

Abstract
We and others have shown that soluble amyloid-β peptide (Aβ) and cerebral amyloid angiopathy (CAA) cause significant cerebrovascular dysfunction in mutant amyloid precursor protein (APP) mice, and that these deficits are greater in aged APP mice having CAA compared to young APP mice lacking CAA. Aβ in young APP mice also increases infarction following focal cerebral ischemia, but the impact of CAA on ischemic brain injury is unknown. To determine this, we assessed cerebrovascular reactivity, cerebral blood flow (CBF), and extent of infarction and neurological deficits following transient middle cerebral artery occlusion (MCAO) in aged APP mice having extensive CAA versus young APP mice lacking CAA (and aged-matched littermate controls). We found that aged APP mice have more severe cerebrovascular dysfunction that is CAA-dependent, have greater CBF compromise during and immediately following MCAO, and develop larger infarctions after MCAO. These data indicate CAA induces a more severe form of cerebrovascular dysfunction than Aβ alone, leading to intra- and post-ischemic CBF deficits that ultimately exacerbate cerebral infarction. Our results shed mechanistic light on human studies identifying CAA as an independent risk factor for ischemic brain injury.
Keywords: Alzheimer's disease, amyloid angiopathy, cerebral ischemia, transgenic mice, cerebrovascular, amyloid beta, cerebral blood flow
Introduction
Alzheimer’s disease and vascular dementia are the two most common forms of cognitive impairment in the elderly. For decades, each was considered a distinct disorder; recent results, however, show that they share many characteristics, including risk factors, neuropathology, and hemodynamics.1 This has led to a new conception that “[Alzheimer’s disease] and [vascular dementia] are at the extremes of a spectrum of pathologies in which vascular and non-vascular factors coexist to varying degrees”2, a shift in thinking that has led to prioritization of investigating vascular contributors to dementia by the American Heart Association and American Stroke Association3 as well as by the National Institute on Neurological Disorders and Stroke (NINDS).4 In vascular dementia, cerebrovascular dysfunction and ischemic brain injury drive cognitive impairment;5 multiple lines of evidence indicate these two factors also contribute to Alzheimer’s disease.2 One common link between these three entities – cerebrovascular dysfunction, ischemic brain injury, and Alzheimer’s disease – is cerebral amyloid angiopathy (CAA).
CAA is characterized by cerebrovascular accumulation of amyloid-β peptide in a fibrillar form. Common in the elderly (~30% in those >60),6 it is also almost universally found in Alzheimer’s patients.7, 8 Experimental evidence in animals9–13 and humans14, 15 shows that CAA significantly perturbs cerebral arteriole function and cerebral blood flow (CBF). In human autopsy studies, CAA is a strong and independent risk factor for both ischemic infarction16–18 and cognitive impairment19, 20. Taken together, these results have led many to postulate that CAA contributes to Alzheimer’s disease by compromising cerebral hemodynamics and promoting infarctions throughout the cerebral hemispheres17, 21. Yet experimental evidence that CAA contributes to ischemic infarction is currently lacking, as is underlying mechanism.
Previously, two groups showed that APP mice having elevated Aβ (but no CAA) develop larger infarcts after focal cerebral ischemia compared to littermate controls. One study implicated Aβ-induced cerebrovascular function,22 while the other linked it to Aβ-induced inflammation.23 Neither examined the effect of CAA on cerebral infarction, as both examined APP mice at ages where CAA is not present. In the current study, we sought to determine the effect of CAA on cerebral ischemia by examining pre-CAA and post-CAA APP mice. We also examined whether CAA-induced alterations in cerebrovascular function and CBF might underlie the observed effect.
Materials and Methods
Animals
All experiments were approved by the Animal Studies Committee at Washington University. All data were collected by experimenters blinded to age and genotype. Tg2576 mice, originally a generous gift from Dr. K. Ashe (University of Minnesota, MN), were bred to B6/SJL wild-type (WT) mice (Taconic Farms, Germantown, NY) and genotyped as described.9, 24 Male Tg2576 mice were used at 6 months (having elevated Aβ but no CAA) and at 15 months (having elevated Aβ and extensive CAA); WT littermates served as controls.
Cerebrovascular reactivity
Cerebrovascular reactivity was assessed as previously described.9 Briefly, mice were anesthetized with isoflurane and a 4-mm right parietal cranial window was made. After 15 hours’ recovery, mice were re-anesthetized and ventilated with a rodent ventilator (Harvard Apparatus, Holliston, MA). Core body temperature was maintained at 37±0.1°C by a thermo-regulated heating pad. Arterial blood pressure and gases were assessed via femoral catheterization. Leptomeningeal arterioles were visualized using Nikon Eclipse ME600 microscope (Nikon Instruments Inc., Melville, NY) and MetaMorph Image Analysis (Molecular Devices, Sunnyvale, CA). The endothelium-dependent vasodilator acetylcholine (ACh; 100 μM) and the endothelium-independent vasodilator S-nitroso-N-acetyl-penicillamine (SNAP; 500 μM) were infused, followed by artificial CSF until baseline vessel diameter returned. Vessel diameters were quantified via Diamtrak (Tim Neild, Monash University, Melbourne, Australia).
Transient focal cerebral ischemia model
Focal ischemia was performed as described.22 Briefly, following anesthesia with isoflurane (4% induction, 1.5% maintenance), burr holes were made to allow placement of laser Doppler flow (LDF) probes. A 6-0 nylon filament was advanced from the left external carotid through the internal carotid to the middle cerebral artery (MCA). After 45 minutes of MCA occlusion (MCAO), the filament was removed to allow reperfusion. Temperature was maintained as above.
Measurement of CBF
CBF was monitored before, during, and after MCAO via LDF as described.22 Probes were placed through burr holes over the ischemic core (3.5 mm lateral, 1 mm caudal to bregma) and penumbra (1.5 mm lateral, 1.7 mm rostral to lambda). Data are presented as a percentage of the pre-occlusion value.
Infarct volume quantification
Infarction was assessed via 2,3,5-triphenyltetrazolium chloride (TTC) staining 72 hours post-MCAO as previously described.25 Briefly, mice were transcardially perfused with heparinized PBS. Brains were sectioned coronally using a 1.5-mm matrix and stained in 2% TTC; following fixation in 10% formalin, imaging was performed with a desktop scanner. Infarct area was assessed using ImageJ (National Institutes of Health, Bethesda, MD) and used to calculate infarct volume via the indirect method.
Neurobehavioral Tests
Neurobehavioral outcome was assessed daily using 8-point sensorimotor scoring as previously described.26
CAA and neuritic plaque quantification
In animals undergoing cerebrovascular reactivity assessment, CAA was quantified as previously described.9 Briefly, mice were administered the Congo red derivative methoxy-X04 (10 mg/kg IP) at installation of the cranial window. Percent CAA coverage was determined for eight 25-µm segments per animal using MetaMorph. In a subset of aged Tg2576 mice (N=4), CAA and neuritic plaque load was quantified post mortem as per our established protocol.27 Briefly, mice were sacrificed 72 hours post-MCAO, followed by brain extraction, fixation (4% paraformaldehyde), equilibration (30% sucrose), and coronal sectioning (50μm). Brain sections were stained with 1μM resorufin (which selectively stains CAA) and 2μM methoxy-X34 (which stains both CAA and neuritic plaques). Fluorescent staining was visualized using Nikon Eclipse ME600 microscope and MetaMorph. CAA and neuritic plaque load was assessed as percent coverage of MCA-territory parietal cortex, contra- and ipsilateral to MCAO, using ImageJ.
Statistical analyses
Data are expressed as means ± SEM. Following determination of normality, comparisons between two groups were performed with a Student’s t-test; among multiple groups, with a one-way omnibus ANOVA followed by Dunnett’s multiple comparison method or with a omnibus repeated measures ANOVA followed by Newman-Keuls method. STATISTICA (StatSoft, Inc., Tulsa, OK) was used. Statistical significance was set at p<0.05.
Results
Aged Tg2576 mice develop extensive CAA and more severe cerebrovascular dysfunction
Young Tg2576 mice had no CAA, while aged Tg2576 mice had extensive CAA covering most leptomeningeal arterioles without interruption (Figure 1A). In young mice, no significant difference in baseline vessel diameter was noted. In aged mice, baseline vessel diameter was significantly greater in Tg2576 mice (Figure 1D; p<0.05, Student’s t-test), which is consistent with our9, 28 and others’10, 29 past results. This difference is consistent with a hypocontractile vascular phenotype, likely the result of CAA-induced smooth muscle cell dysfunction and/or death. In young Tg2576 mice, responses to ACh and SNAP were not significantly different. In aged Tg2576 mice, responses to ACh and SNAP were severely impaired (Figure 1B,C; p<0.05, omnibus ANOVA). These deficits were CAA-dependent, as ACh-induced dilation was normal in vessel segments from aged WT controls, moderately reduced in vessel segments from aged Tg2576 mice having mild CAA (<20%), and absent in vessel segments from aged Tg2576 mice having extensive CAA (>20%) (Figure 1E; p<0.05, omnibus ANOVA).
Figure 1. CAA deposition and cerebrovascular dysfunction in young and aged Tg2576 mice.
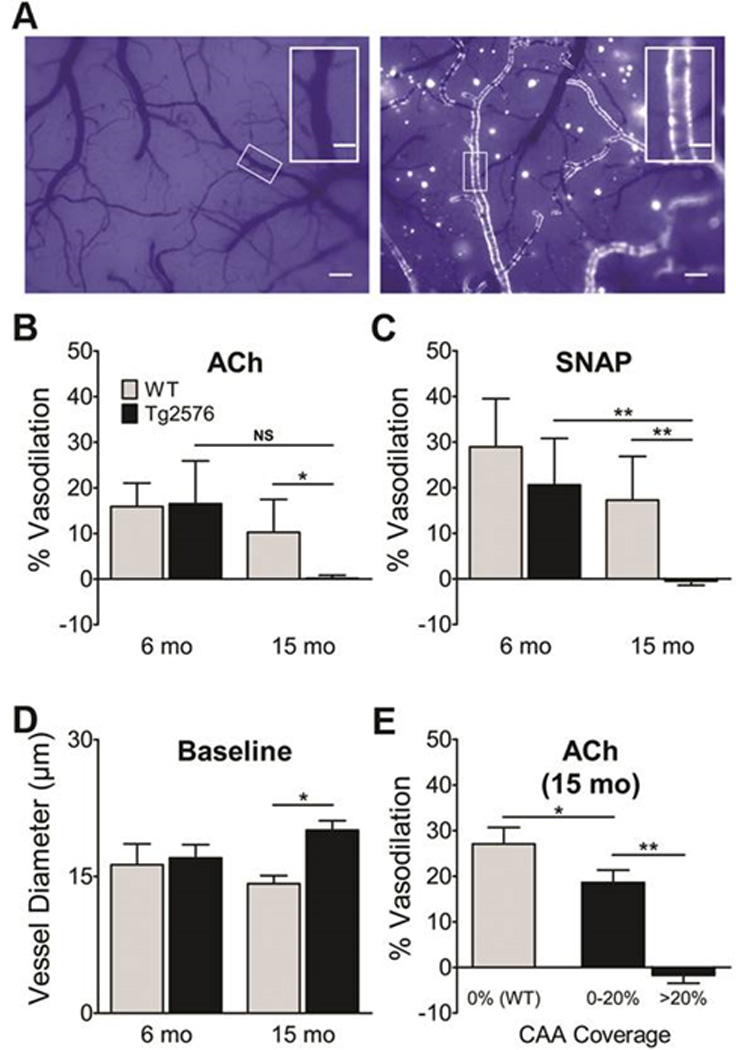
A, CAA deposits in pial arteries of naïve 6 month-old (left) and 15 month-old (right) Tg2576 mice were visualized with the Congo red derivative methoxy-X04 through a closed cranial window. Scale bars: 150 µm (inserts, 50 µm). Vasodilatory responses to ACh (B) and SNAP (C), calculated as percent change in vessel diameter (N=5–7/grp). D, Baseline diameters were similar in young mice; baseline vessel diameter was greater in aged Tg2576 mice. E, Vasodilatory response in aged mice as a function of CAA coverage (N=4 segments/mouse). Data indicate mean ± S.E.M. *: p < 0.05, **: p < 0.01.
Aged Tg2576 mice develop worse CBF deficits during and immediately following focal cerebral ischemia
In young Tg2576 mice, no significant differences in CBF – in core or penumbra, intra- or post-ischemia – were noted (Figure 2A; p>0.05, omnibus repeated measures ANOVA). In aged Tg2576 mice, no significant difference in core CBF was noted during MCAO; however, 15 minutes post-reperfusion it was significantly lower (Figure 2B; p<0.05, omnibus repeated measures ANOVA). Even greater deficits were noted in the ischemic penumbra of aged Tg2576 mice: CBF was lower both during and 15 minutes after MCAO (Figure 2B; p<0.05, omnibus repeated measures ANOVA).
Figure 2. Cerebral blood flow compromise during and after focal cerebral ischemia in aged Tg2576 mice.
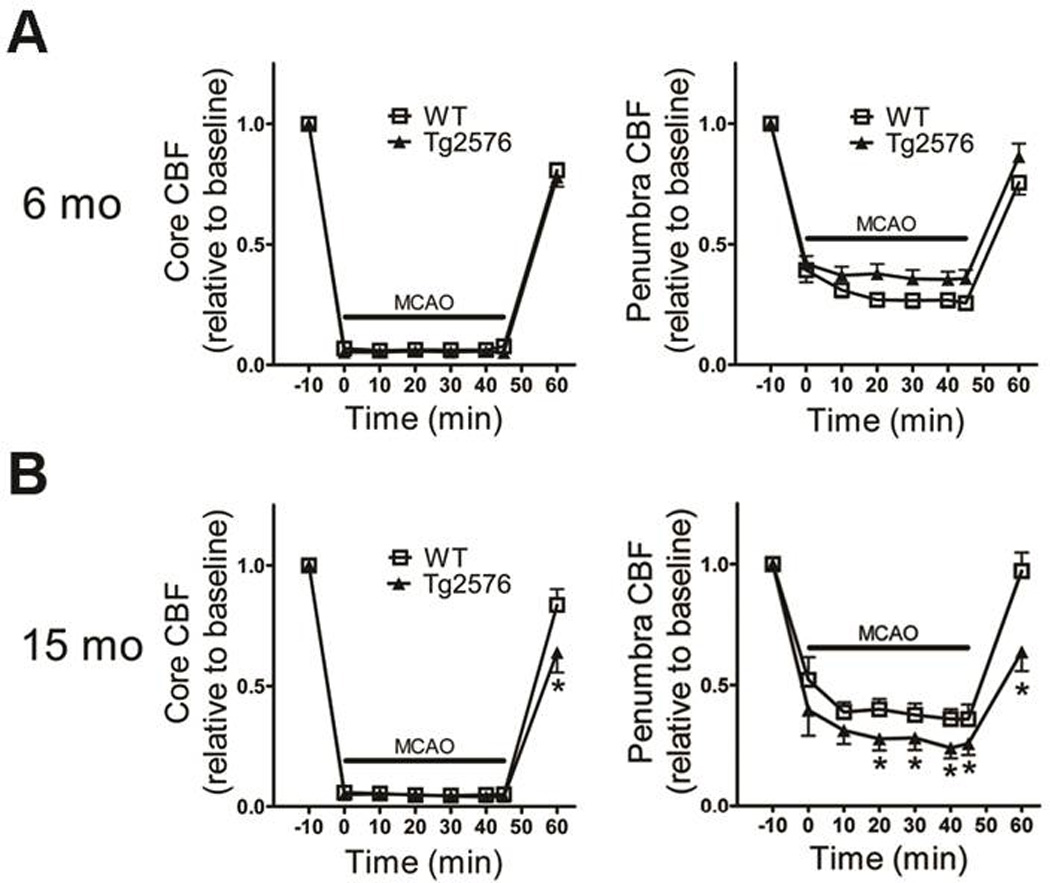
Cerebral blood flow (CBF) in the ischemic core (left) and penumbra (right) during focal ischemia was monitored using laser Doppler flowmetery in 6- and 15 month-old mice (N=13–17/grp) (A–B). Data indicate mean ± S.E.M. *: p < 0.05 vs. WT mice.
Aged Tg2576 mice have increased susceptibility to focal cerebral ischemia
Young Tg2576 mice had infarct volumes that were 46% larger than littermate controls’ (Tg2576: 62±9 mm3 vs. control: 42±7 mm3), while aged Tg2576 mice had infarct volumes that were 84% larger than littermate controls’ (Tg2576: 85±9 mm3 vs. control: 46±9 mm3, p<0.05; omnibus ANOVA). The difference in infarct volumes was statistically significant in aged vs. young Tg2576 mice, but not in WT mice (Figure 3A,B).
Figure 3. Exacerbation of infarction in aged Tg2576 mice.
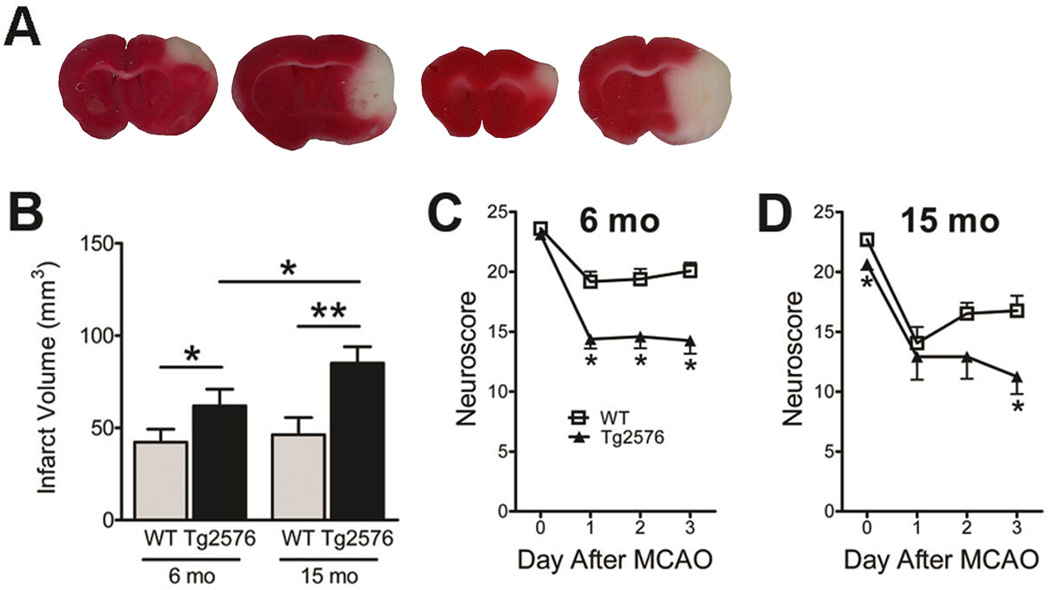
Six- and 15 month-old Tg2576 mice (and WT littermates; N=13–17/grp) underwent MCAO followed by reperfusion for 3 days. A, Representative images of cerebral infarction by TTC staining. B, Infarct volumes (mm3) were calculated by the indirect method. *: p < 0.05, **: p < 0.01. Neurological outcome was assessed via sensorimotor scoring (C–D). Data indicates mean ± S.E.M. *: p<0.05 vs. WT mice.
Aged and young Tg2576 mice develop comparable neurological deficits following focal cerebral ischemia
At baseline (prior to MCAO), no difference in sensorimotor scoring was noted between young Tg2576 and littermate controls, while a small but significant difference was noted between aged Tg2576 and littermate controls (Figure 3D; p<0.05, Student’s t-test). Following MCAO, deficits were greater in Tg2576 mice (Figure 3C,D; p<0.05, omnibus repeated measures ANOVA); however, no significant age-related difference was noted.
Focal cerebral ischemia does not affect CAA or neuritic plaque load
In aged Tg2576 mice, no significant differences in CAA load (0.50±0.16% vs. 0.48±0.13%) or neuritic plaque load (1.37±0.15% vs. 1.24±0.09%; both p>0.05, Student’s t-test) were noted between ischemic and non-ischemic motor cortex 3 days after MCAO (Figure 4).
Figure 4. MCAO has no effect on CAA and neuritic plaque load.
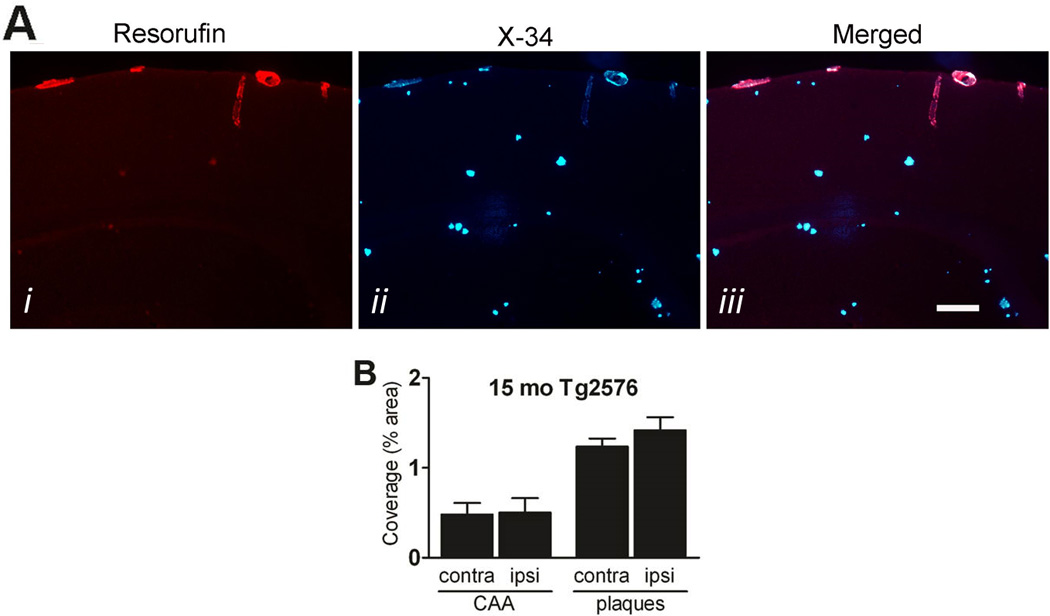
Fifteen month-old Tg2576 (N=4) were sacrificed 3 days after MCAO and subjected to staining with the CAA-selective dye resorufin (Ai) and the Congo red derivative methoxy-X34 (Aii; merged in Aiii). Scale bar: 100 µm. B, Percent coverage of contralateral and ipsilateral cortex was determined. Data indicate mean ± S.E.M.
Discussion
The main findings of our study are: 1) aged Tg2576 mice develop more severe cerebrovascular dysfunction than young Tg2576 mice, and that this is due to the presence of CAA; 2) aged Tg2576 mice develop more severe CBF deficits than young Tg2576 mice during and immediately following focal cerebral ischemia; and 3) infarct volumes (but not neurological deficits) are exacerbated to a greater degree in aged versus young Tg2576 mice. In combination, these findings implicate CAA – via its deleterious effects on cerebrovascular function and CBF – in the heightened susceptibility to cerebral infarction observed in aged Tg2576 mice. They also corroborate numerous human studies identifying CAA as a strong and independent risk factor for ischemic infarction,16–18 and lend mechanistic insight into the pathophysiological underpinnings.
Two previous studies have examined the susceptibility of APP mice to ischemic brain injury. The first utilized 3–4 month-old Tg2576 mice, which have elevated Aβ but no CAA or neuritic plaques. They found that young Tg2576 mice have reduced CBF in the ischemic penumbra immediately following MCAO and develop infarct volumes 32% larger than littermate controls.22 The second utilized 8– and 20 month-old APP751 transgenic mice, which also have elevated Aβ but no CAA or neuritic plaques. They found that APP751 mice develop infarct volumes 34–41% larger than littermate controls.23 Both studies causally linked Aβ to the increased infarct volume noted in APP mice, though the former implicated the deleterious effect of Aβ on cerebrovascular function and the latter its impact on inflammation. Neither, however, ruled out the potential effect of APP over-expression (as opposed to Aβ) as the underlying cause, and neither examined the potential effect of CAA on ischemic brain injury. Our study utilized two distinct ages of Tg2576 mice – one with elevated Aβ alone and one where elevated Aβ is accompanied by extensive CAA – to examine this potential CAA effect.
First, we confirmed what we9 and others10, 11, 30 have reported, that Tg2576 mice develop age-dependent cerebrovascular dysfunction, with the most severe deficits noted in aged Tg2576 mice having extensive CAA. We also found that the severity of cerebrovascular impairment in aged Tg2576 mice depends on the extent of CAA, consistent with our and others’ results that CAA is toxic to vascular smooth muscle cells, causing dysfunction and eventually death in a dose-dependent fashion,9, 31 a process that contributes to the impairment of microvascular function. Second, we found that post-ischemia CBF was modestly but significantly compromised in the ischemic core of aged Tg2576 mice as compared to young Tg2576 mice and littermate controls. We also found that intra-ischemic and post-ischemic CBF was significantly reduced in the ischemic penumbra of aged Tg2576 mice as compared to young Tg2576 mice and littermate controls. These results are consistent with previous studies in AD mice documenting neurovascular uncoupling: astrocyte endfeet swell and retract from CAA-laden vessels (but not from CAA-free vessels) in ArcAβ mice,32 and greater disruption is seen after cerebral ischemia in 3xTg mice than in age-matched wild-type controls.33 Third, we documented that CAA and neuritic plaque load in aged Tg2576 mice was not acutely altered by MCAO. Taken together, these data strongly indicate that CAA’s pathological effect on cerebrovascular function produced worse CBF deficits during and immediately following MCAO, which likely relates to the inability of nearby CAA-laden cerebral arterioles to provide collateral CBF through autoregulatory vasodilation.
Next, we examined what effect CAA-induced cerebrovascular dysfunction has on infarct volume and neurological outcome following MCAO. Consistent with past reports,22, 23 we found that young Tg2576 mice develop larger infarct volumes and worse neurological deficits compared to littermate controls. In addition, we found that aged Tg2576 mice also develop greater infarct volumes and more severe neurological deficits compared to controls, but found that infarct volumes were significantly larger in aged versus young Tg2576 mice. No age-dependent effect on sensorimotor scoring was noted, likely due to baseline neurological deficits associated with aged Tg2576 mice, a floor effect in the post-ischemic deficits seen in this model, or both. Taken together, these data strongly suggest that the heightened vulnerability to ischemic brain injury observed in aged versus young Tg2576 mice is the direct result of CAA and its deleterious effect on cerebrovascular function and CBF.
Our data have several important implications. First, they provide the first experimental support for multiple observational human studies identifying a role for CAA in inducing and/or exacerbating ischemic brain injury. Second, they provide an advanced experimental foundation for future studies designed to examine the efficacy of a growing number of novel CAA-directed therapeutics (e.g., anti-Aβ immunotherapy, beta-secretase inhibitors, etc.). Specifically, study endpoints could comprise not only CAA load and CAA-induced microhemorrhage, but also cerebrovascular function, CBF, and ischemic brain injury. Third, they provide an opportunity to examine whether an entirely new therapeutic approach – targeting vessel function (rather than CAA itself) – may reduce the deleterious effect of CAA on ischemic infarction.
Other interpretations of our results exist. One possibility is that elevated Aβ (rather than CAA) underlies the heightened susceptibility to ischemic brain injury seen in aged versus young Tg2576 mice. This would be substantiated if Aβ levels were higher in aged versus young Tg2576 mice; however, evidence utilizing the most sophisticated techniques for specifically measuring Aβ strongly argues against this. Initial studies examining this issue noted greater levels of biochemically extractable Aβ with increasing age and greater amyloid pathology in APP mice.34–36 The extraction technique employed, however, is confounded by its potential to liberate Aβ from amyloid deposits with tissue homogenization, and therefore it likely overestimates the true level of Aβ present in the extracellular space. To address this concern, several groups turned to in vivo microdialysis of APP mice to directly assess Aβ in the interstitial fluid (ISF). Using this assay, we and others show that, in fact, absolute levels of Aβ decrease with age in several APP mouse models of Alzheimer’s disease and CAA.37–39 Similar decreases in Aβ are seen in CSF of aged APP mice,37, 40 patients with Alzheimer’s,41 and patients with CAA.42 Based on these results, it is therefore highly unlikely that differences in extracellular Aβ in aged versus young APP mice account for the observed heightened susceptibility to ischemic brain injury.
Another possibility is that greater ischemic brain injury is seen in aged Tg2576 mice due to increased neuronal vulnerability that is CAA-independent. It could be that overproduction of mutant human APP and/or exposure to elevated Aβ throughout the lives of Tg2576 mice renders the aging brain less capable of coping with ischemic injury, leading to increased infarction. The fact that extracellular levels of Aβ are actually lower in aged versus young Tg2576 mice, however, argues against this. Finally, the enhanced vulnerability to ischemia could also relate to the presence of neuritic plaque pathology in aged Tg2576 mice. But our observation that aged Tg2576 mice have significant intra- and post-ischemic CBF deficits that appear CAA-induced argues strongly that vascular, not parenchymal, pathology is the primary underlying driver of the heightened susceptibility. To completely exclude this possibility, however, additional experiments would be required, likely using APPDutch mice that exclusively develop CAA, albeit at an extremely advanced age (22–25 months).43
In summary, our work corroborates past results that APP mice having elevated Aβ (but no CAA) have increased vulnerability to ischemic brain injury. In addition, we are the first to show that aged APP mice having extensive CAA and severe CAA-induced cerebrovascular dysfunction develop marked intra- and post-ischemic CBF deficits and greater infarct volumes following MCAO. These data strongly implicate CAA as a key contributor to ischemic brain injury, which substantiates the growing notion that CAA – and its attendant cerebrovascular and CBF deficits – is a key contributor to the ischemic infarcts and cognitive dysfunction found in Alzheimer’s disease and vascular dementia. Whether restoration of vasoreactivity via CAA-directed or vessel function-directed therapeutics ultimately improves CBF, reduces ischemic brain injury, and enhances cognitive function will require further investigation.
Acknowledgements
This work was supported by grants from the American Heart Association (12PRE9040015; E.M.) the National Institutes of Health (R01 NS071011; G.J.Z.), American Health Assistance Foundation (B.H.H. and G.J.Z.), and Harrington/Zhu Alzheimer Research Fund (G.J.Z.). The authors would like to thank Ernesto Gonzales for performing all MCAO surgeries.
Footnotes
Conflict of Interest: The authors declare no competing financial interest.
References
- 1.Iadecola C, Gorelick PB. Converging pathogenic mechanisms in vascular and neurodegenerative dementia. Stroke; a journal of cerebral circulation. 2003;34:335–337. doi: 10.1161/01.str.0000054050.51530.76. [DOI] [PubMed] [Google Scholar]
- 2.Iadecola C. Neurovascular regulation in the normal brain and in alzheimer's disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 3.Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, et al. Vascular contributions to cognitive impairment and dementia: A statement for healthcare professionals from the american heart association/american stroke association. Stroke; a journal of cerebral circulation. 2011;42:2672–2713. doi: 10.1161/STR.0b013e3182299496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vickrey BG, Brott TG, Koroshetz WJ Stroke Research Priorities Meeting Steering C, the National Advisory Neurological D. Stroke C, et al. Research priority setting: A summary of the 2012 ninds stroke planning meeting report. Stroke; a journal of cerebral circulation. 2013;44:2338–2342. doi: 10.1161/STROKEAHA.113.001196. [DOI] [PubMed] [Google Scholar]
- 5.Hachinski VC, Lassen NA, Marshall J. Multi-infarct dementia. A cause of mental deterioration in the elderly. Lancet. 1974;2:207–210. doi: 10.1016/s0140-6736(74)91496-2. [DOI] [PubMed] [Google Scholar]
- 6.Jellinger KA. Alzheimer disease and cerebrovascular pathology: An update. J Neural Transm. 2002;109:813–836. doi: 10.1007/s007020200068. [DOI] [PubMed] [Google Scholar]
- 7.Mandybur TI. The incidence of cerebral amyloid angiopathy in alzheimer's disease. Neurology. 1975;25:120–126. doi: 10.1212/wnl.25.2.120. [DOI] [PubMed] [Google Scholar]
- 8.Jellinger KA. The pathology of ischemic-vascular dementia: An update. J Neurol Sci. 2002;203–204:153–157. doi: 10.1016/s0022-510x(02)00282-4. [DOI] [PubMed] [Google Scholar]
- 9.Han BH, Zhou ML, Abousaleh F, Brendza RP, Dietrich HH, Koenigsknecht-Talboo J, et al. Cerebrovascular dysfunction in amyloid precursor protein transgenic mice: Contribution of soluble and insoluble amyloid-beta peptide, partial restoration via gamma-secretase inhibition. J Neurosci. 2008;28:13542–13550. doi: 10.1523/JNEUROSCI.4686-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christie R, Yamada M, Moskowitz M, Hyman B. Structural and functional disruption of vascular smooth muscle cells in a transgenic mouse model of amyloid angiopathy. Am J Pathol. 2001;158:1065–1071. doi: 10.1016/S0002-9440(10)64053-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, et al. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci U S A. 2008;105:1347–1352. doi: 10.1073/pnas.0711568105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tong XK, Nicolakakis N, Kocharyan A, Hamel E. Vascular remodeling versus amyloid beta-induced oxidative stress in the cerebrovascular dysfunctions associated with alzheimer's disease. J Neurosci. 2005;25:11165–11174. doi: 10.1523/JNEUROSCI.4031-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paris D, Humphrey J, Quadros A, Patel N, Crescentini R, Crawford F, et al. Vasoactive effects of a beta in isolated human cerebrovessels and in a transgenic mouse model of alzheimer's disease: Role of inflammation. Neurological research. 2003;25:642–651. doi: 10.1179/016164103101201940. [DOI] [PubMed] [Google Scholar]
- 14.Smith EE, Vijayappa M, Lima F, Delgado P, Wendell L, Rosand J, et al. Impaired visual evoked flow velocity response in cerebral amyloid angiopathy. Neurology. 2008;71:1424–1430. doi: 10.1212/01.wnl.0000327887.64299.a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dumas A, Dierksen GA, Gurol ME, Halpin A, Martinez-Ramirez S, Schwab K, et al. Functional magnetic resonance imaging detection of vascular reactivity in cerebral amyloid angiopathy. Annals of neurology. 2012;72:76–81. doi: 10.1002/ana.23566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okazaki H, Reagan TJ, Campbell RJ. Clinicopathologic studies of primary cerebral amyloid angiopathy. Mayo Clin Proc. 1979;54:22–31. [PubMed] [Google Scholar]
- 17.Jellinger KA, Attems J. Prevalence and pathogenic role of cerebrovascular lesions in alzheimer disease. J Neurol Sci. 2005;229–230:37–41. doi: 10.1016/j.jns.2004.11.018. [DOI] [PubMed] [Google Scholar]
- 18.Greenberg SM, Vonsattel JP, Stakes JW, Gruber M, Finklestein SP. The clinical spectrum of cerebral amyloid angiopathy: Presentations without lobar hemorrhage. Neurology. 1993;43:2073–2079. doi: 10.1212/wnl.43.10.2073. [DOI] [PubMed] [Google Scholar]
- 19.Neuropathology Group. Medical Research Council Cognitive F, Aging S. Pathological correlates of late-onset dementia in a multicentre, community-based population in england and wales. Neuropathology group of the medical research council cognitive function and ageing study (mrc cfas) Lancet. 2001;357:169–175. doi: 10.1016/s0140-6736(00)03589-3. [DOI] [PubMed] [Google Scholar]
- 20.Pfeifer LA, White LR, Ross GW, Petrovitch H, Launer LJ. Cerebral amyloid angiopathy and cognitive function: The haas autopsy study. Neurology. 2002;58:1629–1634. doi: 10.1212/wnl.58.11.1629. [DOI] [PubMed] [Google Scholar]
- 21.Jellinger KA, Attems J. Prevalence and impact of cerebrovascular pathology in alzheimer's disease and parkinsonism. Acta Neurol Scand. 2006;114:38–46. doi: 10.1111/j.1600-0404.2006.00665.x. [DOI] [PubMed] [Google Scholar]
- 22.Zhang F, Eckman C, Younkin S, Hsiao KK, Iadecola C. Increased susceptibility to ischemic brain damage in transgenic mice overexpressing the amyloid precursor protein. J Neurosci. 1997;17:7655–7661. doi: 10.1523/JNEUROSCI.17-20-07655.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koistinaho M, Kettunen MI, Goldsteins G, Keinanen R, Salminen A, Ort M, et al. Beta-amyloid precursor protein transgenic mice that harbor diffuse a beta deposits but do not form plaques show increased ischemic vulnerability: Role of inflammation. Proc Natl Acad Sci U S A. 2002;99:1610–1615. doi: 10.1073/pnas.032670899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 25.Lee JM, Zipfel GJ, Park KH, He YY, Hsu CY, Choi DW. Zinc translocation accelerates infarction after mild transient focal ischemia. Neuroscience. 2002;115:871–878. doi: 10.1016/s0306-4522(02)00513-4. [DOI] [PubMed] [Google Scholar]
- 26.Milner E, Holtzman JC, Friess S, Hartman RE, Brody DL, Han BH, et al. Endovascular perforation subarachnoid hemorrhage fails to cause morris water maze deficits in the mouse. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2014 doi: 10.1038/jcbfm.2014.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han BH, Zhou ML, Vellimana AK, Milner E, Kim DH, Greenberg JK, et al. Resorufin analogs preferentially bind cerebrovascular amyloid: Potential use as imaging ligands for cerebral amyloid angiopathy. Mol Neurodegener. 2011;6:86. doi: 10.1186/1750-1326-6-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dietrich HH, Xiang C, Han BH, Zipfel GJ, Holtzman DM. Soluble amyloid-beta, effect on cerebral arteriolar regulation and vascular cells. Mol Neurodegener. 2010;5:15. doi: 10.1186/1750-1326-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Nostrand WE, Melchor J, Wagner M, Davis J. Cerebrovascular smooth muscle cell surface fibrillar a beta. Alteration of the proteolytic environment in the cerebral vessel wall. Ann N Y Acad Sci. 2000;903:89–96. doi: 10.1111/j.1749-6632.2000.tb06354.x. [DOI] [PubMed] [Google Scholar]
- 30.Shin HK, Jones PB, Garcia-Alloza M, Borrelli L, Greenberg SM, Bacskai BJ, et al. Age-dependent cerebrovascular dysfunction in a transgenic mouse model of cerebral amyloid angiopathy. Brain. 2007;130:2310–2319. doi: 10.1093/brain/awm156. [DOI] [PubMed] [Google Scholar]
- 31.Van Nostrand WE, Melchor JP, Ruffini L. Pathologic amyloid beta-protein cell surface fibril assembly on cultured human cerebrovascular smooth muscle cells. Journal of neurochemistry. 1998;70:216–223. doi: 10.1046/j.1471-4159.1998.70010216.x. [DOI] [PubMed] [Google Scholar]
- 32.Merlini M, Meyer EP, Ulmann-Schuler A, Nitsch RM. Vascular beta-amyloid and early astrocyte alterations impair cerebrovascular function and cerebral metabolism in transgenic arcabeta mice. Acta neuropathologica. 2011;122:293–311. doi: 10.1007/s00401-011-0834-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hawkes CA, Michalski D, Anders R, Nissel S, Grosche J, Bechmann I, et al. Stroke-induced opposite and age-dependent changes of vessel-associated markers in co-morbid transgenic mice with alzheimer-like alterations. Experimental neurology. 2013;250:270–281. doi: 10.1016/j.expneurol.2013.09.020. [DOI] [PubMed] [Google Scholar]
- 34.Shankar GM, Leissring MA, Adame A, Sun X, Spooner E, Masliah E, et al. Biochemical and immunohistochemical analysis of an alzheimer's disease mouse model reveals the presence of multiple cerebral abeta assembly forms throughout life. Neurobiology of disease. 2009;36:293–302. doi: 10.1016/j.nbd.2009.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, csf, and plasma amyloid (beta) protein in the tg2576 transgenic mouse model of alzheimer's disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, et al. Amyloid precursor protein processing and a beta42 deposition in a transgenic mouse model of alzheimer disease. Proc Natl Acad Sci U S A. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cirrito JR, May PC, O'Dell MA, Taylor JW, Parsadanian M, Cramer JW, et al. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci. 2003;23:8844–8853. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, et al. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hong S, Quintero-Monzon O, Ostaszewski BL, Podlisny DR, Cavanaugh WT, Yang T, et al. Dynamic analysis of amyloid beta-protein in behaving mice reveals opposing changes in isf versus parenchymal abeta during age-related plaque formation. J Neurosci. 2011;31:15861–15869. doi: 10.1523/JNEUROSCI.3272-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maia LF, Kaeser SA, Reichwald J, Hruscha M, Martus P, Staufenbiel M, et al. Changes in amyloid-beta and tau in the cerebrospinal fluid of transgenic mice overexpressing amyloid precursor protein. Science translational medicine. 2013;5:194re192. doi: 10.1126/scitranslmed.3006446. [DOI] [PubMed] [Google Scholar]
- 41.Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid abeta42 in humans. Annals of neurology. 2006;59:512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 42.Verbeek MM, Kremer BP, Rikkert MO, Van Domburg PH, Skehan ME, Greenberg SM. Cerebrospinal fluid amyloid beta(40) is decreased in cerebral amyloid angiopathy. Annals of neurology. 2009;66:245–249. doi: 10.1002/ana.21694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD, et al. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nature neuroscience. 2004;7:954–960. doi: 10.1038/nn1302. [DOI] [PubMed] [Google Scholar]