibility criteria included confirmed histological diagnosis of supratentorial GBM in adults (aged ≥18 years), a KPS ≥ 60%, any extent of resection, and successful completion of standard chemoradiation without tumor progression. Pseudoprogression (PsP) was allowed and was defined as image-worsening (increased enhancement and/or edema within the radiation field) in the first scan after chemoradiation without clinical progression. Adequate hematological, renal, and hepatic function was required (absolute neutrophil count ≥1500/mm3, platelet count ≥100,000/mm3, serum glutamic pyruvic transaminase and alkaline phosphatase <2 times the upper limit of institutional normal [ULN], bilirubin <1.5 times ULN), blood urea nitrogen and creatinine <1.5 times ULN). Patients with a history of any other cancer (except nonmelanoma skin cancer or carcinoma in situ of the cervix) were not eligible unless in complete remission and off all therapy for ≥3 years. Patients with serious intercurrent medical illness, peptic ulcer disease, or active/recent GI bleeding (eg, ≤3 months), and history of allergic reactions to sulfa drugs and nonsteroidal anti-inflammatory drugs were excluded. All patients enrolled to thalidomide arms were registered in the S.T.E.P.S® program. All patients enrolled to isotretinoin arms were enrolled in the iPLEDGE™ program, starting in March 2006 as mandated by the FDA. Patients of childbearing potential were required to use adequate contraception. All patients provided informed consent, indicating that they were aware of the investigational nature of this study. The protocol was approved by the MD Anderson Cancer Center Institutional Review Board.
Participants were accrued at MD Anderson Cancer Center and 11 other institutions from the Brain Tumor Trials Collaborative group and MD Anderson Community Clinical Oncology Program (Supplementary Data S1).
Study Design
This study had 8 treatment arms (Fig. 1). Arm 1 consisted of single-agent, ddTMZ, arms 2, 3, and 4 (“doublet therapy”) combined ddTMZ with 1 agent (isotretinoin, celecoxib, or thalidomide), arms 5, 6, and 7 (“triplet therapy”) combined ddTMZ with 2 agents, and arm 8 combined ddTMZ with all 3 agents (“quadruplet therapy”). Patients with suspected tumor progression after chemoradiation (imaging worsening accompanied by clinical progression, or new lesions outside of the radiation field), prior to initiating study treatment, were removed from the study before randomization and replaced. Patients with suspected PsP (defined as above) were allowed to participate.
Fig. 1.

Study design.
All participants who received any study-related treatment were considered evaluable for efficacy and toxicity in an intent-to-treat analysis. Participants lost to follow-up were censored at their last clinic visit. PFS was calculated from date of randomization (ie, post chemoradiation) to the date of progression or death, whichever occurred first, or from the date of randomization to the date of last clinic visit for participants who were alive and without progression. OS was a secondary efficacy endpoint and was measured from date of randomization.
Treatment Plan
After maximal safe surgical resection, all participants were required to have undergone external beam radiation therapy over a period of 6 weeks to a total dose of 60 Gy (2-Gy fractions) and concurrent daily TMZ (75 mg/m2/day). No other chemotherapy was allowed during radiation. Four weeks after chemoradiation, a gadolinium-diethylenetriamine penta-acetic acid (GD-DPTA) MRI of the brain was performed. If there was no evidence of progression, participants were randomized to one of the 8 arms of adjuvant chemotherapy (ddTMZ alone [150 mg/m2/day, week-on/week-off] or with isotretinoin, celecoxib, and/or thalidomide ) following the combinations outlined in Fig. 1. Participants were registered and randomized within 5 weeks of the last dose of radiotherapy.
Participants were treated for a maximum of 12 cycles as long as progressive disease did not occur and side effects were tolerable; each cycle was defined as 28 days. The thalidomide dose was 400 mg orally daily (starting at 200 mg daily and escalating weekly by 100 mg); the isotretinoin dose was 40 mg/m2 orally twice daily, days 1-21; and the celecoxib dose was 400 mg orally twice daily.
Participants on thalidomide received 1 mg of warfarin daily. Those receiving celecoxib were not allowed to take other nonsteroidal anti-inflammatory drugs. Dose modifications were performed when grade 3–4 toxicities developed. Based on the toxicity, the dose of the most likely offending agent was lowered. Participants with evidence of progression were taken off the study and treated at the discretion of the attending physician.
Evaluations During Study
Complete blood count with differential was performed prior to randomization and every 2 weeks during treatment. Electrolyte levels, hepatic and renal function, and anticonvulsant levels (where appropriate) were performed within 14 days prior to randomization and before each cycle. For participants receiving celecoxib, stools were evaluated for occult blood and creatinine clearance before treatment and every 2 cycles thereafter. For participants receiving isotretinoin, serum cholesterol and triglycerides were assessed before treatment and every 2 cycles. Among participants receiving thalidomide, prothrombin time and international normalized ratio were determined prior to the initiation of treatment and before each cycle. Toxicity was assessed at every clinic visit and initially graded according to the National Cancer Institute's Common Toxicity Criteria (CTC) version 3.0; version 4.0 was used after September 2010. Evaluation of treatment response was performed every 2 cycles by serial brain-imaging studies, most commonly with GD-DPTA MRI. Radiological response and progression were evaluated using Macdonald criteria.23
Statistical Methods
The accrual goal was 20 participants per arm (160 in total). To ensure adequate accrual of evaluable patients, the total accrual was increased by 10%. Participants were randomly assigned to arms 1 to 8 at the time of completion of chemoradiation, with equal probability. All participants received ddTMZ; half of the arms received thalidomide (n = 80; arms 2, 5, 6, and 8), half received isotretinoin (n = 80; arms 3, 5, 7, and 8), and half received celecoxib (n = 80; arms 4, 6, 7 and 8).
The primary objective was to compare the efficacy of adding one of 3 agents (isotretinoin, celecoxib, thalidomide) to adjuvant ddTMZ, measured by PFS from the time of randomization (post chemoradiation). Secondary objectives included comparisons between doublet versus triplet therapy and between individual arms, as well as OS analysis and feasibility of conducting a randomized phase II trial with factorial design in newly diagnosed GBM participants in a cooperative group trial.
Median PFS and OS were estimated using the Kaplan-Meier method from time of randomization to time of progression, death, or last follow-up. Toxicity and survival analyses included all participants enrolled in the study. There was no stratification due to the small number of participants in each arm. A Cox proportional hazard model was performed to account for imbalances regarding age, performance status (KPS), and extent of resection among treatment arms. KPS and age were included as continuous variables in the Cox model. Extent of resection was modeled as gross total resection versus other. Violations of the proportional hazards assumption were assessed using rescaled Schoenfeld residuals. Analyses were performed using S+ 8.2 for Windows (TIBCO Software).
Power calculations were based on accrual rate of 10 per month, a 3-month postaccrual follow-up, and 20 participants per arm. This would provide ∼80% power for the agent versus no agent comparisons to detect a change in median PFS from 3 months to 5 months (ie, a hazard ratio of 0.6) for a 2-sided 5% alpha rate. The primary comparisons specified in the protocol were the 3 main effects (isotretinoin vs not isotretinoin, celecoxib vs not celecoxib, and thalidomide vs not thalidomide). No adjustment was made to the type I error rate for testing 3 hypotheses. We performed computer simulations based on the exponential distribution to assess the power of various comparisons in a 2 × 2 × 2 factorial design with a time-to-event endpoint. Our power to detect synergy between 2 agents varied considerably, depending not only on the magnitude of the synergy but also on the remaining separate and joint effects of the 3 treatments.
Results
Participants
This phase I/II study was open from September 2005 to September 2010. Overall phase II accrual was 178 patients, of which 155 were randomized and treated (see Supplementary Data, Fig. S5, CONSORT Diagram). Twenty-three patients were randomized but not treated either because of consent withdrawal or difficulties obtaining insurance coverage. A total of 146 patients were accrued at MD Anderson Cancer Center in Houston and 32 at the remaining participating sites. The patients accrued at other sites were evenly distributed among the 8 treatment arms, with 2–4 participants per arm except for ddTMZ + isotretinoin (5 participants) and ddTMZ + thalidomide (7 participants).
Patient characteristics are summarized in Table 1 and Table S3. Median age was 53 years (range, 18–84 y); median KPS was 90% (range, 60%-100%). Arms were well balanced for age except for an excess of participants older than aged 60 years in the arms combining ddTMZ + thalidomide and ddTMZ + thalidomide/isotretinoin. Gross total resection was achieved in 99 participants, 53 underwent subtotal resection, and 21 biopsy only; the extent of resection was unknown in 3. Median time from diagnosis to study registration (ie, post chemoradiation) was similar in all arms (3.0 to 3.4 months). The highest drop-out rate was in arm 8 (quadruplet combination), where 7 patients withdrew consent or insurance denied coverage for one or more of the agents.
Table 1.
Patient characteristics
| Total | |
|---|---|
| Total enrolled | 178 |
| Total treated | 155 |
| Age | |
| 10 to 19 years | 1 |
| 20 to 39 years | 27 |
| 40 to 59 years | 103 |
| ≥ 60 years | 47 |
| Sex | |
| Female | 55 |
| Male | 123 |
| Race/ethnicity | |
| White | 160 |
| Non-white | 16 |
| Unknown | 2 |
| KPS | |
| 60%–70% | 16 |
| 80%–100% | 144 |
| Unknown | 18 |
| Extent of Resection | |
| Gross total | 99 |
| Subtotal | 53 |
| Biopsy | 21 |
| Unknown | 3 |
Efficacy
For the phase II component of the study, the overall median PFS for all arms combined was 11.6 months, and the overall 6-month PFS rate was 73%.
Primary Endpoint (Agent vs No Agent Analysis)
There were ∼80 participants who received each of the 3 cytostatics and 80 participants who did not receive that specific agent. None of the agents demonstrated particular benefit regarding PFS when compared with others (Fig. 2). The arms containing isotretinoin demonstrated worse PFS than the arms not containing this agent, although the difference did not reach statistical significance (median PFS, 6.6 months vs 9.1 months; HR, 1.3; 95% CI, 0.9–1.8; P = .18). The arms containing celecoxib showed a median PFS of 8.3 months compared with 7.4 months for arms not containing celecoxib (HR, 0.8; 95% CI, 0.6–1.2; P = .32). The arms containing thalidomide showed a median PFS of 7.6 months compared with 8.7 months for arms without this agent (HR, 1.2; 95% CI, 0.8–1.7; P = .33). Three-way interaction between isotretinoin, celecoxib, and thalidomide was not significant (P = .15 for PFS). The three 2-way interactions were not significant (P = .97 for isotretinoin vs celecoxib; (P = .37 for isotretinoin vs thalidomide; P = .54 for celecoxib vs thalidomide). Thus, the 3 main effects (comparisons of isotretinoin vs not isotretinoin, celecoxib vs not celecoxib, and thalidomide vs not thalidomide) are valid.
Fig. 2.
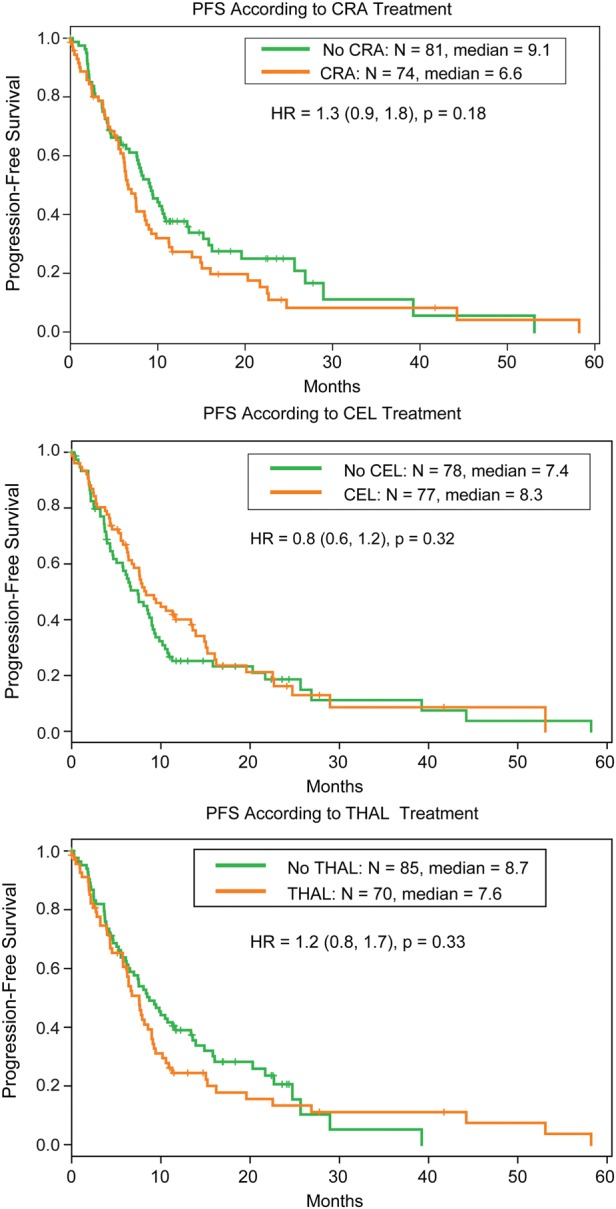
Progression-free survival (PFS) by agent versus no agent analysis.
Secondary Endpoints
Doublet Versus Triplet Therapy Analysis
There were ∼60 participants who received doublet therapy (ddTMZ + 1 cytostatic) and 60 participants who received triplet therapy (ddTMZ + 2 cytostatics). There were no statistically significant differences in PFS between these 2 groups, with a median PFS of 8.3 months for doublet and 8.2 months for triplet therapy (HR, 0.9; 95% CI, 0.6–1.3; P = .45) (Fig. 3).
Fig. 3.
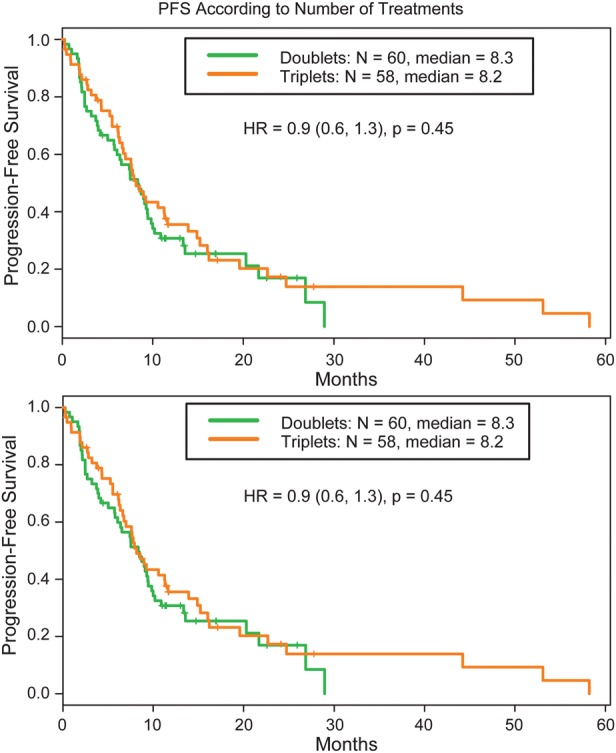
Progression-free Survival (PFS) according to the number of agents (doublet versus triplet therapy).
Individual Treatment Arms
Data on PFS according to treatment regimen is summarized in Fig. 4 and Table 2. Combination of ddTMZ + isotretinoin was inferior to ddTMZ alone (6.5 months vs 10.5 months; HR, 2.0; 95% CI, 1.0–4.0; P = .043). Differences in PFS in the remaining arms did not reach statistical significance and ranged from 6.2 months (ddTMZ + isotretinoin/thalidomide) to 13.4 months (ddTMZ + celecoxib) (Table 2). Cox analysis with age and KPS treated as continuous covariates confirmed similar results (data not shown). A 30-unit increase in KPS was a highly significant factor for PFS (HR, 0.2; 95% CI, 0.1–0.5; P < .0001), whereas age (50-year increase) and extent of resection (gross total resection vs other) were not significant (HR, 2.0; 95% CI, 0.8–4.6; P = .11 and HR, 0.8; 95% CI, 0.5–1.2; P = .27, respectively).
Fig. 4.
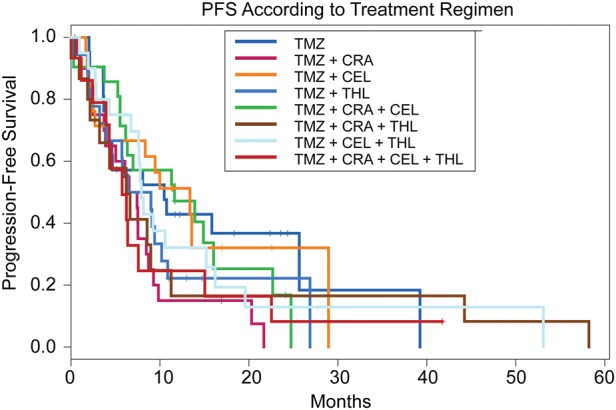
Progression-free survival (PFS) according to treatment regimen (individual arms).
Table 2.
Progression-free survival (PFS)a according to treatment regimen (individual arms)
| Regimen | No. | Median PFS (months) | HR (95% CI) | P Value |
|---|---|---|---|---|
| TMZ | 22 | 10.5 | 1 | – |
| TMZ + CRA | 21 | 6.5 | 2.0 (1.0–4.0) | .043 |
| TMZ + Cel | 21 | 13.4 | 1.0 (0.5–2.1) | .97 |
| TMZ + Thal | 18 | 7.7 | 1.6 (0.8–3.3) | .21 |
| TMZ + CRA + Cel | 21 | 11.6 | 1.1 (0.5–2.2) | .80 |
| TMZ + CRA + Thal | 17 | 6.2 | 1.4 (0.6–3.1) | .39 |
| TMZ + Cel + Thal | 20 | 7.9 | 1.2 (0.6–2.5) | .54 |
| TMZ + CRA + Cel + Thal | 15 | 5.8 | 1.7 (0.8–3.7) | .16 |
Abbreviations: Cel, celecoxib; CRA, isotretinoin; Thal, thalidomide; TMZ, temozolomide.
aPFS was calculated from date of randomization (postchemoradiation) to the date of progression or death.
Overall Survival Analysis
On agent versus no agent analysis, the arms containing isotretinoin trended below the non-isotretinoin arms, but the difference was not statistically significant (median OS, 17.1 months vs 19.9 months; HR, 1.2; 95% CI, 0.8–1.8; P = .26). The arm combining ddTMZ + isotretinoin showed worse OS compared with ddTMZ alone (11.7 months vs 21.2 months; HR, 2.2; 95% CI, 1.1–4.6; P = .037). When comparing doublet versus triplet therapy, no significant differences were seen in OS (17.0 months vs 20.1 months; HR, 0.7; 95% CI, 0.5–1.1; P = .15) (Supplementary Data; Figs. S6, S7 and S8). Cox analysis adjusted for covariates (age, KPS, extent of resection) confirmed similar results (data not shown). Older age was a significant factor determining OS (HR, 3.2; 95% CI, 1.2–8.0; P = .015).
Efficacy Analysis of Pooled Data (Phase I and Phase II Participants)
All participants in the Phase I component were treated at the target dose and de-escalated if dose-limiting toxicity occurred. Since no participants were treated at doses considered to be subtherapeutic, additional efficacy analysis was performed by pooling the data from both components of the study. A total of 196 participants were available for analysis. Again, the ddTMZ + isotretinoin doublet arm performed worse in regard to both PFS and OS (median PFS, 7 months; HR, 1.9; 95% CI, 1.1–3.4; P = .029 and median OS, 12 months; HR, 2.2; 95% CI, 1.2–4.1; P = .011). Cox analysis adjusted for age and KPS confirmed similar results (HR for OS, 2.0; 95% CI, 1.1–3.8; P = .027; HR for PFS, 1.8; 95% CI, 1.0–3.2; P = .055). PFS in the remaining arms ranged from 7 to 13 months, and OS ranged from 17 to 23 months. None of these differences reached statistical significance. Agent versus no agent and doublet therapy versus triplet therapy analysis did not demonstrate any statistically significant differences (Supplementary Data, Tables S4 and S5).
Toxicity
Overall, treatment in all arms was well tolerated. A total of 4 grade 5 adverse events occurred during the study. One was felt to be probably related to ddTMZ + isotretinoin/celecoxib (pneumonia), and one was possibly related to ddTMZ + thalidomide/isotretinoin (bilateral pulmonary embolism). Grade 3-4 toxicities felt to be related to each treatment arm (definite, probable, and possible) are summarized in Table S6 (Supplementary Data). Among the grade 4 adverse events, 2 were definitely related to ddTMZ + isotretinoin (leukopenia, neutropenia), one was definitely related to ddTMZ + thalidomide/isotretinoin/celecoxib (erythema multiforme), and one was definitely related to ddTMZ + celecoxib (thrombocytopenia). Among the grade 3 toxicities, 6 were definitely related to ddTMZ + celecoxib (2 thrombocytopenia, 2 leukopenia, 2 rash), one was definitely related to ddTMZ + isotretinoin (leukopenia), and 3 were definitely related to ddTMZ + thalidomide/isotretinoin (pruritus, rash, somnolence). No treatment-related deaths were specified in the data.
The highest rate of lymphopenia was seen in the arm containing ddTMZ alone. Leukopenia was more frequently seen in the arm combining ddTMZ + thalidomide/celecoxib. Regarding nonhematological toxicities, the thalidomide arms showed a slight increase in the incidence of thromboembolic events.
Discussion
Novel clinical trial designs are critical for testing combinations of targeted agents more efficiently in patients with malignant gliomas. We present the results of the first phase II randomized trial following a factorial design in patients with newly diagnosed GBM, proving the feasibility of this novel biostatistical tool in this disease and paving the way for similar trials in the future.
At the time our study was designed, the use of alternative dosing schemas of temozolomide in malignant gliomas was an area of active research, and a dose-dense schema (ddTMZ) was chosen as the backbone of this adjuvant factorial trial. However, despite the modest activity demonstrated in recurrent gliomas,21,24–26 dose intensification did not translate into improvement of overall survival when tested as first-line therapy. The RTOG 0525 study was a prospective randomized phase III trial comparing standard-dose TMZ (150–200 mg/m2 for 5 days every 28 days) to ddTMZ (75 mg/m2 for 21 days every 28 days) in participants with newly diagnosed GBM.27 The results of RTOG 0525 were reported after this factorial trial completed accrual and showed that participants received no additional benefit from ddTMZ as compared with standard-dose TMZ.
Our study did not demonstrate clear benefit from the addition of isotretinoin, celecoxib or thalidomide when combined with adjuvant ddTMZ. In addition, there was no benefit associated with triplet or quadruplet combinations compared with doublet combinations. Treatment was well tolerated, although there was an unexpectedly high rate of lymphopenia in the ddTMZ alone arm. Intriguingly, the study also suggested a negative impact of adding isotretinoin for adjuvant treatment of GBM. This finding is consistent with a recent phase II study that evaluated the addition of isotretinoin to standard chemoradiation.28 However, earlier studies suggested potential therapeutic benefit for isotretinoin when used alone or added sequentially to TMZ.14,16,29 Overall, these data suggest the possibility of a negative interaction between cytotoxic therapy and the cytostatic effect of isotretinoin that may reflect the overlap of week on/week off dosing of TMZ with the typical isotretinoin 21-day cycle. The interpretation of this result in our trial is also limited by the nature of the factorial design (not powered for comparison arm to arm) and the lack of molecular data such as MGMT promoter methylation or IDH mutation status in the different treatment arms, which may have resulted in an imbalance of these known prognostic factors working against the isotretinoin-containing arms. As a consequence, caution is warranted in interpreting these data.
Since the introduction of chemoradiation as standard therapy for GBM, there has been increased awareness of the occurrence of PsP. However, precise identification of PsP is challenging and is often only possible in retrospect. In addition, the time of occurrence and its impact on the clinical outcome remain poorly defined. In our trial, a potentially incorrect diagnosis of PsP may have impacted the results in several ways. First, it is possible that some of the participants with image worsening classified as having PsP and randomized to receive adjuvant therapy had, in fact, true tumor progression. Theoretically, the unbalanced inclusion of participants with true tumor progression in isotretinoin-containing treatment arms may have contributed to worse PFS and OS. However, the exclusion of participants with symptomatic worsening and new lesions outside of the radiation field likely minimized the risk of inclusion of participants with true progression in all arms. Second, the unbalanced inclusion of participants with true PsP in certain arms may have contributed to better outcome, as suggested in the literature.30 However, the role of PsP as a prognostic indicator still remains under debate.31 Finally, late development of PsP (after the first post-chemoradiation scan but still within the first few months after chemoradiation) may have led to declaration of progressive disease and discontinuation of therapy, thereby confounding the determination of PFS but not the determination of OS.
A factorial design provides certain advantages over conventional phase II studies, but there are shortcomings that limit interpretation of results. Whereas multiple combinations of agents can be tested with fewer participants per arm than a traditional phase II trial, arm-to-arm comparison is limited by low statistical power. In contrast, the contribution of each agent can be analyzed by comparing the results in the arms containing this specific agent versus the treatment arms that do not. Furthermore, the impact of multiagent treatment can be determined by comparing doublet with triplet regimens.
The limitations of the factorial design also include possible therapeutic and statistical interactions. Therapeutic interactions between the agents need to be recognized and incorporated into the statistical modeling. These include pharmacokinetic or pharmacodynamic interactions that may alter the efficacy of each individual agent. Statistical interactions typically refer to the incongruity between absolute versus relative changes in treatment efficacy.32,33
Despite failure to demonstrate superiority of isotretinoin, celecoxib, or thalidomide over ddTMZ alone, our study confirms the feasibility of using the factorial design for future trials to explore different drug combinations targeting multiple signaling pathways in malignant gliomas. Furthermore, the implementation of novel and complex trial designs is feasible in a cooperative group setting. In view of the infrequent rate or “actionable” molecular and genetic alterations in malignant gliomas, collaborative efforts are critical for testing new molecularly driven hypotheses. The implementation of further trials with factorial design in neuro-oncology remains to be explored. Potential future directions include the development of trials targeting several critical and complementary pathways with selection of participants with actionable molecular and genetic alterations of those pathways.
Supplementary Material
Funding
This work was supported by a research grant from Merck & Co. Inc. [M.J.F.], an MD Anderson Community Clinical Oncology Program Research Base grant from the National Cancer Institute [U10 CA045809 to M.J.F.], and a Cancer Center Support grant [P30 CA016672 to K.R.H].
Supplementary Material
Acknowledgments
Kathy L. Hunter, Supervisor, Research Nurse, Neuro-Oncology; Be Lian Pei, Senior Research Nurse, Neuro-Oncology. Sandra Ictech, Data Manager, Neuro-Oncology. Vivien Liu, Research Nurse Manager, Neuro-Oncology; Marlys M. Harden-Harrison, Supervisor, Research Nurse, General Oncology, MD Anderson's Community Clinical Oncology program (CCOP) staff.
Conflict of interest statement. Marta Penas-Prado, Kenneth R. Hess, Michael J. Fisch, Lore W. Lagrone, Victor A. Levin, Howard Colman: honoraria from Merck. Gena Volas-Redd, Pierre Giglio, Michael E. Salacz, Justin D. Floyd, Monica E. Loghin, Sigmund H. Hsu, Javier Gonzalez: consultant and honoraria from Biogen Idec, Teva Pharmaceutical. Eric L. Chang, Charles A. Conrad, Shiao Y. Woo, Anita Mahajan, Kenneth D. Aldape: No conflicts to disclose. Morris D. Groves received research support from GSK and Merck, honoraria from Merck and Foundation Medicine. He is a consultant in Sanofi Aventis and Foundation medicine and received speakers' bureau from Foundation Medicine. John F. De Groot: honoraria from Merck. Vinay K. Puduvalli: research funding from Merck and Genentech. W. K. Alfred Yung: consultant and honoraria from Merck, Actelion, Novartis. Mark R. Gilbert: consultant, honoraria, and research funding from Merck.
Previous Presentations
This study was presented at the ASCO Annual Meeting 2012 in Chicago, USA.
References
- 1.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Chang SM, Butowski NA, Sneed PK, et al. Standard treatment and experimental targeted drug therapy for recurrent glioblastoma multiforme. Neurosurg Focus. 2006;20(4):E4. [PubMed] [Google Scholar]
- 3.Chowdhary S, Chamberlain M. Bevacizumab for the treatment of glioblastoma. Expert Rev Neurother. 2013;13(8):937–949. doi: 10.1586/14737175.2013.827414. [DOI] [PubMed] [Google Scholar]
- 4.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 5.Cloughesy TF, Cavenee WK, Mischel PS. Glioblastoma: from molecular pathology to targeted treatment. Ann Rev Pathol. 2014;9:1–25. doi: 10.1146/annurev-pathol-011110-130324. [DOI] [PubMed] [Google Scholar]
- 6.Brandsma D, van den Bent MJ. Molecular targeted therapies and chemotherapy in malignant gliomas. Curr Opin Oncol. 2007;19(6):598–605. doi: 10.1097/CCO.0b013e3282f0313b. [DOI] [PubMed] [Google Scholar]
- 7.Penas-Prado M, Gilbert MR. Molecularly targeted therapies for malignant gliomas: advances and challenges. Expert Rev Anticancer Ther. 2007;7(5):641–661. doi: 10.1586/14737140.7.5.641. [DOI] [PubMed] [Google Scholar]
- 8.Bozic I, Reiter JG, Allen B, et al. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife. 2013;2:e00747. doi: 10.7554/eLife.00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woodcock J, Griffin JP, Behrman RE. Development of novel combination therapies. N Engl J Med. 2011;364(11):985–987. doi: 10.1056/NEJMp1101548. [DOI] [PubMed] [Google Scholar]
- 11.Montgomery AA, Peters TJ, Little P. Design, analysis and presentation of factorial randomised controlled trials. BMC Med Res Methodol. 2003;3:26. doi: 10.1186/1471-2288-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Short SC, Traish D, Dowe A, et al. Thalidomide as an anti-angiogenic agent in relapsed gliomas. J Neurooncol. 2001;51(1):41–45. doi: 10.1023/a:1006414804835. [DOI] [PubMed] [Google Scholar]
- 13.Fine HA, Figg WD, Jaeckle K, et al. Phase II trial of the antiangiogenic agent thalidomide in patients with recurrent high-grade gliomas. J Clin Oncol. 2000;18(4):708–715. doi: 10.1200/JCO.2000.18.4.708. [DOI] [PubMed] [Google Scholar]
- 14.Jaeckle KA, Hess KR, Yung WK, et al. Phase II evaluation of temozolomide and 13-cis-retinoic acid for the treatment of recurrent and progressive malignant glioma: a North American Brain Tumor Consortium study. J Clin Oncol. 2003;21(12):2305–2311. doi: 10.1200/JCO.2003.12.097. [DOI] [PubMed] [Google Scholar]
- 15.Yung WK, Lotan R, Lee P, et al. Modulation of growth and epidermal growth factor receptor activity by retinoic acid in human glioma cells. Cancer Res. 1989;49(4):1014–1019. [PubMed] [Google Scholar]
- 16.Yung WK, Kyritsis AP, Gleason MJ, et al. Treatment of recurrent malignant gliomas with high-dose 13-cis-retinoic acid. Clin Cancer Res. 1996;2(12):1931–1935. [PubMed] [Google Scholar]
- 17.Shono T, Tofilon PJ, Bruner JM, et al. Cyclooxygenase-2 expression in human gliomas: prognostic significance and molecular correlations. Cancer Res. 2001;61(11):4375–4381. [PubMed] [Google Scholar]
- 18.Joki T, Heese O, Nikas DC, et al. Expression of cyclooxygenase 2 (COX-2) in human glioma and in vitro inhibition by a specific COX-2 inhibitor, NS-398. Cancer Res. 2000;60(17):4926–4931. [PubMed] [Google Scholar]
- 19.Kang SG, Kim JS, Park K, et al. Combination celecoxib and temozolomide in C6 rat glioma orthotopic model. Oncol Rep. 2006;15(1):7–13. [PubMed] [Google Scholar]
- 20.Tolcher AW, Gerson SL, Denis L, et al. Marked inactivation of O6-alkylguanine-DNA alkyltransferase activity with protracted temozolomide schedules. Br J Cancer. 2003;88(7):1004–1011. doi: 10.1038/sj.bjc.6600827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wick W, Steinbach JP, Kuker WM, et al. One week on/one week off: a novel active regimen of temozolomide for recurrent glioblastoma. Neurology. 2004;62(11):2113–2115. doi: 10.1212/01.wnl.0000127617.89363.84. [DOI] [PubMed] [Google Scholar]
- 22.Gilbert MR, Gonzalez J, Hunter K, et al. A phase I factorial design study of dose-dense temozolomide alone and in combination with thalidomide, isotretinoin, and/or celecoxib as postchemoradiation adjuvant therapy for newly diagnosed glioblastoma. Neuro Oncol. 2010;12(11):1167–1172. doi: 10.1093/neuonc/noq100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Macdonald DR, Cascino TL, Schold SC, Jr., et al. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol. 1990;8(7):1277–1280. doi: 10.1200/JCO.1990.8.7.1277. [DOI] [PubMed] [Google Scholar]
- 24.Taal W, Segers-van Rijn JM, Kros JM, et al. Dose dense 1 week on/1 week off temozolomide in recurrent glioma: a retrospective study. J Neurooncol. 2012;108(1):195–200. doi: 10.1007/s11060-012-0832-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wick W, Platten M, Weller M. New (alternative) temozolomide regimens for the treatment of glioma. Neuro Oncol. 2009;11(1):69–79. doi: 10.1215/15228517-2008-078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perry JR, Belanger K, Mason WP, et al. Phase II trial of continuous dose-intense temozolomide in recurrent malignant glioma: RESCUE study. J Clin Oncol. 2010;28(12):2051–2057. doi: 10.1200/JCO.2009.26.5520. [DOI] [PubMed] [Google Scholar]
- 27.Gilbert MR, Wang M, Aldape KD, et al. Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol. 2013;31(32):4085–4091. doi: 10.1200/JCO.2013.49.6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Butowski N, Prados MD, Lamborn KR, et al. A phase II study of concurrent temozolomide and cis-retinoic acid with radiation for adult patients with newly diagnosed supratentorial glioblastoma. Int J Radiat Oncol Biol Phys. 2005;61(5):1454–1459. doi: 10.1016/j.ijrobp.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 29.See SJ, Levin VA, Yung WK, et al. 13-cis-retinoic acid in the treatment of recurrent glioblastoma multiforme. Neuro Oncol. 2004;6(3):253–258. doi: 10.1215/S1152851703000607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brandes AA, Franceschi E, Tosoni A, et al. MGMT promoter methylation status can predict the incidence and outcome of pseudoprogression after concomitant radiochemotherapy in newly diagnosed glioblastoma patients. J Clin Oncol. 2008;26(13):2192–2197. doi: 10.1200/JCO.2007.14.8163. [DOI] [PubMed] [Google Scholar]
- 31.Kruser TJ, Mehta MP, Robins HI. Pseudoprogression after glioma therapy: a comprehensive review. Expert Rev Neurother. 2013;13(4):389–403. doi: 10.1586/ern.13.7. [DOI] [PubMed] [Google Scholar]
- 32.McAlister FA, Straus SE, Sackett DL, et al. Analysis and reporting of factorial trials: a systematic review. JAMA. 2003;289(19):2545–2553. doi: 10.1001/jama.289.19.2545. [DOI] [PubMed] [Google Scholar]
- 33.Green S, Liu PY, O'Sullivan J. Factorial design considerations. J Clin Oncol. 2002;20(16):3424–3430. doi: 10.1200/JCO.2002.03.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.