

Abstract
Interest in finding small RNAs (sRNAs) in bacteria has significantly increased in recent years due to their regulatory functions. Development of high-throughput methods and more sophisticated computational algorithms has allowed rapid identification of sRNA candidates in different species. However, given their various sizes (50 to 500 nucleotides [nt]) and their potential genomic locations in the 5′ and 3′ untranslated regions as well as in intergenic regions, identification and validation of true sRNAs have been challenging. In addition, the evolution of bacterial sRNAs across different species continues to be puzzling, given that they can exert similar functions with various sequences and structures. In this study, we analyzed the enrichment patterns of sRNAs in 13 well-annotated bacterial species using existing transcriptome and experimental data. All intergenic regions were analyzed by WU-BLAST to examine conservation levels relative to species within or outside their genus. In total, more than 900 validated bacterial sRNAs and 23,000 intergenic regions were analyzed. The results indicate that sRNAs are enriched in intergenic regions, which are longer and more conserved than the average intergenic regions in the corresponding bacterial genome. We also found that sRNA-coding regions have different conservation levels relative to their flanking regions. This work provides a way to analyze how noncoding RNAs are distributed in bacterial genomes and also shows conserved features of intergenic regions that encode sRNAs. These results also provide insight into the functions of regions surrounding sRNAs and into optimization of RNA search algorithms.
INTRODUCTION
Recently, small noncoding RNAs (sRNAs) have been under closer scrutiny as mediators and regulators of gene expression (1–5). This class of RNAs has been found to play a variety of roles in important cell functions (6, 7). Typically composed of 50 to 500 nucleotides, sRNAs are known to control plasmid replication, bacterial virulence, and various stress responses (8–11).
An interesting aspect of sRNAs is the wide diversity of their functional mechanisms. sRNAs can repress or stimulate gene expression posttranscriptionally by pairing their targets through base complementarity; a target can be, but is not limited to, an mRNA or a protein. sRNAs that regulate other RNAs can be cis encoded or trans encoded. A cis-encoded sRNA is typically encoded adjacent to its regulatory target on the same strand as a riboswitch or on the opposite strand to an antisense sRNA. In most cases, they will base pair to their targets or change the secondary structure to inhibit ribosome binding (12–14). In contrast, a trans-encoded sRNA is encoded away from its target, has a lower base complementarity to its target, and can potentially bind multiple targets (15).
With advances in high-throughput sequencing technologies (16), it is now possible to sequence gigabases of nucleotides in a matter of hours (17). Aided by sRNA prediction algorithms, these large data sets are paving the way for continual sRNA discovery (12, 18, 19). However, sRNA validation as well as determination of mechanistic function remains elusive. This is mainly due to the complexity of sRNA regulatory mechanisms. As a result, a plethora of computational approaches for sRNA prediction have gained popularity (20, 21). Some of the most widely used methods include eQRNA (22), RNAz (23), sRNAPredict3/SIPHT (24), and nucleic acid phylogenetic profiling (NAPP) (25). These methods rely on searches for a variety of patterns: compensatory mutations consistent with base-paired secondary structure, thermodynamic stability and structural conservation, regions of primary sequence conservation followed by transcriptional termination signals, and noncoding sequence clusters based on cross-genome conservation profiles. While different computational methods of sRNA identification include a multitude of criteria, even the most popularly applied methods tend to have low precision and sensitivity. Indeed, a previous study reported a mean precision between 4% and 12% for eQRNA, RNAz, sRNAPredict3, and NAPP across 10 data sets (20). Thus, a significant challenge stems from the fact that computational approaches tend to generate a large bank of potential sRNA sequences that result in only a few accurate hits.
Various approaches are routinely used to complement computational sRNA identification; these include cloning, high-throughput sequencing, Northern blotting, and microarray analysis. While microarray analysis has been the most common method for transcriptome analysis (26–28), this method is limited by indirect recording of expression levels and by typically not encompassing the entire transcriptome. Most recently, RNA sequencing (RNA-seq) has become a powerful technique (29–31). However, RNA-seq also has drawbacks, one of the major limitations being that certain sRNAs expressed during a particular cellular condition may not be present during cellular harvesting for RNA preparation. For the most part, Northern blotting has become an accepted method for verification of potential sRNA candidates that stem from prediction techniques and RNA-seq data. Even so, a significant amount of RNA is required for detection by Northern blotting, and sRNAs with lower copy numbers can be difficult to detect.
In this study, we performed a genome-wide analysis of conservation and length distribution patterns for all the intergenic regions in 13 selected species that have well-annotated genomes and experimental RNA-seq analysis with significant genome coverage (all greater than 50%). Using highly stringent criteria, we compared the query genomes to species both inside and outside their genera and determined the conservation level of all intergenic regions. Previous studies that have focused on the analysis of only the sRNA-coding regions do not indicate a consistent trend in sRNA conservation levels (24, 32–35). In this study, we took a different approach by considering the entire intergenic region where an sRNA is housed. We also analyzed the lengths of the intergenic regions where experimentally observed sRNAs were found in their native genomes. This large-scale study encompasses 13 different species for analysis of a total of more than 900 validated bacterial sRNAs and of more than 23,000 total intergenic regions. Our genome-wide analysis has yielded trends that provide clues to various questions regarding (i) how distant and/or independently trans-acting sRNAs have evolved from coding regions, (ii) how large intergenic regions that encode sRNAs are relative to the average size of intergenic regions in their native genomes, and (iii) how conserved sRNAs are relative to the intergenic regions where they are found.
This study takes advantage of detailed transcriptomic work that has now been completed in a diverse set of bacterial species with sequenced genomes. As such, this analysis contributes to our understanding of conservation patterns in sRNA-encoding intergenic regions and of sRNA evolution among bacterial species of various phylogenetic distances. This contributes new insights to possible refinement strategies that can improve current identification of transcribed intergenic sRNA sequences.
MATERIALS AND METHODS
Targeted bacterial species.
In this study, we selected 13 bacterial species: Bacillus subtilis 168, Chlamydia trachomatis L2b/UCH-1/proctitis, Enterococcus faecalis V583, Escherichia coli K-12 strain MG1655, Helicobacter pylori 26695, Listeria monocytogenes EGD, Mycobacterium bovis BCG Pasteur, Pseudomonas aeruginosa PAO1, Salmonella enterica subsp. enterica serovar Typhi Ty2, Staphylococcus aureus N315, Streptococcus pneumoniae TIGR4, Streptococcus pyogenes MGAS5005, and Vibrio cholerae El Tor. These species were selected due to the availability of detailed transcriptome analysis data that have been reported for their genomes using high-throughput sequencing or other traditional methods. The list of species, along with the Gram stain results, pathogenicity, and reference to the corresponding published transcriptome study, is given in Table S1 in the supplemental material.
Genome-wide extraction of intergenic and extended intergenic region sequences.
Data for all the sample genomes were found in the J. Craig Venter Institute (JCVI) database or in the National Center for Biotechnology Information (NCBI) genome database (36). To prevent conservation bias due to the presence of protein-coding sequences, the analysis of sRNA candidates was limited to sequences that were completely intergenic (as determined by the most recent genome annotations) and showed negligible overlap with nearby annotated open reading frames. Sequences that had up to a 10-nucleotide (nt) overlap upstream and/or downstream of the candidate sRNAs were included in the analysis, to accommodate for any potential annotation errors. The list of “extended intergenic regions” was generated by including a part of the upstream and downstream coding regions along with each intergenic region sequence. An intergenic region with a length of n nucleotides was extended for n nucleotides upstream and downstream. As a result, extended intergenic regions were three times the length of the original intergenic regions.
Conservation analysis of different genomes by BLAST.
WU-BLAST (BLASTN 2.0MP-WashU [4 May 2006]) (W. Gish, personal communication) was used to perform the sequence conservation analysis of intergenic and extended intergenic regions. Intergenic sequences with a minimum length of 60 nt were used to avoid spurious hits. However, the conservative expectation value (E value) established for the WU-BLAST analysis rarely returned hits for short (<60-nt) sequences when used with genome-sized databases. WU-BLAST outputs were filtered with a PERL script to a stringent threshold of at least 50% query sequence coverage with 50% identity in the conserved regions. The filtering restricted the hits for the search of homologous sequences to ones with a “high-to-extreme similarity” regime. These parameters were selected according to search criteria that have been developed to analyze conservation levels of protein-encoding sequences, where the expected level of conservation is much higher (37).
Two measures of conservation were used: “within-genus” and “outside-genus.” For the within-genus criterion, the homology of a specific sRNA candidate and/or intergenic region was determined relative to a list of specific genomes of species within the genus. For instance, for all C. trachomatis intergenic region sequences, homology was analyzed relative to Chlamydia psittaci, C. pneumonia, C. pecorum, and C. muridarum (members of the genus Chlamydia). The full list of genomes that apply to each species is included in Table S2 in the supplemental material. A measure of within-genus homology was obtained by counting the number of organisms within the genus where homology was observed. The length of all the query sequences, the resulting hit score, and the E value were summarized in Table S3 in the supplemental material.
For the outside-genus criterion, the homology of a specific sRNA candidate and/or intergenic region was determined relative to any species within a specific list of the following genera: Agrobacterium, Bacillus, Bacteroides, Bordetella, Borrelia, Brucella, Burkholderia, Chlamydia, Clostridium, Deinococcus, desulfobacteria, Enterobacter, Enterococcus, Escherichia, Geobacter, Haemophilus, Helicobacter, Lactobacillus, Listeria, Mycobacterium, Mycoplasma, Neisseria, Pseudomonas, Rhizobium, Rhodobacter, Rhodococcus, Rickettsia, Shigella, Salmonella, Streptococcus, Streptomyces, Staphylococcus, Synechococcus, Thermotoga, Vibrio, Xanthomonas, Yersinia, and Zymomonas. This list was generated as a way to further control the searches conducted for all sample species in a way that broadly sampled across all bacterial species. The length of all query sequences, the resulting hit score, and the E value were recorded as for the outside-genus analysis. The BLAST data are presented in Table S3 in the supplemental material.
The NCBI BLASTn discontiguous Megablast tool was used to determine sequence conservation of sRNA-coding regions (sRCR) and an adjacent random sequence in the same intergenic region (RIGR). Stringent conservation parameters were used: an E value of <0.001, ≥50% query coverage, and ≥50% identity. Using discontiguous Megablast, each sRNA-coding region and a random selected region of the respective intergenic region were analyzed. The number of hits returned from species of the same genus (within-genus group) and the number of hits returned of genera that differed from the target species (outside-genus group) are summarized in Table S4 in the supplemental material.
Collection of experimentally observed sRNAs from published works.
For each species analyzed with WU-BLAST, coordinates of experimentally observed sRNAs were collected from online databases or published reports (all sources used are listed in Tables S1 and S5 in the supplemental material). All pooled sRNAs were identified either by experimental techniques, such as Northern blotting or cloning, or by transcriptome sequencing techniques, such as RNA-seq or microarray analysis.
Phylogenetic distance calculation.
Phylogenetic distances were estimated by MEGA5 (Molecular Evolutionary Genetics Analysis), a tool for aligning sequences and computing nucleotide pairwise distances (38). 16S RNA sequences were retrieved from the NCBI database and aligned by ClustalW (a MEGA5 built-in algorithm). The P-distance model was used to estimate the phylogenetic distance between each species.
Comparisons of all intergenic regions with experimentally observed sRNAs.
The list of all intergenic regions generated from the JCVI or NCBI database was compared to all sRNAs that have been experimentally observed (see Table S1 in the supplemental material). Any intergenic region within the genome that contained one or more experimentally observed sRNAs was identified as an sRNA-coding intergenic region. Further criteria were applied to the data to explore any possible correlations between the likelihood of intergenic regions being sRNA-containing regions and the length or conservation level of those regions.
A survey of the longest intergenic regions is shown in Table S6 in the supplemental material, where the top 20% longest regions of all intergenic regions within a species were defined as “long intergenic regions.” Conservation data from the WU-BLAST analysis were also used to verify correlations between conserved intergenic regions and sRNA-coding intergenic regions. An intergenic region was considered conserved if the hit number returned by WU-BLAST was at least 1 and was higher than the hit number of the extended region.
RESULTS
Analysis of intergenic regions in different species shows conservation of a large number of intergenic regions.
To confirm the orthology of analyzed intergenic regions, we compared the conservation level of intergenic regions and extended intergenic regions. This eliminates the possibility that the intergenic region is coconserved with the adjacent protein-coding region, or potential untranslated regions (UTR) that are not annotated.
WU-BLAST was used to analyze the conservation level of all intergenic regions in the 13 selected bacterial species in this study (see Table S1 in the supplemental material). Our selection of the 13 species used for this study ensures that identification of sRNAs has been exhaustive since these species have all been well characterized at the transcriptome level. A list of “extended intergenic regions” for each species was created as a control for conservation analysis; Fig. 1 illustrates how extended intergenic regions were determined. The extended region includes parts of the upstream and downstream coding regions that are the same size as the original intergenic region along with the intergenic sequence, so that the combined region is three times the size of the original intergenic region. The parameters and criteria for conservation analysis are described in Materials and Methods. In brief, an intergenic or extended intergenic region is considered conserved in a species if a hit is returned by WU-BLAST and satisfies the filter criteria. Conservation levels of within-genus and outside-genus regions were calculated separately. The within-genus criterion refers to all species that are in the same genus as the analyzed species, and the outside-genus criterion refers to a group that includes 38 species from different genera (see Table S2 in the supplemental material for the full list). Since some genera have a limited number of species, such as Escherichia, this analysis can broaden the diversity of species for WU-BLAST and provide insight into how phylogenetic distance can affect the results of conservation analysis. Only intergenic regions with a conservation level equal to or higher than that of the extended intergenic regions are defined as “conserved intergenic regions.”
FIG 1.
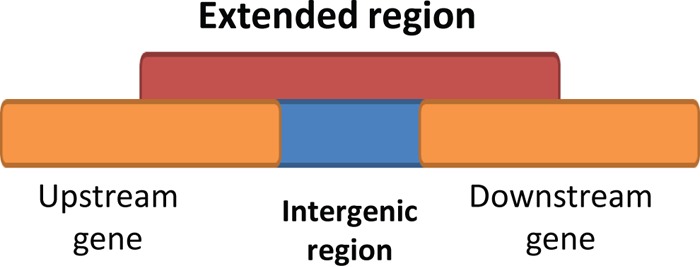
Extended intergenic region. The orange bars indicate the upstream and downstream protein-coding regions, the blue bar indicates the intergenic region, and the red bar indicates the extended intergenic region, which includes the intergenic region along with a part of the upstream and downstream region that equals the length of the intergenic region. The intergenic regions and extended regions were analyzed with WU-BLAST and compared with each other for conservation-level analysis.
Figure 2 shows representative within-genus and outside-genus conservation patterns of three selected species (others are shown in Fig. S1 in the supplemental material). All intergenic regions were grouped into nonconserved, equally conserved, and more highly conserved (where conservation levels are lower than, equal to, or higher than those of extended intergenic regions, respectively). Results show that while most intergenic regions (68 to 82%) are not conserved within-genus or outside-genus among the species studied, a large enough fraction of all intergenic regions are either equally conserved or highly conserved relative to the surrounding (gene-carrying) regions; the latter cases were of the most interest, as we aimed to analyzed enrichment patterns of sRNAs in exceptionally conserved intergenic regions. Raw data from WU-BLAST can be found in Table S3 in the supplemental material. To fully understand how heterogeneously distributed conserved intergenic regions were among species surveyed, we tabulated the distribution of all conserved intergenic regions among all with within-genus species. According to unique conservation patterns that were observed, we classified the target species into two categories: group 1 includes the majority of all species analyzed, where the main characteristic is that most intergenic regions are conserved in a way that is not specific to a single species. In contrast, group 2 is characterized by having most of its intergenic regions (>50%) conserved in a limited set of species. The two species that fall into this category are M. bovis, which has a 63.8% of its conserved intergenic regions conserved only in Mycobacterium tuberculosis, and C. trachomatis, which has 83.4% of its conserved intergenic regions conserved only in C. muridarum.
FIG 2.

Conservation patterns of intergenic regions in selected species. The figure shows the conservation level distribution of intergenic regions in three selected species (see Fig. S1 in the supplemental material for all other species). The conservation level is the number of within-genus or outside-genus organisms found to have homology of the intergenic region. The intergenic region would be marked as “nonconserved” if its conservation level is less than that of the extended intergenic region or as “equally conserved” or “more highly conserved” if the conservation level is equal to or higher than that of the extended intergenic region. The pie charts show how conservation levels are distributed in the more highly conserved intergenic regions, and the total numbers of within-genus organisms are shown above them.
Conserved intergenic regions are enriched for small RNAs.
To understand if sRNAs were more likely encoded by conserved intergenic regions, we first cross-referenced the reported sRNA coordinates to all the intergenic regions for each species. The pools of experimentally observed sRNAs that were identified via transcriptome or Northern blotting for each species were collected from published works and online databases. References for all sRNAs collected are listed in Table S1 in the supplemental material (39–81). Experimentally observed sRNAs were mapped to their corresponding genomes to identify sRNA encoding regions. The antisense sRNAs are beyond the scope of this study and are excluded from our analysis. An important general observation that stems from this analysis (see Table S1 in the supplemental material) is that a range of ∼2% to 12% of all intergenic regions encode sRNAs. This is close to computational and experimental estimations of sRNAs in bacteria (82).
After mapping all experimentally observed sRNAs to their corresponding intergenic regions, we determined the percentage of within-genus or outside-genus nonconserved, conserved, and higher-conserved intergenic regions that encoded sRNAs. The percentages of sRNA-coding intergenic regions for all 13 species are showed in Fig. 3. For the within-genus analysis, most of the species have a greater percentage of sRNA-coding regions in conserved intergenic regions than in nonconserved intergenic regions. We used Fisher's exact test to test the statistical significance (P < 0.05), and all but one species (M. bovis) show significant sRNA enrichment. Importantly, the more highly conserved intergenic regions in most species are even more enriched for sRNAs than the nonconserved intergenic regions, indicating that sRNAs are more likely to be encoded within highly conserved intergenic regions. Our general findings of sRNA enrichment in conserved intergenic regions in outside-genus species compared to nonconserved intergenic regions (see Fig. S2 in the supplemental material) further support these results
FIG 3.

Enrichment of sRNAs in outside-genus conserved intergenic regions. The percentage is defined as the number of sRNA-coding intergenic regions relative to nonconserved, equally conserved, or more highly conserved (outside-genus) intergenic regions. A conserved intergenic region refers to any intergenic region that has a conservation level equal to or higher than that of the extended intergenic region. A single asterisk denotes statistically significant enrichment of sRNA compared to nonconserved regions by Fisher's exact test (P < 0.05), and double asterisks denote statistically significant enrichment of sRNA compared to the conserved intergenic region.
It is interesting that the enrichment of sRNA-coding regions is not as significant as the within-genus analysis across all species (as determined by Fisher's exact test). This is particularly the case with species that exhibit that fall into the second conservation pattern group, where conservation is observed among only a very limited set of species (i.e., C. trachomatis and M. bovis). It is also possible that many sRNAs remain unidentified in these species. We suspect that this might be the case in species such as C. trachomatis and E. faecalis, where the percentage of all intergenic regions that has been identified as encoding sRNAs remains lower than 3% and the number of reported sRNAs remains low.
Refined conservation analysis based on phylogenetic distance strengthens observations of sRNA enrichment in conserved intergenic regions.
To investigate how phylogenetic distance affects the enrichment of sRNAs in conserved intergenic regions, we selected two species, B. subtilis and S. pneumoniae, and analyzed how intergenic regions were conserved across differently phylogenetically distant sets of species. These species were selected due to the larger number of identified sRNAs and the larger set of within-genus species that has been sequenced and can be used as a basis for conservation analysis. For this analysis, we used MEGA5 to compute the phylogenetic distances between the specific species of interest (e.g., B. subtilis) and the respective within-genus species (e.g., other Bacillus species, listed in Table S2 in the supplemental material). As shown in Fig. 4, a wide variation in evolutionary spread was observed among the species we tested. For instance, the distances between all the within-genus Bacillus species and B. subtilis range from 0.019 (Bacillus amyloliquefaciens) to 0.123 (Bacillus pseudofirmus). In contrast, the phylogenetic spread is lower in S. pneumoniae (0.004 to 0.077) than in other Streptococcus species. As such, the latter genus clusters more closely in terms of phylogenetic distance than the Bacillus species. As a reference, the phylogenetic distance from E. coli (a bacterium from a different genus) was evaluated for the three selected organisms to gain a sense of how these evolutionary measurements could be interpreted. The distance for B. subtilis is 0.231, and that for S. pneumoniae is 0.229.
FIG 4.

sRNA-coding-region enrichments in intergenic regions conserved in species with different phylogenetic distances and sRNAs enrichments with a refined species set. The phylogenetic distances between the within-genus species were calculated for B. subtilis and S. pneumoniae with MEGA5. The percentages of all sRNA-encoding intergenic regions that are conserved in a certain species were also calculated. For instance, in B. subtilis, 9.3% of the intergenic regions that are conserved in B. amyloliquefaciens (dot 1) were found to encode sRNAs. The mean distance (MD) and standard deviation (STD) to the within-genus species were calculated for B. subtilis and S. pneumoniae and marked in each graph. The following species were included in the plot. (Top) 1, B. amyloliquefaciens; 2, B. atrophaeus; 3, B. licheniformis; 4, B. pumilus; 5, B. anthracis; 6, B. cereus; 7, B. halodurans; 8, B. megaterium; 9, B. weihenstephanensis; 10, B. thuringiensis; 11, B. clausii; 12, B. selenitireducens; 13, B. pseudofirmus. (Bottom) 1, S. mitis; 2, S. oralis; 3, S. sanguinis; 4, S. gordonii; 5, S. parasanguinis; 6, S. salivarius; 7, S. constellatus; 8, S. pasteurianus; 9, S. intermedius; 10,S. lutetiensis; 11, S. gallolyticus; 12, S. macedonicus; 13, S. infantarius; 14, S. iniae; 15, S. dysgalactiae; 16, S. agalactiae; 17, S. mutans; 18, S anginosus; 19, S. suis; 20, S. equi; 21, S. uberis; 22, S. parauberis; 23, S. pyogenes. A refined set of organisms was selected by phylogenetic distance. Any species within one standard deviation (within the boxed area)of the mean distance was included in the analysis. The percentages of sRNA-coding intergenic regions in conserved and nonconserved intergenic regions were calculated and compared. The asterisk denotes enrichment of sRNA compared to nonconserved regions as determined by Fisher's exact test (P < 0.05), and the pound sign denotes a statistically significant difference compared to conserved intergenic regions of all included species.
The level of enrichment for intergenic regions that encode sRNAs was analyzed relative to the phylogenetic distance of within-genus species for Bacillus, and Streptococcus (Fig. 4). The sRNA-coding-region percentages were calculated for intergenic regions that were conserved in different species and plotted against the phylogenetic distance for each within-genus species. A positive correlation of sRNA enrichment and phylogenetic distance was observed, indicating that intergenic regions, which are conserved in more distant species, are more likely to encode sRNA. This trend was consistent up to a certain threshold distance, where the species were too distant to have significant homology. Representative data are shown for B. subtilis and S. pneumoniae in Fig. 4.
A pattern observed in some species from our phylogenetic-distance analysis was that certain within-genus species that were analyzed appeared significantly closer (in evolutionary distance) to the species under analysis than the rest of the within-genus species. One example was S. pneumoniae, for which two outlier species (Streptococcus mitis and Streptococcus oralis) were more than two standard deviations farther than the mean distance of all other within-genus species (Fig. 4). The conservation analysis also showed that a large number (71.4%) of all the intergenic regions in S. pneumoniae are also conserved in S. mitis. In this case, we rationalized that intergenic region conservation likely stems from general genome-wide conservation and not from any functional sRNA feature that poses an evolutionary advantage to these species. We reasoned that this could weaken our ability to observe true sRNA enrichment in conserved intergenic regions, since conservation of intergenic regions among organisms that are evolutionarily very close may not possess biological importance.
To evaluate the effect of phylogenetic distance on the sRNA enrichment observed in intergenic regions, we repeated the conservation analysis with a refined set of target organisms. The refined set includes only species with a phylogenetic distance within one standard deviation from the mean phylogenetic distance. As shown in Fig. 5, in the context of S. pneumoniae and B. subtilis, a statistically significant enrichment of sRNAs in conserved intergenic regions was observed after the outliers were removed. These results were consistent with those for other species analyzed, where species that were originally too close or too far were used to determine conservation. These results demonstrated that the refinement of the set of organisms used in the conservation studies to a more appropriate phylogenetic distance results in even higher sRNA enrichment in conserved intergenic regions. Importantly, these data also highlight the importance of selecting an appropriate set of species to make valid conclusions regarding sRNA conservation.
FIG 5.

Illustration of the sRNA-coding region in an intergenic region and comparison of conservation levels. (A) Sketch of an sRNA-coding intergenic region (sRCR) and a randomly selected, nonoverlapping region of the same length as the sRCR in the same intergenic region (RIGR) used for comparison of conservation levels. (B) Comparisons of the conservation levels of sRCRs and corresponding RIGRs. The percentages show how many sRCRs have a conservation level that is greater than, equal to, or lower than that of the respective RIGRs. For instance, 69.2% of the sRCRs have a greater conservation level than the RIGRs, while 15.4% of the sRCRs have a lower conservation level than the respective RIGRs.
Conservation of sRNAs relative to conservation of flanking coding regions.
Since sRNA-encoding intergenic regions were observed to be more conserved than all other intergenic regions, we wanted to test whether conservation was specific to regions encoding sRNAs. This initially caught our attention due to the high number of sRNAs that we observed to be encoded from a small fraction of all intergenic regions, leaving a large fraction of all intergenic regions seemingly idle. For this analysis, we determined the relationship between the conservation levels of sRNA coding regions (sRCRs) and random intergenic regions (RIGRs) (Fig. 5A). A random intergenic region was defined as a segment of the intergenic region of the same length as the sRNA-coding region with no overlapping sequences.
In order to collect statistically meaningful data, we first generated a list of suitable target genomes in which a high number of conserved intergenic regions were more than double the length of the encoding sRNA in that same region. We selected Bacillus subtilis 168 and Streptococcus pyogenes MGAS5005 for this analysis because each species included over 25 conserved intergenic regions that met the set criteria. These species were analyzed for conservation against two groups, the within-genus and outside-genus groups, as described above. Intergenic regions that contained sRNAs with lengths that were <40% of the entire intergenic region were considered for analysis. Figure 5B shows analysis of 61 conserved intergenic regions in B. subtilis and 28 in S. pyogenes, where 65% and 69% of sRNA-encoding regions, respectively, were more conserved than the respective RIGR control (see Table S4 in the supplemental material). Importantly, this result indicates that fragments that encode sRNAs are significantly more conserved than a random region of the same size within the same conserved intergenic region. This interesting result supports our underlying hypothesis that conserved intergenic regions are enriched in sRNAs, as these represent biologically important regions that are beneficial to bacteria.
Isolated genomic regions are enriched in sRNAs.
One last interesting question that we explored concerned the presence of sRNAs in large intergenic regions that were previously thought to be noncoding. We suspect that these large intergenic regions isolated from protein coding regions potentially serve some purpose. After examination of all 13 genomes in this study, we found that the size distributions of all their intergenic regions are highly similar (see Fig. S3 in the supplemental material) despite pronounced differences in their genome sizes (ranging from 1 to 6.8 million nucleotides). We therefore speculated that in addition to conservation, the presence of isolated (long intergenic) regions in the genome could be another signature of the presence of sRNAs. Given the recent findings of a large number of noncoding RNAs in bacterial genomes, it is also informative to determine what percentage of the genome is indeed noncoding. Our analysis of long intergenic regions (as defined by the top 20% longest intergenic regions), showed significant sRNA enrichment for all species analyzed compared to that of all the intergenic regions (Fig. 6).
FIG 6.

Enrichment of sRNAs in long and conserved intergenic regions (IGR). The percentage is defined as the number of sRNA-coding intergenic regions relative to long intergenic regions (top 20% long), long and conserved (within-genus) intergenic regions, and other intergenic regions. A conserved intergenic region refers to any intergenic region that has a conservation level equal to or higher than that of the extended intergenic region. The asterisk denotes statistically significant enrichment of sRNA compared to other regions, as determined by Fisher's exact test (P < 0.05), and double asterisks denote values that are statistically significant relative to those for the long intergenic region.
Figure 6 also shows the combined enrichment effect we observed for long and conserved intergenic regions. For this analysis, we calculated the percentage of sRNAs found in intergenic regions of the longest 20% of regions that are also conserved or highly conserved (within-genus and outside-genus). We consistently observed a significant level of sRNA enrichment in intergenic regions that were both conserved and long.
DISCUSSION
Advances in experimental and computational techniques have led to continual identification of a vast number of sRNAs in bacteria. We now understand that sRNA structures and sequences can be conserved between evolutionarily close organisms (83). However, conservation patterns of functional sRNAs are more complex than those observed in coding regions. For example, some sRNAs are always coconserved adjacent to coding regions, other sRNAs have similar sequences but perform different roles in different organisms, and, even in the same organism, some sRNAs can have multiple genomic copies that have different regulatory functions (84). Thus far, most of the conservation properties of bacterial sRNAs are not well understood. Given this, the evolution of bacterial sRNAs continues to be puzzling; this is particularly intriguing in the case of intergenic sRNAs that have evolved outside genomic coding regions.
For our analysis, we collected data for experimentally observed sRNAs in intergenic regions from 13 different bacterial species that have been widely studied and well annotated. Given the dependency of this analysis on selected species whose sRNAs we used and collected, we collected a vast amount of data to ensure statistical significance. Despite our selection of species that possess a well-annotated genome, have more comprehensive transcriptome data, and are more commonly used in sRNA studies and our use of only experimentally observed sRNAs, our data could be inherently biased based on our current selection of bacteria that have been sequenced and characterized extensively for medical or biotechnological purposes. Moreover, sRNAs that were identified by different techniques could weigh differently, and some regulatory sRNAs may be expressed only under certain environmental conditions. While ideally this study can be done with sRNAs that all come from the same experimental technique (such as Northern blotting), this would yield only a relatively small number of sRNA candidates in some species that lack large-scale Northern blotting confirmation. Given the large numbers of sRNAs and the broad sample of organisms analyzed that validate the trends that we have observed, we believe that these patterns will hold for an even larger and more comprehensive data set. Furthermore, to assess the possible conservation bias from different techniques, we compared the conservation level of intergenic regions that encode sRNAs identified from Northern blotting to that of sRNAs identified with other techniques (microarray, RNA-seq, etc.) and found no significant difference in the conservation levels between these two groups of sRNAs (Table S5 in the supplemental material shows the classification of sRNAs according to how they were experimentally identified).
A second key observation that results from our work is that intergenic regions that are conserved are enriched for sRNAs relative to nonconserved intergenic regions. Since some sRNA might be conserved along with adjacent coding regions, and to eliminate the possibility that high conservation levels of intergenic regions are due to 5′ or 3′ UTRs, we define as conserved only the intergenic regions that have a higher conservation level than flanking regions. Since most intergenic regions carry functional sequences, they are expected to be less conserved than protein-coding regions. This is a different approach from others that have been used in the literature to study conservation of intergenic regions (85, 86). In most of the analyzed species, more than 20% of the intergenic regions have an conservation level equal to or higher than that of the extended region. As a result, it is possible that more functional sequences are yet to be identified in these highly conserved intergenic regions. These results support the hypothesis that intergenic regions that are conserved across multiple species encode functional entities that are important for survival. This is further stressed by our findings that the actual sRNA-encoding regions are even more conserved than random regions within the same intergenic area.
The above results also depend on the technicalities of the WU-BLAST analysis. We used two different groups for WU-BLAST: the within-genus and outside-genus groups. The two groups yielded similar results, indicating that the number of species (outside-genus groups include more species than most genera) is not a critical parameter in this analysis. We hypothesized that by using an optimal phylogenetic distance to select species for WU-BLAST, we could eliminate species that are too close or too far from the interested species and yield more significant results. Our analysis of B. subtilis and S. pneumoniae supports this idea, while it was less significant for other species (data not shown), as these appeared to be evolutionarily clustered within a more optimal distance. Nevertheless, we believe that this approach can be further improved by systematically performing a cross-genus analysis to find the optimal phylogenetic distance applied for all species. The dependency on appropriate phylogenetic distance for conservation analysis is not surprising given that phylogenetic distances that are too close will obscure identification of intergenic regions that are truly conserved due to the potential importance of their encoded function. In contrast, organisms that are phylogenetically too far away will not show enough conservation among intergenic regions for meaningful analysis.
A third observation of our study is that the average sizes and distributions of intergenic-region lengths are very similar among the species analyzed, regardless of their genome size. Furthermore, intergenic areas that are significantly longer than the average are largely enriched in sRNAs. Indeed, this trend was observed to increase as intergenic regions increased in length. This suggests that bacteria use their genome space highly efficiently, without the presence of large “unused regions” that do not encode functional transcripts. Interestingly, not many intergenic regions in our analysis were observed to encode more than one sRNA, and the few intergenic regions that did encode multiple sRNAs (no more than two) were not significantly longer. A more fundamental question is whether these long intergenic regions are long because they encode sRNAs or whether sRNAs are more likely to be encoded in long intergenic regions. Based on this study, we believe that most long intergenic regions could have encoded functional sequences. This is not limited to sRNAs but also applies other functional noncoding transcripts or sequences in other organisms (87, 88). Long intergenic regions have more space to house noncoding RNAs, and it would be interesting to look for unknown sRNAs in long intergenic regions in which no functional transcripts have been found yet.
In summary, the evolution of sRNA in bacteria is an intriguing subject. A major challenge in this field is that some sRNAs with the same function could have different sequences in different organisms, or the same sRNA sequence could have different functions in different organisms. This study provides insight into some critical questions that remain unanswered about sRNA evolution in bacteria. A future approach could incorporate the use of structural homology prediction models in addition to sequence homology methods to better identify and understand sRNA conservation patterns in terms of function (89). An advantage of the strategy we have used is the ability to look at sRNAs in the context of the entire genetic region in which they are found. Consideration of the complete intergenic regions could potentially simplify the identification of sRNAs, since many of these surrounding regions could have additional biological functions that are yet to be understood.
Supplementary Material
ACKNOWLEDGMENTS
We thank Joe Wade for the insightful discussion and Marlene Belfort for supporting the initial collection of data for this work.
We are grateful to the University of Texas at Austin for an undergraduate research fellowship to R.L. This work was supported by the Welch Foundation (F-1756), the Defense Threat Reduction Agency Young Investigator Program (HDTRA1-12-0016), the National Institutes of Health (GM39422), and the National Science Foundation CAREER program (CBET-1254754).
We have no competing interests to declare.
C.-H.T. carried out collection of the transcriptome data, analysis of long intergenic regions, analysis of the conservation data, and overall statistical analysis and drafted the manuscript. R.L. helped C.-H.T. with some of the above-described analysis, carried out the phylogenetic analysis and the comparison of the small-RNA coding region and flanking intergenic region, and helped to draft the manuscript. B.C. helped R.L. with the small-RNA coding region analysis and helped to draft the manuscript. M.P. carried out the WU-BLAST analysis and helped to revise the manuscript. L.M.C. conceived of the study and participated in the statistical analysis and drafting of the manuscript. All authors read and approved the final manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02359-14.
REFERENCES
- 1.Storz G, Vogel J, Wassarman KM. 2011. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 43:880–891. doi: 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Storz G, Haas D. 2007. A guide to small RNAs in microorganisms. Curr Opin Microbiol 10:93–95. doi: 10.1016/j.mib.2007.03.017. [DOI] [Google Scholar]
- 3.Pellin D, Miotto P, Ambrosi A, Cirillo DM, Di Serio C. 2012. A genome-wide identification analysis of small regulatory RNAs in Mycobacterium tuberculosis by RNA-Seq and conservation analysis. PLoS One 7:e32723. doi: 10.1371/journal.pone.0032723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ji L, Chen X. 2012. Regulation of small RNA stability: methylation and beyond. Cell Res 22:624–636. doi: 10.1038/cr.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gottesman S, Storz G. 2011. Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harb Perspect Biol 3:a003798. doi: 10.1101/cshperspect.a003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suzuki T, Ueguchi C, Mizuno T. 1996. H-NS regulates OmpF expression through micF antisense RNA in Escherichia coli. J Bacteriol 178:3650–3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waters LS, Storz G. 2009. Regulatory RNAs in bacteria. Cell 136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiao B, Li W, Guo G, Li B, Liu Z, Jia K, Guo Y, Mao X, Zou Q. 2009. Identification of small noncoding RNAs in Helicobacter pylori by a bioinformatics-based approach. Curr Microbiol 58:258–263. doi: 10.1007/s00284-008-9318-2. [DOI] [PubMed] [Google Scholar]
- 9.Guillier M, Gottesman S. 2006. Remodelling of the Escherichia coli outer membrane by two small regulatory RNAs. Mol Microbiol 59:231–247. doi: 10.1111/j.1365-2958.2005.04929.x. [DOI] [PubMed] [Google Scholar]
- 10.Wagner G, Simons RW. 1994. Antisense RNA control in bacteria, phages, and plasmids. Annu Rev Microbiol 48:713–742. doi: 10.1146/annurev.mi.48.100194.003433. [DOI] [PubMed] [Google Scholar]
- 11.Majdalani N, Cunning C, Sledjeski D, Elliott T, Gottesman S. 1998. DsrA RNA regulates translation of RpoS message by an anti-antisense mechanism, independent of its action as an antisilencer of transcription. Proc Natl Acad Sci U S A 95:12462–12467. doi: 10.1073/pnas.95.21.12462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Backofen R, Hess WR. 2010. Computational prediction of sRNAs and their targets in bacteria. RNA Biol 7:33–42. doi: 10.4161/rna.7.1.10655. [DOI] [PubMed] [Google Scholar]
- 13.Isaacs FJ, Dwyer DJ, Collins JJ. 2006. RNA synthetic biology. Nat Biotechnol 24:545–554. doi: 10.1038/nbt1208. [DOI] [PubMed] [Google Scholar]
- 14.Liang JC, Bloom RJ, Smolke CD. 2011. Engineering biological systems with synthetic RNA molecules. Mol Cell 43:915–926. doi: 10.1016/j.molcel.2011.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Papenfort K, Vogel J. 2009. Multiple target regulation by small noncoding RNAs rewires gene expression at the post-transcriptional level. Res Microbiol 160:278–287. doi: 10.1016/j.resmic.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 16.Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin D, Lu L, Law M. 2012. Comparison of next-generation sequencing systems. J Biomed Biotechnol 2012:251364. doi: 10.1155/2012/251364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacLean D, Moulton V, Studholme DJ. 2010. Finding sRNA generative locales from high-throughput sequencing data with NiBLS. BMC Bioinformatics 11:93. doi: 10.1186/1471-2105-11-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sridhar J, Gunasekaran P. 2013. Computational small RNA prediction in bacteria. Bioinform Biol Insights 7:83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Livny J. 2012. Bioinformatic discovery of bacterial regulatory RNAs using SIPHT, p 3–14. In Keiler KC. (ed), Bacterial regulatory RNA: methods and protocols. Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 20.Lu X, Goodrich-Blair H, Tjaden B. 2011. Assessing computational tools for the discovery of small RNA genes in bacteria. RNA 17:1635–1647. doi: 10.1261/rna.2689811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pichon C, du Merle L, Caliot ME, Trieu-Cuot P, Le Bouguénec C. 2012. An in silico model for identification of small RNAs in whole bacterial genomes: characterization of antisense RNAs in pathogenic Escherichia coli and Streptococcus agalactiae strains. Nucleic Acids Res 40:2846–2861. doi: 10.1093/nar/gkr1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rivas E, Eddy SR. 2001. Noncoding RNA gene detection using comparative sequence analysis. BMC Bioinformatics 2:8. doi: 10.1186/1471-2105-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Washietl S, Hofacker IL. 2004. Consensus folding of aligned sequences as a new measure for the detection of functional RNAs by comparative genomics. J Mol Biol 342:19–30. doi: 10.1016/j.jmb.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 24.Livny J, Waldor MK. 2007. Identification of small RNAs in diverse bacterial species. Curr Opin Microbiol 10:96–101. doi: 10.1016/j.mib.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 25.Marchais A, Naville M, Bohn C, Bouloc P, Gautheret D. 2009. Single-pass classification of all noncoding sequences in a bacterial genome using phylogenetic profiles. Genome Res 19:1084–1092. doi: 10.1101/gr.089714.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akama T, Suzuki K, Tanigawa K, Kawashima A, Wu H, Nakata N, Osana Y, Sakakibara Y, Ishii N. 2009. Whole-genome tiling array analysis of Mycobacterium leprae RNA reveals high expression of pseudogenes and noncoding regions. J Bacteriol 191:3321–3327. doi: 10.1128/JB.00120-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu Z, Zhang A, Storz G, Gottesman S, Leppla SH. 2006. An antibody-based microarray assay for small RNA detection. Nucleic Acids Res 34:e52. doi: 10.1093/nar/gkl142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altuvia S. 2007. Identification of bacterial small non-coding RNAs: experimental approaches. Curr Opin Microbiol 10:257–261. doi: 10.1016/j.mib.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 29.Wilms I, Overlöper A, Nowrousian M, Sharme CM, Narberhaus F. 2012. Deep sequencing uncovers numerous small RNAs on all four replicons of the plant pathogen Agrobacterium tumefaciens. RNA Biol 9:446–457. doi: 10.4161/rna.17212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Z, Gerstein M, Snyder M. 2009. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gelderman G, Contreras LM. 2013. Discovery of Posttranscriptional regulatory RNAs using next generation sequencing technologies. Methods Mol Biol 985:269–295. doi: 10.1007/978-1-62703-299-5_14. [DOI] [PubMed] [Google Scholar]
- 32.Pang KC, Frith MC, Mattick JS. 2006. Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Genome Anal 22:1–5. doi: 10.1016/j.tig.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 33.Voß B, Georg J, Schön V, Ude S, Hess WR. 2009. Biocomputational prediction of non-coding RNAs in model cyanobacteria. BMC Genomics 10:123. doi: 10.1186/1471-2164-10-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Washietl S, Hofacker IL, Stadler PF. 2005. Fast and reliable prediction of noncoding RNAs. Proc Natl Acad Sci U S A 102:2454–2459. doi: 10.1073/pnas.0409169102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A. 2013. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res 41:D226–D232. doi: 10.1093/nar/gks1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davidsen T, Beck E, Ganapathy A, Montgomery R, Zafar N, Yang Q, Madupu R, Goetz P, Galinsky K, White O, Sutton G. 2010. The comprehensive microbial resource. Nucleic Acids Res 38:D340–D345. doi: 10.1093/nar/gkp912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lipman DJ, Souvorov A, Koonin EV, Panchenko AR, Tatusova TA. 2002. The relationship of protein conservation and sequence length. BMC Evol Biol. 2:20. doi: 10.1186/1471-2148-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu JM, Livny J, Lawrence MS, Kimball MD, Waldor MK, Camilli A. 2009. Experimental discovery of sRNAs in Vibrio cholerae by direct cloning, 5S/tRNA depletion and parallel sequencing. Nucleic Acids Res 37:e46. doi: 10.1093/nar/gkp080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davis BM, Waldor MK. 2007. RNase E-dependent processing stabilizes MicX, a Vibrio cholerae sRNA. Mol Microbiol 65:373–385. doi: 10.1111/j.1365-2958.2007.05796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lenz DH, Mok KC, Lilley BN, Kulkarni RV, Wingreen NS, Bassler BL. 2004. The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell 118:69–82. doi: 10.1016/j.cell.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 42.Bradley ES, Bodi K, Ismail AM, Camilli A. 2011. A genome-wide approach to discovery of small RNAs involved in regulation of virulence in Vibrio cholerae. PLoS Pathog 7:e1002126. doi: 10.1371/journal.ppat.1002126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mandin P, Repoila F, Vergassola M, Geissmann T, Cossart P. 2007. Identification of new noncoding RNAs in Listeria monocytogenes and prediction of mRNA targets. Nucleic Acids Res 35:962–974. doi: 10.1093/nar/gkl1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toledo-Arana A, Dussurget O, Nikitas G, Sesto N, Guet-Revillet H, Balestrino D, Loh E, Gripenland J, Tiensuu T, Vaitkevicius K, Barthelemy M, Vergassola M, Nahori M-A, Soubigou G, Régnault B, Coppée J-Y, Lecuit M, Johansson J, Cossart P. 2009. The Listeria transcriptional landscape from saprophytism to virulence. Nature 459:950–956. doi: 10.1038/nature08080. [DOI] [PubMed] [Google Scholar]
- 45.Mraheil MA, Billion A, Mohamed W, Mukherjee K, Kuenne C, Pischimarov J, Krawitz C, Retey J, Hartsch T, Chakraborty T, Hain T. 2011. The intracellular sRNA transcriptome of Listeria monocytogenes during growth in macrophages. Nucleic Acids Res 39:4235–4248. doi: 10.1093/nar/gkr033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Albrecht M, Sharma CM, Reinhardt R, Vogel J, Rudel T. 2010. Deep sequencing-based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res 38:868–877. doi: 10.1093/nar/gkp1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bohn C, Rigoulay C, Chabelskaya S, Sharma CM, Marchais A, Skorski P, Borezée-Durant E, Barbet R, Jacquet E, Jacq A, Gautheret D, Felden B, Vogel J, Bouloc P. 2010. Experimental discovery of small RNAs in Staphylococcus aureus reveals a riboregulator of central metabolism. Nucleic Acids Res 38:6620–6636. doi: 10.1093/nar/gkq462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Geissmann T, Chevalier C, Cros M-J, Boisset S, Fechter P, Noirot C, Schrenzel J, François P, Vandenesch F, Gaspin C, Romby P. 2009. A search for small noncoding RNAs in Staphylococcus aureus reveals a conserved sequence motif for regulation. Nucleic Acids Res 37:7239–7257. doi: 10.1093/nar/gkp668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abu-Qatouseh LF, Chinni SV, Seggewiss J, Proctor RA, Brosius J, Rozhdestvensky TS, Peters G, von Eiff C, Becker K. 2010. Identification of differentially expressed small non-protein-coding RNAs in Staphylococcus aureus displaying both the normal and the small-colony variant phenotype. J Mol Med 88:565–575. doi: 10.1007/s00109-010-0597-2. [DOI] [PubMed] [Google Scholar]
- 50.Beaume M, Hernandez D, Farinelli L, Deluen C, Linder P, Gaspin C, Romby P, Schrenzel J, Francois P. 2010. Cartography of methicillin-resistant S. aureus transcripts: detection, orientation and temporal expression during growth phase and stress conditions. PLoS One 5:e10725. doi: 10.1371/journal.pone.0010725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pichon C, Felden B. 2005. Small RNA genes expressed from Staphylococcus aureus genomic and pathogenicity islands with specific expression among pathogenic strains. Proc Natl Acad Sci U S A 102:14249–14254. doi: 10.1073/pnas.0503838102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saito S, Kakeshita H, Nakamura K. 2009. Novel small RNA-encoding genes in the intergenic regions of Bacillus subtilis. Gene 428:2–8. doi: 10.1016/j.gene.2008.09.024. [DOI] [PubMed] [Google Scholar]
- 53.Rasmussen S, Nielsen HB, Jarmer H. 2009. The transcriptionally active regions in the genome of Bacillus subtilis. Mol Microbiol 73:1043–1057. doi: 10.1111/j.1365-2958.2009.06830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kumar R, Shah P, Swiatlo E, Burgess SC, Lawrence ML, Nanduri B. 2010. Identification of novel non-coding small RNAs from Streptococcus pneumoniae TIGR4 using high-resolution genome tiling arrays. BMC Genomics 11:350. doi: 10.1186/1471-2164-11-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mann B, van Opijnen T, Wang J, Obert C, Wang Y-D, Carter R, McGoldrick DJ, Ridout G, Camilli A, Tuomanen EI, Rosch JW. 2012. Control of virulence by small RNAs in Streptococcus pneumoniae. PLoS Pathog 8:e1002788. doi: 10.1371/journal.ppat.1002788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Acebo P, Martin-Galiano AJ, Navarro S, Zaballos A, Amblar M. 2012. Identification of 88 regulatory small RNAs in the TIGR4 strain of the human pathogen Streptococcus pneumoniae. RNA 18:530–546. doi: 10.1261/rna.027359.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Perez N, Treviño J, Liu Z, Ho SCM, Babitzke P, Sumby P. 2009. A genome-wide analysis of small regulatory RNAs in the human pathogen group A Streptococcus. PLoS One 4:e7668. doi: 10.1371/journal.pone.0007668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shioya K, Michaux C, Kuenne C, Hain T, Verneuil N, Budin-Verneuil A, Hartsch T, Hartke A, Giard J-C. 2011. Genome-wide identification of small RNAs in the opportunistic pathogen Enterococcus faecalis V583. PLoS One 6:e23948. doi: 10.1371/journal.pone.0023948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fouquier d'Hérouel A, Wessner F, Halpern D, Ly-Vu J, Kennedy SP, Serror P, Aurell E, Repoila F. 2011. A simple and efficient method to search for selected primary transcripts: non-coding and antisense RNAs in the human pathogen Enterococcus faecalis. Nucleic Acids Res 39:e46. doi: 10.1093/nar/gkr012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DiChiara JM, Contreras-Martinez LM, Livny J, Smith D, McDonough KA, Belfort M. 2010. Multiple small RNAs identified in Mycobacterium bovis BCG are also expressed in Mycobacterium tuberculosis and Mycobacterium smegmatis. Nucleic Acids Res 38:4067–4078. doi: 10.1093/nar/gkq101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsai C-H, Baranowski C, Livny J, McDonough KA, Wade JT, Contreras LM. 2013. Identification of novel sRNAs in mycobacterial species. PLoS One 8:e79411. doi: 10.1371/journal.pone.0079411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hershberg R, Altuvia S, Hanah M. 2003. A survey of small RNA-encoding genes in Escherichia coli. Nucleic Acids Res 31:1813–1820. doi: 10.1093/nar/gkg297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang A, Wassarman KM, Rosenow C, Tjaden BC, Storz G, Gottesman S. 2003. Global analysis of small RNA and mRNA targets of Hfq. Mol Microbiol 50:1111–1124. doi: 10.1046/j.1365-2958.2003.03734.x. [DOI] [PubMed] [Google Scholar]
- 64.Rivas E, Klein RJ, Jones TA, Eddy SR. 2001. Computational identification of noncoding RNAs in E. coli by comparative genomics. Curr Biol 11:1369–1373. doi: 10.1016/S0960-9822(01)00401-8. [DOI] [PubMed] [Google Scholar]
- 65.Tjaden B, Saxena RM, Stolyar S, Haynor DR, Kolker E, Rosenow C. 2002. Transcriptome analysis of Escherichia coli using high-density oligonucleotide probe arrays. Nucleic Acids Res 30:3732–3738. doi: 10.1093/nar/gkf505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Argaman L, Hershberg R, Vogel J, Bejerano G, Wagner EG, Margalit H, Altuvia S. 2001. Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr Biol 11:941–950. doi: 10.1016/S0960-9822(01)00270-6. [DOI] [PubMed] [Google Scholar]
- 67.Macvanin M, Edgar R, Cui F, Trostel A, Zhurkin V, Adhya S. 2012. Noncoding RNAs binding to the nucleoid protein HU in Escherichia coli. J Bacteriol 194:6046–6055. doi: 10.1128/JB.00961-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Castillo-Keller M, Vuong P, Misra R. 2006. Novel mechanism of Escherichia coli porin regulation. J Bacteriol 188:576–586. doi: 10.1128/JB.188.2.576-586.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kawano M, Oshima T, Kasai H, Mori H. 2002. Molecular characterization of long direct repeat (LDR) sequences expressing a stable mRNA encoding for a 35-amino-acid cell-killing peptide and a cis-encoded small antisense RNA in Escherichia coli. Mol Microbiol 45:333–349. doi: 10.1046/j.1365-2958.2002.03042.x. [DOI] [PubMed] [Google Scholar]
- 70.Chen S, Lesnik EA, Hall TA, Sampath R, Griffey RH, Ecker DJ, Blyn LB. 2002. A bioinformatics based approach to discover small RNA genes in the Escherichia coli genome. Biosystems 65:157–177. doi: 10.1016/S0303-2647(02)00013-8. [DOI] [PubMed] [Google Scholar]
- 71.Boysen A, Møller-Jensen J, Kallipolitis B, Valentin-Hansen P, Overgaard M. 2010. Translational regulation of gene expression by an anaerobically induced small non-coding RNA in Escherichia coli. J Biol Chem 285:10690–10702. doi: 10.1074/jbc.M109.089755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kawano M, Reynolds AA, Miranda-Rios J, Storz G. 2005. Detection of 5′- and 3′-UTR-derived small RNAs and cis-encoded antisense RNAs in Escherichia coli. Nucleic Acids Res 33:1040–1050. doi: 10.1093/nar/gki256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sharma CM, Hoffmann S, Darfeuille F, Reignier J, Findeiss S, Sittka A, Chabas S, Reiche K, Hackermüller J, Reinhardt R, Stadler PF, Vogel J. 2010. The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464:250–255. doi: 10.1038/nature08756. [DOI] [PubMed] [Google Scholar]
- 74.Chao Y, Papenfort K, Reinhardt R, Sharma CM, Vogel J. 2012. An atlas of Hfq-bound transcripts reveals 3′ UTRs as a genomic reservoir of regulatory small RNAs. EMBO J 31:4005–4019. doi: 10.1038/emboj.2012.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sittka A, Lucchini S, Papenfort K, Sharma CM, Rolle K, Binnewies TT, Hinton JCD, Vogel J. 2008. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet 4:e1000163. doi: 10.1371/journal.pgen.1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ferrara S, Brugnoli M, De Bonis A, Righetti F, Delvillani F, Dehò G, Horner D, Briani F, Bertoni G. 2012. Comparative profiling of Pseudomonas aeruginosa strains reveals differential expression of novel unique and conserved small RNAs. PLoS One 7:e36553. doi: 10.1371/journal.pone.0036553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Livny J, Brencic A, Lory S, Waldor MK. 2006. Identification of 17 Pseudomonas aeruginosa sRNAs and prediction of sRNA-encoding genes in 10 diverse pathogens using the bioinformatic tool sRNAPredict2. Nucleic Acids Res 34:3484–3493. doi: 10.1093/nar/gkl453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gómez-Lozano M, Marvig RL, Molin S, Long KS. 2012. Genome-wide identification of novel small RNAs in Pseudomonas aeruginosa. Environ Microbiol 14:2006–2016. doi: 10.1111/j.1462-2920.2012.02759.x. [DOI] [PubMed] [Google Scholar]
- 79.Sonnleitner E, Sorger-Domenigg T, Madej MJ, Findeiss S, Hackermüller J, Hüttenhofer A, Stadler PF, Bläsi U, Moll I. 2008. Detection of small RNAs in Pseudomonas aeruginosa by RNomics and structure-based bioinformatic tools. Microbiology 154:3175–3187. doi: 10.1099/mic.0.2008/019703-0. [DOI] [PubMed] [Google Scholar]
- 80.Chinni SV, Raabe CA, Zakaria R, Randau G, Hoe CH, Zemann A, Brosius J, Tang T. 2010. Experimental identification and characterization of 97 novel npcRNA candidates in Salmonella enterica serovar Typhi. Nucleic Acids Res 38:5893–5908. doi: 10.1093/nar/gkq281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Irnov I, Sharma CM, Vogel J, Winkler WC. 2010. Identification of regulatory RNAs in Bacillus subtilis. Nucleic Acids Res 38:6637–6651. doi: 10.1093/nar/gkq454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li L, Huang D, Cheung MK, Nong W, Huang Q, Kwan HS. 2013. BSRD: a repository for bacterial small regulatory RNA. Nucleic Acids Res 41:D233–D238. doi: 10.1093/nar/gks1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Peer A, Margalit H. 2011. Accessibility and evolutionary conservation mark bacterial small-RNA target-binding regions. J Bacteriol 193:1690–1701. doi: 10.1128/JB.01419-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stauffer LT, Stauffer GV. 2013. Multiple roles for the sRNA GcvB in the regulation of Slp levels in Escherichia coli. ISRN Bacteriol 2013:1–8. doi: 10.1155/2013/918106. [DOI] [Google Scholar]
- 85.Degnan PH, Ochman H, Moran NA. 2011. Sequence conservation and functional constraint on intergenic spacers in reduced genomes of the obligate symbiont Buchnera. PLoS Genet 7:e1002252. doi: 10.1371/journal.pgen.1002252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hupalo D, Kern AD. 2013. Conservation and functional element discovery in 20 angiosperm plant genomes. Mol Biol Evol 30:1729–1744. doi: 10.1093/molbev/mst082. [DOI] [PubMed] [Google Scholar]
- 87.Willment JA, Martin DP, Palmer KE, Schnippenkoetter WH, Shepherd DN, Rybicki EP. 2007. Identification of long intergenic region sequences involved in maize streak virus replication. J Gen Virol 88:1831–1841. doi: 10.1099/vir.0.82513-0. [DOI] [PubMed] [Google Scholar]
- 88.Zhang C, Tintó SC, Li G, Lin N, Chung M, Moreno E, Moberg KH, Zhou L. 16 June 2014. An intergenic regulatory region mediates Drosophila Myc-induced apoptosis and blocks tissue hyperplasia. Oncogene doi: 10.1038/onc.2014.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nawrocki EP, Eddy SR. 2013. Computational identification of functional RNA homologs in metagenomic data. RNA Biol 10:1170–1179. doi: 10.4161/rna.25038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.