

Abstract
Although the enzymes for dissimilatory sulfate reduction by microbes have been studied, the mechanisms for transcriptional regulation of the encoding genes remain unknown. In a number of bacteria the transcriptional regulator Rex has been shown to play a key role as a repressor of genes producing proteins involved in energy conversion. In the model sulfate-reducing microbe Desulfovibrio vulgaris Hildenborough, the gene DVU_0916 was observed to resemble other known Rex proteins. Therefore, the DVU_0916 protein has been predicted to be a transcriptional repressor of genes encoding proteins that function in the process of sulfate reduction in D. vulgaris Hildenborough. Examination of the deduced DVU_0916 protein identified two domains, one a winged helix DNA-binding domain common for transcription factors, and the other a Rossman fold that could potentially interact with pyridine nucleotides. A deletion of the putative rex gene was made in D. vulgaris Hildenborough, and transcript expression studies of sat, encoding sulfate adenylyl transferase, showed increased levels in the D. vulgaris Hildenborough Rex (RexDvH) mutant relative to the parental strain. The RexDvH-binding site upstream of sat was identified, confirming RexDvH to be a repressor of sat. We established in vitro that the presence of elevated NADH disrupted the interaction between RexDvH and DNA. Examination of the 5′ transcriptional start site for the sat mRNA revealed two unique start sites, one for respiring cells that correlated with the RexDvH-binding site and a second for fermenting cells. Collectively, these data support the role of RexDvH as a transcription repressor for sat that senses the redox status of the cell.
INTRODUCTION
The anaerobic process of microbially influenced corrosion is estimated to account for 15% of the total cost of ferrous metal and concrete/stonework corrosion. For the U.S. energy industry, this amounts to approximately $100 billion yearly (1). Most notably, the metabolic processes of the sulfate-reducing microbes (SRM) have been implicated in this observed corrosion (2, 3). Clearly, it would be beneficial to understand the mechanism and the regulation of genes involved in the key processes leading to metal dissolution.
The SRM convert energy by dissimilatory sulfate reduction, where sulfate is used as a terminal electron acceptor in respiration. Although SRM are mainly found in sulfate-rich anoxic environments (4), they are also found in anoxic habitats depleted of sulfate because they are able to use electron acceptors other than sulfate (5, 6). Desulfovibrio vulgaris Hildenborough, a Gram-negative deltaproteobacterium, is a model organism for examining sulfate reduction as it is genetically accessible and is readily cultured in the lab (7, 8). Also, the genome for D. vulgaris Hildenborough has been sequenced (9) and has recently been reannotated (10).
The process of dissimilatory sulfate reduction is carried out by a well-conserved biochemical pathway in heterogeneous SRM (5, 11–13). In brief, sulfate (SO42−) is taken up by the cell and is activated by sulfate adenylyl transferase (Sat, encoded by sat) to adenylyl phosphosulfate (APS) and inorganic pyrophosphate. APS is then reduced to sulfite (SO32−) and AMP by APS reductase (ApsBA, encoded by apsBA). Sulfite is further reduced to hydrogen sulfide (H2S) by dissimilatory sulfite reductase (DsrAB, encoded by dsrAB). Although enzymes for sulfate reduction have been well studied (5, 12, 13) and their crystal structures have been solved (14–17), the mechanism for transcriptional regulation of the encoding genes remains unknown.
In D. vulgaris Hildenborough, genes involved in sulfate reduction are differentially expressed depending on the available nutrients (http://www.microbesonline.org/) (18). For example, apsBA expression was decreased in medium containing sulfite compared to expression in sulfate medium (19). From the genome sequence of D. vulgaris Hildenborough, more than 150 transcriptional regulators have been predicted, several of which have the potential to be responsible for the observed expression changes (http://networks.systemsbiology.net/dvh/search/advanced) (20). DVU_0916 was predicted to encode a Rex protein and was hypothesized to be involved with sulfate reduction by regulating more than 50 genes that are responsible for energy conversion processes (21). From an examination of genome sequences, Rex has been predicted to be present in a wide range of microbes, including Gram-negative and Gram-positive aerobes and anaerobes. To date, Rex proteins have been studied experimentally in several aerobes, including Thermus aquaticus (22–24), Streptomyces coelicolor (25), and Bacillus subtilis (24, 26–28), and the anaerobe Desulfovibrio alaskensis G20 (30). The present study will add to our understanding of the function of Rex in anaerobic SRM.
Functional Rex proteins contain an N-terminal DNA-binding domain, a dimerization domain, and a Rossman fold (22, 24). The latter apparently functions to bind pyridine nucleotides (NADH and NAD+). The consensus binding sequence for Rex, TTTGTGAAATATTTCACAAA, has been compiled from comparisons of more than 100 genomes (21). The sequence contains an inverted repeat (underlined), since Rex functions as a homodimer, and the inverted repeat is the minimum sequence for Rex binding, as observed in S. coelicolor (25).
Targets for Rex typically include genes encoding proteins involved in NADH oxidation, and Rex acts as a transcriptional repressor when the NADH/NAD+ ratio is low (21, 26). Rex from T. aquaticus, expressed in Escherichia coli, was crystallized in the presence of NADH and NAD+ separately, and the structures were solved (22, 24). From these in vitro assays it was clear that Rex underwent structural changes that modulated the binding activity between Rex and a consensus DNA sequence. Specifically, when Rex bound NADH, the resulting conformation of Rex was no longer able to interact within the major groove of DNA and therefore would no longer repress (24). These results were consistent with in vitro protein-DNA interaction assays performed on an upstream DNA sequence of adhE2 in Clostridium acetobutylicum. In these assays, increased expression of adhE2 was observed in a rex deletion strain (31). Examination of expression data in an SRM closely related to D. vulgaris Hildenborough, D. alaskensis G20, revealed increased transcript expression for sat in a rex deleted strain; however, minimal differences were observed for apsBA and dsrABD (30). The increase in expression for sat, encoding the first enzyme in the process of sulfate reduction, but not apsBA and dsrABD provides support for the role of Rex as a transcriptional repressor for early steps in sulfate reduction. Interestingly, rnfC, which is predicted to be within the Rex regulon, was decreased in the Rex mutant, which was interpreted to mean that Rex acts not only as a repressor but as an activator as well.
The properties described for Rex (i.e., a regulon that contains genes that encode proteins responsible for energy conversion and a regulator that senses the NADH/NAD+ ratio) would be those expected for a regulator of genes that encode proteins that function in sulfate reduction in D. vulgaris Hildenborough, since sulfate reduction is the primary mechanism for energy conversion in D. vulgaris Hildenborough. Therefore, a Rex homolog in D. vulgaris Hildenborough (RexDvH) would be a reasonable candidate regulator. By differentially binding NAD+ and NADH and repressing genes for NADH oxidation when NADH is low, Rex may maintain the NADH/NAD+ ratio (26). Using this paradigm, we proposed that Rex might be involved in the switch between respiration and fermentation in SRM and be necessary for adaptation to fluctuating electron acceptor concentrations.
We sought to confirm that the protein encoded by DVU_0916 was a functional Rex homolog in D. vulgaris Hildenborough. To accomplish this, the growth kinetics of a deletion strain for rex were determined, and transcript differences for sat—encoding the enzyme that activates sulfate for the first step in its reduction—were analyzed. All results were consistent with the conclusion that RexDvH is a redox-responsive transcriptional repressor for sat.
MATERIALS AND METHODS
Culture, media, and sample collection.
The growth of all cultures was measured in Balch tubes by determining the optical density at 600 nm (OD600) with a Spectronic Genesys 20 spectrophotometer (Thermo Fisher Scientific, Waltham, MA). E. coli strains were grown at 37°C aerobically in 5 ml of LC medium containing (per liter) 10 g of tryptone, 5 g of sodium chloride, and 5 g of yeast extract (pH 7.0). D. vulgaris Hildenborough strains were started from freezer stocks that contained 10% (vol/vol) glycerol in growth medium and frozen at −80 °C. Growth medium was generally MO medium (19) supplemented with 0.1% (wt/vol) yeast extract and 60 mM lactate with 30 mM sulfate (MOYLS4). The medium was reduced with 1.2 mM sodium thioglycolate, and the pH was adjusted to pH 7.2 with 12 M HCl. The inhibitors for D. vulgaris Hildenborough strains were as follows: Geneticin (G418; 400 μg/ml), spectinomycin (Sp; 100 μg/ml), kanamycin (Km; 50 μg/ml), ampicillin (100 μg/ml) or 5-fluorouracil (5-FU; 40 μg/ml) obtained from Thermo Fisher Scientific, Sigma-Aldrich (St. Louis, MO), or Gold Biotechnologies (St. Louis, MO). D. vulgaris Hildenborough cultures were grown in an anaerobic chamber (Coy Laboratory Product, Inc., Grass Lake, MI) overnight until reaching the stationary phase (OD600 > 1). The atmosphere of the chamber was ca. 95% N2 and 5% H2. A 2% (vol/vol) inoculum was used to start 5-ml cultures in defined MOLS4 (MOYLS4 lacking yeast extract) or pyruvate fermentation medium (MO medium with 60 mM pyruvate, supplemented with 0.1% [wt/vol] yeast extract and 0.5 mM cysteine [MOYPyr]). For pyruvate-fermenting cultures, cysteine was provided as a sulfur source and reductant. D. vulgaris Hildenborough did not grow on yeast extract alone in the absence of pyruvate (data not shown). For mRNA studies, 4-ml samples were spun down anaerobically at 34°C (10 min at 5,600 × g) and resuspended in 1 ml of TRI reagent (Sigma-Aldrich).
Genomic DNA (Wizard Genomic DNA purification kit; Promega, Madison, WI), plasmid (GeneJET plasmid kit; Thermo Fisher Scientific), and DNA fragments (Wizard SV Gel and PCR Clean-Up System [Promega]) were purified according to the manufacturer's protocol. Sequence analysis was performed by University of Missouri DNA Core Facility. Oligonucleotides necessary for these studies were purchased from Integrated DNA Technologies (Coralville, IA) (see Table S1 in the supplemental material). The DNA and RNA concentrations (and A260/A280 ratio) were calculated from NanoDrop ND-1000 spectrophotometric readings (Thermo Fisher Scientific).
Strain and plasmid constructions.
All of the strains used in the present study are listed in Table 1. All plasmid constructs were made by the SLIC procedure (32). The protocol used to generate the strain lacking the sat promoter (ΔPsat), JW9293, conferring kanamycin resistance and 5-FU sensitivity, has previously been described (33). Briefly, 150 bp upstream of sat (−150 to −1) was replaced with a cassette conferring resistance to kanamycin and a counterselectable marker (upp, the gene encoding uracil phosphoribosyltransferase that confers 5-FU sensitivity to the 5-FUr parental strain). To accomplish the cassette insertion, a delivery plasmid (pMO9292) was constructed that contained the selectable cassette flanked by chromosomal regions from either side of the promoter sequence to be deleted. This plasmid was electroporated into the parental strain, JW710, and selected for kanamycin resistance and screened for 5-FU sensitivity. The successful recombinant was designated JW9293. For the construction of the subsequent Psat mutants with alterations to the RexDvH-binding site (JW9312, JW9314, JW9316, JW9318, and JW9320), a protocol similar to that described by Parks et al. (34) was followed. JW9293 was transformed with nonreplicating plasmids with individual site-specific mutations in the 150-bp fragment flanked by DNA homologous to that on either side of the integrated cassette. These Psat mutant or restored strains were selected as 5-FUr and screened for Km sensitivity. The accuracy of the constructs was confirmed by sequencing both strands of a PCR-amplified product across the mutation site. The deletion strain and promoter mutation strains that prevented sat transcription were expected not to grow with sulfate as an electron acceptor, and therefore 20 mM sulfite was used in medium to recover these promoter mutations.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Genotype and/or relevant characteristicsa | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| α-Select (Silver Efficiency) | deoR endA1 recA1 relA1 gyrA96 hsdR17(rK− mK+) supE44 thi-1 Δ(lacZYA-argFV169) ϕ80dlacZΔM15 F− | Bioline |
| BL21(DE3) competent cells | B F− dcm ompT hsdS(rB− mB−) gal λ (DE3) | Agilent |
| D. vulgaris Hildenborough | ||
| ATCC 29579 | Wild type; 5-FUs | ATCC |
| JW710 | WT Δupp; 5-FUr (used as a WT control for D. vulgaris Hildenborough growth kinetics in this study; parent strain for deletions; retains pDV1 present in WT) | 8 |
| JW3311 | JW710 ΔDVU_0916::(npt upp); Kmr; 5-FUs (Δrex marker exchange) | 33 |
| JW9293 | JW710 Δ−150–1 Psat::(npt upp); Kmr; 5-FUs (Psat disruption) | This study |
| JW9312 | JW710; Kms; 5-FUr (sat promoter restored) | This study |
| JW9314 | JW9293 G−147A Psat; Kms; 5-FUr (G −147 A) | This study |
| JW9316 | JW9293 GTA−147–145ACG Psat; Kms; 5-FUr (IR1) | This study |
| JW9318 | JW9293 CAC−136–134TGT Psat; Kms; 5-FUr (IR2) | This study |
| JW9320 | JW9293 GTA−147–145ACG Psat CAC−136–134TGT Psat; Kms; 5-FUr (IR1and2) | This study |
| JW9011 | JW710 ΔDVU_2547::(npt upp); Kmr; 5-FUs (ΔhcpR marker exchange) | 50 |
| GZ0481 | Genome position 2680507::Tn5-RL27; insertion 273 bp from predicted AUG start codon within DVU_2567; Kmr (LysX mutant) | Wall laboratory |
| Plasmids | ||
| pET14b | 6×His tag fusion protein vector with T7 promoter | Novagen |
| pMO719 | pCR8/GW/TOPO containing SRB replicon (pBG1); Spr; source of Spr and pUC ori fragment; for marker exchange suicide plasmid construction | 8 |
| pMO746 | Source of upp in artificial operon with npt and Apr-pUC ori; Pnpt-npt-upp; Kmr; 5-FUs; for marker exchange suicide plasmid construction | 34 |
| pMO3312 | pET14b plus rex (without start codon, 642 bp); Apr; for Rex expression in BL21(DE3) competent cells | This study |
| pMO3313 | pMO9075 with DVU_0916 (rex) constitutively expressed from Pnpt | 33 |
| pMO9075 | pMO719 containing Pnpt for constitutive expression of complementation constructs; pBG1 stable SRB replicon; Spr | 7 |
| pMO9292 | Spr and pUC ori from pMO719 plus 383-bp upstream and 319-bp downstream DNA regions from Psat (−150–1) flanking the artificial operon of Pnpt-npt-upp from pMO746; for marker exchange mutagenesis; Spr and Kmr | This study |
| pMO9311 | Spr and pUC ori from pMO719 plus 403-bp upstream and 467-bp downstream DNA regions from Psat (−150); wild-type sequence; Spr; for site-directed mutagenesis | This study |
| pMO9313 | Spr and pUC ori from pMO719 plus 403-bp upstream and 467-bp downstream DNA regions from Psat (−150); G−147A Psat; Spr; for site-directed mutagenesis | This study |
| pMO9315 | Spr and pUC ori from pMO719 plus 403-bp upstream and 467-bp downstream DNA regions from Psat (−150); GTA−147–145ACG Psat; Spr; for site-directed mutagenesis | This study |
| pMO9317 | Spr and pUC ori from pMO719 plus 403-bp upstream and 467-bp downstream DNA regions from Psat (−150); CAC−136–134TGT Psat; Spr; for site-directed mutagenesis | This study |
| pMO9319 | Spr and pUC ori from pMO719 plus 403-bp upstream and 467-bp downstream DNA regions from Psat (−150); GTA−147–145ACG Psat CAC−136–134TGT Psat; Spr; for site-directed mutagenesis | This study |
Km, kanamycin; Sp, spectinomycin; Ap, ampicillin; 5-FU, 5-fluorouracil; superscript “r” or “s,” resistance or sensitivity, respectively.
Quantitative reverse transcriptase-PCR (qRT-PCR). (i) Initial optimization.
Primers were manually designed to amplify an approximately 100- to 150-bp region at the 3′ end of the transcript for each gene (see Table S1 in the supplemental material). qPCRs were set up according to the manufacturer's protocol with SsoFast EvaGreen Supermix (Bio-Rad, Hercules, CA) on a CFX96 and analyzed with CFX Manager (version 3.1; Bio-Rad), with the curve fit to regression (35–37). The amplification protocol was initiated with a 3-min preincubation at 95°C and processed through 40 cycles of denaturation (30 s at 95°C) and annealing/extension (30 s at 65°C). The fluorescent signal was acquired at the end of each annealing/extension step. The melting curve protocol included annealing at the annealing/extension temperature (65°C) and melting at a ramp rate of 0.5°C/5 s up to 95°C, with the fluorescent signal acquired continuously during the melting curve. The genes to be used as internal (reference) controls were rplS (DVU_0835) and rpmC (DVU_1311) (38) because they are expressed at levels similar to those of the genes to be assessed in the present study and their expression levels were not found to change during exposure to environmental stresses (http://www.microbesonline.org/; data not shown). To validate these reference genes, the strategy of Hellemans et al. was implemented (35), and the genes were shown to have minimal transcriptional differences among the strains, mutants, and conditions tested, with a mean coefficient of variance (CV) of <0.25 and mean (M) of <0.5.
(ii) Sample preparation.
Samples were initially resuspended in 1 ml of TRI reagent (Sigma-Aldrich) and kept frozen until RNA isolation by phenol extraction (39, 40). The RNA quality was assessed visually in an agarose-denaturing gel, and an A260/A280 ratio of >1.8 was required. RNA samples were treated with Turbo DNase (Life Technologies, Carlsbad, CA) according to the manufacturer's protocol and confirmed by PCR to be free of genomic DNA. For each gene to be analyzed, a standard curve (6 logs, serial dilution from 100 ng/μl stock cDNA) was performed to calculate efficiency. For each transcript, the relative abundance was normalized by the reference gene transcripts in the specified sample.
Transcriptional start site (TSS) determination by 5′-RACE (rapid amplification of cDNA ends).
Samples were initially collected from DNase-treated RNA samples used for qRT-PCR. The procedure used was adapted from that used by Scotto-Lavino et al. (41). In brief, 100 ng of DNase-treated RNA was reverse transcribed to single-stranded cDNA (iScript Select cDNA synthesis kit; Bio-Rad) with primer DVU1295-sat-GSP1, degraded with RNase A/T1 mix and RNase H (Thermo Fisher Scientific), and the cDNA was purified by column purification. With terminal deoxynucleotidyltransferase (Thermo Fisher Scientific), an adenosine tail was added to the cDNA, followed by the generation of the second DNA strand with iScript and the primer RACE-2nd Strand. The RACE-2nd Strand primer was adapted from primer AUAP (Invitrogen 5′ RACE system, v2.0) to include additional T residues and a “V” at the 3′ position (“V” as A/G/C but not T) to allow for better anchoring to the modified cDNA. Double-stranded DNA (dsDNA) was purified, diluted 1:1,000, and amplified by PCR (PCR1). Thirty cycles of amplification were carried out (30 s at 94°C, 30 s at 58°C, and 1 min at 72°C), followed by a final extension for 5 min at 72°C with Taq DNA polymerase (NEB, Ipswich, MA). An additional PCR (PCR2) followed, which was performed according to the same protocol used for PCR1, but with 1:1,000-diluted PCR1 as the template and nested primers. Fragments were purified, sequenced, and mapped to the genome.
In vitro protein-DNA interaction assays.
To obtain RexDvH for protein-DNA interaction studies, a 6×His tag was added to RexDvH by cloning rex into pET-14b (Novagen, Madison, WI) and transforming this into BL21(DE3) competent cells (Agilent, Santa Clara, CA). After induction with IPTG (isopropyl-β-d-thiogalactopyranoside; Gold Biotechnologies), the tagged protein was purified by using a His60 Ni gravity column purification kit (Clontech, Mountain View, CA). Eluted 1-ml fractions were analyzed for protein (42), and fractions were pooled together and passed over a PD-10 desalting column (GE Healthcare Biosciences, Piscataway, NJ). SDS-PAGE was performed to ensure the purity of the RexDvH monomer (∼25 kDa) (data not shown). The presence of NADH bound to RexDvH was checked by measuring the absorbance at 340 nm to ensure the purity of the protein from additional cofactors.
The protocol by Brekasis and Paget (25) was used for DNA template generation of the relevant RexDvH-binding site (>100 bp). For smaller DNA fragments (40 bp), reverse complemented primers were annealed (Sigma-Aldrich). These sequences were 5′ end labeled with T4 polynucleotide kinase (Promega) and [γ-32P]ATP (Perkin-Elmer, Waltham, MA) according to the manufacturer's protocol. The unlabeled nucleotides were removed by using a QIAquick nucleotide removal kit (Qiagen, Valencia, CA).
EMSA.
Electrophoretic mobility shift assays (EMSAs) were performed according to the method of Ravcheev et al. (21). In brief, dsDNA fragments (0.1 nM) were incubated with RexDvH at specified concentrations (0 to 2,000 nM) in a final volume of 30 μl. The binding buffer contained 20 mM Tris-HCl (pH 8.0), 10% (vol/vol) glycerol, 1 mM MgCl2, and 40 mM KCl. Samples were incubated at 37°C for 25 min, placed on ice for 2 min, and then separated (90 V, 70 min, 4°C) on a 5% (wt/vol) native Tris-borate-EDTA (TBE) polyacrylamide gel that was preincubated (200 V, 30 min at 4°C) in 0.5× TBE buffer (Bio-Rad). When pyridine nucleotides were examined, the desired concentrations were added after the initial incubation and then incubated for an additional 10 min at 37°C. The gel was removed from the apparatus, wrapped in plastic wrap, and exposed to a Kodak Imaging Screen K (Bio-Rad) typically for 15 to 60 min in a sealed cassette, followed by imaging with a personal molecular imager (Bio-Rad).
Fluorescent polarization assay (FPA).
Fluorescent polarization was performed as described previously (43). The binding assay was performed in 96-well black plate (VWR, Radnor PA) with 1 nM fluorescently labeled (6-FAM) oligonucleotides. Different concentrations of protein (10, 25, 50, 100, 250, 500, and 1,000 nM) were incubated with 1 nM labeled oligonucleotides in 100-μl reaction mixture in the binding buffer (20 mM Tris-HCl [pH 7.5], 100 mM NaCl, 0.3 mg of bovine serum albumin/ml, 1 μg of herring sperm DNA). The fluorescence reading was taken on a Beckman multimode plate reader (DTX 880) with excitation and emission filters at 495 and 520 nm. The background fluorescence from buffer was subtracted and the fluorescence polarization values were defined as follows: Pmp = [(Iparallel − G-factor) × Iperpendicular]/[(Iparallel + G-factor) × Iperpendicular] × 1,000, where Iparallel and Iperpendicular are the fluorescence intensity in the parallel and perpendicular orientation respective to the orientation of the excitation polarizer. The G-factor is an experimental correction for the polarization bias of the detection system (44).
RESULTS
Deletion of rex.
To examine the role of RexDvH, a marker-exchange deletion of rex and a complemented deletion strain were constructed (33). These two strains, in addition to the parental strain, were grown by sulfate respiration in MOLS4 or by pyruvate fermentation in MOYPyr. The former was expected to differ from the latter by changes in the ratio of NADH/NAD+ proposed to be a signal for Rex regulation. It was assumed that fermenting cultures lacking an inorganic terminal electron acceptor would exhibit an increase in NADH (45). Examination of the growth in either medium revealed no significant differences among the three strains (see Table S2 in the supplemental material). In parental cells, qRT-PCR analysis of sat expression (Table 2) showed that sat transcription had a 7-fold increase in fermenting cultures compared to cultures respiring sulfate. In the RexDvH mutant, sat expression was minimally increased when growing fermentatively but was already 11-fold increased from the parental strain while respiring sulfate, a finding consistent with a repressor role for Rex. When this strain was complemented with a plasmid copy of rex that was transcribed from a constitutive promoter, partially restored (decreased) levels of sat transcription were observed. To eliminate the possibility that growth modes were affecting the expression of rex, transcription of that gene was examined and found to be unchanged in the parental strain and was undetected in the RexDvH mutant as expected. Therefore, these studies provided support that RexDvH is a transcriptional repressor for sat.
TABLE 2.
Transcript analysis of parental, Δrex, and complement of rex strains grown by sulfate respiration or pyruvate fermentationa
| Medium and strain | Description or genotype | Mean transcript level ± SEM for: |
|
|---|---|---|---|
| sat | rex | ||
| MOLS4 | |||
| JW710 | Parental | 1.0 ± 0.2 | 1.0 ± 0.0 |
| JW3311 | Δrex | 11.2 ± 2.3 | ND |
| JW3311(pMO3313) | Complement of rex | 5.9 ± 0.8 | 10.9 ± 0.0 |
| MOYPyr | |||
| JW710 | Parental | 7.3 ± 1.2 | 1.7 ± 0.0 |
| JW3311 | Δrex | 13.6 ± 3.7 | ND |
| JW3311(pMO3313) | Complement of rex | 6.4 ± 0.6 | 24.9 ± 0.2 |
The OD600 was monitored through growth, and samples were collected for analysis by qRT-PCR at the early exponential phase. Approximately 100 ng of Turbo DNase-treated RNA was converted to cDNA, and 1 μl of cDNA (5 ng of RNA) was used per qRT-PCR. Each gene was assessed individually and normalized with respect to the reference genes rplS and rpmC. The efficiency for each gene was determined as follows: rplS = 92.6%, rpmC = 95.5%, sat = 91.3%, and rex = 90.2%. Samples were normalized to JW710 MOLS4. ND, not detected.
Rex binding site in the sat promoter.
Since the expression levels for sat were increased by the deletion of rex, we sought to determine whether the regulation was direct or indirect. There is a putative Rex binding site located at positions −150 to −131 (TTTGTAAATTTTTTCACAAG) relative to the translational start codon for sat (21). Therefore, RexDvH was purified for protein-DNA interaction studies. Four dsDNA fragments were examined for interaction with RexDvH (Fig. 1): one upstream and one downstream of the predicted Rex binding site, and two of different sizes that contained the motif. The two fragments that contained the Rex binding site (fragments C and D in Fig. 1) shifted in electrophoretic mobility when the putative RexDvH protein was present, whereas the other two fragments did not. These results confirm a direct interaction between RexDvH protein and the putative RexDvH-binding site upstream of sat.
FIG 1.
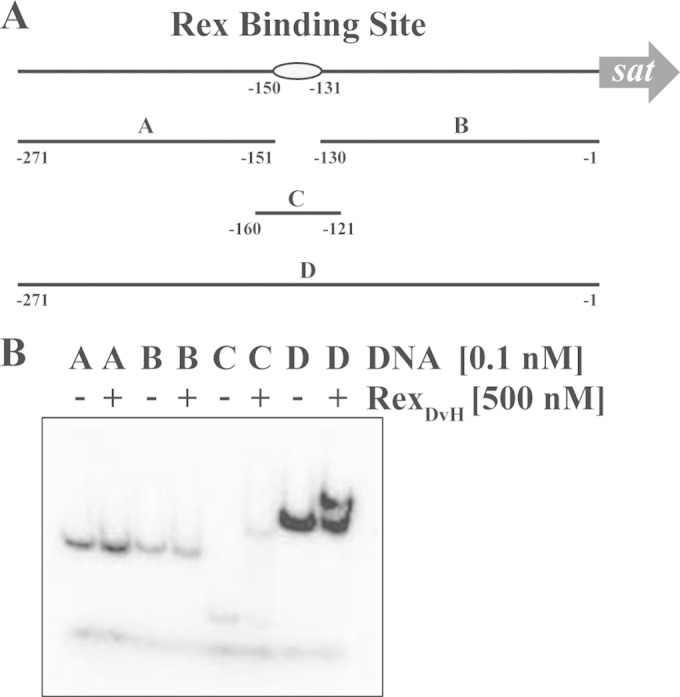
EMSA demonstrating specific interaction between RexDvH and the predicted RexDvH-binding site within the sat promoter. (A) Schematic representation of the sat promoter region drawn approximately to scale. The predicted RexDvH-binding site is annotated by an oval. Fragments (A, B, C, and D) used in EMSA are shown with their positions noted. Fragments A (121 bp), B (130 bp), and D (271 bp) were PCR amplified, while fragment C (40 bp) was generated by annealing two oligonucleotides. (B) Native polyacrylamide gel of individual DNA fragments (A, B, C, and D; 1 nM stock prior to column purification) without (−) or with (+) RexDvH. An equal concentration of DNA was labeled and then passed over a column to separate the fragments from the rest of the components, i.e., unlabeled nucleotides. Fragment C, the smallest fragment, is below the size cutoff for the column (∼100 bp), and so only a small amount of this fragment is actually recovered compared to the other three larger fragments. Each fragment was eluted in the same volume of buffer, and so the concentration of this smaller fragment is considerably lower than the rest. Therefore, the band intensity for fragment C is less than the others. The lowest band common in all lanes is the dye front.
Transcriptional start sites for sat.
With a physical interaction observed in vitro between RexDvH and the RexDvH-binding site upstream of sat, determination of the relative proximity of this motif to the transcription start site (TSS) for sat might provide a logical mechanism for regulation. This analysis could provide evidence that RexDvH repressed by occluding the polymerase from interaction with the promoter region of sat. Therefore, RNA samples were isolated from parental and RexDvH strains grown by sulfate respiration or pyruvate fermentation and assessed for the TSS of sat (Fig. 2). To determine the 5′ end of the transcript, the RACE technique was applied. Examination of the TSS for the parental strain revealed two unique sites: one that was identified from cells growing by sulfate respiration at bp −122 relative to the assumed translational start codon and one from pyruvate-fermenting cells at bp −64. A previous study performed by 5′-RNA-seq analysis also identified bp −122 as the TSS of sat for D. vulgaris Hildenborough grown by sulfate respiration (10). The RexDvH-binding site (bp −150 to −131) is less than 10 bp upstream of the 5′ end of the mRNA and therefore supports the occlusion of RNA polymerase binding for repression by RexDvH during respiration. Furthermore, a potential −35 site of a σ70 promoter was identified in close proximity to this region (Fig. 2). Interestingly, examination of the TSS for the RexDvH mutant growing either by respiration or by fermentation revealed the same sites as those identified for the parental strain. Because two sites were observed and deletion of rex did not reveal a change, these results suggest that factors in addition to RexDvH are involved in determining the TSS.
FIG 2.
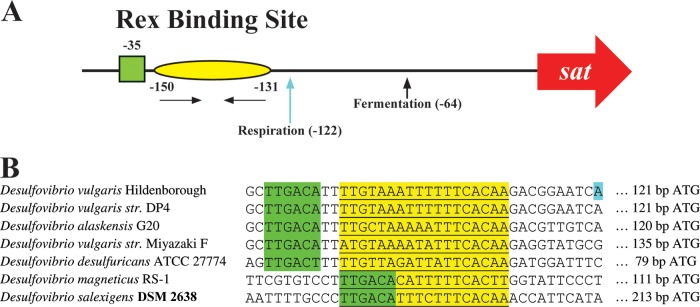
Two transcription start sites (TSSs) for sat identified dependent on growth substrates. Parental and RexDvH mutant strains were grown in medium that would allow for sulfate respiration or pyruvate fermentation. Samples at the early-exponential-growth phase were analyzed for the TSS of sat by 5′-RACE. (A) Schematic representation of sat promoter. The predicted RexDvH-binding site is annotated with a yellow oval with nucleotide positions listed relative to the assumed ATG translational start codon of sat in D. vulgaris Hildenborough. Horizontal arrows denote the half-sites (inverted repeats) within the RexDvH-binding site. The predicted −35 site is indicated by a green box. TSSs are identified with vertical arrows (and positions) for each sample tested. (B) The Rex-binding site (underlined, highlighted in yellow) and the surrounding region is shown for the promoter sequence of sat of Desulfovibrio strains, with the predicted −35 site displayed (TTGACA, highlighted in green). A TSS (respiration) for D. vulgaris Hildenborough is highlighted in blue.
Effect of NADH concentrations on RexDvH function.
Rex proteins contain a pyridine nucleotide binding domain that has been shown in other bacteria to interact with NAD+ or NADH and influence regulation (22, 46). To determine the role of pyridine nucleotide interaction with RexDvH, DNA-binding assays were performed with purified RexDvH protein in the presence of NAD+, NADH, NADP+, or NADPH at 0.1 or 1.0 mM (Fig. 3) and a DNA fragment containing the RexDvH-binding site (fragment C, Fig. 1). The addition of NAD+ appeared to have little effect on RexDvH binding at either concentration compared to purified RexDvH and DNA alone, whereas NADH disrupted the interaction at both concentrations tested. NADP+ did not appear to have any effect on the binding event, and NADPH disrupted binding only at high, presumably nonphysiological, concentrations. Concentration gradient assays conducted for each of the pyridine nucleotides substantiated these results (data not shown), which are similar to the findings of the Rex homolog in T. aquaticus (22).
FIG 3.
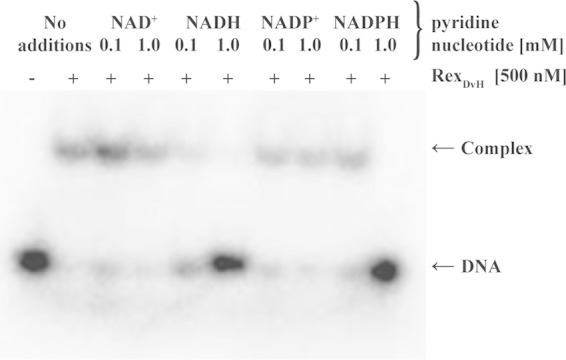
NADH disrupts interaction between RexDvH and the RexDvH-binding site. The results of an electrophoretic assay demonstrate the effect of the pyridine nucleotide on RexDvH binding. Fragment C (40 bp, including the RexDvH-binding site upstream of sat) at 0.1 nM (1 nM prior to column purification) was incubated with RexDvH in the presence of a low or high concentration of the specified pyridine nucleotide. The location of the DNA or protein-DNA complex is indicated.
Confirmation of essential nucleotides in the Rex binding motif.
To determine the key base pair(s) recognized by RexDvH for binding, a number of mutations within the motif upstream of sat were altered and binding studies were performed (Fig. 4A and Table 1). The strategy for the base alterations was to make transitional mutations (i.e., purine to purine [A↔G] or pyrimidine to pyrimidine [C↔T]) for the most conserved bases of the predicted RexDvH-binding sites in D. vulgaris Hildenborough (see sequence logo, Fig. 4A). From the JW9293 deletion strain (ΔPsat) lacking the Rex motif, five additional strains were constructed: (i) a restored promoter sequence with the exact promoter region upstream of sat as the parental strain (restored Psat), (ii) a promoter with the position −147 conserved “G” residue altered to an “A” (G−147A), (iii) a promoter with the distal inverted repeat “GTA” altered to “ACG” (−147 to −145) (IR1), (iv) a promoter with the well-conserved proximal inverted repeat “CAC” altered to “TGT” (−135 to −133) (IR2), and (v) a promoter with mutations to both inverted repeat sequences in a single strain (IR1and2). The mutants along with the restored promoter and parental strains were grown by sulfate respiration (Fig. 4B) and pyruvate fermentation (Fig. 4C). Growth was similar for all strains with the exception of that deleted for the promoter (ΔPsat), which was unable to grow by sulfate respiration and consistently grew slightly more efficiently by pyruvate fermentation.
FIG 4.

Characterization of alterations to the RexDvH-binding site within the sat promoter. (A) List of strains and alterations made to the RexDvH-binding site upstream of sat, with a sequence logo of the predicted RexDvH-binding site shown above. The RexDvH-binding site is underlined, with the alterations made shown in red. An alignment of sequences is shown relative to the assumed translational start codon for sat. The fragments used for EMSA are displayed, along with the estimated Kd (nM) for each 40-bp fragment. N.A., not assessed. The strains examined included the parental strain (JW710), the sat promoter exchange deletion strain (ΔPsat), the restored promoter strain (Psat), the conserved “G−147” altered to “A” strain (G−147A), the distal inverted repeat “GTA” altered to “ACG” strain (IR1), the proximal inverted repeat “CAC” altered to “TGT” strain (IR2), and the strain with alterations to both inverted repeat sites (IR1and2). The relative growth of three replicates of mutants and the parental strain by sulfate respiration (B) or pyruvate fermentation (C) was examined. (D) An electrophoretic assay demonstrated RexDvH binding to native (fragment C) and altered (CI, CII, CIII, and CIV) RexDvH-binding sites. RexDvH was added with increasing concentrations (0, 10, 100, 250, 500, 750, 1,000, and 2,000 nM) to each DNA fragment (0.1 nM; 1 nM prior to column purification). The estimated Kd is shown. The location of the DNA or protein-DNA complex is noted. Triangles represent increasing RexDvH concentration across the lanes.
It was predicted that the modifications to the RexDvH-binding site might prevent RexDvH repression of sat and therefore increase expression of sat. Therefore, samples early in exponential growth were analyzed for sat and rex expression (Table 3) (see Table S2 in the supplemental material). All strains with sequence changes in the RexDvH-binding site had increased sat expression levels relative to the parental and restored strains. dsDNA fragments of 40 bp containing the same alterations that were introduced into the genome were assayed for interactions with increasing RexDvH concentrations (0 to 2,000 nM) (Fig. 4D). For three of the altered fragments tested, G−147A, IR2, and IR1and2 (CI, CIII, and CIV), no shift was observed (dissociation constant [Kd] > 2,000 nM). However, the fragment that contained the three-base alteration to the distal inverted repeat sequence, IR1 (CII), did shift (Kd ∼ 500 nM), although not to the same extent as the wild-type sequence (Kd ∼ 100 nM). This result was rather interesting because this sequence also contained the G−147A alteration that appeared to completely disrupt the binding. As expected, no sat transcription was detected for the strain deleted for the sat promoter grown by pyruvate fermentation (Table 3). The expression of rex was also examined to verify that an unexpected decrease in RexDvH caused by a transcriptional change was not a factor for the observed differences in sat expression. Across the Psat strains, rex expression was not significantly different. Therefore, alterations to the RexDvH-binding site within the promoter sequence of sat increased sat expression confirming the importance of the highly conserved base pairs in the motif.
TABLE 3.
Transcript analysis of parental and RexDvH-binding site alteration strains grown by sulfate respiration or pyruvate fermentationa
| Medium and strain | Description or genotype | Mean transcript level ± SEM for: |
|
|---|---|---|---|
| sat | rex | ||
| MOLS4 | |||
| JW710 | Parental | 1.0 ± 0.1 | 1.0 ± 0.1 |
| JW9293 | ΔPsat | ||
| JW9312 | Restored Psat | 0.9 ± 0.6 | 0.9 ± 0.5 |
| JW9314 | G−147A | 2.7 ± 0.2 | 1.2 ± 0.1 |
| JW9316 | IR1 | 1.8 ± 0.1 | 0.9 ± 0.1 |
| JW9318 | IR2 | 4.0 ± 0.3 | 1.0 ± 0.1 |
| JW9320 | IR1and2 | 4.6 ± 0.6 | 1.4 ± 0.3 |
| MOYPyr | |||
| JW710 | Parental | 1.0 ± 0.1 | 1.0 ± 0.1 |
| JW9293 | ΔPsat | ND | 0.6 ± 0.1 |
| JW9312 | Restored Psat | 1.0 ± 0.3 | 1.6 ± 0.7 |
| JW9314 | G−147A | 2.9 ± 0.7 | 0.9 ± 0.2 |
| JW9316 | IR1 | 3.7 ± 0.7 | 0.9 ± 0.1 |
| JW9318 | IR2 | 2.8 ± 0.5 | 1.1 ± 0.1 |
| JW9320 | IR1and2 | 2.3 ± 0.1 | 1.5 ± 0.1 |
The OD600 was monitored through growth, and samples were collected for analysis by qRT-PCR at the early exponential phase. Approximately 100 ng of Turbo DNase-treated RNA was converted to cDNA, and 1 μl of cDNA (5 ng of RNA) was used per qRT-PCR. Analysis of each gene was conducted separately for each medium tested. For MOLS4, the gene efficiencies were as follows: rplS = 91.8%, rpmC = 84.2%, sat = 92.5%, and rex = 83.6%. For MOYPyr, the gene efficiencies were as follows: rplS = 90.7%, rpmC = 106.1%, sat = 97.7%, and rex = 89.2%. Each gene was assessed individually and normalized to the reference genes rplS and rpmC. JW9293 (ΔPsat) was unable to be grown by sulfate respiration and therefore no data are provided. Transcript values obtained with MOLS4 and MOYPyr were normalized to the corresponding value obtained for JW710. ND, not detected.
RexDvH interacts in vitro with all predicted RexDvH-binding motifs in the D. vulgaris Hildenborough genome.
To explore potential RexDvH regulation of other target genes, a more high-throughput DNA-binding assay was used, fluorescent polarization assay (FPA). Twelve operons with putative Rex motifs in their upstream regions, predicted at the time of the present study (21), were analyzed (see Table S3 in the supplemental material). Exact 20-bp predicted RexDvH-binding sites were created with five “G's” at the 5′ end to improve annealing and with four “G's” with a “T” at the 3′ end to which the fluorophore would be attached. This approach takes advantage of the fact that the degree of polarization of a fluorophore is inversely related to its molecular rotation. Thus, the change in fluorescence of a fast-moving small unbound DNA fragment compared to a larger RexDvH-bound DNA fragment is evidence of protein-DNA interaction. By increasing the protein concentration over a range of values (0 to 1,000 nM), a dissociation constant was determined for RexDvH with each 6-FAM-labeled dsDNA fragment. Dissociation constants (Kd) of ca. 40 to 105 nM were determined and were similar to previously published values for Rex (i.e., Kd ≈ 1 to 100 nM) (21, 22, 47). The two techniques used in the present study to calculate protein-DNA interaction between RexDvH and the RexDvH-binding site upstream of sat were similar (EMSA, Kd ≈ 100 nM; FPA, Kd ≈ 90 nM). In conclusion, RexDvH protein was determined to interact in vitro with all predicted RexDvH-binding sites, and the calculated Kd values were similar.
DISCUSSION
The sulfate reduction gene sat has been shown to be altered in expression depending on the available electron acceptor (18). Bioinformatic predictions for the sulfate reduction pathway, including coexpression studies of the genes, as well as looking for conserved motifs upstream of these genes in multiple Desulfovibrio species, proposed regulatory contributions from Rex (encoded by DVU_0916) (21), HcpR (encoded by DVU_2547) (48), LysX (encoded by DVU_2567), and other DNA-binding proteins (encoded by DVU_0057, DVU_0744, DVU_1142, DVU_2275, DVU_2690, DVU_2799, and DVU_2802) (20). Preliminary transcript studies examining sat expression were performed on parental D. vulgaris Hildenborough cells, on a marker-exchange deletion of hcpR, and on a transposon insertion mutant of lysX. No observable differences of >2-fold were found among strains growing on 60 mM lactate with 30 mM sulfate or 20 mM sulfite (see Table S4 in the supplemental material). Therefore, these candidates were no longer pursued as major regulators of the sulfate activation and reduction steps for the present study. However, these proteins may still be regulators under other physiological conditions or regulators of other genes in the complete reduction of sulfate. In particular, annotated transcriptional regulators DVU_0744, DVU_2802, and DVU_2275 have been examined further by others and found to play a role in expression of sulfate reduction targets during respiration (20).
Preliminary transcript studies examining sat expression of a RexDvH mutant had shown that the deletion of rex increased sat expression relative to a parental strain (data not shown). These findings are consistent with the role of RexDvH as a repressor of sat. This transcription factor has been proposed to be responsible for the redox poise of the cell through the NADH/NAD+ ratio and to alter cellular metabolism to reestablish the pyridine nucleotide balance (21). To confirm the role of RexDvH, the RexDvH mutant was cultured in two media proposed to alter the redox status of the cell, and sat transcription was measured (Table 2). Examination of the RexDvH mutant revealed increased sat transcripts compared to the parental strain for both growth modes. Furthermore, the differential increase in transcript levels observed in the absence of sulfate for the parental strain was not maintained in the RexDvH mutant. We interpreted these observations to mean that RexDvH is a transcriptional repressor for sat responding to redox status in the cell.
The rex gene encoded on a plasmid under a npt promoter was introduced into the Rex mutant to test restoration of sat repression. When this complementation construct was respiring sulfate, sat repression was only partially restored. However, rex was transcribed >10-fold higher than in the wild type (Table 2). Interestingly, for the fermenting culture the expression of sat was restored to wild-type levels. The increased transcription of rex, and therefore we assume the protein RexDvH, in the complemented strain might lead to higher-than-normal levels of repression of other targets of RexDvH not yet studied. This aberrant expression of rex may adversely affect the overall metabolism of the cell. However, a comparison of growth curves showed little difference in rate or extent of sulfate respiration by the parent, rex mutant and complement under the conditions examined in the present study.
Interestingly, there is a conserved hypothetical gene located 33 bp downstream of rex, DVU_0915, that was originally predicted to be in the same operon. However, the expression of DVU_0915 is quite low and, based on a large battery of transcriptome data publicly available for D. vulgaris Hildenborough (http://www.microbesonline.org/), these genes do not appear to be coregulated. Examination of the intergenic region revealed several strong hairpins that might function as transcriptional regulators consistent with a separate operonal structure for the adjacent genes. Alternatively, the deletion of the DNA sequence of rex may have eliminated regulatory elements responsible for the proper expression of DVU_0915. Preliminary studies examining DVU_0915 expression in the three strains revealed that DVU_0915 expression was indeed elevated in the Rex mutant and complemented strain compared to the parental strain (data not shown). Therefore, the increased abundance of DVU_0915 may contribute to the inconsistencies in the expression of sat in the complemented strain, although no function is known for the protein encoded by this gene.
To date, it has been assumed that RexDvH blocks transcription by preventing the polymerase from binding to promoter DNA, limiting the expression of the downstream gene. Interestingly, based on the identified TSS for respiring cultures the −10 position should be within the RexDvH-binding site, specifically overlapping with the proximal inverted repeat (IR2). However, no conventional −10 site could be identified, but a site similar to a classical −35 consensus (TTCACA) was apparent just upstream of the RexDvH-binding position. This was interpreted to mean that the RNA polymerase binding site of a respiring culture is within the RexDvH-binding site, and therefore the proposed RNA polymerase occlusion mechanism for Rex seems plausible during respiration.
However, this RNA polymerase occlusion mechanism does not explain the second site of transcript initiation apparently functioning when D. vulgaris Hildenborough is grown by fermentation and even more sat transcript is produced. When sulfate becomes limiting and NADH is plentiful, increased Sat could possibly scavenge low sulfate as a terminal electron acceptor and/or provide reduced sulfur for cell biosynthesis. Preliminary work suggests that the levels of Sat may be linked to sulfate uptake (G. M. Zane et al., unpublished data). Therefore, it would be reasonable, when the preferred electron acceptor is limiting (e.g., sulfate), that the cell might increase the expression of a gene that encodes a protein that may facilitate sulfate uptake. A second possibility is that there is an alternate regulator that recognizes a specific ligand (e.g., sulfate or sulfite) and that the absence of this ligand is the signal for the selection of the second start site of transcription. It would seem plausible that sulfite, which at high concentrations is toxic to D. vulgaris Hildenborough, may be a ligand for controlling the overall expression of sat as Sat activates sulfate for reduction to sulfite. This latter interpretation might also explain why sat expression is relatively low when sulfite is the electron acceptor and much higher with sulfate.
It was predicted that the deletion of rex should resemble the state at which Rex should be removed from the promoter (i.e., fermentation). Therefore, when the RexDvH mutant had the same TSS pattern for sat as the parental strain, it was clear that additional regulators are likely involved and that the order of addition of these other factors might be critical for regulation. Therefore, the mechanism proposed above, regarding another potential regulator that responds to sulfite levels, seems reasonable. Sulfite concentrations and not the protein RexDvH could be signaling the selection of the transcriptional start site. Overall, sat is still subject to transcriptional control by RexDvH in the fermentative condition since transcription is derepressed to even higher levels when RexDvH is deleted and pyruvate is being fermented.
Because sat expression was increased significantly as a result of a deletion of rex, we sought to examine the interaction of RexDvH with the promoter of sat (Fig. 4). To confirm the specific bases required for interaction, in vitro assays were performed on short DNA sequences of the promoter region. All alterations constructed in the consensus binding site disrupted the interaction between RexDvH and the DNA in vitro. Interestingly, when a highly conserved base (G−147) was altered to an “A” there was a complete loss of detectable interaction (fragment CI, G−147A, “GTA” altered to “ATA”); however, a triple mutation at IR1 (fragment CII, “GTA” altered to “ACG”), which included the G−147A mutation, caused only a slight decrease in the interaction and not the complete loss that was observed for the single base mutation. Closer examination of the sequence revealed that the modified sequence for fragment CII (“ACG”), for which a “G” is now in the third position, may still resemble the consensus site (“GTG”), but that the single base mutation does not. Therefore, a partially restored binding may occur, but only when the second half site, “CAC,” is present as in IR1 (fragment CII). However, additional mutation studies would be required to further characterize the specific contribution of each base on RexDvH binding.
Strains containing the altered promoter sequences discussed above were then constructed and grown by sulfate respiration or pyruvate fermentation and compared to the parental strain (Fig. 4). In addition, a strain that was deleted for 150 bp upstream of sat (ΔPsat) was also assayed. As expected, sat expression increased for any mutation that limited RexDvH binding to the promoter region but was eliminated in the promoter deletion strain. Interestingly, ΔPsat grew to a higher cell density while fermenting pyruvate. This phenomenon has been observed for strains with the genes in the sulfate reduction step of the respiratory pathway in D. vulgaris Hildenborough deleted (e.g., quinone-interacting membrane-bound oxidoreductase, qmoABCD [data not shown] or a tetraheme cytochrome TpIc3, cycA [49]). The consistent increase in growth on pyruvate may result from a block in sulfate reduction that prevents flux through the activation step, functioning in the parental strain, that requires two ATP equivalents. Although 0.5 mM cysteine is the sulfur source during our growth experiments, it should be noted that nickel (2.3 μM) is added as a trace element with sulfate as a counterion in our preparation (NiSO4).
In a recent study (21), RexDvH was predicted to interact with the promoter region for 12 operons, including many that encode proteins functioning in other steps of sulfate reduction in D. vulgaris Hildenborough (see Table S3 in the supplemental material). Interestingly, most of the targets predicted to be regulated by RexDvH are not involved with NADH oxidation directly (except the Rnf complex) as they are for other bacteria (21).
Since we examined here only a subset of potential electron donors and acceptors for RexDvH regulation of sat, the effect of other substrates (e.g., pyruvate, formate, H2, sulfite, or thiosulfate) might reveal additional features of Rex regulation. In fact, we recently reported that the RexDvH mutant was inhibited for growth with thiosulfate as the terminal electron (33), an observation that deserves examination. In addition, RexDvH has been predicted to regulate more than 50 genes (21). Recently, one of those genes, rnfC, has been shown to decrease in a strain deleted for rex in D. alaskensis G20 (30), suggesting Rex as an activator as well. Finally, more than 150 potential regulators have been predicted in D. vulgaris Hildenborough and should be considered either for their interaction with RexDvH or for their role in regulating genes that encode for proteins responsible for sulfate reduction. Interestingly, the binding site for RexDvH is similar to several known FNR family transcription factors that might compete with RexDvH for binding. Much additional work is needed to identify the transcriptional regulators that signal cellular nutrient and energy status that are integrated at the level of control of sulfate reduction.
Supplementary Material
ACKNOWLEDGMENTS
This research, conducted by ENIGMA-Ecosystems and Networks Integrated with Genes and Molecular Assemblies (http://enigma.lbl.gov/), a Scientific Focus Area Program at Lawrence Berkeley National Laboratory, was supported by the Office of Science, Office of Biological and Environmental Research, of the U.S. Department of Energy under contract DE-AC02-05CH11231.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02083-14.
REFERENCES
- 1.Enning D, Garrelfs J. 2014. Corrosion of iron by sulfate-reducing bacteria: new views of an old problem. Appl Environ Microbiol 80:1226–1236. doi: 10.1128/AEM.02848-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee W, Lewandowski Z, Nielson PH, Hamilton HA. 1995. Role of sulfate-reducing bacteria in corrosion of mild steel: a review. Biofouling 8:165–194. doi: 10.1080/08927019509378271. [DOI] [Google Scholar]
- 3.Hamilton WA. 2003. Microbially influenced corrosion as a model system for the study of metal microbe interactions: a unifying electron transfer hypothesis. Biofouling 19:65–76. doi: 10.1080/0892701021000041078. [DOI] [PubMed] [Google Scholar]
- 4.Cypionka H. 2000. Oxygen respiration by Desulfovibrio species. Annu Rev Microbiol 54:827–848. doi: 10.1146/annurev.micro.54.1.827. [DOI] [PubMed] [Google Scholar]
- 5.Thauer RK, Stackebrandt E, Hamilton WA. 2007. Energy metabolism and phylogenetic diversity of sulphate-reducing bacteria, p 1–38. In Barton L, Hamilton WA (ed), Sulphate-reducing bacteria: environmental and engineered systems. Cambridge University Press, Cambridge, United Kingdom. [Google Scholar]
- 6.Muyzer G, Stams AJ. 2008. The ecology and biotechnology of sulphate-reducing bacteria. Nat Rev Microbiol 6:441–454. doi: 10.1038/nrmicro1892. [DOI] [PubMed] [Google Scholar]
- 7.Keller KL, Wall JD, Chhabra S. 2011. Methods for engineering sulfate reducing bacteria of the genus Desulfovibrio. Methods Enzymol 497:503–517. doi: 10.1016/B978-0-12-385075-1.00022-6. [DOI] [PubMed] [Google Scholar]
- 8.Keller KL, Bender KS, Wall JD. 2009. Development of a markerless genetic exchange system for Desulfovibrio vulgaris Hildenborough and its use in generating a strain with increased efficiency. Appl Environ Microbiol 75:7682–7691. doi: 10.1128/AEM.01839-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heidelberg JF, Seshadri R, Haveman SA, Hemme CL, Paulsen IT, Kolonay JF, Eisen JA, Ward N, Methe B, Brinkac LM, Daugherty SC, Deboy RT, Dodson RJ, Madupu R, Nelson WC, Sullivan SA, Fouts D, Haft DH, Selengut J, Peterson JD, Davidsen TM, Zafar N, Zhou L, Radune D, Dimitrov G, Hance M, Tran K, Khouri H, Gill J, Utterback RT, Feldblyum TV, Wall JD, Voordouw G, Fraser CM. 2004. The genome sequence of the anaerobic, sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. Nat Biotechnol 22:554–559. doi: 10.1038/nbt959. [DOI] [PubMed] [Google Scholar]
- 10.Price MN, Deutschbauer AM, Kuehl JV, Liu H, Witkowska HE, Arkin AP. 2011. Evidence-based annotation of transcripts and protein in the sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. J Bacteriol 193:5716–5727. doi: 10.1128/JB.05563-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barton L, Hamilton WA. 2007. Sulphate-reducing bacteria: environmental and engineered systems. Cambridge University Press, Cambridge, United Kingdom. [Google Scholar]
- 12.Peck HD, LeGall J. 1994. Inorganic microbial sulfur metabolism. Academic Press, Inc, San Diego, CA. [Google Scholar]
- 13.Dahl C, Friedrich CG. 2008. Microbial sulfur metabolism. Springer-Verlag, Berlin, Germany. [Google Scholar]
- 14.Ullrich TC, Blaesse M, Huber R. 2001. Crystal structure of ATP sulfurylase from Saccharomyces cerevisiae, a key enzyme in sulfate activation. EMBO J 20:316–329. doi: 10.1093/emboj/20.3.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fritz G, Roth A, Schiffer A, Buchert T, Bourenkov G, Bartunik HD, Huber H, Stetter KO, Kroneck PM, Ermler U. 2002. Structure of adenylylsulfate reductase from the hyperthermophilic Archaeoglobus fulgidus at 1.6-Å resolution. Proc Natl Acad Sci U S A 99:1836–1841. doi: 10.1073/pnas.042664399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schiffer A, Parey K, Warkentin E, Diederichs K, Huber H, Stetter KO, Kroneck PM, Ermler U. 2008. Structure of the dissimilatory sulfite reductase from the hyperthermophilic archaeon Archaeoglobus fulgidus. J Mol Biol 379:1063–1074. doi: 10.1016/j.jmb.2008.04.027. [DOI] [PubMed] [Google Scholar]
- 17.Oliveira TF, Vonrhein C, Matias PM, Venceslau SS, Pereira IAC, Archer M. 2008. The crystal structure of Desulfovibrio vulgaris dissimilatory sulfite reductase bound to DsrC provides novel insights into the mechanism of sulfate respiration. J Biol Chem 283:34141–34149. doi: 10.1074/jbc.M805643200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wall JD, Arkin AP, Balci NC, Rapp-Giles BJ. 2008. Genetics and genomics of sulfate respiration in Desulfovibrio, p 1–12. In Dahl C, Friedrich CG (ed), Microbial sulfur metabolism. Springer-Verlag, Berlin, Germany. [Google Scholar]
- 19.Zane GM, Yen HCB, Wall JD. 2010. Effect of the deletion of qmoABC and the promoter-distal gene encoding a hypothetical protein on sulfate reduction in Desulfovibrio vulgaris Hildenborough. Appl Environ Microbiol 76:5500–5509. doi: 10.1128/AEM.00691-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turkarslan S, Wurtmann EJ, Wu WJ, Jiang N, Bare JC, Foley K, Reiss DJ, Novichkov P, Baliga NS. 2014. Network portal: a database for storage, analysis and visualization of biological networks. Nucleic Acids Res 42:D184–D190. doi: 10.1093/nar/gkt1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ravcheev DA, Li X, Latif H, Zengler K, Leyn SA, Korostelev YD, Kazakov AE, Novichkov PS, Osterman AL, Rodionov DA. 2012. Transcriptional regulation of central carbon and energy metabolism in bacteria by redox-responsive repressor Rex. J Bacteriol 194:1145–1157. doi: 10.1128/JB.06412-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sickmier EA, Brekasis D, Paranawithana Shanthi Bonanno JB, Paget MSB, Burley SK, Kielkopf CL. 2005. X-ray structure of a Rex-family repressor/NADH complex insights into the mechanism of redox sensing. Structure 13:43–54. doi: 10.1016/j.str.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 23.Du X, Pène JJ. 1999. Identification, cloning and expression of p25, an AT-rich DNA-binding protein from the extreme thermophile, Thermus aquaticus YT-1. Nucleic Acids Res 27:1690–1697. doi: 10.1093/nar/27.7.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McLaughlin KJ, Strain-Damerell CM, Xie K, Brekasis D, Soares AS, Paget MSB, Kielkopf CL. 2010. Structural basis for NADH/NAD+ redox sensing by a Rex family repressor. Mol Cell 38:563–575. doi: 10.1016/j.molcel.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 25.Brekasis D, Paget MSB. 2003. A novel sensor of NADH/NAD+ redox poise in Streptomyces coelicolor A3(2). EMBO J 22:4856–4865. doi: 10.1093/emboj/cdg453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gyan S, Shiohira Y, Sato I, Takeuchi M, Sato T. 2006. Regulatory loop between redox sensing of the NADH/NAD+ ratio by Rex (YdiH) and oxidation of NADH by NADH dehydrogenase Ndh in Bacillus subtilis. J Bacteriol 188:7062–7071. doi: 10.1128/JB.00601-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang E, Bauer MC, Rogstam A, Linse S, Logan DT, von Wachenfeldt C. 2008. Structural and functional properties of the Bacillus subtilis transcriptional repressor Rex. Mol Microbiol 69:466–478. doi: 10.1111/j.1365-2958.2008.06295.x. [DOI] [PubMed] [Google Scholar]
- 28.Larsson JT, Rogstam A, von Wachenfeldt C. 2005. Coordinated patterns of cytochrome bd and lactate dehydrogenase expression in Bacillus subtilis. Microbiology 151(Pt 10):3323–3335. [DOI] [PubMed] [Google Scholar]
- 29.Reference deleted.
- 30.Kuehl JV, Price MN, Ray J, Wetmore KM, Esquivel Z, Kazakov AE, Nguyen M, Kuehn R, Davis RW, Hazen TC, Arkin AP, Duetschbauer A. 2014. Functional genomics with a comprehensive library of transposon mutants for the sulfate-reducing bacterium Desulfovibrio alaskensis G20. mBio 5:e01041–14. doi: 10.1128/mBio.01041-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wietzke M, Bahl H. 2012. The redox-sensing protein Rex, a transcriptional regulator of solventogenesis in Clostridium acetobutylicum. Appl Microbiol Biotechnol 96:749–761. doi: 10.1007/s00253-012-4112-2. [DOI] [PubMed] [Google Scholar]
- 32.Li MZ, Elledge SJ. 2007. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat Methods 4:251–256. doi: 10.1038/nmeth1010. [DOI] [PubMed] [Google Scholar]
- 33.Korte HL, Fels SR, Christensen GA, Price MN, Kuehl JV, Zane GM, Deutschbauer AM, Arkin AP, Wall JD. 2014. Genetic basis for nitrate resistance in Desulfovibrio strains. Front Microbiol 5:1–12. doi: 10.3389/fmicb.2014.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parks JM, Johs A, Podar M, Bridou R, Hurt RA, Smith SD, Tomanicek SJ, Qian Y, Brown SD, Brandt CC, Palumbo AV, Smith JC, Wall JD, Elias DA, Liang L. 2013. The genetic basis for bacterial mercury methylation. Science 339:1332–1335. doi: 10.1126/science.1230667. [DOI] [PubMed] [Google Scholar]
- 35.Hellemans J, Mortier G, De Paepe A, Speleman F, Vandesompele J. 2007. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 8:R19. doi: 10.1186/gb-2007-8-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. 2002. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:research0034.1–research0034.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou L, Lim QE, Wan G, Too HP. 2010. Normalization with genes encoding ribosomal proteins but not GAPDH provides an accurate quantification of gene expressions in neuronal differentiation of PC12 cells. BMC Genomics 11:75. doi: 10.1186/1471-2164-11-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chomczynski P, Sacchi N. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 40.Chomczynski P, Sacchi N. 2006. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nat Protoc 1:581–585. doi: 10.1038/nprot.2006.83. [DOI] [PubMed] [Google Scholar]
- 41.Scotto-Lavino E, Du G, Frohman MA. 2007. 5′ end cDNA amplification using classic RACE. Nat Protoc 1:2555–2562. doi: 10.1038/nprot.2006.480. [DOI] [PubMed] [Google Scholar]
- 42.Noble JE, Bailey MJ. 2009. Quantitation of protein. Methods Enzymol 463:73–95. doi: 10.1016/S0076-6879(09)63008-1. [DOI] [PubMed] [Google Scholar]
- 43.Novichkov PS, Li X, Kuehl JV, Deutschbauer AM, Arkin AP, Price MN, Rodionov DA. 2014. Control of methionine metabolism by the SahR transcriptional regulator in proteobacteria. Environ Microbiol 16:1–8. doi: 10.1111/1462-2920.12273. [DOI] [PubMed] [Google Scholar]
- 44.Titolo S, Brault K, Majewski J, White PW, Archambault J. 2003. Characterization of the minimal DNA-binding domain of the human papillomavirus E1 helicase: fluorescence anisotropy studies and characterization of a dimerization-defective mutant protein. J Virol 77:5178–5191. doi: 10.1128/JVI.77.9.5178-5191.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Williamson DH, Lund P, Krebs HA. 1967. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem J 103:514–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pagels M, Fuchs S, Pané-Farré J, Kohler C, Menschner L, Hecker M, McNamarra PJ, Bauer MC, von Wachenfeldt C, Liebeke M, Sander G, von Eiff C, Proctor RA, Engelmann S. 2010. Redox sensing by a Rex-family repressor is involved in the regulation of anaerobic gene expression in Staphylococcus aureus. Mol Microbiol 76:1142–1161. doi: 10.1111/j.1365-2958.2010.07105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang E, Ikonen TP, Knappila M, Svergun D, Logan DT, von Wachenfeldt C. 2011. Small-angle X-ray scattering study of a Rex family repressor: conformational response to NADH and NAD+ binding in solution. J Mol Biol 408:670–683. doi: 10.1016/j.jmb.2011.02.050. [DOI] [PubMed] [Google Scholar]
- 48.Rodionov DA, Dubchak I, Arkin A, Alm E, Gelfand MS. 2004. Reconstruction of regulatory and metabolic pathways in metal-reducing δ-proteobacteria. Genome Biol 5:R90. doi: 10.1186/gb-2004-5-11-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keller KL, Rapp-Giles BJ, Semkiw ES, Porat I, Brown SD, Wall JD. 2014. New model for electron flow for sulfate reduction in Desulfovibrio alaskensis G20. Appl Environ Microbiol 80:855–868. doi: 10.1128/AEM.02963-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou A, Chen YI, Zane GM, He Z, Hemme CL, Joachimiak MP, Baumohl JK, He Q, Fields MW, Arkin AP, Wall JD, Hazen TC, Zhou J. 2012. Functional characterization of Crp/Fnr-Type global transcriptional regulators in Desulfovibrio vulgaris Hildenborough. Appl Environ Microbiol 78:1168–1177. doi: 10.1128/AEM.05666-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.