

Abstract
Rotation of the polar flagellum of Vibrio alginolyticus is driven by a Na+-type flagellar motor. FliG, one of the essential rotor proteins located at the upper rim of the C ring, binds to the membrane-embedded MS ring. The MS ring is composed of a single membrane protein, FliF, and serves as a foundation for flagellar assembly. Unexpectedly, about half of the Vibrio FliF protein produced at high levels in Escherichia coli was found in the soluble fraction. Soluble FliF purifies as an oligomer of ∼700 kDa, as judged by analytical size exclusion chromatography. By using fluorescence correlation spectroscopy, an interaction between a soluble FliF multimer and FliG was detected. This binding was weakened by a series of deletions at the C-terminal end of FliF and was nearly eliminated by a 24-residue deletion or a point mutation at a highly conserved tryptophan residue (W575). Mutations in FliF that caused a defect in FliF-FliG binding abolish flagellation and therefore confer a nonmotile phenotype. As data from in vitro binding assays using the soluble FliF multimer correlate with data from in vivo functional analyses, we conclude that the C-terminal region of the soluble form of FliF retains the ability to bind FliG. Our study confirms that the C-terminal tail of FliF provides the binding site for FliG and is thus required for flagellation in Vibrio, as reported for other species. This is the first report of detection of the FliF-FliG interaction in the Na+-driven flagellar motor, both in vivo and in vitro.
INTRODUCTION
Many motile bacteria can swim in liquid environments by means of a motility organelle, the flagellum. Bacteria propel themselves by rotating a helical flagellar filament to move forward, and flagellar rotation is driven by a reversible rotary motor at its base. The flagellum is divided into three parts: the filament (screw), the hook (universal joint), and the basal body (motor). About 50 gene products are required for flagellar assembly and function (1, 2). The energy source for the flagellar motor is the electrochemical gradient of protons or, in some species, sodium ions. The ion flux through stator units that are incorporated around the rotary part of the motor (rotor) is coupled with the generation of torque. The flagellar motor can rotate up to 1,700 revolutions per second (rps) (in the case of the Na+-driven Vibrio motor) and can switch its rotational direction within a millisecond, properties which identify it as an elaborate biological nanomachine (3). However, the key question, how is the rotor-stator interaction coupled to ion flux to generate motor torque, has remained a mystery.
Genetic, biochemical, and structural analyses identified key proteins that are most closely involved in torque generation: the stator complex and FliG in the rotor (Fig. 1A) (4). The stator is composed of two membrane proteins (MotA and MotB or their orthologs) that form an ion-conducting force-generating unit (5, 6). In the H+-driven Escherichia coli or Salmonella motor, MotA, with 4 transmembrane (TM) segments, and MotB, with a single TM segment, form the stator complex with an A4B2 stoichiometry (7–10), whereas in the Na+-driven Vibrio motor, the Mot orthologs PomA and PomB form the stator complex (11, 12). In the functional motor, multiple stator units assemble around the rotor ring (13–16), and the ion flux through the stator channel is coupled to a rotor-stator interaction, presumably involving the cytoplasmic region of the A subunit and the C-terminal globular domain of FliG, as described below. When incorporated into the motor, the stator is anchored to the peptidoglycan (PG) layer via the PG-binding domain in the periplasmic region of the B subunit (17). Determination of the crystal structure and subsequent functional analyses revealed that the assembly-coupled conformational change in this region is required for stator anchoring and activation of the ion-conducting activity of the stator channel (18).
FIG 1.
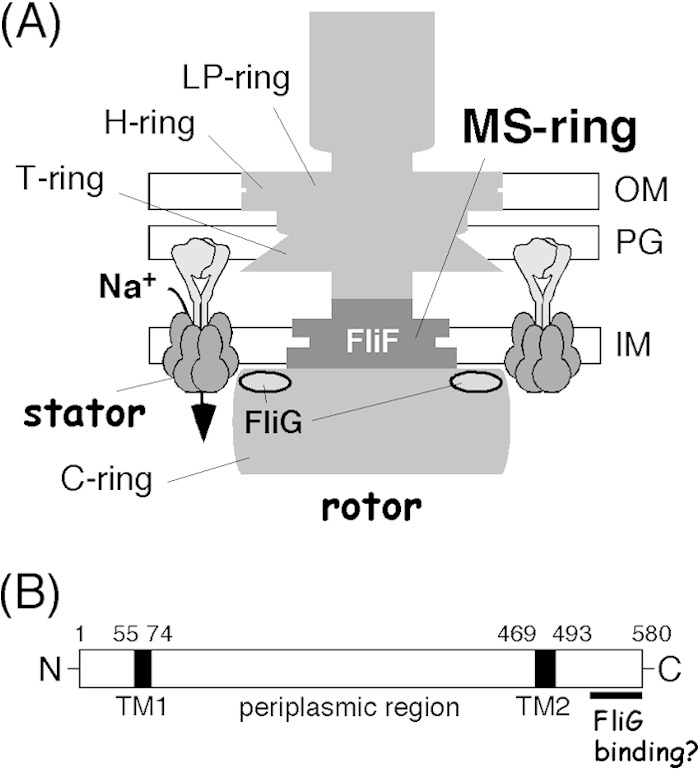
Flagellar basal body protein FliF of Vibrio alginolyticus. (A) Schematic representation of the polar flagellar motor of V. alginolyticus. Its basal body is composed of several rings, including the H ring and T ring, which are not observed in the well-characterized Salmonella and E. coli basal bodies. FliF forms the MS ring of the basal body, and the C-ring protein FliG binds to FliF at the cytoplasmic face of the MS ring. The rotor-stator interactions that couple Na+ influx through the stator channel generate torque. OM, outer membrane; IM, inner membrane. (B) Primary structure of V. alginolyticus FliF. It has a large periplasmic region and two cytoplasmic tails at the N and C termini. The putative FliG-binding site is located at the very end of the C terminus. TM, transmembrane segment. Amino acid residue numbers are shown at the top.
FliG is located in the upper outer rim of the C-ring structure and has 3 domains: the N-terminal (FliGN), the middle (FliGM), and the C-terminal (FliGC) domains. FliGN and FliGM are important for flagellar assembly and rotational switching (19), whereas FliGC is primarily responsible for torque generation (20). The atomic structure of FliG has been reported for each domain (21–25) as well as for the full-length protein (26). Structural analysis showed that FliGC forms a single globular domain with a cluster of charged residues aligned to form a “V”-like ridge on the surface. These charged residues are highly conserved, and genetic studies have revealed that they are involved in functionally important electrostatic interactions with conserved charged residues in the cytoplasmic region of the stator A subunit (27). Recent studies revealed that these electrostatic interactions are also responsible for stator assembly around the rotor (28, 29). However, the detailed mechanism of how such interactions at the rotor-stator interface produce torque has remained unsolved. Many efforts to detect the rotor-stator interaction in vitro have been made but so far have not been successful. The difficulty is derived mainly from the nature of this interaction; it has to be transient but successive and therefore completed within a very short period of time. Otherwise, the motor would be stuck and would not rotate, being jammed by the long period of rotor-stator binding.
Experiments involving the overexpression of FliF, as documented here, unexpectedly revealed that roughly half of the overproduced FliF, the MS-ring component with two TM segments (Fig. 1B) (30, 31), partitioned in the soluble fraction. About 26 FliF molecules form the MS ring (32), and MS-ring assembly is a very early step of flagellar formation (33). FliG then binds to FliF from the cytoplasmic side of the membrane (34, 35) to initiate C-ring formation, which is also a critical step of flagellar assembly. FliF is known to bind tightly to FliGN at the very C-terminal portion (36). Thus, we decided to use this FliF in the soluble fraction to assess FliG-FliF binding using fluorescence correlation spectroscopy (FCS).
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
Bacterial strains and plasmids used in this study are listed in Table 1. Routine DNA manipulations were carried out according to standard protocols. Plasmid pTY502 carries the Vibrio alginolyticus fliF gene under an arabinose-inducible promoter. For overproduction of V. alginolyticus FliG from the cold shock expression vector, a DNA fragment that harbors an open reading frame of fliG was cloned into the NdeI and XhoI sites of pCold I to generate pRAY201. Similarly, V. alginolyticus fliF was cloned into the NdeI and BamHI sites of pCold I to generate pRO101. In both constructs, 16 amino acids, including a His6 tag and a factor Xa cleavage site (underlined) (MNHKVHHHHHHIEGRH) from the vector sequence, were attached at the N terminus of FliF or FliG. We chose the cold shock promoter system for overproduction, because low-temperature induction suppresses the expression of host E. coli proteins and enables the overproduction of only the plasmid-borne gene product. We have already succeeded in the overproduction of several recombinant proteins from V. alginolyticus using the pCold system, including membrane proteins. V. alginolyticus was cultured in VC medium (0.5% [wt/vol] Bacto tryptone, 0.5% [wt/vol] yeast extract, 0.4% [wt/vol] K2HPO4, 3% [wt/vol] NaCl, 0.2% [wt/vol] glucose) or in VPG medium (1% [wt/vol] Bacto tryptone, 0.4% [wt/vol] K2HPO4, 3% [wt/vol] NaCl, 0.5% [wt/vol] glycerol). E. coli was cultured in LB broth (1% [wt/vol] Bacto tryptone, 0.5% [wt/vol] yeast extract, 0.5% [wt/vol] NaCl). Chloramphenicol was added to final concentrations of 2.5 μg/ml for V. alginolyticus and 25 μg/ml for E. coli. Ampicillin was added to a final concentration of 100 μg/ml for E. coli. Arabinose was added to a final concentration of 0.02% or 0.2% (wt/vol) for V. alginolyticus. Isopropyl-β-d-thiogalactopyranoside (IPTG) was added to a final concentration of 0.5 mM. Transformation of V. alginolyticus was carried out by electroporation, as described previously (37).
TABLE 1.
Bacterial strains and plasmidsa
| Strain or plasmid | Description | Reference or source |
|---|---|---|
| Strains | ||
| V. alginolyticus | ||
| VIO5 | Rifr Pof+ Laf− | 44 |
| NMB196 | Rifr Pof+ Laf− ΔfliF | 45 |
| E. coli | ||
| BL21(DE3) | Host for overexpression | |
| JM109 | Recipient for cloning experiments | |
| Plasmids | ||
| pCold I | Ampr; cold shock expression vector | TaKaRa |
| pBAD33 | Cmr; PBAD | 46 |
| pTY57 | Cmr; PBAD, with a multicloning site of pBAD24 | 47 |
| pTY502 | pTY57/FliF | This study |
| pRAY201 | pCold I/FliG | This study |
| pRO101 | pCold I/FliF | This study |
| pRO102 | pCold I/FliF(557Δ) (deletion of residues 557–580) | This study |
| pRO103 | pCold I/FliF(573Δ) (deletion of residues 573–580) | This study |
| pRO104 | pCold I/FliF(577Δ) (deletion of residues 577–580) | This study |
| pRO201 | pTY57/FliF(557Δ) (deletion of residues 557–580) | This study |
| pRO202 | pTY57/FliF(561Δ) (deletion of residues 561–580) | This study |
| pRO202 | pTY57/FliF(565Δ) (deletion of residues 565–580) | This study |
| pRO203 | pTY57/FliF(569Δ) (deletion of residues 569–580) | This study |
| pRO204 | pTY57/FliF(573Δ) (deletion of residues 573–580) | This study |
| pRO205 | pTY57/FliF(577Δ) (deletion of residues 577–580) | This study |
Rifr, rifampin resistant; Pof+, normal polar flagellar formation; Laf−, defective in lateral flagellar formation; Ampr, ampicillin resistant; Cmr, chloramphenicol resistant; PBAD, araBAD promoter.
Mutagenesis.
Point mutations in fliF were introduced into pTY502 or pRO101 by the QuikChange site-directed mutagenesis method according to instructions provided by Stratagene. C-terminal deletion derivatives of FliF were constructed by introducing a stop codon at the desired position. Each mutation was confirmed by DNA sequencing.
Preparation of whole-cell extracts and subcellular fractions.
Vibrio cells were grown in VPG medium containing 2.5 μg/ml chloramphenicol and 0.02% (wt/vol) arabinose at 30°C until the late log phase was reached (typically 3.5 h). Cells were then harvested and resuspended in SDS loading buffer by using a cell concentration equivalent to an optical density at 660 nm (OD660) of 5; whole-cell extracts were loaded. Subcellular fractionation of Vibrio cells was carried out by resuspending the cultured cells in V buffer (50 mM Tris-HCl [pH 8.0], 300 mM NaCl, 5 mM MgCl2) containing 0.5 mM phenylmethylsulfonyl fluoride (PMSF) and then disrupting them by sonication. After removal of unbroken cells by centrifugation (3,440 × g for 5 min), the supernatant was ultracentrifuged (112,000 × g for 30 min). The pellets (insoluble fraction) were resuspended in V buffer containing 0.5 mM PMSF with the same volume of supernatant to prepare soluble and insoluble (membrane) fractions. Subcellular fractionation of E. coli cells overproducing Vibrio FliF was performed similarly except that cells were resuspended in HisTrap buffer (see below).
Immunoblotting.
Protein samples were mixed with SDS loading buffer and were boiled at 95°C for 5 min. Proteins were then separated by SDS-PAGE, and immunoblotting was performed as described previously (38), using an antiserum against Vibrio FliF (FliFB0079). The FliFB0079 antiserum was raised against a mixture of peptide 1 (S23 to K41), peptide 2 (K308 to Y326), and peptide 3 (A564 to G580) of Vibrio alginolyticus FliF and was produced by Biogate Co., Ltd. (Gifu, Japan).
Motility assay on soft-agar plates.
VPG semisolid agar (0.25% [wt/vol] Bacto agar), including 2.5 μg/ml chloramphenicol and 0.02 or 0.2% (wt/vol) arabinose, was used for motility assays of V. alginolyticus. A 2-μl aliquot of a culture grown overnight in VC medium at 30°C was spotted onto VPG semisolid agar and was incubated at 30°C. After 6 h, the diameter of the motility ring was measured. The ring size of each mutant strain was normalized to that of the wild-type strain with a vector plasmid (VIO5/pBAD33) obtained from the same soft-agar plate, and the relative ring size was computed. All measurements were repeated 3 times, and average values and standard deviations are reported.
Overproduction and purification of Vibrio FliG and soluble FliF.
For overproduction and purification of Vibrio FliG and soluble FliF, the following procedure was used. E. coli BL21(DE3) cells harboring pRAY201 (fliG) or pRO101 (fliF) were grown at 37°C in 30 ml LB medium containing ampicillin. When the cell density reached an OD660 of ∼0.8, the entire culture was inoculated in 1 liter of fresh LB medium containing ampicillin and grown at 37°C for ∼2 h until the OD660 reached ∼0.4. The culture was then cooled to 16°C on ice, followed by the addition of IPTG to a final concentration of 0.5 mM, and cultured overnight at 16°C. Cells were harvested by centrifugation (8,000 × g for 10 min) and resuspended in HisTrap buffer (50 mM Tris-HCl [pH 8.0], 20 mM imidazole, 150 mM NaCl) containing one tablet of protease inhibitor cocktail (Roche Diagnostics). The suspension was frozen at −80°C and thawed on ice. Cells were then disrupted with a French press (Ohtake Works). Unbroken cells were removed by low-speed centrifugation at 4°C, and the supernatant was ultracentrifuged at 100,000 × g for 60 min at 4°C. The supernatant (soluble fraction) was loaded onto a 5-ml HisTrap column (GE Healthcare) at 4°C. The column was washed extensively with HisTrap buffer, and His-tagged proteins were eluted with a linear 20 to 500 mM gradient of imidazole in HisTrap buffer. Peak fractions were collected, diluted 5-fold with HiTrap Q buffer (50 mM Tris-HCl [pH 8.0], 10 mM NaCl, 1 mM EDTA), and then loaded onto a 5-ml anion-exchange column (HiTrap Q; GE Healthcare). The column was extensively washed with HiTrap Q buffer, and proteins were eluted with a linear 10 to 1,000 mM gradient of NaCl. Peak fractions were collected, concentrated by ultrafiltration when necessary, and stored at 4°C until use. For FCS analysis, purified FliG and FliF were dialyzed against phosphate-buffered saline (PBS) (pH 7.4), as described below for protein labeling. Protein concentrations were determined with a bicinchoninic acid (BCA) protein assay kit (Thermo Scientific), according to the protocol supplied by the manufacturer. Although purified FliG is stable at 4°C for at least 1 week, purified FliF aggregated easily. Therefore, after dialysis, aggregated proteins were removed by ultracentrifugation (100,000 × g for 60 min) immediately before the FCS analysis. Mutant FliF proteins were prepared by using the same protocol as that used for the wild-type protein.
Analytical size exclusion column chromatography.
Analytical size exclusion chromatography was performed with a Superdex 200 HR10/300 column (GE Healthcare) connected to an AKTA system (GE Healthcare). The column was equilibrated with buffer C (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA) and eluted with the same buffer at a flow rate of 0.6 ml/min. Blue dextran 2000 (for void volume), thyroglobulin (669 kDa), ferritin (440 kDa), aldolase (158 kDa), ovalbumin (43 kDa), and RNase A (13.7 kDa) were used as molecular mass standards. For data shown in Fig. S2 in the supplemental material, a Superdex 200 Increase GL 10/300 column (GE Healthcare) was used at a flow rate of 0.75 ml/min.
Labeling of FliG with fluorescence dye.
Purified FliG was dialyzed against PBS buffer (8 mg/ml NaCl, 0.2 mg/ml KCl, 1.44 mg/ml Na2PO4, 0.24 mg/ml KH2PO4 [pH 7.4]) by using a Slide-A-Lyzer device (Thermo Scientific) in a cold room for 6 h, with buffer exchanges every 2 h. FliG was labeled with the fluorescent dye ATTO633 by using the Protein Labeling kit (Olympus) according to the protocol provided by the manufacturer. Briefly, 4.5 μl of a 1 M bicarbonate solution was added to 45 μl of the protein solution (2.23 mg/ml) and mixed. One microliter of ATTO633–N-hydroxysuccinimide (NHS), dissolved in dimethyl sulfoxide, was added to the protein solution, mixed, and then incubated at 25°C for 1 h in the dark. The reaction was terminated by the addition of 5 μl Stop reagent, provided in the kit, and the mixture was incubated at room temperature for 30 min. Next, 445 μl of filtration buffer (PBS based; included in the kit) was added to the reaction mixture, which was then loaded onto an Ultrafree-0.5 10K centrifugal filter device (Millipore). Ultrafiltration and multiple washes (at least 4 times) with filtration buffer were carried out on this device to remove unattached dye. By using this protocol, >95% of FliG was labeled, and residual unattached free dye was <10% of the total fluorescence in the final preparation.
Fluorescence correlation spectroscopy.
FCS is a powerful tool that examines molecular interactions in solution. FCS measures fluorescence fluctuations within a defined small area (∼1 fl). These signals contain information about the diffusion time, which is the average time required for a particle to cross the area. The FCS value can be computed by correlation function analysis. The diffusion time is dependent on the size of the particle; a fluorescently labeled particle that binds to another molecule(s) exhibits a longer diffusion time than a free particle (no binding), so information can be obtained for protein interactions in solution. FCS analysis was carried out by using an MF-20 instrument (Olympus) according to the procedure supplied by the manufacturer. FCS samples (50 μl) contained PBS buffer (pH 7.4), 10 nM ATTO633-labeled FliG, and various concentrations (1 to 1,000 nM, as indicated in the figures) of purified soluble FliF. The analysis was carried out by using a 384-well glass-bottomed microplate. Note that the FliF concentration was calculated for the monomer, and purified soluble FliF, immediately after ultracentrifugation to remove aggregated proteins, was used for the FCS analysis. For the detection of ATTO633-labeled particles, a He-Ne laser (633 nm; 100 μW) was used. For each condition, an FCS measurement with a data acquisition time of 10 s was repeated 10 times at room temperature to obtain a single data set, and each autocorrelation function was calculated by the system. From this autocorrelation function, the diffusion time of the fluorescently labeled particles was obtained. We conducted three individual experiments, and all three data sets obtained showed basically the same profile, so only one of the data sets, which reflects typical results, is shown in Fig. 3, 5, and 6D with averages and standard deviations (10 measurements for each data point). To obtain the fraction of unbound FliG, two-component analysis was performed. Because the diffusion time of labeled FliG at 1 nM FliF is 700 μs (almost no binding) and that at >250 nM FliF is 1,700 μs (saturated binding), we assumed that there were two states of labeled FliG: free FliG, which exhibits a diffusion time of 700 μs, and FliF-bound FliG, which exhibits a diffusion time of 1,700 μs. Under each experimental condition, these two components exist as a mixture at various ratios. Using this assumption, the fraction of free FliG was obtained by estimating how much the fraction of labeled free particles contributed to the measured diffusion time. Two-component analysis was conducted by using the software provided with the MF-20 system.
FIG 3.

FCS analyses of FliF binding to ATTO633-labeled FliG. (A) Plots of FCS diffusion time against FliF protein concentration. Exponential curve fitting was performed to aid in visualization. A 10 nM sample of ATTO633-labeled FliG was mixed with various concentrations of purified FliF, and diffusion times of labeled FliG particles were obtained by FCS analysis using the MF-20 system. Three independent experiments were performed, and typical data are shown. (B) Plots of the fraction of unbound FliG against the FliF concentration. Two-component analysis was performed for the data set shown in panel A. Exponential curve fitting was performed to aid in visualization. Details are described in Materials and Methods.
FIG 5.
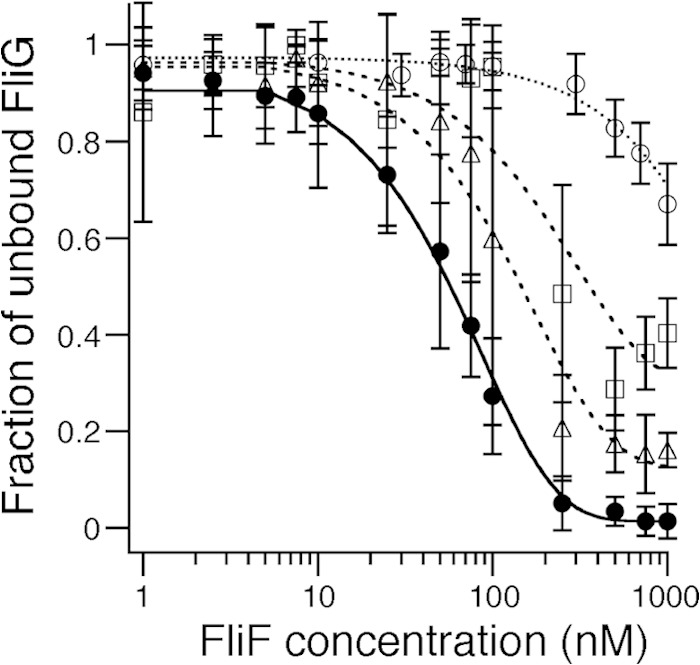
FCS analyses of FliF mutant binding to ATTO633-labeled FliG. A total of 10 nM ATTO633-labeled FliG was mixed with various concentrations of purified FliF mutant proteins, and the diffusion times of labeled FliG particles were obtained by FCS analysis using the MF-20 system, as described in the legend to Fig. 3A. Two-component analysis was then performed as described in the legend to Fig. 3B. Three independent experiments were performed, and typical data are shown. Filled circles, full-length FliF (same data as in Fig. 3B); open triangles, FliF(577Δ); open squares, FliF(573Δ); open circles, FliF(557Δ). Exponential curve fitting was performed to aid in visualization.
FIG 6.

Alanine-scanning analyses of the C-terminal end of FliF. (A) Motility of cells on soft-agar plates. NMB196 cells expressing mutant FliF proteins were inoculated onto soft-agar plates containing 0.02% arabinose and were incubated at 30°C for 6 h. (B) Relative motility ring sizes of NMB196 (ΔfliF) cells expressing Ala-replaced FliF mutant proteins. Motility on soft-agar plates was analyzed in the presence 0.02% of arabinose, and ring sizes were measured and normalized to those formed by VIO5 cells harboring the vector pTY57. WT, wild type (full-length FliF expressed from plasmid pTY502). (C) Expression of mutant FliF proteins with a single Ala replacement in the C-terminal region. Whole-cell samples (top) and membrane fractions (bottom) of NMB196 (ΔfliF) cells expressing wild-type or mutant FliF proteins from the plasmid were prepared from the same cell cultures, analyzed by SDS-PAGE, and immunoblotted with an anti-FliF antibody. Arrowheads show the position of FliF. The vector was pBAD33. (D) FCS analyses of FliF(W575A) binding to ATTO633-labeled FliG. A total of 10 nM ATTO633-labeled FliG was mixed with various concentrations of purified FliF(W575A), and diffusion times of labeled FliG particles were obtained by FCS analysis using the MF-20 system, as described in the legend to Fig. 3A. Two-component analysis was then performed as described in the legend to Fig. 3B. Three independent experiments were performed, and typical data are shown. Filled circles, full-length FliF (same data as in Fig. 3B); open circles, FliF(W575A). Exponential curve fitting was performed to aid in visualization.
RESULTS
Properties of overproduced, soluble Vibrio alginolyticus FliF and FliG.
We constructed an overproduction system for V. alginolyticus FliF and FliG using the cold shock expression vector pCold I and E. coli host strain BL21(DE3). Vibrio FliF (here simply FliF) was overproduced at a high level with this system, and we detected FliF on Coomassie brilliant blue (CBB)-stained gels, even in whole-cell extracts (data not shown). Unexpectedly, we found that FliF overproduced in E. coli fractionated approximately equally into the soluble and insoluble fractions after disruption of the cells by sonication (Fig. 2A). FliF is a membrane protein that has two TM segments near both ends of the protein, with a large periplasmic region in between (Fig. 1B) (31). We assume that the unnaturally rapid protein synthesis in our expression system did not allow proper membrane insertion of FliF and caused its mislocalization into the cytoplasm. The low temperature used for induction is probably not the only reason for the failure of all the FliF to be incorporated into the membrane, as we still detected a small but significant amount of FliF in the soluble fraction when we expressed FliF at 30°C from an arabinose-inducible promoter (PBAD) at a relatively high level in E. coli BL21(DE3) cells (see Fig. S1 in the supplemental material).
FIG 2.
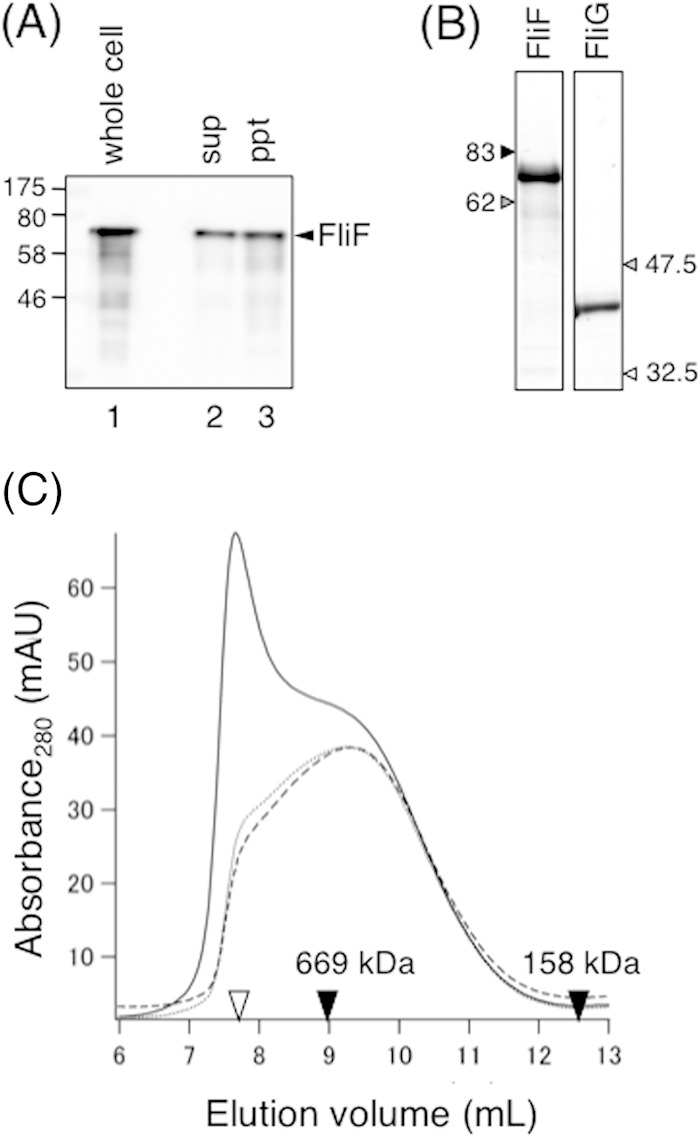
Size exclusion analysis of purified soluble FliF. (A) Subcellular fractionation of Vibrio FliF overproduced in E. coli BL21(DE3) cells. Cells were disrupted by sonication and then fractionated by ultracentrifugation. The same volume of the supernatant (sup) and pellet (ppt) was analyzed by SDS-PAGE followed by immunoblotting with an anti-FliF antibody. The whole-cell sample was analyzed as a control. (B) Purified FliG and soluble FliF analyzed by SDS-PAGE. The gel was stained with CBB. (C) Elution profiles of purified soluble FliF by analytical size exclusion chromatography using a Superdex 200 column. Purified soluble FliF (1 μM) was subjected to ultracentrifugation, and the supernatant was incubated at 4°C for 24 h. Samples before ultracentrifugation (solid line), immediately after ultracentrifugation (broken line), and after incubation at 4°C overnight (dotted line) were loaded onto the column and analyzed. Open triangle, void volume of the column; closed triangles, elution volumes of the size standards thyroglobulin (669 kDa) and aldolase (158 kDa). mAU, milli-absorbance units.
Because we obtained large amounts of the soluble form of FliF, we purified it without using a detergent by running the soluble fraction of the cell lysate through a His tag affinity column followed by anion-exchange chromatography. SDS-PAGE of the purified soluble form of FliF followed by staining with CBB yielded a single band at the calculated molecular mass of the FliF monomer (68 kDa) (Fig. 2B). However, analytical size exclusion chromatography revealed that the soluble form of FliF tends to aggregate (Fig. 2C) and eluted shortly after the void volume (Fig. 2C, solid line). These aggregated proteins were removed by ultracentrifugation, as indicated by the disappearance of the peak at the void volume (Fig. 2C, broken line) and the emergence of a relatively broad but significant peak at an elution volume of 9.3 ml, which, if the FliF multimer is globular, corresponds to ∼700 kDa, as estimated by comparison with the size calibration markers. A small shoulder at the void volume appeared in the gel filtration profile after incubation at 4°C overnight (Fig. 2C, dotted line), indicating that aggregation occurs during storage at 4°C. Therefore, we ultracentrifuged freshly purified FliF before FCS analysis, as described below.
Vibrio FliG was also expressed at a high level in E. coli after induction by cold shock. We obtained a sufficient amount of highly purified FliG from the cytoplasmic fraction using the same purification protocol as that used for FliF (Fig. 2B). Unlike soluble FliF, purified soluble FliG did not aggregate and was stable at 4°C for at least a week.
FliF-FliG interaction measured by FCS.
To investigate whether soluble FliF retains the ability to interact with FliG, we conducted in vitro FCS analysis using a purified soluble FliF multimer and FliG. To carry out the FCS analysis, the protein with the lower molecular mass had to be labeled. Since purified soluble FliF forms a multimer and is much larger than purified FliG, we labeled FliG with the fluorescent dye ATTO633 via an NHS-ester in the dye.
FCS measurements were conducted with a series of samples containing a constant amount of labeled FliG (10 nM) and various concentrations (1 to 1,000 nM, as a monomer concentration) of FliF (Fig. 3). The diffusion time of labeled FliG at 1 nM FliF was ∼700 μs and showed a dose-dependent increase, reaching a plateau of 1,700 μs at ∼250 nM FliF, presumably because the labeled FliG forms a complex with the FliF multimer (Fig. 3A). These results suggest that at 1 nM FliF, very few FliF molecules bind to labeled FliG, whereas at 250 nM FliF, most of the labeled FliG molecules bind to FliF.
The ratio of the diffusion time (τ) for the dye to that for the sample (τdye/τsample) is roughly proportional to the cube root of the ratio of molecular weight (MW) of the dye to that of the sample [3√(MWdye/MWsample)] (39, 40). Assuming that a diffusion time of 700 μs reflects unbound FliG particles and that a diffusion time of 1,700 μs reflects the FliF/FliG complex and by using the parameters of the ATTO633 dye (the molecular mass is 652 Da, and the diffusion time obtained at a laser power of 100 μW is 150 μs), we estimated the molecular masses of FliG and the FliF/FliG complex in solution. This estimation shows that the molecular masses of the FliG particles are ∼60 kDa and that the molecular mass of the FliF/FliG complex is ∼900 kDa. According to size estimation by gel filtration analysis (Fig. 2C), purified soluble FliF forms a multimer of ∼700 kDa, so our results from FCS analysis are consistent with the idea that some numbers of labeled FliG particles bind to a FliF multimer of 700 kDa. We conclude that soluble FliF retains the ability to bind FliG in vitro.
Assuming the two states of FliG, one unbound (diffusion time of 700 μs) and the other bound to the FliF multimer (diffusion time of 1,700 μs), we can estimate the fraction of unbound FliG from the FCS data (for details of the two-component analysis, see Materials and Methods). From the same data set as that shown in Fig. 3A, we calculated the fraction of unbound FliG at each FliF concentration and obtained a dose-dependent FliF-FliG binding profile (Fig. 3B). The concentration of FliF that gave half-maximal FliG binding was ∼50 nM, indicative of a quite high binding affinity. Note here that the concentration of FliF is calculated as a monomer, although we do not know how many of the FliF monomers in the multimer are accessible to FliG.
Deletion analysis of the C-terminal region of FliF.
Previous studies showed that the C-terminal region of FliF of enteric bacteria is required for flagellar assembly and FliG binding (36, 41). To investigate the C-terminal region of V. alginolyticus FliF in the interaction with FliG, we first carried out sequence alignments of FliF proteins from six species of bacteria. The secondary structure in this region of V. alginolyticus FliF predicted by the PSIPRED program (42) is shown together with the alignments in Fig. 4A. According to this program, the C-terminal end of FliF has two α-helices (helices 1 and 2) that contain a cluster of conserved amino acids. Pro and Trp at positions 565 and 575 (V. alginolyticus numbering), respectively, are completely conserved. Using the sequence alignments as a guide, we constructed a series of C-terminally truncated FliF mutants and expressed them in V. alginolyticus ΔfliF strain NMB196. All these mutant proteins were expressed and inserted into the membrane (Fig. 4B), although mutants that lack the C-terminal 12 residues (569Δ) or 8 residues (573Δ) were detected at much lower levels than the others. Cells expressing the FliG variants with C-terminal truncations, except for the shortest 4-residue deletion (577Δ), were nonmotile on soft-agar plates (Fig. 4C). Nonmotile cells had no flagella, as judged by observations by dark-field microscopy (data not shown), indicating that the C-terminal region of FliF is required for flagellation, as has been found for other bacteria.
FIG 4.

Function and expression of mutant FliF proteins with a series of C-terminal deletions. (A) Schematic representation of the C-terminal end of V. alginolyticus FliF and sequence alignments of FliF from various species. Segments deleted in this study are shown as lines, and Ala-substituted residues are indicated by closed triangles. Residues shown in black (or gray) boxes are completely (or partially) conserved among these six species. Aligned FliF sequences are from Vibrio alginolyticus, Vibrio cholerae, Salmonella enterica serovar Typhimurium, Yersinia enterocolitica, Shewanella oneidensis, and Caulobacter crescentus. (B) Expression of mutant FliF proteins with various C-terminal deletions. Whole-cell samples (top) and membrane fractions (bottom) of NMB196 (ΔfliF) cells expressing FliF proteins from the plasmid were prepared from the same cell cultures, analyzed by SDS-PAGE, and immunoblotted with an anti-FliF antibody. Closed triangles show the position of FliF. The vector was pBAD33. (C) Motility of cells on soft-agar plates. NMB196 cells expressing mutant FliF proteins were inoculated on soft-agar plates containing 0.02% arabinose and incubated at 30°C for 6 h. (D) Relative motility ring sizes of the wild-type strain expressing FliF mutants. Motility on soft-agar plates was analyzed for VIO5 cells expressing mutant FliF proteins in the presence of 0.02% (black bars) or 0.2% (white bars) arabinose. Ring sizes were measured and normalized to those of VIO5 cells harboring the vector pTY57. WT, wild type (full-length FliF expressed from plasmid pTY502).
We next examined the effect of multicopy mutant fliF genes expressed in the wild-type strain in order to assess the dominant/recessive phenotype associated with the mutated gene products. As shown in Fig. 4D, moderate overproduction of the mutant FliF protein from the arabinose-inducible plasmid (0.02% arabinose) in wild-type strain VIO5 (filled bars) caused a slight reduction of motility. The multicopy effect was more severe when a higher concentration of arabinose (0.2%) (Fig. 4D, open bars) was used for induction, especially for longer deletions that completely lack helix 2 (557Δ, 561Δ, and 565Δ). These results suggest that the mutant FliF protein retains the ability to interact with endogenous FliF to interfere with the proper formation of the MS ring. Note that the multicopy effect was also observed for the plasmid-borne wild-type fliF gene (Fig. 4D), presumably because excess FliF molecules create an inappropriate stoichiometry for the basal body proteins.
Three deletion derivatives of FliF, 557Δ, 573Δ, and 577Δ, were overproduced in E. coli. As observed for wild-type FliF, all three mutant proteins were found in the soluble fraction. We purified them by using the same protocol as that used for the wild-type protein, and the yield and purity were comparable to those for the wild-type protein. FCS analysis was then performed under the same conditions as those used for wild-type FliF. We found that the diffusion time of labeled FliG increased with increasing concentrations of the mutant FliF proteins, although it did not reach 1,700 μs and was always shorter than that with wild-type FliF. We estimated the fraction of unbound FliG with each FliF mutant protein by two-component analysis (Fig. 5). Although data for the mutant FliF protein contain large deviations, especially at high concentrations of FliF, the fraction of unbound FliG was higher than that of the wild type for all mutant proteins. The nonfunctional FliF mutant (573Δ) showed weaker binding than the functional FliF mutant (577Δ), and the mutant with the largest deletion (557Δ) showed almost no binding, with unbound FliG remaining the major form even at the saturated concentration for wild-type FliF. These results suggest that deletion of the C-terminal region of FliF abolishes FliG binding and thereby prevents flagellation. Because the effect of the deletions observed in vivo (defects in motility and flagellation) correlated with that observed in vitro (reduced binding), we conclude that the C-terminal region of the soluble form of FliF in the multimeric structure still retains the ability to bind FliG, perhaps as in the in vivo FliF ring.
Conserved tryptophan at the C-terminal end of FliF is critical for function.
Since the 573Δ FliF protein is nonfunctional but the 577Δ FliF protein is functional, we converted the four residues between positions 573 and 577 and the well-conserved Pro565 to Ala (Fig. 4A and 6). The mutant proteins were expressed in ΔfliF strain NMB196, and the motility of cells expressing them as their sole source of FliF was examined on soft-agar plates (Fig. 6A). The K573A, N574A, and M576A residue substitutions reduced motility slightly, whereas the W575A substitution resulted in severely reduced motility. The P565A substitution had no discernible effect on motility. When we observed W575A mutant cells under high-intensity dark-field microscopy, most did not move and lacked flagella. The mutant proteins were expressed and inserted into the membrane at the wild-type level, as judged by immunoblotting of whole-cell lysates and membrane fractions (Fig. 6C).
The W575A mutant protein overproduced in E. coli was also detected in the soluble fraction. We purified the soluble form of the W575A FliF protein and performed FCS analysis (Fig. 6D). Two-component analysis of the FCS data showed that the fraction of unbound FliG remained at a high and almost constant level, even at high concentrations of the mutant FliF protein. The FCS profile was similar to that of the nonfunctional 557Δ protein (Fig. 5), so the in vitro and in vivo data were correlated. These results suggest that the conserved tryptophan located in the putative α-helix (helix 2) at the very C terminus of FliF is important for the interaction with FliG.
The 557Δ and W575A mutations do not cause severe aggregation of the soluble form of FliF.
The purified mutant FliF proteins may not interact with FliG because the mutations affect the overall structure of FliF and render it nonfunctional. To test this possibility, we analyzed the purified mutant FliF multimers by size exclusion chromatography. The wild-type and 557Δ and W575A mutant proteins were purified in the same day, concentrated, and stored overnight at 4°C. The next morning, purified samples were ultracentrifuged to remove aggregated species and then analyzed on a size exclusion column. Results are shown in Fig. S2 in the supplemental material. Wild-type FliF showed an elution profile similar to the one presented in Fig. 2C, with a relatively broad peak at 9.87 ml. The 557Δ FliF protein also showed a broad elution profile, with the peak being shifted toward a larger size at 8.95 ml. The W575A FliF protein had an elution profile similar to that of the wild-type protein, but the peak was shifted toward a slightly larger size (9.56 ml). All three proteins eluted within the same volume range (8.5 ml to ∼11 ml), and the elution profiles were mostly overlapping. Blue dextran 2000, the marker for the void volume of the column, showed a sharp peak at 8.62 ml and eluted significantly earlier than all three of the FliF variants. This result indicates that both the 557Δ and W575A mutant proteins did not form very large aggregates and thus indicates that the multimeric composition of soluble FliF was retained for both mutant proteins. Therefore, we conclude that the mutations do not cause global structural changes in the soluble FliF multimer.
DISCUSSION
In this study, we found that a certain fraction of Vibrio FliF is soluble when overproduced in E. coli. Using this soluble form of FliF, we used FCS analysis to monitor the interaction between FliF and FliG. We found that the extreme C-terminal end of FliF is required for FliG binding to the Na+-driven motor of Vibrio alginolyticus. Our results are consistent with previous studies with the Caulobacter crescentus (41) and Thermotoga maritima (36) FliF proteins. Our results strengthen this conclusion because we directly related phenotypic effects with the results of in vitro binding measurements. This is the first report detecting the FliF-FliG interaction in the Na+-driven flagellar motor, both in vivo and in vitro.
We found that about half of the Vibrio FliF overproduced at a low temperature in E. coli was found in the soluble fraction, where it formed oligomers with a broad range of sizes with a tendency to aggregate. The fraction of purified FliF that we used in this study contained multimeric structures with an estimated molecular mass of ∼700 kDa, corresponding to ∼10 molecules of FliF. The MS ring contains ∼26 copies of FliF (32), so the soluble FliF multimer does not appear to be a functional MS ring. The structure of the soluble FliF multimer is unknown, but it still retains the ability to bind FliG, as discussed below.
Because FliF has two TM domains and a large periplasmic region, it is likely that the rapid protein synthesis under our overexpression conditions does not allow the proper membrane insertion of FliF and causes its mislocalization in the cytoplasm. The large periplasmic region may contribute to its soluble nature. It is also plausible that FliF forms multimeric structures in which hydrophobic TM domains interact with each other to reduce the total hydrophobic area of the proteins so that the soluble form of FliF remains stable in the cytoplasm. Because FliG could bind to this multimer, and because defects in binding to FliG are clearly related to the flagellation/motility phenotype, as discussed below, the C terminus of the soluble FliF molecule remains intact in the multimer.
MS-ring formation is believed to occur at an early step in flagellar assembly. Because the MS ring can form when Salmonella FliF is overproduced in E. coli (30), MS-ring formation by FliF could be the initial step in flagellar assembly. However, a recent report demonstrated that in E. coli, FlhA, a flagellar export apparatus protein, together with FliG promote the efficient formation of the MS ring (43). It is possible that besides Salmonella, FliF proteins of other species also need additional components to complete the proper formation of the MS ring. If so, the co-overproduction of Vibrio FliF with FlhA and FliG in E. coli may facilitate MS-ring formation. Such efforts are now in progress.
Multiple sequence alignments and secondary structure prediction showed that Vibrio FliF is very likely to have two α-helices at its extreme C terminus. FliG binding to this region was reported previously for the C. crescentus and T. maritima proteins (36, 41). Our motility assay showed that deletion of the C-terminal 8 residues of FliF, which removes about half of the residues of helix 2, impairs flagellation to the extent of producing a nonmotile phenotype. A similar deletion in C. crescentus FliF also resulted in a nonflagellated phenotype.
Overproduction of truncated FliF variants in the wild-type strain caused some inhibition of motility (Fig. 4D), suggesting that the mutant FliF protein could be incorporated into the MS-ring structure. Therefore, we assume that mutant variants still retain the ability to form the MS ring in vivo but that a C-terminal truncation impairs binding to FliG, which is a crucial step of flagellar assembly. This multicopy effect was more obvious for mutants that completely lacked helix 2 (Fig. 4D), suggesting that this helix is essential for FliG binding. This idea was confirmed by an in vitro binding assay using the FCS method. The FliF-FliG interaction was greatly reduced for the 557Δ FliF variant, which lacks all of helix 2 and half of helix 1, whereas the 573Δ variant, in which two-thirds of helix 2 remained, showed a less severe reduction of the FliF-FliG interaction. It should be noted that the 569Δ and 573Δ proteins were detected in whole-cell lysates at lower levels than wild-type FliF and other FliF mutant proteins, indicating that deletion of helix 2 destabilizes the FliF protein in Vibrio cells.
In helix 2, a highly conserved tryptophan (W575) was the key residue for FliG binding, as FliF with the W575A substitution greatly reduced motility and flagellation as well as FliG binding in vitro. The results shown here are consistent with recently reported data on FliF-FliG binding of Thermotoga proteins in vitro (36). Levenson et al. demonstrated that a FliF peptide that contains only helix 2 can bind FliGN although at least 10 times more weakly than a FliF peptide that contains both helices 1 and 2. Those researchers also showed that upon binding of the C-terminal peptide of FliF to the FliGN fragment, the unstructured region of FliGN became structured, which resulted in a strong and stable 1:1 heterodimeric interaction of the FliF peptide and FliGN, with a dissociation constant of ∼40 nM. This value is similar to the concentration of FliF that gives half-maximal FliG binding (∼50 nM) obtained by our FCS analysis of Vibrio proteins. Moreover, the intrinsic fluorescence of the conserved tryptophan in Thermotoga FliF(W527) upon FliGN binding indicates that this residue moves from a solvated to an unsolvated environment when interacting with the N-terminal region of FliG. This result supports the concept that the region containing the conserved tryptophan is directly involved in FliG binding, which contributes to the formation of the stable structure of FliGN. Considering our results presented here and other previously reported results, we conclude that Caulobacter, Thermotoga, and Vibrio share a mode of FliF-FliG interaction, which represents an initial step in flagellar assembly. It should be noted that the C-terminal mutations in Vibrio FliF apparently do not abolish FliG binding by causing global structural changes in the mutant FliF proteins. Future work includes a detailed structural characterization of the soluble FliF multimers, which will allow a comparison with the purified MS ring and will help us to understand the nature of FliF-FliG binding and its role in flagellar assembly.
Supplementary Material
ACKNOWLEDGMENTS
We thank Tomohiro Yorimitsu for construction of plasmid pTY502 and Sachi Tatematsu and Mayumi Taniguchi for technical assistance. We also thank Noriko Kato at Olympus for her technical advice and discussions about FCS analysis.
This work was supported by Ministry of Education, Culture, Sports, Science and Technology (MEXT) KAKENHI grant number 20051009 to S.K. and Japan Society for the Promotion of Science (JSPS) KAKENHI grant numbers 24117004 and 23247024 to M.H.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02271-14.
REFERENCES
- 1.Berg HC. 2003. The rotary motor of bacterial flagella. Annu Rev Biochem 72:19–54. doi: 10.1146/annurev.biochem.72.121801.161737. [DOI] [PubMed] [Google Scholar]
- 2.Kojima S, Blair DF. 2004. The bacterial flagellar motor: structure and function of a complex molecular machine. Int Rev Cytol 233:93–134. doi: 10.1016/S0074-7696(04)33003-2. [DOI] [PubMed] [Google Scholar]
- 3.Li N, Kojima S, Homma M. 2011. Sodium-driven motor of the polar flagellum in marine bacteria Vibrio. Genes Cells 16:985–999. doi: 10.1111/j.1365-2443.2011.01545.x. [DOI] [PubMed] [Google Scholar]
- 4.Blair DF. 2003. Flagellar movement driven by proton translocation. FEBS Lett 545:86–95. doi: 10.1016/S0014-5793(03)00397-1. [DOI] [PubMed] [Google Scholar]
- 5.Dean GD, Macnab RM, Stader J, Matsumura P, Burks C. 1984. Gene sequence and predicted amino acid sequence of the motA protein, a membrane-associated protein required for flagellar rotation in Escherichia coli. J Bacteriol 159:991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stader J, Matsumura P, Vacante D, Dean GE, Macnab RM. 1986. Nucleotide sequence of the Escherichia coli MotB gene and site-limited incorporation of its product into the cytoplasmic membrane. J Bacteriol 166:244–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braun TF, Al-Mawsawi LQ, Kojima S, Blair DF. 2004. Arrangement of core membrane segments in the MotA/MotB proton-channel complex of Escherichia coli. Biochemistry 43:35–45. doi: 10.1021/bi035406d. [DOI] [PubMed] [Google Scholar]
- 8.Kojima S, Blair DF. 2004. Solubilization and purification of the MotA/MotB complex of Escherichia coli. Biochemistry 43:26–34. doi: 10.1021/bi035405l. [DOI] [PubMed] [Google Scholar]
- 9.Zhou J, Fazzio RT, Blair DF. 1995. Membrane topology of the MotA protein of Escherichia coli. J Mol Biol 251:237–242. doi: 10.1006/jmbi.1995.0431. [DOI] [PubMed] [Google Scholar]
- 10.Chun SY, Parkinson JS. 1988. Bacterial motility: membrane topology of the Escherichia coli MotB protein. Science 239:276–278. doi: 10.1126/science.2447650. [DOI] [PubMed] [Google Scholar]
- 11.Asai Y, Kojima S, Kato H, Nishioka N, Kawagishi I, Homma M. 1997. Putative channel components for the fast-rotating sodium-driven flagellar motor of a marine bacterium. J Bacteriol 179:5104–5110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sato K, Homma M. 2000. Functional reconstitution of the Na+-driven polar flagellar motor component of Vibrio alginolyticus. J Biol Chem 275:5718–5722. doi: 10.1074/jbc.275.8.5718. [DOI] [PubMed] [Google Scholar]
- 13.Blair DF, Berg HC. 1988. Restoration of torque in defective flagellar motors. Science 242:1678–1681. doi: 10.1126/science.2849208. [DOI] [PubMed] [Google Scholar]
- 14.Block SM, Berg HC. 1984. Successive incorporation of force-generating units in the bacterial rotary motor. Nature 309:470–472. doi: 10.1038/309470a0. [DOI] [PubMed] [Google Scholar]
- 15.Reid SW, Leake MC, Chandler JH, Lo CJ, Armitage JP, Berry RM. 2006. The maximum number of torque-generating units in the flagellar motor of Escherichia coli is at least 11. Proc Natl Acad Sci U S A 103:8066–8071. doi: 10.1073/pnas.0509932103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sowa Y, Rowe AD, Leake MC, Yakushi T, Homma M, Ishijima A, Berry RM. 2005. Direct observation of steps in rotation of the bacterial flagellar motor. Nature 437:916–919. doi: 10.1038/nature04003. [DOI] [PubMed] [Google Scholar]
- 17.De Mot R, Vanderleyden J. 1994. The C-terminal sequence conservation between OmpA-related outer membrane proteins and MotB suggests a common function in both gram-positive and gram-negative bacteria, possibly in the interaction of these domains with peptidoglycan. Mol Microbiol 12:333–334. doi: 10.1111/j.1365-2958.1994.tb01021.x. [DOI] [PubMed] [Google Scholar]
- 18.Kojima S, Imada K, Sakuma M, Sudo Y, Kojima C, Minamino T, Homma M, Namba K. 2009. Stator assembly and activation mechanism of the flagellar motor by the periplasmic region of MotB. Mol Microbiol 73:710–718. doi: 10.1111/j.1365-2958.2009.06802.x. [DOI] [PubMed] [Google Scholar]
- 19.Irikura VM, Kihara M, Yamaguchi S, Sockett H, Macnab RM. 1993. Salmonella typhimurium fliG and fliN mutations causing defects in assembly, rotation, and switching of the flagellar motor. J Bacteriol 175:802–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lloyd SA, Tang H, Wang X, Billings S, Blair DF. 1996. Torque generation in the flagellar motor of Escherichia coli: evidence of a direct role for FliG but not for FliM or FliN. J Bacteriol 178:223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brown PN, Hill CP, Blair DF. 2002. Crystal structure of the middle and C-terminal domains of the flagellar rotor protein FliG. EMBO J 21:3225–3234. doi: 10.1093/emboj/cdf332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lam KH, Ip WS, Lam YW, Chan SO, Ling TK, Au SW. 2012. Multiple conformations of the FliG C-terminal domain provide insight into flagellar motor switching. Structure 20:315–325. doi: 10.1016/j.str.2011.11.020. [DOI] [PubMed] [Google Scholar]
- 23.Lloyd SA, Whitby FG, Blair DF, Hill CP. 1999. Structure of the C-terminal domain of FliG, a component of the rotor in the bacterial flagellar motor. Nature 400:472–475. doi: 10.1038/22794. [DOI] [PubMed] [Google Scholar]
- 24.Minamino T, Imada K, Kinoshita M, Nakamura S, Morimoto YV, Namba K. 2011. Structural insight into the rotational switching mechanism of the bacterial flagellar motor. PLoS Biol 9:e1000616. doi: 10.1371/journal.pbio.1000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vartanian AS, Paz A, Fortgang EA, Abramson J, Dahlquist FW. 2012. Structure of flagellar motor proteins in complex allows for insights into motor structure and switching. J Biol Chem 287:35779–35783. doi: 10.1074/jbc.C112.378380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee LK, Ginsburg MA, Crovace C, Donohoe M, Stock D. 2010. Structure of the torque ring of the flagellar motor and the molecular basis for rotational switching. Nature 466:996–1000. doi: 10.1038/nature09300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou J, Lloyd SA, Blair DF. 1998. Electrostatic interactions between rotor and stator in the bacterial flagellar motor. Proc Natl Acad Sci U S A 95:6436–6441. doi: 10.1073/pnas.95.11.6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morimoto YV, Nakamura S, Hiraoka KD, Namba K, Minamino T. 2013. Distinct roles of highly conserved charged residues at the MotA-FliG interface in bacterial flagellar motor rotation. J Bacteriol 195:474–481. doi: 10.1128/JB.01971-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morimoto YV, Nakamura S, Kami-Ike N, Namba K, Minamino T. 2010. Charged residues in the cytoplasmic loop of MotA are required for stator assembly into the bacterial flagellar motor. Mol Microbiol 78:1117–1129. doi: 10.1111/j.1365-2958.2010.07391.x. [DOI] [PubMed] [Google Scholar]
- 30.Ueno T, Oosawa K, Aizawa S. 1992. M ring, S ring and proximal rod of the flagellar basal body of Salmonella typhimurium are composed of subunits of a single protein, FliF. J Mol Biol 227:672–677. doi: 10.1016/0022-2836(92)90216-7. [DOI] [PubMed] [Google Scholar]
- 31.Ueno T, Oosawa K, Aizawa S. 1994. Domain structures of the MS ring component protein (FliF) of the flagellar basal body of Salmonella typhimurium. J Mol Biol 236:546–555. doi: 10.1006/jmbi.1994.1164. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki H, Yonekura K, Namba K. 2004. Structure of the rotor of the bacterial flagellar motor revealed by electron cryomicroscopy and single-particle image analysis. J Mol Biol 337:105–113. doi: 10.1016/j.jmb.2004.01.034. [DOI] [PubMed] [Google Scholar]
- 33.Kubori T, Shimamoto N, Yamaguchi S, Namba K, Aizawa S. 1992. Morphological pathway of flagellar assembly in Salmonella typhimurium. J Mol Biol 226:433–446. doi: 10.1016/0022-2836(92)90958-M. [DOI] [PubMed] [Google Scholar]
- 34.Oosawa K, Ueno T, Aizawa S. 1994. Overproduction of the bacterial flagellar switch proteins and their interactions with the MS ring complex in vitro. J Bacteriol 176:3683–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kubori T, Yamaguchi S, Aizawa S. 1997. Assembly of the switch complex onto the MS ring complex of Salmonella typhimurium does not require any other flagellar proteins. J Bacteriol 179:813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levenson R, Zhou H, Dahlquist FW. 2012. Structural insights into the interaction between the bacterial flagellar motor proteins FliF and FliG. Biochemistry 51:5052–5060. doi: 10.1021/bi3004582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawagishi I, Okunishi I, Homma M, Imae Y. 1994. Removal of the periplasmic DNase before electroporation enhances efficiency of transformation in a marine bacterium Vibrio alginolyticus. Microbiology 140:2355–2361. doi: 10.1099/13500872-140-9-2355. [DOI] [Google Scholar]
- 38.Kojima S, Nonoyama N, Takekawa N, Fukuoka H, Homma M. 2011. Mutations targeting the C-terminal domain of FliG can disrupt motor assembly in the Na+-driven flagella of Vibrio alginolyticus. J Mol Biol 414:62–74. doi: 10.1016/j.jmb.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 39.Eigen M, Rigler R. 1994. Sorting single molecules: application to diagnostics and evolutionary biotechnology. Proc Natl Acad Sci U S A 91:5740–5747. doi: 10.1073/pnas.91.13.5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schroers A, Hecht O, Kallen KJ, Pachta M, Rose-John S, Grotzinger J. 2005. Dynamics of the gp130 cytokine complex: a model for assembly on the cellular membrane. Protein Sci 14:783–790. doi: 10.1110/ps.041117105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grunenfelder B, Gehrig S, Jenal U. 2003. Role of the cytoplasmic C terminus of the FliF motor protein in flagellar assembly and rotation. J Bacteriol 185:1624–1633. doi: 10.1128/JB.185.5.1624-1633.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones DT. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 43.Li H, Sourjik V. 2011. Assembly and stability of flagellar motor in Escherichia coli. Mol Microbiol 80:886–899. doi: 10.1111/j.1365-2958.2011.07557.x. [DOI] [PubMed] [Google Scholar]
- 44.Okunishi I, Kawagishi I, Homma M. 1996. Cloning and characterization of motY, a gene coding for a component of the sodium-driven flagellar motor in Vibrio alginolyticus. J Bacteriol 178:2409–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yorimitsu T, Mimaki A, Yakushi T, Homma M. 2003. The conserved charged residues of the C-terminal region of FliG, a rotor component of the Na+-driven flagellar motor. J Mol Biol 334:567–583. doi: 10.1016/j.jmb.2003.09.052. [DOI] [PubMed] [Google Scholar]
- 46.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li N, Kojima S, Homma M. 2011. Characterization of the periplasmic region of PomB, a Na+-driven flagellar stator protein in Vibrio alginolyticus. J Bacteriol 193:3773–3784. doi: 10.1128/JB.00113-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.