

Abstract
The established association between inflammatory bowel disease and colorectal cancer underscores the importance of inflammation in colon cancer development. Based on evidence that hemostatic proteases are powerful modifiers of both inflammatory pathologies and tumor biology, gene-targeted mice carrying low levels of prothrombin were used to directly test the hypothesis that prothrombin contributes to tumor development in colitis-associated colon cancer (CAC). Remarkably, imposing a modest 50% reduction in circulating prothrombin in fII+/− mice, a level that carries no significant bleeding risk, dramatically decreased adenoma formation following an azoxymethane/dextran sodium sulfate challenge. Similar results were obtained with pharmacological inhibition of prothrombin expression or inhibition of thrombin proteolytic activity. Detailed longitudinal analyses showed that the role of thrombin in tumor development in CAC was temporally associated with the antecedent inflammatory colitis. However, direct studies of the antecedent colitis showed that mice carrying half-normal prothrombin levels were comparable to control mice in mucosal damage, inflammatory cell infiltration and associated local cytokine levels. These results suggest that thrombin supports early events coupled to inflammation-mediated tumorigenesis in CAC that are distinct from overall inflammation-induced tissue damage and inflammatory cell trafficking. That prothrombin is linked to early events in CAC was strongly inferred by the observation that prothrombin deficiency dramatically reduced the formation of very early, pre-cancerous aberrant crypt foci. Given the importance of inflammation in the development of colon cancer, these studies suggest that therapeutic interventions at the level of hemostatic factors may be an effective means to prevent and/or impede colitis-associated colon cancer progression.
INTRODUCTION
Multiple studies have suggested that hemostatic factors in general, and the central hemostatic protease thrombin, are major determinants of tumor biology, particularly late stage processes such as metastasis (1-4). In addition to supporting tumor cell dissemination, thrombin has been suggested to play a role in tumor proliferation and stroma formation (5). However, the contribution of thrombin to tumor growth appears to be perplexingly variable and seemingly context dependent, as the growth of multiple transplantable tumors has been shown to be unaffected in mice with specific deficits in prothrombin or mice lacking selected downstream thrombin targets (e.g., fibrinogen, platelet function, factor XIII) (1, 3, 6-9). Equally enigmatic is the precise contribution of thrombin and other hemostatic factors to early events leading to cancer development, including tumor cell initiation events and events driving tumor promotion. Previous studies linking tissue factor expression by transformed cells to tumorigenesis infer a role for circulating hemostatic factors in these early events (10, 11), but the role of hemostatic factors downstream of tissue factor in cancer development, if any, remains to be determined. Drawing together the independent evidence that: i) cancer risk is linked to inflammatory processes (12-14), ii) hemostatic factors control inflammatory events (15-17), and iii) the malignant phenotype is linked to procoagulant functions (1), an attractive hypothesis is that a major contribution of thrombin to initial tumor development would be most manifest in inflammation-driven cancers.
Chronic inflammation is a major risk factor for the development of multiple malignancies, including cancers of the lung, liver, prostate, pancreas and colon (18). There is equally impressive evidence that thrombin and multiple thrombin substrates drive the inflammatory response in diverse experimental settings (e.g., bacterial infection, arthritis, neuroinflammatory disease) (15-17, 19-21). However, the contribution of thrombin to inflammation-driven cancers, particularly early events involved in tumor development, has never been formally explored. A prime example of the strong association between inflammation and cancer is colitis-associated colon cancer (CAC). Patients with inflammatory bowel disease (IBD) have a 10- to 20-fold increased risk of colon cancer (22). Multiple indirect observations are compatible with the hypothesis that thrombin is a determinant of CAC pathogenesis. Patients with IBD or colon cancer have a significantly increased risk of thromboembolism (23, 24). Elevated biomarkers of hemostatic system activation correlate with a poor response to therapy in colitis and a poor prognosis in colon cancer (25, 26). Epidemiological studies suggesting that IBD is less common in patients with inherited bleeding disorders infer a role for hemostatic factors in IBD pathogenesis (27). Conversely, other studies have shown a strong correlation between some prothrombotic mutations (e.g., homozygosity for factor VLeiden) and increased colon cancer risk (28). Studies of animal models of colitis and CAC also suggest that hemostatic system components actively contribute to these pathologies. The immunological blockade of tissue factor was shown to diminish colitis associated inflammation and mucosal damage in mice (29). Fibrin(ogen)-mediated engagement of the leukocyte integrin αMβ2 has been shown to drive colitis pathology and support the development and growth of colitis-associated adenomas (30). While these studies support the view that thrombin contributes to CAC pathogenesis, a direct analysis of the potential contribution(s) of thrombin to CAC has never been done.
To directly test the hypothesis that prothrombin supports tumor formation in the setting of inflammation-driven colon cancer, comparative analyses of CAC were done in cohorts of control mice and mice with germline- or pharmacologically-induced quantitative deficits in prothrombin. We report that a modest decrease in circulating prothrombin (just 50% of normal) dramatically diminished adenoma formation. Interestingly, prothrombin diminution had no obvious impact on the severity of the antecedent colitis. However, lowering circulating prothrombin levels either constitutively or just during the inflammatory phase of the disease prevented the development of aberrant crypt foci and subsequent adenoma formation. These studies suggest that thrombin plays a significant role in very early events involved in tumorigenesis, at least in the context of inflammation-driven carcinogenesis.
MATERIALS AND METHODS
Transgenic mice
Mice heterozygous for a prothrombin (F2) null allele (fII+/−) have been previously described (31, 32). All study group mice were 8-12 week old males that were C57BL/6-inbred at least 6 generations. Wildtype and fII+/− mice were generated within the same breeding colony and housed in identical conditions. Study protocols were approved by the Cincinnati Children's Hospital Institutional Animal Care and Use Committee in accordance with NIH guidelines.
Antisense oligonucleotide-mediated prothrombin depletion and hirudin treatment
Prothrombin synthesis was suppressed in wildtype C57BL/6 mice using a previously described 20-mer antisense oligonucleotide (ASO) “gapmer” (2). Cohorts were injected subcutaneously with 5 mg/kg fII-specific ASO, or an equivalent amount of a control irrelevant oligonucleotide of similar chemistry with no significant homology in the mouse transcriptome. Hepatic prothrombin mRNA and plasma prothrombin activity was measured as previously described (31). In separate experiments, 10 mg/kg once daily hirudin (Lepirudin; Baxter) or saline carrier was injected i.p. as specified in the text.
Induction of colitis and colon cancer
CAC was induced as previously described using a single i.p. injection of 10 μg/kg azoxymethane (Midwest Research Institute) on day 0, followed by two courses of 1.5% DSS (days 7-14 and 28-35) (30). Histological analyses were performed on each lesion dissected, confirming each as an adenoma. Acute colitis was induced using 1.5% DSS (MP Biomedicals) in drinking water for 7 days. Chronic colitis was induced by administering DSS on days 1-7 and 21-28. Colonic adenomas were generated without DSS exposure by 6 weekly i.p. injections of 10 μg/kg AOM (33). Here, colons were analyzed 18 weeks after AOM initiation.
Histologic analysis, cytokine measurements, and statistical analyses
Colonic tissue was fixed in Carnoy's solution and processed for histology. Immunostaining for fibrin(ogen), PCNA, MPO, phosphorylated RelA/p65 and Iba1 were performed essentially as previously described (30, 34). Colitis severity was assessed histologically using a semiquantitative histopathological scoring system based on the following: edema (0-3), percent ulceration (0-4), percent crypt loss (0-4), and inflammatory cell infiltrate (0-3) (30, 34). Colonic cytokine concentrations were determined using Luminex technology on a BioPlex analyzer (Bio-Rad, Hercules, CA) from flash frozen distal colons homogenized in PBS with a protease inhibitor cocktail (Sigma-Adlrich). Statistical analyses were generated using Graphpad Prism software. Unless otherwise indicated, data shown represent mean with SEM, and P values were generated with a Mann-Whitney U test.
RESULTS
Prothrombin supports adenoma development/outgrowth in CAC
To test the hypothesis that prothrombin supports tumor development in inflammation-driven cancers, we explored the impact of imposing a simple two-fold diminution in circulating prothrombin on colonic adenoma development in mice with CAC. Cohorts of mice heterozygous for a prothrombin null allele (fII+/−) and wild-type (WT) controls were challenged in parallel with AOM/DSS as previously described (29). As expected, evaluation of colonic tissues collected 12 weeks after AOM injection revealed uniform development of adenomas in all WT mice, most with multiple adenomas (Fig. 1A). In contrast, fII+/− mice developed significantly fewer colonic adenomas (Fig. 1A). Impressively, a substantial fraction (~30%) of the fII+/− mice enrolled had no discernable adenomas (P<0.05, Fisher's exact test) whereas WT mice exhibited 100% phenotypic penetrance of adenoma formation. The data shown represent the combined results of two experiments with similar outcomes.
Figure 1. Prothrombin supports colitis-associated adenoma formation.

(A) Quantitation of the total number of adenomas formed per animal after AOM/DSS challenge. Note that mice with a genetically imposed diminution in prothrombin levels to ~50% of normal (fII+/−) developed significantly fewer adenomas. (Horizontal bars represent median values.) Shown are representative H&E stained sections of adenoma tissue harvested from WT mice (B) and fII+/−mice (C). The high-powered insets show the loss of epithelial cell polarity, nuclear pleomorphism, cell piling (*) and frequent mitotic figures (arrowheads) typical of adenomas. (D) A section of unchallenged colonic tissue cut in the same plane is shown for comparison. Size bars represent 20 μm or 10 μm (insets).
Although adenoma formation in CAC was significantly reduced in mice carrying low prothrombin levels, lowering prothrombin had no significant impact on clinical metrics associated with the antecedent colitis, such as rectal bleeding and weight loss. We anticipated that the AOM/DSS challenge in fII+/− mice would be easily tolerated with regard to bleeding risk based on prior studies showing that fII+/− mice exhibited plasma coagulation parameters (i.e., PT, aPTT) that were similar to WT mice, never exhibited spontaneous hemorrhagic events, survived well into adulthood, and exhibited tail tip bleeding times comparable to control animals (31). Consistent with these expectations, both WT and fII+/− mice challenged with AOM/DSS developed some DSS-induced rectal hemorrhage, but this was modest and indistinguishable between genotypes. The degree of DSS-induced weight loss was also similar between genotypes. After two courses of DSS, WT mice had lost an average of 4.7% of their body mass (range −10.3% to +11.5%) compared to a mean loss of body mass of 3.4% (range −13.1% to +8.2%) in fII+/− mice (P=0.7).
While significantly fewer in number, those adenomas that did form in mice with low prothrombin levels were histologically indistinguishable from those that formed in mice with normal prothrombin levels. The adenomas harvested from both genotypes included a similar spectrum of low-grade and high-grade lesions (see Figs. 1B&C for representative adenoma histology). Low-grade lesions consisted of thickened mucosa with elongated colonic glands and modest nuclear pleomorphism, but no loss of mucosal polarity. High-grade lesions had significant loss of polarity with some glands characterized by piling of epithelial cells (Figs. 1B&C). Colonocytes in these lesions tended to have large vesicular nuclei with nucleoli. Mitotic figures were readily apparent in adenomas harvested from both genotypes, and were particularly common in high-grade lesions, regardless of genotype. The adenomas that formed in fII+/− mice were also similar in size to those harvested from control animals. The mean adenoma mass for tumors harvested from WT mice was 15 mg (range 0.8-130 mg), compared to a mean of 8.9 mg (range 0.2-39 mg) for tumors harvested from fII+/− mice (P=0.4).
As a complementary approach to explore the role of prothrombin in CAC, hepatic prothrombin expression was pharmacologically reduced using a previously described, highly-selective murine prothrombin ASO “gapmer” (2). Here, cohorts of WT mice received weekly s.c. injections of a prothrombin-specific ASO or an equivalent dose of a biologically irrelevant control oligonucleotide beginning 2 weeks prior to the initiation of AOM/DSS challenge. This dosing regimen was established to decrease circulating prothrombin levels to ~50% of normal based on quantitation of hepatic mRNA and functional protein assays (Figs. 2A&B). Paralleling our prior observations in mice with germline-based alterations in prothrombin, mice treated with prothrombin-specific ASO developed significantly fewer adenomas than control mice challenged in parallel (Fig. 2C). Also paralleling findings in fII+/− mice, ASO-mediated diminution in prothrombin had no discernable effect on the qualitative degree of DSS-induced diarrhea, intestinal bleeding and weight loss. At the conclusion of the second DSS course, animals treated with a control ASO had lost an average of 2.3% of their body mass (range −9% to +3.4%) and fII ASO treated mice had lost an average of 1.8% of their body mass (range −5.1% to +0.7%, P=0.8). While adenoma formation was quantitatively reduced in mice treated with the prothrombin-specific ASO, the mass of adenomas harvested in those mice were comparable to those observed in control mice. The mean adenoma mass for tumors harvested from control mice was 5 mg (range 0.1-62.1 mg), compared to a mean of 5.9 mg (range 0.1-27.1 mg) for adenomas harvested from fII ASO treated mice (P=0.7). The histological features of adenomas were also similar between cohorts treated with a control oligonucleotide or fII ASO and indistinguishable from those harvested from WT and fII+/− mice in separate experiments (data not shown). These results reveal that prothrombin constitutes a seminal determinant of tumor development/outgrowth in the setting of colitis-associated cancer. However, lowering prothrombin did not appreciably influence the features of adenomas, once developed, including overall growth and histological features.
Figure 2. Pharmacologically decreasing prothrombin (fII) expression significantly limits adenoma formation in CAC.
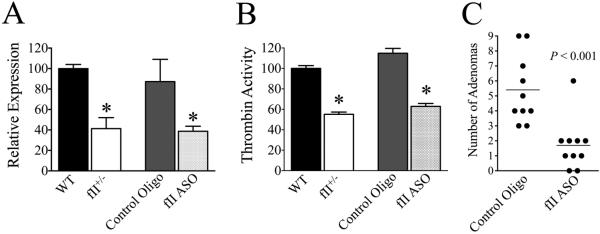
Shown are quantitation of hepatic prothrombin mRNA (A) and chromogenic plasma prothrombin activity (B) obtained following 3 weekly doses of prothrombin-specific ASO (5 mg/kg/dose) or a control oligonucleotide, as well as parallel analyses in unchallenged WT and fII+/− mice (*P < 0.05, n = 4 per group). (C) Weekly treatment with a prothrombin-specific ASO at this dose significantly limits adenoma formation following AOM/DSS challenge relative to a control, irrelevant oligonucleotide.
Decreasing circulating prothrombin does not significantly alter mucosal damage, inflammatory cell accumulation, or local cytokine production in DSS-induced colitis
Previous studies have shown that thrombin and multiple thrombin targets (e.g., PARs, fibrinogen, factor XIII, protein C, osteopontin) are coupled to the inflammatory response in several experimental settings (15-17, 19-21). To determine if a modest decrease in circulating prothrombin influences the severity of DSS-induced colitis, we challenged cohorts of fII+/− mice and controls in parallel with a single 7 day course of DSS. Consistent with observations in mice challenged with AOM and multicourse DSS, the degree of diarrhea and rectal bleeding observed in fII+/− mice was qualitatively similar to that observed in WT mice, as was the degree of weight loss. Wildtype mice lost an average of 10.8% of their body mass (range −27.6% to +6.1%) compared to a mean loss of body mass of 11.7% (range −25.7% to −3.0%) in fII+/− mice (P=0.9). Histological analyses of colonic tissue harvested immediately after 7 days of DSS exposure revealed significant areas of inflammatory cell infiltration, edema, crypt loss and ulceration that were similar between genotypes (Fig. 3A). As expected, analyses of colons harvested from unchallenged WT and fII+/− mice (i.e., proximal to any DSS challenge) appeared normal and indistinguishable (Fig. 3A). Detailed comparisons of colonic tissue using an established multiparameter semiquantitative histological analysis of DSS-induced colitis severity did not reveal any significant differences between genotypes in any of the parameters assessed, including the degree of edema, crypt loss, ulceration, and inflammatory cell infiltration (Fig. 3B). Similar results were obtained in complementary comparisons of colitis severity following 7 days of DSS exposure in cohorts of mice treated with either a prothrombin-specific ASO or a control oligonucleotide (See Supplemental Material).
Figure 3. A modest decrease in prothrombin expression does not overtly affect the severity of DSS-induced mucosal damage, inflammatory cell accumulation, or local inflammatory cytokine production.

(A) Shown are representative photomicrographs of unchallenged colonic tissue from WT and fII+/− mice and colonic sections harvested from WT and fII+/− mice following 7 days of DSS challenge. Note that DSS-challenged colons from mice of both genotypes revealed similar degrees of significant mucosal ulcerations (arrows) and inflammatory edema (*). (B) Detailed microscopic analyses using a multiparameter histopathological scoring system confirmed that DSS-challenged colons harvested from WT mice (n = 8) and fII+/− mice (n = 9) showed similar degrees of disease severity for each parameter analyzed. (P values were not significant for each comparison.) (C) Immunohistochemical staining revealed qualitatively similar fibrin(ogen) deposits (brown staining) in both genotyopes that was primarily associated with areas of inflammatory edema. Immunohistochemical staining for neutrophils (α-MPO) and macrophages (α-Iba1) revealed qualitatively similar infiltration with both these cell types in DSS-challenged colons from both genotypes. Size bars in each photomicrograph represent 50 μm. (D) Shown are the levels of inflammatory cytokines measured in colonic homogenates harvested from WT and fII+/− mice immediately following 7 days of DSS challenge (n = 7 per group) as well as levels in colons harvested from unchallenged mice (n = 4). Note that levels of each cytokine measured were significantly elevated in DSS-challenged colons relative to those from unchallenged mice, but prothrombin genotype had no significant effect on local cytokine levels. (P values were not significant for each comparison between DSS-challenged WT and fII+/− cohorts.)
Immunohistochemical staining of colons for fibrin(ogen) showed qualitatively similar degrees of fibrin(ogen) deposition in colons harvested from WT and fII+/− mice following 7 days of DSS exposure. Fibrin(ogen) deposits were primarily associated with areas of significant mucosal edema (Fig. 3C). The inflammatory infiltrates present in DSS-challenged colons were primarily composed of macrophages and neutrophils. Immunostaining for myeloperoxidase (neutrophils) and Iba1 (macrophages) revealed similar degrees of infiltration with both these cell types in DSS-challenged WT and fII+/− mice (Fig. 3C). Consistent with these more qualitative histological findings, separate quantitative comparisons of local cytokines previously shown to drive colitis and CAC (TNFα, IL-6, KC, IFNγ, and IL-1β) (35, 36) in colonic homogenates harvested from WT and fII+/− mice immediately following 7 days of DSS exposure were similar between genotypes (Fig. 3D). Analyses of local cytokines in colonic homogenates from prothrombin-specific ASO and control oligonucleotide treated mice following 7 days of DSS also showed no significant differences associated with lowering prothrombin levels to ~50% of normal (See Supplemental Material).
To determine if decreasing prothrombin levels affects the degree of mucosal damage and inflammatory cell infiltration associated with multicourse DSS exposure, we challenged WT and fII+/− mice with 2 courses of DSS analogous to that used to induce adenoma formation. Paralleling observations detailed above, the qualitative degree of waxing and waning colonic bleeding and diarrhea, as well as weight loss following DSS challenge/withdrawal were similar between genotypes (Fig. 4A). Detailed comparisons of the histopathology of colons harvested immediately following the second DSS course revealed similar degrees of inflammatory cell infiltration, edema, crypt loss and ulceration in WT and fII+/− mice (Figs. 4B&C).
Figure 4. Prothrombin deficiency does not significantly alter the clinical severity or mucosal damage associated with multicourse DSS challenge.

(A) The waxing and waning weight loss associated with multicourse DSS challenge is similar in WT and fII+/− mice (n = 9 per group). (B) Shown are photomicrographs of representative H&E stained sections of colonic tissue harvested from WT and fII+/− mice at the end of the second course of DSS (i.e. day 28). Note that DSS-challenged colons from mice of both genotypes revealed similar degrees of significant mucosal ulcerations (arrows) and inflammatory edema (*). Size bars represent 50 μm. (C) Detailed microscopic analyses using a multiparameter histopathological scoring system confirmed that DSS-challenged colons harvested from WT mice (n = 8) and fII+/− mice (n = 9) showed similar degrees of disease severity for each parameter analyzed. (P values were not significant for each comparison between DSS-challenged WT and fII+/− cohorts.)
Thrombin promotes adenoma formation/outgrowth in CAC primarily through mechanisms temporally associated with, and dependent on inflammatory colitis
To determine whether the impact of thrombin on adenoma development/outgrowth is formally dependent on the antecedent colitis, we explored whether a modest reduction in prothrombin expression would limit adenoma formation in an experimental setting not driven by overt inflammation. Here, we induced adenomas in fII+/− and WT mice in parallel using an established protocol consisting of 6 weekly doses of AOM without DSS exposure. Contrasting experiments where adenoma formation was dependent on DSS-induced colitis, the decrease in prothrombin expression conferred by the fII+/− genotype had no effect on the number of adenomas formed after multicourse AOM (Fig. 5A). Collectively, these results suggest that although limiting prothrombin does not overtly alter initial tissue damage and secondary inflammatory cell infiltration in AOM/DSS-challenged mice, the contribution of prothrombin to adenoma formation appears to be dependent on an inflammatory microenvironment.
Figure 5. Prothrombin supports adenoma formation in CAC primarily through mechanisms dependent on the antecedent inflammatory colitis.

(A) Genetically imposed prothrombin deficiency had no significant impact on the number of colonic adenomas formed following multiple i.p. injections of azoxymethane without DSS exposure. (B) Treatment during the antecedent inflammatory colitis with either a fII-specific ASO at doses that lower prothrombin levels to ~50% of normal or daily i.p. hirudin injections (10 mg/kg/dose) significantly limits adenoma formation following AOM/DSS. (C) Multiparameter histological analyses of colonic tissue harvested from hirudin or saline vehicle treated mice (n = 8 per group) immediately following 7 days of DSS exposure revealed no significant differences in any of the parameters assessed. (P values were not significant for each comparison between saline vehicle and hirudin treated mice.) (D) In contrast to the significant diminution in adenoma formation observed when prothrombin expression or thrombin proteolytic function was targeted during the antecedent DSS challenge, treatment with either fII ASO or hirudin following DSS withdrawal did not have a significant impact on the number of adenomas formed following AOM/DSS challenge.
To better define when in the course of disease pathogenesis prothrombin sufficiency is vital to adenoma development in CAC, two experimental designs were employed whereby ASO-mediated suppression of prothrombin levels was imposed either early (i.e., 1 week prior to initiation of the AOM/DSS challenge and terminated at the time of withdrawal of the second DSS course; see Fig. 5B) or late (i.e., 1 week after the withdrawal of the second DSS course and continued until the time of sacrifice 12 weeks post-AOM initiation; see Fig. 5D). Weekly s.c. injections of prothrombin-specific ASO were given to reduce and subsequently maintain circulating prothrombin at approximately half-normal (the new steady state approached within about 5 days of initiating ASO based on the prothrombin half-life of ~60 hours). Lowering prothrombin levels during the early phase of disease pathogenesis resulted in a major reduction in the number of adenomas formed 12 weeks after AOM injection (Fig. 5B), paralleling previous results observed in fII+/− mice and mice receiving continuous ASO treatment. Also paralleling previous results, the adenomas that did form in mice with low prothrombin levels were similar in size and histologically indistinguishable from those that formed in mice with normal prothrombin levels (data not shown). Similar results were obtained when AOM/DSS challenged mice were treated with a protein-level inhibitor of thrombin proteolytic activity during the antecedent DSS-induced colitis (days 7 through 35). Here, mice receiving daily i.p. injections of the highly specific, albeit short-lived, thrombin inhibitor, hirudin, during this early timeframe developed significantly fewer adenomas than saline vehicle treated mice challenged in parallel (Fig. 5B). Consistent with findings in mice with low prothrombin levels, daily hirudin treatment had no significant effect on the degree of colonic mucosal damage or inflammatory cell infiltration observed following 7 days of DSS exposure (Fig. 5C)
In contrast to the significant diminution in adenoma formation observed when selectively limiting prothrombin expression or thrombin catalytic activity during the early effector phase characterized by epithelial damage and colitis, the same interventions later in disease pathogenesis had no significant impact on adenoma formation. Here, ASO-mediated suppression of circulating prothrombin to half-normal starting one week after the withdrawal of the second DSS course (i.e., day 42) and continuing until sacrifice (i.e., day 84) had no significant effect on adenoma formation (Fig. 5D). Similarly, daily hirudin treatment initiated immediately after completion of the second DSS course (i.e., day 35) and continuing until sacrifice did not significantly affect adenoma formation (Fig. 5D). Taken together, these results suggest that the decrease in colitis-induced adenomas observed in mice with diminished circulating prothrombin is due to a direct action of thrombin-mediated proteolysis that is most relevant during the inflammatory phase of the disease process.
Prothrombin promotes tumorigenesis in CAC
In order to directly determine if prothrombin supports early transformation events preceding gross tumor outgrowth in CAC, we analyzed colonic tissue harvested from AOM/DSS-challenged mice treated with prothrombin-specific ASO or control oligonucleotide just 1 week after withdrawal of the second course of DSS (i.e., day 42). While the degree of residual mucosal damage and local inflammatory markers (see Supplemental Material) at this early time point were not significantly affected by diminished prothrombin levels, there were 4-fold fewer aberrant crypt foci (ACF) within colonic epithelium harvested from prothrombin ASO treated animals relative to control mice (Fig. 6A). These foci exhibited complex glandular structure with tortuous, branched lumens. The epithelial cells in these lesions showed loss of polarity with mitotic figures and nuclear atypia, including vesicular chromatin and nucleoli (Fig. 6B). In contrast, the mucosa of colons harvested from mice with an ASO-mediated decrease in prothrombin expression was generally normal in appearance with significantly fewer ACF (Fig. 6B). However, the few abnormal lesions that were present in this cohort were histologically indistinguishable from those observed in colons harvested from mice with normal prothrombin levels (Fig. 6C). A separate independent analysis of identical experimental design was completed with similar results.
Figure 6. Prothrombin promotes the development of aberrant crypt foci in CAC.

(A) Quantitation of dysplastic foci early AOM/DSS driven CAC performed 1 week after withdrawal of the second DSS course showed that lowering prothrombin levels with a prothrombin ASO to ~50% of normal significantly decreased the number of ACF observed per animal. (B) Shown are representative colonic sections from these same tissues. Dysplastic mucosal lesions consistent with aberrant crypt foci (ACF) were frequently found in mice treated with a control oligonucleotide, whereas the colonic mucosa in fII-specific ASO treated mice was largely healed and appeared normal. Immunostaining revealed significant fibrin(ogen) deposits (brown staining) and PCNA+ colonic epithelial cells (brown staining) in ACF. In contrast, PCNA staining cells were few in number and localized to colonic crypts, and fibrin(ogen) staining was generally scant in more normal appearing mucosa prevalent in fII ASO treated mice. (C) The few ACF that did develop in fII ASO treated mice appeared qualitatively similar to those observed in control mice and exhibited similar degrees of staining for PCNA and fibrin(ogen). (D) Shown is a quantitation of the number of colonic epithelial cell nuclei staining positive for phosphorylated RelA/p65 as a function of the number of intact, properly oriented crypts present within multiple random microscope fields for each mouse. Each data point represents one field and approximately 6 40X fields were assessed per animal. Size bars represent 50 μm.
Immunohistochemical staining of colonic tissue harvested from mice with normal and low prothrombin levels 1 week after withdrawal of the second DSS course revealed significant fibrin(ogen) deposition associated with ACF (Fig. 6B). The ACF that were observed in mice with low prothrombin levels, although significantly fewer in number, exhibited fibrin(ogen) deposits comparable to those observed in ACF seen in control mice (Fig. 6B). Fibrin(ogen) deposition was rarely observed in areas of normal appearing colonic tissue, which predominated in mice with low prothrombin levels. Comparisons of apoptotic cells in these same tissues performed using a cleaved caspase 3 antibody revealed a similar degree of cellular apoptosis in cohorts with normal and low prothrombin levels (data not shown). Apoptotic nuclei were noted in superficial epithelial cells in intact appearing crypts and occasionally in ACF, regardless of prothrombin ASO treatment. Immunohistochemical staining for the marker of cellular proliferation, PCNA, revealed strongly positive nuclear staining in epithelial cells in ACF (Fig. 6B). While the overall number of PCNA positive cells was lower in prothrombin ASO treated mice, this was a function of the fewer total ACF in this cohort. The ACF that did form in prothrombin ASO treated animals were generally composed of epithelial cells strongly positive for nuclear PCNA and histologically similar to ACF observed in control mice (Fig. 6B). The more normal appearing colonic mucosa had only a few PCNA positive cells localized in the base of the colonic crypts, a pattern consistent with normal and/or healing mucosa (Fig. 6B). Immunohistochemical staining for phosphorylated RelA/p65, a marker of NF-κB activation, revealed a similar degree of positive staining cells in ACF, regardless of prothrombin levels (data not shown). Cells staining positive for this marker were uncommon in normal-appearing crypts. However, detailed quantitation of phosphorylated RelA/p65 staining cells in intact, non-ACF epithelia was significantly greater in control mice relative to mice with low prothrombin levels (Fig. 6C), consistent with the view that thrombin-mediated events fundamentally alter colonic epithelial cell functions in this context.
DISCUSSION
These studies establish, for the first time, that thrombin can play a key role in the development of pre-cancerous lesions and subsequent tumor formation in the context of inflammation-driven colon cancer. Taken together with prior reports showing that pro/thrombin is critical to metastasis in vivo (1, 2, 4, 8, 37, 38), these studies show that this central hemostatic protease can drive both tumorigenesis and tumor dissemination. A remarkable facet of these studies was the finding that even a very modest 50% reduction in circulating prothrombin, imposing no significant bleeding risk (31), was sufficient to dramatically decrease tumor formation in CAC. Thrombin-mediated proteolysis was important in driving adenoma formation during the early, initiating phase of mucosal inflammation-associated tumorigenesis, although a modest reduction in thrombin activity affected neither the overall severity of mucosal damage, nor the accumulation of inflammatory cells and associated cytokines following DSS challenge. Nevertheless, these studies demonstrate that inflammatory processes underlie the mechanism linking thrombin and adenoma formation, as decreasing prothrombin levels had no effect on colonic adenoma formation elicited by repeated carcinogen exposure in the absence of investigator-imposed inflammation. Together these results suggest thrombin supports colitis-associated adenoma formation by regulating local inflammatory cell functions and/or epithelial cell responses to inflammation rather than altering overall tissue damage or inflammatory cell trafficking. Although the mechanistic details remain to be fully resolved, it is clear that prothrombin constitutes an important, but never previously appreciated, modifier of early events in CAC and that modest interventions at the level of prothrombin can dramatically reduce the formation of pre-cancerous lesions and subsequent adenoma formation.
The finding that thrombin promotes tumorigenesis in CAC through mechanisms that appear to depend on antecedent inflammation raises the possibility that thrombin-mediated events alter inflammatory functions in ways that are important for tumorigenesis but do not significantly affect inflammatory cell accumulation or mucosal damage. The local elaboration of inflammatory products has been suggested to directly support the progression of CAC through several independent mechanisms. For example, the local release of cytokines and growth factors has been proposed to support the survival and proliferation of pre-malignant and transformed cells (12, 18, 35, 39, 40). A separate attractive concept is that the local release of potentially mutagenic reactive oxygen and nitrogen species increases the risk of transforming mutations in epithelial cells (13). A link between thrombin, inflammatory control mechanisms, and adenoma formation is consistent with a substantial body of prior work illustrating the biological importance thrombin and thrombin substrates (e.g., PARs, protein C, factor XIII, fibrinogen) play in other inflammatory pathologies (15-17, 20, 41). These findings support a working hypothesis whereby diminished thrombin activity results in profound differences in adenoma formation by modifying specific leukocyte activities (e.g., ROS production, elaboration of proteases, production of key cytokines or other effector molecules) rather than overtly affecting colonic mucosal damage or inflammatory cell infiltration. The overall view that thrombin promotes the formation of a microenvironment compatible with tumor formation in CAC is consistent with previous reports proposing that hemostatic system components contribute to the formation of a provisional cancer stem cell niche (42, 43). The view that thrombin-mediated inflammatory events promote alterations in epithelial cell functions important in tumorigenesis is compatible with the data presented here showing that prothrombin influences NF-κB activation in colonic epithelial cells in CAC.
Several thrombin targets have been specifically implicated in supporting inflammation in the context of colitis and/or CAC, including fibrin(ogen) and platelets. Platelets have been shown to play a role in leukocyte recruitment in colitis (44). As mice with prothrombin levels of 50% have no significant spontaneous bleeding risk, any alteration in platelet function resulting from this degree of prothrombin deficiency does not significantly affect hemostasis. However, local platelet activation at sites of mucosal damage in colitis would be expected to result in the release of a plethora of platelet-derived cytokines and growth factors capable of altering local inflammatory functions as well as supporting the proliferation and/or early survival of transformed epithelia (45). Previous studies from our laboratory have shown that fibrinogen is one thrombin target that clearly supports tumorigenesis in CAC through mechanisms directly related to inflammatory cell functions. Specifically, these studies showed that fibrin(ogen) promotes the development of adenomas in CAC by mechanisms coupled to engagement of the leukocyte integrin αMβ2 (30). Paralleling the decrease in adenoma formation observed with decreasing prothrombin expression, both the genetic elimination of fibrinogen and the expression of a mutant form of fibrinogen lacking the αMβ2 binding motif but retaining normal clotting function significantly limited the formation of adenomas following AOM/DSS (30). In contrast to the current studies where lowering prothrombin expression had no significant effect on DSS-induced mucosal damage or leukocyte accumulation, mice expressing a mutant form of fibrinogen lacking the αMβ2 binding motif developed significantly attenuated inflammatory changes following DSS challenge, including less mucosal damage and lower local levels of numerous inflammatory cytokines relative to mice expressing wild-type fibrinogen (30). Therefore, fibrin(ogen)-mediated αMβ2 engagement promotes early inflammatory events in colitis that are coupled to the later development of adenomas. Given the critical importance of fibrin(ogen) in tumorigenesis in the context of CAC, it is possible that even a very modest decrease in local fibrin polymer formation stemming from lowered prothrombin expression, one that would not necessarily be detectable by immunostaining, could limit events crucial for tumorigenesis in this setting. It is also possible that modestly decreasing prothrombin expression limits fibrin polymer formation during a narrow, but critically important time period in tumorigenesis in CAC that ultimately limits adenoma formation. Of course, the fact that our analyses of experimental colitis in mice with lowered prothrombin expression did not precisely phenocopy analyses in mice with alterations in fibrinogen also raises the possibility that thrombin promotes tumorigenesis in CAC through mechanisms independent of fibrin polymerization.
Another model compatible with the data showing that thrombin drives tumorigenesis in CAC is that thrombin-mediated signaling events within tumor cell precursors dependent on an inflammatory milieu support tumorigenesis, but these events do not fundamentally alter mucosal damage or inflammatory events. In this regard, signaling through the thrombin-activatable receptor, PAR-1, could directly influence cell-intrinsic properties important in early tumor development (5, 37). Intestinal epithelial cells have been shown to upregulate PAR-1 expression during times of cellular stress, such as inflammatory colitis (46). Previous studies have suggested that tumor cell-associated PAR-1 activation supports tumor cell proliferation and limits apoptosis (5, 37). Studies showing that PAR-1 is dispensable for tumorigenesis in the context of oncogene-driven breast cancer in mice demonstrate that this thrombin receptor is not critically required for tumorigenesis (47). However, the role of PAR-1 in tumorigenesis has not been investigated in the setting of non-sterile, inflammation-associated cancer such as CAC in which any survival/proliferative advantage conferred by PAR-1 signaling in epithelial cells could be a decisive factor in tumor formation. Dissecting the precise contribution, if any, of thrombin-mediated PAR-1 activation to colonic epithelial cell transformational changes is complicated by the fact that this receptor is expressed on numerous cell types capable of influencing CAC pathogenesis through distinct, and potentially opposing, mechanisms. In fact, constitutional PAR-1 deletion was shown to augment the inflammatory response and subsequent inflammatory damage in experimental colitis mediated by local oxazalone administration as well as H. pylori driven gastritis (42, 43), suggesting that PAR-1 activation has an anti-inflammatory role in colitis. It remains to be resolved whether PAR-1 signaling in colonic epithelia underlies the present finding of diminished colonic adenoma formation in low prothrombin mice following CAC challenge. Specifically determining if colonic epithelial cell-associated PAR-1, or other PAR-1 expressing cellular compartments in the colon, plays a role in tumor formation in CAC would require the ability to manipulate PAR-1 expression in a cell type-specific fashion.
The data presented here reveal a role for thrombin in tumorigenesis in the context of colitis-driven colon cancer. However, it is conceivable that hemostatic factors may also play a role in the pathogenesis of colon cancer in contexts not associated with overt colitis. Clinical studies have shown that persistent elevation of markers of hemostatic system activation (i.e., elevated plasma levels of prothrombin fragment 1+2 and fibrinopeptide A) as well as certain prothrombotic risk factors (i.e., factor VLeiden) are coupled to a significantly increased risk of developing cancers of the digestive tract (28, 48, 49), suggesting that thrombin generation promotes the development of spontaneous colorectal cancer. Emerging evidence suggests that commensal bacteria as well as dietary and host factors (e.g., obesity) regulate the mucosal immune microenvironment and may influence colon cancer pathogenesis (36). Similarly, it is possible that thrombin mediates highly localized inflammatory events affecting immune and/or epithelial cell types that could play a broad role in colon cancer pathogenesis. Although lowering prothrombin did not affect adenoma formation following multicourse AOM exposure, this approach may not model all aspects of the complex genetic and environmental/immunological factors that lead to spontaneous colon cancer in humans (50).
In summary, these studies clearly indicate that the role of thrombin in cancer pathogenesis is not limited to late events, such as metastasis. Thrombin can also strongly influence tumorigenesis in the context of inflammation-driven colon cancer. Given that inflammation has been strongly implicated in the development of multiple malignancies (e.g., cervical, lung, gastric, ovarian, and prostate cancers) (12-14), it is possible that the importance of thrombin in tumor development is not limited to colon cancer. These studies also imply that therapies designed to limit prothrombin expression or thrombin function may represent attractive options for preventing colon cancer in high-risk individuals without bleeding risk.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Dr. Jay L. Degen for critically reviewing the manuscript. This work was supported by the National Institutes of Health grant R01-HL085545 and a grant from the American Society of Hematology.
Footnotes
Conflict of interest disclosure: The authors declare no competing financial interests or conflicts of interest with regard to this work.
REFERENCES
- 1.Palumbo JS. Mechanisms linking tumor cell-associated procoagulant function to tumor dissemination. Semin Thromb Hemost. 2008;34:154–60. doi: 10.1055/s-2008-1079255. [DOI] [PubMed] [Google Scholar]
- 2.Horowitz NA, Blevins EA, Miller WM, Perry AR, Talmage KE, Mullins ES, et al. Thrombomodulin is a determinant of metastasis through a mechanism linked to the thrombin binding domain but not the lectin-like domain. Blood. 2011;118:2889–95. doi: 10.1182/blood-2011-03-341222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palumbo JS, Kombrinck KW, Drew AF, Grimes TS, Kiser JH, Degen JL, et al. Fibrinogen is an important determinant of the metastatic potential of circulating tumor cells. Blood. 2000;96:3302–9. [PubMed] [Google Scholar]
- 4.DeFeo K, Hayes C, Chernick M, Ryn JV, Gilmour SK. Use of dabigatran etexilate to reduce breast cancer progression. Cancer Biol Ther. 2010;10:1001–8. doi: 10.4161/cbt.10.10.13236. [DOI] [PubMed] [Google Scholar]
- 5.Green D, Karpatkin S. Role of thrombin as a tumor growth factor. Cell cycle (Georgetown, Tex) 2010;9:656–61. doi: 10.4161/cc.9.4.10729. [DOI] [PubMed] [Google Scholar]
- 6.Palumbo JS, Barney KA, Blevins EA, Shaw MA, Mishra A, Flick MJ, et al. Factor XIII transglutaminase supports hematogenous tumor cell metastasis through a mechanism dependent on natural killer cell function. J Thromb Haemost. 2008;6:812–9. doi: 10.1111/j.1538-7836.2008.02938.x. [DOI] [PubMed] [Google Scholar]
- 7.Palumbo JS, Potter JM, Kaplan LS, Talmage K, Jackson DG, Degen JL. Spontaneous hematogenous and lymphatic metastasis, but not primary tumor growth or angiogenesis, is diminished in fibrinogen-deficient mice. Cancer Res. 2002;62:6966–72. [PubMed] [Google Scholar]
- 8.Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, et al. Tumor cell-associated tissue factor and circulating hemostatic factors cooperate to increase metastatic potential through natural killer cell-dependent and-independent mechanisms. Blood. 2007;110:133–41. doi: 10.1182/blood-2007-01-065995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, et al. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood. 2005;105:178–85. doi: 10.1182/blood-2004-06-2272. [DOI] [PubMed] [Google Scholar]
- 10.Milsom C, Magnus N, Meehan B, Al-Nedawi K, Garnier D, Rak J. Tissue factor and cancer stem cells: is there a linkage? Arterioscler Thromb Vasc Biol. 2009;29:2005–14. doi: 10.1161/ATVBAHA.108.177444. [DOI] [PubMed] [Google Scholar]
- 11.Milsom CC, Yu JL, Mackman N, Micallef J, Anderson GM, Guha A, et al. Tissue factor regulation by epidermal growth factor receptor and epithelial-to-mesenchymal transitions: effect on tumor initiation and angiogenesis. Cancer research. 2008;68:10068–76. doi: 10.1158/0008-5472.CAN-08-2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer. 2003;3:276–85. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- 14.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 15.Davalos D, Akassoglou K. Fibrinogen as a key regulator of inflammation in disease. Seminars in immunopathology. 2012;34:43–62. doi: 10.1007/s00281-011-0290-8. [DOI] [PubMed] [Google Scholar]
- 16.Ma L, Dorling A. The roles of thrombin and protease-activated receptors in inflammation. Seminars in immunopathology. 2012;34:63–72. doi: 10.1007/s00281-011-0281-9. [DOI] [PubMed] [Google Scholar]
- 17.Petaja J. Inflammation and coagulation. An overview. Thrombosis research. 2011;127(Suppl 2):S34–7. doi: 10.1016/S0049-3848(10)70153-5. [DOI] [PubMed] [Google Scholar]
- 18.Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–45. doi: 10.1016/S0140-6736(00)04046-0. [DOI] [PubMed] [Google Scholar]
- 19.Adams RA, Bauer J, Flick MJ, Sikorski SL, Nuriel T, Lassmann H, et al. The fibrin-derived gamma377-395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J Exp Med. 2007;204:571–82. doi: 10.1084/jem.20061931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flick MJ, Du X, Degen JL. Fibrin(ogen)-alpha M beta 2 interactions regulate leukocyte function and innate immunity in vivo. Exp Biol Med (Maywood) 2004;229:1105–10. doi: 10.1177/153537020422901104. [DOI] [PubMed] [Google Scholar]
- 21.Flick MJ, LaJeunesse CM, Talmage KE, Witte DP, Palumbo JS, Pinkerton MD, et al. Fibrin(ogen) exacerbates inflammatory joint disease through a mechanism linked to the integrin alphaMbeta2 binding motif. J Clin Invest. 2007;117:3224–35. doi: 10.1172/JCI30134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dyson JK, Rutter MD. Colorectal cancer in inflammatory bowel disease: what is the real magnitude of the risk? World journal of gastroenterology : WJG. 2012;18:3839–48. doi: 10.3748/wjg.v18.i29.3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee AY, Levine MN. Venous thromboembolism and cancer: risks and outcomes. Circulation. 2003;107:I17–21. doi: 10.1161/01.CIR.0000078466.72504.AC. [DOI] [PubMed] [Google Scholar]
- 24.Solem CA, Loftus EV, Tremaine WJ, Sandborn WJ. Venous thromboembolism in inflammatory bowel disease. Am J Gastroenterol. 2004;99:97–101. doi: 10.1046/j.1572-0241.2003.04026.x. [DOI] [PubMed] [Google Scholar]
- 25.Kilic M, Yoldas O, Keskek M, Ertan T, Tez M, Gocmen E, et al. Prognostic value of plasma D-dimer levels in patients with colorectal cancer. Colorectal Dis. 2008;10:238–41. doi: 10.1111/j.1463-1318.2007.01374.x. [DOI] [PubMed] [Google Scholar]
- 26.Kumar S, Ghoshal UC, Aggarwal R, Saraswat VA, Choudhuri G. Severe ulcerative colitis: prospective study of parameters determining outcome. J Gastroenterol Hepatol. 2004;19:1247–52. doi: 10.1111/j.1440-1746.2004.03486.x. [DOI] [PubMed] [Google Scholar]
- 27.Thompson NP, Wakefield AJ, Pounder RE. Inherited disorders of coagulation appear to protect against inflammatory bowel disease. Gastroenterology. 1995;108:1011–5. doi: 10.1016/0016-5085(95)90197-3. [DOI] [PubMed] [Google Scholar]
- 28.Vossen CY, Hoffmeister M, Chang-Claude JC, Rosendaal FR, Brenner H. Clotting factor gene polymorphisms and colorectal cancer risk. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:1722–7. doi: 10.1200/JCO.2010.31.8873. [DOI] [PubMed] [Google Scholar]
- 29.Anthoni C, Russell J, Wood KC, Stokes KY, Vowinkel T, Kirchhofer D, et al. Tissue factor: a mediator of inflammatory cell recruitment, tissue injury, and thrombus formation in experimental colitis. J Exp Med. 2007;204:1595–601. doi: 10.1084/jem.20062354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Steinbrecher KA, Horowitz N, Blevins EA, Barney KA, Shaw MA, Harmel-Laws E, et al. Colitis-associated cancer is dependent on the interplay between the hemostatic and inflammatory systems and supported by integrin αMβ2 engagement of fibrinogen. Cancer research. 2010;70:2634–43. doi: 10.1158/0008-5472.CAN-09-3465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mullins ES, Kombrinck KW, Talmage KE, Shaw MA, Witte DP, Ullman JM, et al. Genetic elimination of prothrombin in adult mice is not compatible with survival and results in spontaneous hemorrhagic events in both heart and brain. Blood. 2009;113:696–704. doi: 10.1182/blood-2008-07-169003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun WY, Witte DP, Degen JL, Colbert MC, Burkart MC, Holmback K, et al. Prothrombin deficiency results in embryonic and neonatal lethality in mice. Proc Natl Acad Sci U S A. 1998;95:7597–602. doi: 10.1073/pnas.95.13.7597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tuominen I, Al-Rabadi L, Stavrakis D, Karagiannides I, Pothoulakis C, Bugni JM. Diet-induced obesity promotes colon tumor development in azoxymethane-treated mice. PloS one. 2013;8:e60939. doi: 10.1371/journal.pone.0060939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steinbrecher KA, Harmel-Laws E, Sitcheran R, Baldwin AS. Loss of epithelial RelA results in deregulated intestinal proliferative/apoptotic homeostasis and susceptibility to inflammation. J Immunol. 2008;180:2588–99. doi: 10.4049/jimmunol.180.4.2588. [DOI] [PubMed] [Google Scholar]
- 35.Fantini MC, Pallone F. Cytokines: from gut inflammation to colorectal cancer. Curr Drug Targets. 2008;9:375–80. doi: 10.2174/138945008784221206. [DOI] [PubMed] [Google Scholar]
- 36.Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology. 2010;138:2101–14. e5. doi: 10.1053/j.gastro.2010.01.058. [DOI] [PubMed] [Google Scholar]
- 37.Nierodzik ML, Karpatkin S. Thrombin induces tumor growth, metastasis, and angiogenesis: Evidence for a thrombin-regulated dormant tumor phenotype. Cancer Cell. 2006;10:355–62. doi: 10.1016/j.ccr.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 38.Zigler M, Kamiya T, Brantley EC, Villares GJ, Bar-Eli M. PAR-1 and thrombin: the ties that bind the microenvironment to melanoma metastasis. Cancer Res. 2011;71:6561–6. doi: 10.1158/0008-5472.CAN-11-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grivennikov SI, Karin M. Inflammatory cytokines in cancer: tumour necrosis factor and interleukin 6 take the stage. Ann. 2010;70:i104–8. doi: 10.1136/ard.2010.140145. [DOI] [PubMed] [Google Scholar]
- 41.Ichinose A. Factor XIII is a key molecule at the intersection of coagulation and fibrinolysis as well as inflammation and infection control. International journal of hematology. 2012;95:362–70. doi: 10.1007/s12185-012-1064-3. [DOI] [PubMed] [Google Scholar]
- 42.Milsom C, Yu J, May L, Magnus N, Rak J. Diverse roles of tissue factor-expressing cell subsets in tumor progression. Seminars in thrombosis and hemostasis. 2008;34:170–81. doi: 10.1055/s-2008-1079257. [DOI] [PubMed] [Google Scholar]
- 43.Milsom C, Yu J, May L, Meehan B, Magnus N, Al-Nedawi K, et al. The role of tumor-and host-related tissue factor pools in oncogene-driven tumor progression. Thrombosis research. 2007;120(Suppl 2):S82–91. doi: 10.1016/S0049-3848(07)70135-4. [DOI] [PubMed] [Google Scholar]
- 44.Vowinkel T, Wood KC, Stokes KY, Russell J, Tailor A, Anthoni C, et al. Mechanisms of platelet and leukocyte recruitment in experimental colitis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1054–60. doi: 10.1152/ajpgi.00350.2007. [DOI] [PubMed] [Google Scholar]
- 45.Senzel L, Gnatenko DV, Bahou WF. The platelet proteome. Curr Opin Hematol. 2009;16:329–33. doi: 10.1097/MOH.0b013e32832e9dc6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sekiguchi F, Takaoka K, Kawabata A. Proteinase-activated receptors in the gastrointestinal system: a functional linkage to prostanoids. Inflammopharmacology. 2007;15:246–51. doi: 10.1007/s10787-007-1591-3. [DOI] [PubMed] [Google Scholar]
- 47.Versteeg HH, Schaffner F, Kerver M, Ellies LG, Andrade-Gordon P, Mueller BM, et al. Protease-activated receptor (PAR) 2, but not PAR1, signaling promotes the development of mammary adenocarcinoma in polyoma middle T mice. Cancer research. 2008;68:7219–27. doi: 10.1158/0008-5472.CAN-08-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miller GJ, Bauer KA, Howarth DJ, Cooper JA, Humphries SE, Rosenberg RD. Increased incidence of neoplasia of the digestive tract in men with persistent activation of the coagulant pathway. J Thromb Haemost. 2004;2:2107–14. doi: 10.1111/j.1538-7836.2004.01011.x. [DOI] [PubMed] [Google Scholar]
- 49.Mozsik G, Rumi G, Domotor A, Figler M, Gasztonyi B, Papp E, et al. Involvement of serum retinoids and Leiden mutation in patients with esophageal, gastric, liver, pancreatic, and colorectal cancers in Hungary. World journal of gastroenterology : WJG. 2005;11:7646–50. doi: 10.3748/wjg.v11.i48.7646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luceri C, De Filippo C, Caderni G, Gambacciani L, Salvadori M, Giannini A, et al. Detection of somatic DNA alterations in azoxymethane-induced F344 rat colon tumors by random amplified polymorphic DNA analysis. Carcinogenesis. 2000;21:1753–6. doi: 10.1093/carcin/21.9.1753. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.