

Abstract
Lactobacillus plantarum is a lactic acid bacterium that produces a racemic mixture of l- and d-lactate from sugar fermentation. The interconversion of lactate isomers is performed by a lactate racemase (Lar) that is transcriptionally controlled by the l-/d-lactate ratio and maximally induced in the presence of l-lactate. We previously reported that the Lar activity depends on the expression of two divergently oriented operons: (i) the larABCDE operon encodes the nickel-dependent lactate racemase (LarA), its maturases (LarBCE), and a lactic acid channel (LarD), and (ii) the larR(MN)QO operon encodes a transcriptional regulator (LarR) and a four-component ABC-type nickel transporter [Lar(MN), in which the M and N components are fused, LarQ, and LarO]. LarR is a novel regulator of the Crp-Fnr family (PrfA group). Here, the role of LarR was further characterized in vivo and in vitro. We show that LarR is a positive regulator that is absolutely required for the expression of Lar activity. Using gel retardation experiments, we demonstrate that LarR binds to a 16-bp palindromic sequence (Lar box motif) that is present in the larR-larA intergenic region. Mutations in the Lar box strongly affect LarR binding and completely abolish transcription from the larA promoter (PlarA). Two half-Lar boxes located between the Lar box and the −35 box of PlarA promote LarR multimerization on DNA, and point mutations within one or both half-Lar boxes inhibit PlarA induction by l-lactate. Gel retardation and footprinting experiments indicate that l-lactate has a positive effect on the binding and multimerization of LarR, while d-lactate antagonizes the positive effect of l-lactate. A possible mechanism of LarR regulation by lactate enantiomers is proposed.
INTRODUCTION
Proteins of the Crp-Fnr family are highly versatile transcriptional regulators that respond to a broad spectrum of signals, such as cyclic AMP (cAMP), anoxia, redox state, oxidative and nitrosative stress, nitric oxide, carbon monoxide, 2-oxoglutarate, or temperature. They are involved in the control of multiple processes, including virulence, stress response, nitrogen fixation, photosynthesis, and various catabolic pathways (1). Members of the Crp-Fnr family are characterized by the presence of two domains: a C-terminally located helix-turn-helix (HTH) DNA binding domain that usually recognizes a 14- to 22-bp palindromic DNA sequence (1) and an N-terminal β-barrel domain that is responsible for dimerization and signal integration (2). Signal integration can involve prosthetic groups attached to the protein (1).
A gene encoding a transcriptional regulator of the Crp-Fnr family, larR, has been discovered in the lactate racemization (lar) locus of Lactobacillus plantarum, a homofermentative lactic acid bacterium that produces lactate (Lac) as the main fermentation product (3). The lar locus is composed of two divergent operons: (i) the larABCDE (larA-E) operon encodes the lactate racemase (LarA), its accessory proteins (LarBCE), and a lactic acid channel (LarD), and (ii) the larR(MN)QO (larR-O) operon encodes the LarR regulator and an ABC-type nickel transporter composed of a substrate binding protein [Lar(MN), in which the M and N components are fused], a permease (LarQ), and an ATP binding protein (LarO) (Fig. 1A) (3). The lactate racemase is a nickel-dependent enzyme requiring activation by the accessory protein LarE, which itself requires activation by the accessory proteins LarB and LarC and nickel (3). LarD is an aquaglyceroporin that was shown to transport both lactate isomers (4). This lactic acid channel is involved in the racemization of extracellular lactate and lactate metabolism (4). The Lar(MN)QO product was proposed to be a nickel transporter since its inactivation abolished lactate racemization, which could be restored by nickel supplementation in the extracellular medium (3). Concerning LarR, it was suspected to be involved in the transcriptional control of lar genes, a function that was not yet investigated (3).
FIG 1.

In vivo LarR regulation. (A) Schematic representation of the lar locus of Lactobacillus plantarum, which comprises the larA-E operon, responsible for the lactate racemization activity, and the larR-O operon, coding for LarR and for an ABC-type nickel transporter. (B) Specific lactate racemase (Lar) activity of the L. plantarum ΔldhL mutant (control), the double ΔlarR ΔldhL mutant, and the ΔlarR ΔldhL mutant complemented with larR (PnisA-larR) or StrepII-larR (PnisA-strep-tag-larR). Complementations were performed in the presence of the nisin inducer. The activity was measured after induction with 200 mM NaCl (no Lac), 200 mM Na-dl-Lac (dl-Lac), or 200 mM Na-l-Lac (l-Lac), as described in Materials and Methods. (C and D) Specific β-glucoronidase (Gus) activities (C) and specific lactate racemase (Lar) activities (D) of the L. plantarum ΔldhL mutant expressing the larA promoter-gusA fusion (PlarA-gusA) measured after induction with 200 mM Na-Lac at different l-/d-Lac ratios, as described in Materials and Methods. (E to H) Specific Gus activities (E and G) and specific Lar activities (F and H) of the L. plantarum ΔldhL mutant expressing PlarA-gusA after induction with 200 mM Na-l-Lac (E and F) or 50 mM Na-d-Lac (G and H) supplemented with 0, 50, or 200 mM Na-d-Lac (E and F) or Na-l-Lac (G and H). The values shown are the mean results of 3 repetitions from 1 significant experiment out of 2 experiments showing similar results. The error bars indicate the confidence intervals at 95% (Student's t test).
LarR belongs to the PrfA group (see Fig. S1 in the supplemental material), a branch of the Crp-Fnr family mostly linked to pathogenicity (1). The lead member of the group, PrfA from Listeria monocytogenes, binds a 14-bp palindromic sequence (5′-TTAACANNTGTTAA-3′), the PrfA box (5). The effector that activates PrfA is currently unknown but has been proposed to be a host-derived small-molecule second messenger (6). Other members of the PrfA group are the streptococcal regulator of virulence Srv (7), the putative virulence factor Ers of Enterococcus faecalis (8), and the acid stress responsive factor RcfB of Lactococcus lactis (see Fig. S1) (9). Although the effector of these transcriptional regulators remains unknown, Srv and Ers have been shown to bind DNA in vitro (10, 11). The last known member of the PrfA group is the redox regulator of Bacillus subtilis, FnrBac, which responds to the oxygen tension (12).
The presence of LarR in the lar locus suggests its involvement in the regulation of the lactate racemase (Lar) activity. In L. plantarum, the lactate racemase is used as a rescue pathway for the production of d-lactate (d-Lac), which is essential for peptidoglycan biosynthesis (13). The Lar activity of L. plantarum has been shown to depend on the l-lactate (l-Lac)/d-Lac ratio: l-Lac induces Lar activity, while the dl-Lac racemic mixture does not, which suggests repression by d-Lac (13). In this study, we investigate the enantioselective regulation of lactate racemization by the LarR regulator in vivo using genetic fusions and in vitro by analyzing regulator interactions with its DNA targets. We show that LarR is an activator that controls the transcription of Lar-encoding genes by responding differentially to lactate enantiomers: l-Lac acts as a positive effector, while d-Lac antagonizes its effect.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains and plasmids used in the present study are listed in Table 1. Plasmid constructions were performed in Escherichia coli TOP10 for pUC18Ery (14) and pSIP409 derivatives (15) and in L. lactis NZ3900 (16) for pNZ8048 derivatives (17). E. coli was grown in Luria broth (LB) at 37°C with aeration, Lactococcus lactis was grown in M17 medium (Merck, Germany) supplemented with 0.5% glucose at 28°C at 120 rpm, and Lactobacillus plantarum was grown in MRS (De Man-Rogosa-Sharpe) broth (Difco Laboratories, Inc., Detroit, MI) at 28°C without shaking. When appropriate, chloramphenicol and erythromycin were added to the medium at 10 μg ml−1 for L. lactis and L. plantarum, and erythromycin and ampicillin were added at 250 μg ml−1 for E. coli. For the induction of genes under the control of the nisA expression signals in L. lactis, nisin A was added during the early log phase (optical density at 600 nm [OD600] of 0.2 to 0.3) at a concentration of 1 µg liter−1, and the cells were collected 4 h later.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Characteristic(s)a | Source or reference |
|---|---|---|
| L. plantarum strains | ||
| NCIMB8826 | Wild type | NCIMBb |
| TF101 | NCIMB8826 ΔldhL | 27 |
| LR0002 | NCIMB8826 ΔldhL ΔlarR nisRK | This study |
| L. lactis NZ3900 | MG1363 derivative | 19 |
| E. coli TOP10 | F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 nupG recA1 araD 139 Δ(ara-leu)7697 galE15 galK16 rpsL(Strr) endA1 λ− | Invitrogen |
| Plasmids | ||
| pMEC10 | Emr Ampr; pMC1 derivative for integration of the nisRK genes at tRNASer | 28 |
| pUC18Ery | Emr Ampr; pUC18 derivative with a 1.1-kb insert containing the erm gene | 17 |
| pGIZ850 | Emr Ampr Cmr; pUC18Ery with a 4.215-kb insert containing the loxD gene disrupted by a cat gene under the control of the P32 promoter | 26 |
| pNZ5348 | Emr; pGID023 derivative containing cre under the control of the lp_1144 promoter | 23 |
| pGIR002 | Emr Ampr; pUC18Ery with a 4.32-kb insert containing the lox66-P32-cat-lox71 cassette from pNZ5319 surrounded by the larR upstream and downstream regions | This study |
| pBADHisA | Ampr ParaB | Invitrogen |
| pGIR991 | Ampr; pBADHisA with a 0.82-kb insert containing the ParaB-StrepII-larR translational fusion | This study |
| pNZ8048 | Cmr PnisA | 20 |
| pGIR090 | Cmr; pNZ8048 derivative with a 0.78-kb insert containing the PnisA-larR translational fusion | This study |
| pGIR091 | Cmr; pNZ8048 derivative with a 0.82-kb insert containing the PnisA-StrepII-larR translational fusion | This study |
| pSIP409 | Emr; low-copy-no. gusA reporter plasmid | 18 |
| pGIR003 | Emr; pSIP409 derivative with the PlarA-gusA translational fusion | This study |
| pGIR003B | pGIR003 harboring one point mutation in the Lar box (mutation RRb) | This study |
| pGIR003C | pGIR003 harboring one point mutation in the half-Lar box 1 (PlarA side 1, mutation RRc) | This study |
| pGIR003D | pGIR003 harboring two point mutations in half-Lar boxes 1 and 2 (PlarA side 1 and 2, mutations RRc and RRd) | This study |
Emr, Ampr, and Cmr indicate resistance to erythromycin, ampicillin, and chloramphenicol, respectively.
NCIMB, National Collections of Industrial and Marine Bacteria, Ltd., Aberdeen, Scotland.
General DNA techniques and transformation.
General molecular biology techniques were performed according to the instructions given by Sambrook and Russell (18). Electrotransformation of E. coli, L. plantarum, and L. lactis were performed as described by Dower et al. (19), Lambert et al. (20), and Holo and Nes (21), respectively. PCR amplifications were performed with Phusion high-fidelity DNA polymerase according to the manufacturer's instructions (Finnzymes, Espoo, Finland). The primers used in this study were purchased from Eurogentec (Seraing, Belgium) and are listed in Table S1 in the supplemental material. Primer extension was performed with primer LP104PE4 as previously described (22).
Plasmid and mutant construction.
The larR deletion vector pGIR002 was constructed by cloning the upstream and downstream regions of the larR gene from L. plantarum strain NCIMB8826 and the lox66-P32-cat-lox71 cassette from plasmid pGIZ850 (23) in plasmid pUC18Ery (see Table S2 in the supplemental material). This suicide vector was used to delete larR in L. plantarum strain TF101 (24) through a two-step homologous recombination, as previously described (20). The excision of the cat gene was then performed by transforming plasmid pNZ5348 harboring the cre recombinase, as described previously (20). Confirmation of the deletion was performed by PCR amplification with primers LP096UP1 and LP105B1 (see Table S1). Strain TF101 ΔlarR was then transformed with pMEC10 in order to integrate the nisRK genes at the tRNASer locus (25), generating strain LR0002.
Plasmid pGIR090 (PnisA-larR fusion) was constructed by cloning the larR gene from L. plantarum NCIMB8826, which was amplified by PCR with the primers Lp1031 and Lp1032 (see Table S1 in the supplemental material), digested with NcoI and KpnI, and ligated into a similarly digested pNZ8048. The resulting plasmid was transformed into L. lactis strain NZ3900. Plasmid pGIR991 (ParaB-StrepII-larR fusion) was constructed by cloning the larR gene from L. plantarum NCIMB8826 fused to the genetic material encoding the 8-residue minimal peptide sequence (Trp-Ser-His-Pro-Gln-Phe-Glu-Lys) called Strep-tag II (referred to herein as StrepII) at the N terminus, which was amplified by PCR with primers LP103OXA1 and LP103OXB1 (see Table S1), digested with NcoI and EcoRI, and ligated into a similarly digested pBADHisA. The resulting plasmid was transformed into E. coli TOP10. The plasmid pGIR091 (PnisA-StrepII-larR fusion) was constructed by the insertion of the StrepII-larR fusion from pGIR991 between the NcoI and HindIII restriction sites of pNZ8048. E. coli TOP10 containing plasmid pGIR991 was used for StrepII-tagged LarR (referred to herein as rLarR) purification. Plasmids pGIR090 (PnisA-larR fusion) and pGIR091 (PnisA-StrepII-larR fusion) were electrotransformed into strain LR002 for complementation studies. L. lactis NZ3900 harboring plasmid pGIR090 (PnisA-larR fusion) was used to prepare total cell extracts containing the untagged version of LarR for electrophoretic mobility shift assays. The plasmids were confirmed by sequencing the PnisA-larR and PnisA-StrepII-larR fusions with primer UP_PNZ8048 (see Table S1).
Plasmid pSIP409 with gusA as a reporter gene was used as a backbone for promoter regulation studies. Plasmid pGIR003 (PlarA-gusA fusion) was constructed by cloning the larR-larA intergenic region from L. plantarum NCIMB8826, which was amplified with primers PSIP103-104A1/PSIP103-104B2 (see Table S1 in the supplemental material), digested with NcoI and BamHI, and ligated into a pSIP409 plasmid digested with NcoI and BglII. The plasmids were then purified from E. coli TOP10 and electrotransformed into L. plantarum TF101 (ΔldhL mutant, exclusive d-Lac producer). Mutations of the larR-larA intergenic region were performed with the QuikChange protocol on plasmid pGIR003 (26), generating plasmids pGIR003B, pGIR003C, and pGIR003D. Mutations were confirmed by sequencing PlarA-gusA fusions with primers CAT1, GUSA2, and GUSB2 (see Table S1).
Complementation studies.
Cells from a 10-ml culture of L. plantarum TF101 (ΔldhL), L. plantarum LR0002 (ΔldhL ΔlarR) harboring pNZ8048 (empty vector), L. plantarum LR0002 harboring pGIR090 (PnisA-larR), or L. plantarum LR0002 harboring pGIR091 (PnisA-StrepII-larR) were grown until reaching an OD600 of 0.5. For larR induction, nisin A (Sigma-Aldrich, Belgium) was added at a final concentration of 50 ng ml−1. After 1 h of incubation in the presence of nisin, NaCl (control), l-Lac, or dl-Lac (lactate sodium salts; Sigma-Aldrich, Belgium) was added to a final concentration of 200 mM. After 4 h of additional incubation, cells were collected by centrifugation at 5,000 × g for 10 min and washed twice with 10 ml of 60 mM Tris-maleate buffer, pH 6.0 (TM buffer), using the same centrifugal conditions.
Electrophoretic mobility shift assays (EMSA).
32P-radiolabeled primers were obtained by incubating 0.25 nmol of oligonucleotide with 0.4 nmol of [γ-32P]ATP and 20 units of T4 polynucleotide kinase (Roche Diagnostics, Belgium) in 1× T4 polynucleotide kinase buffer for 1 h at 37°C. The reaction was stopped by an incubation of 20 min at 70°C, and primers were purified from the excess [γ-32P]ATP with MicroSpin G-25 columns (GE Healthcare, France). Short DNA probes (30 and 56 bp) were constructed by annealing complementary pairs of radiolabeled oligonucleotides as describe by Sambrook and Russell (18). Longer DNA probes (127 or 256 bp) were obtained by PCR amplification using 32P-radiolabeled primers and genomic DNA from L. plantarum NCIMB8826 as a template (see Table S1 in the supplemental material). Mapping of the different probes on the DNA sequence of the larR-larA intergenic region is shown in Fig. S2 in the supplemental material. PCR products were purified using the QIAquick PCR purification kit (Qiagen, France). Binding reactions were conducted at room temperature for 15 min in TE buffer (100 mM Tris-HCl, pH 7.5, and 1 mM EDTA) in the presence of various amounts of rLarR or untagged LarR and the radiolabeled probe (∼30 pmol). Nonspecific DNA [sonicated salmon sperm DNA or poly(dI-dC)] was not incorporated in the binding reaction mixtures since it has no impact on the formation of rLarR-DNA complexes. For the specificity experiments, 15 ng of rLarR were incubated with a 32P-radiolabeled probe (127 bp) of the larR-larA intergenic region of L. plantarum mixed with either the same cold probe (specific DNA) or a 137-bp probe of the larA-larB intergenic region of L. plantarum (nonspecific DNA), obtained by PCR amplification using oligonucleotides LarA-A_A and LarB-B_B (see Table S1). Bound products were separated from free DNA on 6% native polyacrylamide gel (40% acrylamide–Bis-acrylamide 19:1 solution; Bio-Rad) in 1× Tris-borate-EDTA (TBE) buffer. Gels were run at 200 V for 2 to 3 h at 4°C. The gels were revealed with an Imaging Screen-K (Bio-Rad, Belgium) and read on a Pharos FX Plus (Bio-Rad, Belgium). The quantification of the complexes was performed by densitometry using Quantity One software (Bio-Rad, Belgium).
DNase I footprinting assays.
Binding reactions were performed at room temperature for 15 min in 100 mM Tris-HCl, pH 7.5, 150 mM NaCl, 25 mM MgCl2 buffer. Three units of DNase I (Sigma-Aldrich, Belgium) were added, and the binding reaction mixtures were incubated for 5 min at 30°C. To stop the reaction, 1.5 volumes of stop buffer (20 mM EDTA, 1% SDS, 200 mM NaCl) was added to the reaction mixture. The DNA was precipitated with ethanol, and the pellet was washed three times with 70% ethanol and resuspended in 5 μl loading buffer (80% formamide, 5 M urea, 0.1% bromophenol blue, 0.1% xylene cyanol). The reaction mixtures were heated for 10 min at 100°C, loaded on a 6% denaturing sequencing gel containing 5 M urea, and separated at 75 W for 2 h at room temperature. Marker lanes were loaded with Maxam and Gilbert sequencing reaction mixtures, prepared from the DNA probe as described by Sambrook and Russell (18). The gel was revealed with an Imaging Screen-K (Bio-Rad, Belgium) and read on a Pharos FX Plus (Bio-Rad, Belgium).
Lactate racemase specific activity.
Cells were resuspended in 0.5 ml of TM buffer (60 mM Tris-maleate buffer, pH 6.0) and lysed with 0.17- to 0.18-mm glass beads (Sartorius Mechatronics, Belgium) in a FastPrep-24 (MP, Belgium), using 2 runs of 1 min at 6.5 m/s. After lysis, the supernatant was collected by centrifugation at 13,000 × g at 4°C for 15 min.
The Lar activity was assayed by incubating cell extracts diluted 50-fold with 20 mM d- or l-Lac (sodium salts) in 60 mM morpholineethanesulfonic acid (MES) buffer, pH 6, at 35°C for 10 min. The reaction was stopped by incubating the mixture for 10 min at 90°C. Lactate conversion was measured using the d-lactic acid/l-lactic acid UV test (R-Biopharm, Germany). The protocol was adapted to 100-μl reaction mixture volumes. The lactate conversion was monitored in 96-well, half-area microplates (Greiner, Alphen a/d Rjin, the Netherlands) by reading the absorbance at 340 nm with a Varioskan Flash multimode reader (ThermoScientific). The protein concentrations were measured with a commercial Bradford protein assay (Bio-Rad, Norway). One unit of Lar activity is defined as 1 μmol of lactate converted in 1 min.
β-Glucuronidase activity.
Cells from 10-ml cultures of L. plantarum TF101 carrying pGIR003 (PlarA-gusA) were grown until reaching an OD600 of 0.7. The cells were then collected by centrifugation at 5,000 × g for 10 min and resuspended in fresh MRS medium containing 200 mM lactate at different l-/d-Lac ratios. After 4 h of additional incubation, the culture was split in two, and half of the cells were collected by centrifugation at 5,000 × g for 10 min and washed twice with 10 ml of TM buffer using the same centrifugal conditions. These cells were assayed for their Lar activity, as described above. The other half of the cells were collected by centrifugation at 5,000 × g for 10 min and washed twice with 10 ml of 50 mM Pi buffer, pH 7.5, containing 10 mM β-mercaptoethanol (PB buffer), using the same centrifugal conditions. Cells were resuspended in 0.5 ml PB buffer and lysed with glass beads as described above. After lysis, the supernatant was collected by centrifugation at 13,000 × g at 4°C for 15 min. The β-glucuronidase (Gus) specific activity was measured by incubation of L. plantarum cell extracts diluted 2-fold in Gus buffer (2 mM p-nitrophenyl-β-d-glucuronide [Sigma-Aldrich, Belgium] in PB buffer) (27). The reaction was monitored by reading the absorbance at 405 nm every 5 min for 50 min with a Varioskan Flash multimode reader. The protein concentration was measured as described above. One unit of Gus activity is defined as 1 μmol of p-nitrophenyl-β-d-glucuronide hydrolyzed in 1 min.
Purification of StrepII-LarR.
One liter of E. coli TOP10 cells expressing the StrepII-larR fusion were induced by adding l-arabinose at a final concentration of 0.2% at the early log phase (OD600 of 0.3 to 0.4). Cells were collected 3 h later by centrifugation at 5,000 × g for 10 min and washed twice with wash buffer (150 mM Tris-HCl, pH 8.0, 300 mM NaCl, and 1 mM EDTA). Cells were then resuspended in 9 ml wash buffer containing 0.1 mg ml−1 lysozyme, incubated for 30 min at 37°C, and lysed by sonication 6 times for 10 s, with a 50-s rest between each run, in a Bioruptor (Diagenode, Belgium). Cell extracts were loaded on a Strep-Tactin high-affinity column (IBA, Germany), and the purification of StrepII-LarR (rLarR) was performed as described previously (28). Purified rLarR fractions contained a major form and a minor form of higher electrophoretic mobility (see Fig. S3 in the supplemental material). Both forms were identified as LarR by matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) analysis (see Table S2), suggesting that the minor form resulted from proteolytic cleavage of rLarR.
Bioinformatics analyses.
LarR homology searches were performed with BLASTP against the nonredundant protein sequence database from NCBI (release 193) using default parameters. A cutoff E value of 10−20 was used in order to only select sequences most similar to LarR of L. plantarum. When LarR homologues were found in different strains within one species, only one representative strain was retained. The adjacent genes were then searched for homologous lar genes using BLASTP. The most probable LarR candidates controlling lactate racemization were selected based on their high similarity with LarR (>40% sequence identity) and on their synteny with the larA gene, encoding the lactate racemase. Data referring to LarR homologues and their genetic analyses are summarized in Table S3 in the supplemental material.
For the in silico analysis of LarR binding sites, larR-larA intergenic regions were retrieved from the genome sequences of the 20 identified species where larR and larA genes are adjacent. Sequences similar to the PrfA box (5) were searched visually and are referred to as Lar boxes (see Fig. S4 in the supplemental material). The sequences of the 20 identified Lar boxes were used to define a consensus Lar box sequence, which was then used to identify half-Lar boxes in the regions adjacent to the initial Lar box (see Fig. S4). Logos were generated with WebLogo 3 (http://weblogo.berkeley.edu/) using the 20 Lar boxes identified previously, the 7 PrfA boxes of the core PrfA regulon (29), and 4 ACiD boxes (RcfB binding sites), two previously reported (9) and two identified in Lactococcus garvieae (see Table S3).
The genome of L. plantarum was searched for instances of the Lar box using the Patser software (http://rsat.ulb.ac.be/patser_form.cgi) (30). The position-specific scoring matrix used as the query was the frequency matrix constructed with the 20 identified Lar boxes. Only putative Lar boxes located in intergenic regions with scores higher than 10 [ln(P) < −12, where P is the probability value] were considered.
RESULTS
LarR is a positive regulator of the lactate racemase activity in L. plantarum.
Since previous transcriptome analyses showed that the larR-O and larA-E operons are induced by l-Lac (3), we hypothesized that the transcriptional regulator LarR that is present in the larR-O operon may be directly involved in this regulation (Fig. 1A).
To test the relationship between LarR and Lar activity, the deletion of larR was achieved in L. plantarum strain TF101, which lacks the ldhL gene (ΔldhL) (31). This genetic background was selected because it exclusively produces d-Lac, resulting in no detectable basal Lar activity; however, high Lar activity is obtained upon induction with l-Lac (Fig. 1B) (13). This l-Lac-dependent induction of the Lar activity was totally lost in the double ΔlarR ΔldhL mutant (Fig. 1B). To rule out possible polar effects that may affect Lar activity by acting on downstream genes, the ΔlarR ΔldhL mutant was complemented with an extrachromosomal copy of larR under the control of the nisA promoter (PnisA-larR fusion) (Fig. 1B). Induction of larR expression with nisin in the complemented strain totally restored the l-Lac-inducible Lar activity (Fig. 1B). This demonstrates (i) that LarR is a positive regulator of the expression of the lar locus and (ii) that its presence is absolutely required for the expression of the lactate racemase activity in L. plantarum.
The larR-larA intergenic region contains the cis elements necessary for larA-E regulation.
As a first step toward identifying the DNA target of LarR, the larR-larA intergenic region of L. plantarum was inserted upstream from a gusA reporter gene (PlarA-gusA fusion) and β-glucuronidase (Gus) activity was measured under different l-/d-Lac ratios in strain TF101 (ΔldhL) (Fig. 1C). Concomitantly, we measured the Lar activity to validate the stereoselectivity of the response from the native lar locus (Fig. 1D). The Gus activity from the PlarA-gusA fusion and the native Lar activity were strongly dependent on the l-/d-Lac ratio (Fig. 1C and D): both activities were below the detection level (0.2 mU mg−1 and 0.1 U mg−1, respectively) when the l-/d-Lac ratio was, respectively, ≤2:1 or ≤1:1 and increased with the l-/d-Lac ratio, until the maximal activity was achieved at 100% l-Lac (ratio of 1:0) (Fig. 1C and D). l-Lac alone could not account for this regulation, since the addition of increasing amounts of d-Lac at a constant concentration of 200 mM l-Lac showed a drastic decrease of induction (Fig. 1E and F). Conversely, the relief of both Gus and Lar activities could be achieved in the presence of a constant concentration of d-Lac by the addition of increasing amounts of l-Lac (Fig. 1G and H). However, since the mutant strain produces substantial amounts of d-Lac during growth, this reverse experiment was performed with a lower concentration of d-Lac (50 mM). This shows that the regulation is strictly dependent on the l-/d-Lac ratio and that the two lactate isomers are acting in opposite ways, l-Lac as an activator and d-Lac as an inhibitor. Moreover, the similarity of the induction pattern of Gus to that of Lar indicates that the larR-larA intergenic region contains all required cis elements for the control of the larA-E operon by l- and d-Lac.
LarR recognizes a palindromic sequence of 16 bp.
To identify the DNA binding motif of LarR, we retrieved the closest homologues of L. plantarum LarR and searched for the presence of genes homologous to larA in their vicinity. The larA and larR genes were found to be syntenic in 20 species, mostly belonging to Lactobacillales but also to Clostridiales and Coriobacteriales (see Table S3 in the supplemental material). Strikingly, the genetic organization is fully conserved among all species. The larA and larR genes are adjacent and divergently oriented, suggesting a common control in lactate racemization. The in silico analysis of the 20 larR-larA intergenic regions revealed the presence of a conserved palindromic sequence of 16 bp (Fig. 2A; see also Fig. S4), whose consensus sequence is 5′-GTTAACA(T/A)(T/A)TGTTAAC-3′. We therefore named this sequence the Lar box. The consensus sequence of the Lar box is almost identical to the consensus sequences of the PrfA box (5) and the ACiD box that is recognized by RcfB, another member of the PrfA group (Fig. 2A) (9).
FIG 2.
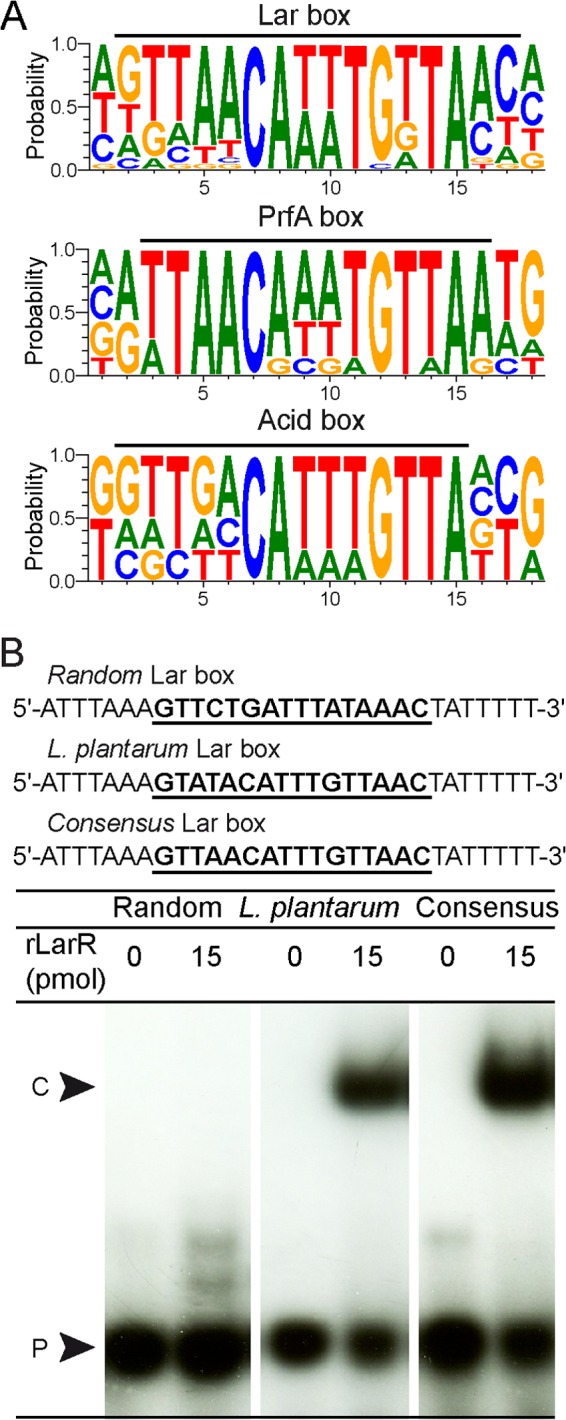
Lar box identification. (A) Logos of the Lar box, the PrfA box (29), and the ACiD (RcfB) box (9) drawn with WebLogo 3 (http://weblogo.berkeley.edu/). The black bars above the logos correspond to the Lar box consensus identified in this study and the previously reported PrfA box (29) and ACiD (RcfB) box (9). (B) EMSA of 30-bp 32P-radiolabeled probes containing the randomly shuffled Lar box (random), the Lar box from L. plantarum (L. plantarum), or a consensus Lar box (consensus) in the absence or presence of purified rLarR. The DNA sequences of the probes are shown, with the corresponding boxes in boldface and underlined. The rLarR-DNA complex (C) and unbound probe (P) are indicated.
To investigate the capacity of LarR to bind the Lar Box, an N-terminally StrepII-tagged LarR variant (referred to herein as rLarR) was constructed and purified to homogeneity by affinity chromatography (see Fig. S3 in the supplemental material) (28). To assess the effect of the StrepII tag on LarR activity, the ability of rLarR to complement the ΔlarR mutant phenotype was evaluated as reported above for the wild-type LarR. StrepII-tagged LarR was able to restore l-Lac induction of the Lar activity in the LarR-deficient background. However, repression by d-Lac was lost (Fig. 1B). This shows that the StrepII tag at the N terminus of LarR impairs d-Lac inhibition without affecting the ability of LarR to respond to l-Lac. In an attempt to circumvent this issue, the StrepII tag was fused to the C terminus of LarR, but the complementation of the ΔlarR mutant resulted in constitutive Lar activity, suggesting that both l-Lac activation and d-Lac repression were impaired (data not shown).
To examine rLarR's binding capacity, we performed electrophoretic mobility shift assays (EMSA) with radiolabeled probes of 30 bp containing the Lar box of L. plantarum (5′-GTATACATTTGTTAAC-3′), the consensus Lar box (5′-GTTAACATTTGTTAAC-3′), or a randomly shuffled Lar box sequence (5′-GTTCTGATTTATAAAC-3′) (Fig. 2B). The presence of rLarR was able to slow the migration of both the Lar box of L. plantarum and the consensus Lar box but not that of the randomly shuffled sequence (Fig. 2B). To further confirm the identity of the Lar box, EMSA were performed with probes containing mutated L. plantarum Lar boxes. Four positions [G→C(LRa)TATAC→G(LRb)ATTTG→C(RRb)TTAAC→G(RRa)] were either mutated individually (LRa and LRb, left repeat mutants, and RRb and RRa, right repeat mutants) or pairwise (LRa+RRa and LRb+RRb) (see Fig. S5 in the supplemental material). All mutations significantly reduced rLarR binding to the Lar box (2- to 5-fold) compared to its binding to the nonmutated probe (see Fig. S5). Finally, the specificity of rLarR binding to the larR-larA intergenic region of L. plantarum was assessed by adding increasing amounts of specific DNA cold probe and nonspecific DNA probe to the binding reaction mixtures (Fig. 3). As expected for a specific interaction between LarR and the larR-larA region, the formation of the LarR-DNA complex was negatively affected by unlabeled DNA carrying the larR-larA intergenic region, whereas nonspecific DNA had no effect (Fig. 3). Altogether, these results show that rLarR specifically binds to the Lar box motif, which consists of a palindromic sequence of 16 bp.
FIG 3.

Specificity of rLarR interaction with the larR-larA intergenic region. Electrophoretic mobility shift assay (EMSA) of a 127-bp 32P-radiolabeled probe of the larR-larA intergenic region of L. plantarum in the presence of purified rLarR and increasing amounts of nonspecific DNA (ns DNA; larA-larB intergenic region, 137 bp) or specific DNA corresponding to the same unlabeled probe (s DNA). The LarR-DNA complex (C) and unbound probe (P) are indicated.
LarR multimerizes on the larR-larA intergenic region.
To characterize the larA promoter, the transcriptional start (+1) was mapped by primer extension (data not shown), and the upstream putative −35 and −10 boxes corresponding to a vegetative promoter were localized (Fig. 4A). We also identified two half-Lar boxes in the region between the Lar box and the −35 box of PlarA (PlarA side 1 and 2). Two additional half-Lar boxes were identified in the presumed larR promoter (PlarR side 1 and 2) (Fig. 4A and B). Half-sites may have a significant regulatory role, especially for the multimerization of regulators on their target sites (32). In order to identify possible high-molecular-weight complexes (HMWC) which would result from such a multimerization, we performed EMSA with increasing concentrations of purified rLarR and a radiolabeled probe containing the entire larR-larA intergenic region (see Fig. S2 in the supplemental material for the 256-bp radiolabeled probe). The addition of rLarR at low concentrations retarded the electrophoretic mobility of the DNA probe as a single band (from 0 to 15 pmol) (Fig. 4C), but multiple discrete bands of lower electrophoretic mobility gradually appeared as the amount of rLarR was increased (from 22.5 to 45 pmol of LarR) (Fig. 4C). The specificity of these HMWC was confirmed by a competition experiment with increasing amounts of a specific DNA cold probe in the binding reaction mixtures (data not shown).
FIG 4.

LarR multimerization. (A) DNA sequence of the central part of the L. plantarum larR-larA intergenic region. The putative −35 and −10 boxes of PlarR and PlarA, the Lar box, and the mapped transcriptional start (+1) of larA are highlighted in black. Protected regions in footprinting assays (lines) and sites displaying enhanced sensitivity to DNase I upon binding of LarR (V), such as are displayed in Fig. 6C, are indicated above the sequence. The half-Lar boxes surrounding the central Lar box are shown by arrows above the sequence. (B) Comparison of the half-Lar box sequences with the consensus Lar box sequence. (C) EMSA of a 256-bp 32P-radiolabeled probe of the larR-larA intergenic region of L. plantarum in the presence of increasing amounts of purified rLarR. The lower-molecular-weight rLarR-DNA complex (C1), unbound probe (P), and high-molecular-weight complexes (HMWC) are indicated.
To evaluate whether these HMWC may result from rLarR binding to the half-Lar boxes within the DNA probe, a mutagenesis study of the larR-larA intergenic region was performed (Fig. 5A). The effects of point mutations on the formation of the different LarR-DNA complexes were studied using EMSA (Fig. 5B and C). Whereas a point mutation in a highly conserved residue of the Lar box itself (Fig. 5A, RRb mutant) strongly decreased the formation of all complexes compared to the results for the native DNA probe (wild type [WT]) (Fig. 5B and C), mutations at the corresponding position in one or both of the half-Lar box motifs located at the larA side (RRc and RRc+RRd mutants) (Fig. 5A) only had a minor effect on rLarR binding (Fig. 5B and C). However, a significantly lower abundance of HMWC (complexes 2 and 3, called C2+3) was observed with the mutated probes RRc and RRc+RRd than with the WT (Fig. 5B and C), while the abundance of the main, lower-molecular-weight complex (named C1), was increased (Fig. 5B and C). The ratios between HMWC and complex C1 decreased strongly when half-Lar box motifs were mutated (Fig. 5D). This analysis suggests that complex C1 corresponds to LarR binding to the central Lar box, while more retarded bands arise from occupation of the flanking half-Lar boxes. In addition, these results show that the formation of all rLarR-DNA complexes is primarily dependent on the central Lar box, whereas half-Lar boxes enhance the formation of HMWC but are not required for the formation of the initial complex C1.
FIG 5.

Mutagenesis of the larR-larA intergenic region. (A) Native (WT) and mutated DNA sequences (RRb, RRc, and RRc+RRd) of the region, including the Lar box and the −35 box of PlarA. The inverted repeat of the Lar box (black arrows) and the two half-Lar boxes on the PlarA side (gray arrows) are shown below the sequence. The mutations in the right repeats are highlighted in black. The arrow corresponding to a mutated right repeat was removed to indicate its alteration. (B) EMSA of 127-bp 32P-radiolabeled probes of the wild type (WT) and mutated larR-larA intergenic regions of L. plantarum in the presence of increasing amounts of purified rLarR. The lower-molecular-weight rLarR-DNA complex (C1), unbound probe (P), and high-molecular-weight complexes C2 and C3 (C2+3) are indicated. (C) Relative abundances of P, C1, and C2+3 in EMSA such as that shown in panel B. Data are from 4 replicates run in parallel on the same gel from one representative experiment. The error bars represent the standard deviations. (D) Ratios of C2+3 to C1 at 30 pmol of LarR in EMSA such as that shown in panel B. Data are from 4 replicates run in parallel on the same gel from one representative experiment. The error bars represent the standard deviations. Significance of the results was determined by Student's t test: **, P < 0.01. (E) Gus activities measured with the mutated larA promoters after induction by 200 mM dl-Lac or l-Lac. ND, not detected (<0.1 mU). The values shown are the mean results of 3 repetitions from 1 significant experiment out of 2 experiments showing similar results. The error bars give the confidence intervals at 95% (Student's t test).
To investigate whether multimerization of LarR on the intergenic region is required for the regulation of the larA-E operon in vivo, mutated promoters were inserted upstream from the gusA reporter gene for measurements of transcriptional induction by l-Lac. Mutations of the conserved residue of the Lar box totally suppressed the l-Lac-induced Gus activity, in agreement with a central role for the Lar box in the transcriptional regulation of expression of lar genes (Fig. 5E, RRb). A similar effect was observed in the presence of mutations affecting the half-Lar boxes located at the PlarA side (Fig. 5E, RRc and RRc+RRd), demonstrating that LarR multimerization via the half-Lar boxes is required for induction by l-Lac.
l-Lac enhances binding and multimerization of LarR.
We first investigated the effect of l-Lac alone on the binding activity of rLarR (Fig. 6). EMSA showed that l-Lac stimulates the formation of the lower-molecular-weight complex C1 in the presence of a small amount of rLarR (Fig. 6A and B) and enhances HMWC at a higher rLarR concentration (Fig. 6A). To investigate whether enhanced HMWC formation in the presence of l-Lac results from rLarR binding to different sites along the intergenic region, we performed a DNase I footprinting experiment on a 127-bp radiolabeled PCR product which contains the region between the transcription start sites of PlarR and PlarA (+1 of PlarR was deduced from sequence analysis) (Fig. 6C and D; see also Fig. S2 in the supplemental material). When this probe was incubated in the presence of rLarR, the Lar box region was protected from DNase I degradation. With increasing concentrations of rLarR, the protection extended toward regions adjacent to the Lar box. Although protection was already observed in the absence of l-Lac, the addition of l-Lac to the reaction mixtures resulted in a significant extension of the protected regions, nearly spanning the entire intergenic region (Fig. 6C, 30 pmol LarR plus l-Lac, far right lane). Several positions, indicated by arrowheads in Fig. 6C, exhibited enhanced sensitivity to DNase I in the presence of rLarR, possibly reflecting rLarR-induced changes in the local DNA structure (33). One such sensitive position was found in the Lar box motif, as could be expected from rLarR binding at this position (34). Additional sensitive positions were observed in the adjacent regions (Fig. 6C). The rLarR-protected regions in footprinting assays and the positions of sites of enhanced susceptibility to DNase I treatment correlate well with the positions of the 4 putative half-Lar boxes surrounding the Lar box that were reported above (Fig. 4A and 6C). Together, the results of these in vitro experiments show the enhancing effect of l-Lac on the DNA binding activity of rLarR not only on the Lar box itself but also on adjacent regions containing half-Lar boxes, reinforcing the importance of multimerization for LarR activity.
FIG 6.

Effect of l-Lac on rLarR DNA binding. (A) EMSA of a 127-bp 32P-radiolabeled probe of the larR-larA intergenic region of L. plantarum in the presence of purified rLarR without or with 200 mM l-Lac (L). The lower-molecular-weight LarR-DNA complex (C1), unbound probe (P), and high-molecular-weight complexes (HMWC) are indicated. (B) Mean values of complexed DNA/free DNA ratios from EMSA such as that shown in panel A. Data are from 6 replicates from 1 significant experiment out of 3 experiments showing similar results. The error bars represent the standard deviations. Significance was determined by Student's t test: **, P < 0.01 compared to the results for the control. (C) DNase I footprint of a 127-bp 32P-radiolabeled probe of the larR-larA intergenic region of L. plantarum with increasing amounts of rLarR without or with 200 mM l-Lac (L). The C+T bands correspond to the Maxam and Gilbert sequencing reaction. A schematic representation of the larR-larA intergenic region is shown on the left. The lines on the right represent the protected regions, and the arrowheads indicate the enhancements. (D) Density measurements (A.U., arbitrary units) of the DNase I footprint shown in panel C. The areas under the curves of the data series have been standardized in order to be equal. Data are from 1 representative experiment out of 3 experiments showing similar results.
d-Lac antagonizes the l-Lac stimulation effect on LarR binding.
Since the d-Lac repression effect was lost with rLarR, we used Lactococcus lactis whole-cell extracts expressing the wild-type LarR (no StrepII tag) to investigate the effect of the l-/d-Lac ratio on LarR binding activity in vitro (Fig. 7). At a fixed concentration of 200 mM total lactate (Fig. 7A and B), a positive effect of l-Lac on LarR binding to the larR-larA intergenic region was observed, as reported above for rLarR, whereas d-Lac alone and the dl-Lac racemic mixture did not stimulate binding (Fig. 7A and B). EMSA performed at a fixed concentration of l-Lac (200 mM) in the presence of increasing amounts of d-Lac (0, 50, or 200 mM) demonstrated that the stimulatory effect of l-Lac on LarR binding is inhibited by d-Lac (Fig. 7C and D).
FIG 7.
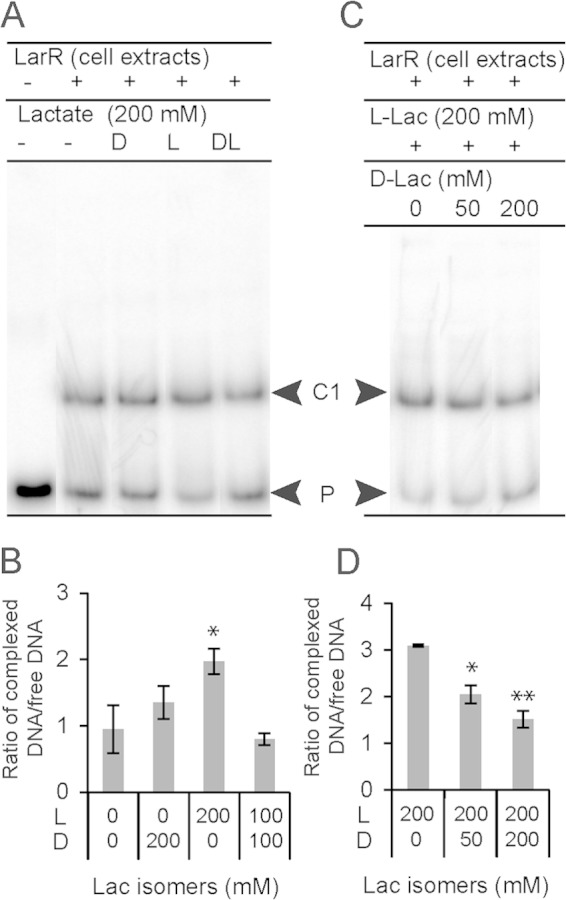
Effects of lactate isomers on untagged LarR DNA binding. (A) EMSA of a 127-bp 32P-radiolabeled probe of the larR-larA intergenic region of Lactobacillus plantarum in the presence of Lactococcus lactis cellular extracts (2 μg) containing untagged LarR without or with 200 mM d-Lac (D), l-Lac (L), or dl-Lac (DL, equimolar ratio). The LarR-DNA complex (C) and unbound probe (P) are indicated. (B) Mean values of complexed DNA/free DNA ratios from EMSA such as that shown in panel A. (C) EMSA of a 127-bp 32P-radiolabeled probe of the larR-larA intergenic region of L. plantarum in the presence of L. lactis cellular extracts (2 μg) containing untagged LarR with 200 mM Na-l-Lac supplemented with 0, 50, or 200 mM Na-d-Lac. (D) Mean values of complexed DNA/free DNA ratios from EMSA as shown in panel B. Data are from 3 replicates from 1 significant experiment out of 2 experiments showing similar results. The error bars represent the standard deviations. Significance was determined by Student's t test: *, P < 0.05; **, P < 0.01. Effects of lactate isomers are compared to the effect of the control (no lactate).
These in vitro assays performed with cell extracts containing the untagged version of LarR suggest that d-Lac may directly or indirectly act as an antagonist of l-Lac in its ability to stimulate LarR binding.
DISCUSSION
Because of the reaction it catalyzes, the lactate racemase of L. plantarum only has to be active when there is an imbalance between the two lactate isomers. Accordingly, a previous study demonstrated that Lar activity in L. plantarum is only transiently induced upon the addition of exogenous l-Lac and disappears as soon as the excess enantiomer (l-Lac) has been converted into d-Lac. The addition of exogenous d-Lac, however, is unable to induce Lar activity (13). In the present study, we unequivocally demonstrated that the l-/d-Lac ratio is the actual physiological signal that controls Lar expression, as a result of the inducing effect of l-Lac being counteracted by d-Lac (Fig. 1).
The regulation of Lar activity by the d-/l-Lac ratio was shown to occur at the transcriptional level and involves the transcriptional activator LarR, a new member of the PrfA group of regulators belonging to the Crp-Fnr family (see Fig. S1 in the supplemental material). To our knowledge, this is the first report of a member of this family being involved in lactate-dependent regulation. Since the binding of purified LarR was affected by l-Lac in vitro without the presence of any additional factor, l-Lac is most probably a direct effector molecule of LarR. Therefore, LarR would be the first regulator of the PrfA group whose effector has been identified. All previously characterized lactate-dependent transcriptional regulators belong either to the GntR (LldR regulators) or LysR (LlpR from Shewanella oneidensis) families. Among all lactate-dependent regulators, the regulation pattern of LarR is unique in that one isomer (l-Lac) acts as a positive effector whereas the other (d-Lac) antagonizes the positive effect of the first. Although some LldR regulators have been described to respond differentially to d- and l-Lac, none were demonstrated to display an antagonistic regulation by the two isomers: in Escherichia coli and Corynebacterium glutamicum, only l-Lac acts as an inducer of gene expression, whereas d-Lac has no effect (35, 36). Only LlpR from S. oneidensis was reported to display a regulation pattern reminiscent of that driven by LarR, with a positive regulation by l-Lac but no induction with dl-Lac; however, the molecular mechanism of this differential regulation was not investigated (37).
All necessary cis DNA elements for the LarR-driven regulation of Lar activity were found to be present in the larA-E intergenic region. A conserved DNA motif—the Lar box—was identified in this region, which was shown to be essential for the lactate-dependent regulation of PlarA via the binding of LarR. The Lar box consensus sequence is nearly identical to those of the PrfA and ACiD (RcfB) boxes (Fig. 2A). As suggested by the palindromic nature of the Lar box, LarR probably binds this motif as a dimer, like most members of the Crp-Fnr family (1). In addition, LarR was also found to bind secondary sites (half-Lar boxes) located between the Lar box and the −35 sequence of the larA promoter, resulting in a multimerization of LarR on the promoter region (Fig. 5 and 6). Half-Lar boxes surrounding the Lar box were identified in the larR-larA intergenic regions of other species where larR and larA are syntenic (see Fig. S4 in the supplemental material), suggesting that multimerization may be generalized to other LarR regulators.
The differential roles of the two lactate isomers in Lar activity in vivo could be reproduced in our in vitro DNA binding experiments: l-Lac stimulated the binding of LarR to the Lar box, whereas d-Lac counteracted this stimulatory effect (Fig. 6 and 7). l-Lac was also demonstrated to stimulate the multimerization of LarR on PlarA. Nevertheless, although the l-/d-Lac ratio had a dramatic effect on the Lar activity in vivo, the l-Lac-dependent stimulation of LarR binding and multimerization in vitro was relatively weak. Moreover, LarR was found to bind the Lar box DNA motif, even in the absence of its effector, and d-Lac did not negatively affect binding. If the only effect of l-Lac was to stimulate the binding and multimerization of LarR, one should expect that the overproduction of LarR alone would activate PlarA, even in the absence of l-Lac; however, such an effect was not observed in our complementation experiments, where LarR was overproduced using the nisin-inducible expression system. Altogether, these observations indicate that additional regulatory mechanisms must be involved. This situation is analogous to the behavior of the nitrogen starvation regulator NtcA, a well-characterized Crp-Fnr member that requires 2-oxoglutarate for its activation (38). NtcA binding to its DNA target sequence was previously reported to be only weakly stimulated by 2-oxoglutarate; rather, it was demonstrated that the effector is required for the activation of transcription initiation by NtcA (39). In most cases, regulators of the Crp-Fnr family bind a palindromic motif centered around position −41 relative to the transcriptional start (1), as classically found in class II Crp-dependent promoters (2). Similarly, if both half-Lar boxes at the larA side are occupied by LarR, the most downstream LarR dimer would also be centered around position −41 (Fig. 4A). This could allow the direct activation of the RNA polymerase.
Based on our results and on the current knowledge of the activation mechanism of Crp/Fnr members (using NtcA as a reference), we postulate the following model for PlarA activation by LarR (Fig. 8). At a high l-/d-Lac ratio, l-Lac would activate LarR through a conformational change, resulting in a stimulation of LarR binding on the Lar box as well as LarR multimerization on the half-Lar boxes. l-Lac would also promote the direct interaction of the proximal LarR dimer with the RNA polymerase, resulting in transcriptional activation. At a low l-/d-Lac ratio, the presence of d-Lac would maintain LarR in an inactive form—for instance, by competing with l-Lac for the same binding site—and, hence, limit LarR binding, multimerization, and interaction with the RNA polymerase.
FIG 8.

Hypothetical model of PlarA regulation by LarR. In the presence of l-Lac, activated LarR binds to the Lar box motif and multimerizes on the half-Lar boxes. This will promote direct interaction of one LarR dimer with the RNA polymerase, resulting in transcriptional activation of the PlarA (productive binding). In the presence of d-Lac, d-Lac could block LarR activation, for instance, by impairing l-Lac recognition as illustrated here, which will result in limited LarR binding and multimerization and absence of transcriptional activation (unproductive binding).
Regulation of gene expression is one mechanism employed by living organisms to tune energy expenses according to their physiological requirements in changing environmental conditions. Therefore, the regulation pattern of genes associated with a given function can shed light on its physiological role. For L. plantarum, we previously published expression data corresponding to the specific regulation pattern performed by LarR (induction by l-Lac but not by dl-Lac) (3). In addition to the larA-E and larR-Q operons, the accD2-A2 locus coding for the acetyl-coenzyme A (CoA) carboxylase was found to be similarly regulated (3). We performed an in silico search for Lar boxes in the L. plantarum genome using the Patser tool (see Materials and Methods), but we failed to identify a LarR binding site upstream from this locus, suggesting that it is not directly under the control of LarR. Five putative Lar boxes were found in intergenic regions outside the lar locus (data not shown). However, all of them are located upstream from genes that are unrelated to lactate or carbon metabolism (data not shown), and their LarR binding ability remains to be demonstrated. From these analyses, the function of LarR seems restricted to the induction of Lar in the presence of l-Lac but not in the presence of dl-Lac. Therefore, this unique regulation pattern of Lar implies that only the conversion of l- to d-Lac is physiologically relevant. The role of Lar activity may be linked to the absolute dependency of the peptidoglycan biosynthesis machinery toward d-Lac (13): lactate racemization would ensure d-Lac production even under conditions where the d-Lac dehydrogenase (LdhD) activity is impaired. A possible alternative role of Lar consistent with its regulation pattern would be the substitution of LdhD for the utilization of d-Lac as a carbon and energy source. In agreement with both possible roles, it has been reported that conditions of very slow growth in L. plantarum result in a lower abundance of ldhD transcripts and higher abundance of larA-E transcripts (40).
To conclude, we demonstrated that LarR is a positive regulator that controls Lar expression by sensing the l-/d-Lac ratio. We mapped its DNA binding sites and showed that its multimerization is required for transcription activation. Finally, we showed that both lactate isomers affect its binding, yet in an antagonistic way, which is a unique example of a regulatory circuit where two enantiomers of the same molecule are performing opposite roles on the same regulator. Future work will be dedicated to deciphering the molecular mechanisms involved in the recognition of lactate isomers by LarR.
Supplementary Material
ACKNOWLEDGMENTS
We warmly thank Lars Axelsson for providing plasmid pSIP409.
This work was supported by grants from the Belgian National Fund for Scientific Research (FNRS), the Interuniversity Attraction Poles Programme–Belgian Science Policy, the Communauté Française de Belgique—Actions de Recherches Concertées, and the UCL—Fonds Speciaux de Recherche. B.D., L.F., B.H., and P.H. are, respectively, a Research Fellow, Postdoctoral Researcher, Honorary Research Associate, and Senior Research Associate at FNRS.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02192-14.
REFERENCES
- 1.Korner H, Sofia HJ, Zumft WG. 2003. Phylogeny of the bacterial superfamily of Crp-Fnr transcription regulators: exploiting the metabolic spectrum by controlling alternative gene programs. FEMS Microbiol Rev 27:559–592. doi: 10.1016/S0168-6445(03)00066-4. [DOI] [PubMed] [Google Scholar]
- 2.Busby S, Ebright RH. 1999. Transcription activation by catabolite activator protein (CAP). J Mol Biol 293:199–213. doi: 10.1006/jmbi.1999.3161. [DOI] [PubMed] [Google Scholar]
- 3.Desguin B, Goffin P, Viaene E, Kleerebezem M, Martin-Diaconescu V, Maroney MJ, Declercq JP, Soumillion P, Hols P. 2014. Lactate racemase is a nickel-dependent enzyme activated by a widespread maturation system. Nat Commun 5:3615. doi: 10.1038/ncomms4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bienert GP, Desguin B, Chaumont F, Hols P. 2013. Channel-mediated lactic acid transport: a novel function for aquaglyceroporins in bacteria. Biochem J 454:559–570. doi: 10.1042/BJ20130388. [DOI] [PubMed] [Google Scholar]
- 5.Vega Y, Dickneite C, Ripio MT, Bockmann R, Gonzalez-Zorn B, Novella S, Dominguez-Bernal G, Goebel W, Vazquez-Boland JA. 1998. Functional similarities between the Listeria monocytogenes virulence regulator PrfA and cyclic AMP receptor protein: the PrfA* (Gly145Ser) mutation increases binding affinity for target DNA. J Bacteriol 180:6655–6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xayarath B, Volz KW, Smart JI, Freitag NE. 2011. Probing the role of protein surface charge in the activation of PrfA, the central regulator of Listeria monocytogenes pathogenesis. PLoS One 6:e23502. doi: 10.1371/journal.pone.0023502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reid SD, Montgomery AG, Musser JM. 2004. Identification of srv, a PrfA-like regulator of group A streptococcus that influences virulence. Infect Immun 72:1799–1803. doi: 10.1128/IAI.72.3.1799-1803.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giard JC, Riboulet E, Verneuil N, Sanguinetti M, Auffray Y, Hartke A. 2006. Characterization of Ers, a PrfA-like regulator of Enterococcus faecalis. FEMS Immunol Med Microbiol 46:410–418. doi: 10.1111/j.1574-695X.2005.00049.x. [DOI] [PubMed] [Google Scholar]
- 9.Madsen SM, Hindre T, Le Pennec JP, Israelsen H, Dufour A. 2005. Two acid-inducible promoters from Lactococcus lactis require the cis-acting ACiD-box and the transcription regulator RcfB. Mol Microbiol 56:735–746. doi: 10.1111/j.1365-2958.2005.04572.x. [DOI] [PubMed] [Google Scholar]
- 10.Doern CD, Holder RC, Reid SD. 2008. Point mutations within the streptococcal regulator of virulence (Srv) alter protein-DNA interactions and Srv function. Microbiology 154:1998–2007. doi: 10.1099/mic.0.2007/013466-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Riboulet-Bisson E, Sanguinetti M, Budin-Verneuil A, Auffray Y, Hartke A, Giard JC. 2008. Characterization of the Ers regulon of Enterococcus faecalis. Infect Immun 76:3064–3074. doi: 10.1128/IAI.00161-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reents H, Munch R, Dammeyer T, Jahn D, Hartig E. 2006. The Fnr regulon of Bacillus subtilis. J Bacteriol 188:1103–1112. doi: 10.1128/JB.188.3.1103-1112.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goffin P, Deghorain M, Mainardi JL, Tytgat I, Champomier-Verges MC, Kleerebezem M, Hols P. 2005. Lactate racemization as a rescue pathway for supplying d-lactate to the cell wall biosynthesis machinery in Lactobacillus plantarum. J Bacteriol 187:6750–6761. doi: 10.1128/JB.187.19.6750-6761.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Kranenburg R, Marugg JD, van Swam II, Willem NJ, de Vos WM. 1997. Molecular characterization of the plasmid-encoded eps gene cluster essential for exopolysaccharide biosynthesis in Lactococcus lactis. Mol Microbiol 24:387–397. doi: 10.1046/j.1365-2958.1997.3521720.x. [DOI] [PubMed] [Google Scholar]
- 15.Sorvig E, Mathiesen G, Naterstad K, Eijsink VG, Axelsson L. 2005. High-level, inducible gene expression in Lactobacillus sakei and Lactobacillus plantarum using versatile expression vectors. Microbiology 151:2439–2449. doi: 10.1099/mic.0.28084-0. [DOI] [PubMed] [Google Scholar]
- 16.de Ruyter PG, Kuipers OP, de Vos WM. 1996. Controlled gene expression systems for Lactococcus lactis with the food-grade inducer nisin. Appl Environ Microbiol 62:3662–3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kleerebezem M, Quadri LE, Kuipers OP, de Vos WM. 1997. Quorum sensing by peptide pheromones and two-component signal-transduction systems in Gram-positive bacteria. Mol Microbiol 24:895–904. doi: 10.1046/j.1365-2958.1997.4251782.x. [DOI] [PubMed] [Google Scholar]
- 18.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 19.Dower WJ, Miller JF, Ragsdale CW. 1988. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res 16:6127–6145. doi: 10.1093/nar/16.13.6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lambert JM, Bongers RS, Kleerebezem M. 2007. Cre-lox-based system for multiple gene deletions and selectable-marker removal in Lactobacillus plantarum. Appl Environ Microbiol 73:1126–1135. doi: 10.1128/AEM.01473-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holo H, Nes IF. 1989. High-frequency transformation, by electroporation, of Lactococcus lactis subsp. cremoris grown with glycine in osmotically stabilized media. Appl Environ Microbiol 55:3119–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Derzelle S, Hallet B, Francis KP, Ferain T, Delcour J, Hols P. 2000. Changes in cspL, cspP, and cspC mRNA abundance as a function of cold shock and growth phase in Lactobacillus plantarum. J Bacteriol 182:5105–5113. doi: 10.1128/JB.182.18.5105-5113.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goffin P, Lorquet F, Kleerebezem M, Hols P. 2004. Major role of NAD-dependent lactate dehydrogenases in aerobic lactate utilization in Lactobacillus plantarum during early stationary phase. J Bacteriol 186:6661–6666. doi: 10.1128/JB.186.19.6661-6666.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferain T, Hobbs JN Jr, Richardson J, Bernard N, Garmyn D, Hols P, Allen NE, Delcour J. 1996. Knockout of the two ldh genes has a major impact on peptidoglycan precursor synthesis in Lactobacillus plantarum. J Bacteriol 178:5431–5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pavan S, Hols P, Delcour J, Geoffroy MC, Grangette C, Kleerebezem M, Mercenier A. 2000. Adaptation of the nisin-controlled expression system in Lactobacillus plantarum: a tool to study in vivo biological effects. Appl Environ Microbiol 66:4427–4432. doi: 10.1128/AEM.66.10.4427-4432.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu H, Naismith JH. 2008. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol 8:91. doi: 10.1186/1472-6750-8-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Griffith KL, Wolf RE Jr. 2002. Measuring β-galactosidase activity in bacteria: cell growth, permeabilization, and enzyme assays in 96-well arrays. Biochem Biophys Res Commun 290:397–402. doi: 10.1006/bbrc.2001.6152. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt TG, Skerra A. 2007. The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat Protoc 2:1528–1535. doi: 10.1038/nprot.2007.209. [DOI] [PubMed] [Google Scholar]
- 29.Scortti M, Monzo HJ, Lacharme-Lora L, Lewis DA, Vazquez-Boland JA. 2007. The PrfA virulence regulon. Microbes Infect 9:1196–1207. doi: 10.1016/j.micinf.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 30.Hertz GZ, Stormo GD. 1999. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics 15:563–577. doi: 10.1093/bioinformatics/15.7.563. [DOI] [PubMed] [Google Scholar]
- 31.Ferain T, Garmyn D, Bernard N, Hols P, Delcour J. 1994. Lactobacillus plantarum ldhL gene: overexpression and deletion. J Bacteriol 176:596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leuze MR, Karpinets TV, Syed MH, Beliaev AS, Uberbacher EC. 2012. Binding motifs in bacterial gene promoters modulate transcriptional effects of global regulators CRP and ArcA. Gene Regul Syst Bio 6:93–107. doi: 10.4137/GRSB.S9357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hampshire AJ, Rusling DA, Broughton-Head VJ, Fox KR. 2007. Footprinting: a method for determining the sequence selectivity, affinity and kinetics of DNA-binding ligands. Methods 42:128–140. doi: 10.1016/j.ymeth.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 34.Lawson CL, Swigon D, Murakami KS, Darst SA, Berman HM, Ebright RH. 2004. Catabolite activator protein: DNA binding and transcription activation. Curr Opin Struct Biol 14:10–20. doi: 10.1016/j.sbi.2004.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aguilera L, Campos E, Gimenez R, Badia J, Aguilar J, Baldoma L. 2008. Dual role of LldR in regulation of the lldPRD operon, involved in l-lactate metabolism in Escherichia coli. J Bacteriol 190:2997–3005. doi: 10.1128/JB.02013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Georgi T, Engels V, Wendisch VF. 2008. Regulation of l-lactate utilization by the FadR-type regulator LldR of Corynebacterium glutamicum. J Bacteriol 190:963–971. doi: 10.1128/JB.01147-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brutinel ED, Gralnick JA. 2012. Preferential utilization of d-lactate by Shewanella oneidensis. Appl Environ Microbiol 78:8474–8476. doi: 10.1128/AEM.02183-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muro-Pastor MI, Reyes JC, Florencio FJ. 2001. Cyanobacteria perceive nitrogen status by sensing intracellular 2-oxoglutarate levels. J Biol Chem 276:38320–38328. doi: 10.1074/jbc.M105297200. [DOI] [PubMed] [Google Scholar]
- 39.Tanigawa R, Shirokane M, Maeda Si S, Omata T, Tanaka K, Takahashi H. 2002. Transcriptional activation of NtcA-dependent promoters of Synechococcus sp. PCC 7942 by 2-oxoglutarate in vitro. Proc Natl Acad Sci U S A 99:4251–4255. doi: 10.1073/pnas.072587199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goffin P, van de Bunt B, Giovane M, Leveau JH, Hoppener-Ogawa S, Teusink B, Hugenholtz J. 2010. Understanding the physiology of Lactobacillus plantarum at zero growth. Mol Syst Biol 6:413. doi: 10.1038/msb.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.