

Abstract
Cationic liposomes (CLs) are studied worldwide as carriers of DNA and short interfering RNA (siRNA) for gene delivery and gene silencing, and related clinical trials are ongoing. Optimization of transfection efficiency and silencing efficiency by cationic liposome carriers requires a comprehensive understanding of the structures of CL–nucleic acid complexes and the nature of their interactions with cell membranes as well as events leading to release of active nucleic acids within the cytoplasm. Synchrotron x-ray scattering has revealed that CL–nucleic acid complexes spontaneously assemble into distinct liquid crystalline phases including the lamellar, inverse hexagonal, hexagonal, and gyroid cubic phases, and fluorescence microscopy has revealed CL–DNA pathways and interactions with cells. The combining of custom synthesis with characterization techniques and gene expression and silencing assays has begun to unveil structure–function relations in vitro. As a recent example, this review will briefly describe experiments with surface-functionalized PEGylated CL–DNA nanoparticles. The functionalization, which is achieved through custom synthesis, is intended to address and overcome cell targeting and endosomal escape barriers to nucleic acid delivery faced by PEGylated nanoparticles designed for in vivo applications.
Introduction
Liposomes consist of closed assemblies of bilayers of lipid molecules with polar head groups and hydrophobic tails (Fig. 1a). A. D. Bangham and R. W. Horne discovered liposomes (also referred to as unilamellar or multilamellar vesicles) during their electron microscopy investigations of phospholipids.1 The immediate significance of the discovery was the realization by these authors that the structural resemblance, between liposomes and cell membranes, provided direct confirmation that the dominant component of biological membranes consists of lipid assemblies. To date, liposomes remain a vital component of model membrane studies aimed at elucidating the biological functions of membrane-associated proteins.2 Bangham’s and Horne’s work also showed that liposomes naturally confine hydrophobic molecules within their bilayer (Fig. 1a) and form a permeability barrier for molecules entrapped within their aqueous interior.
Fig. 1.
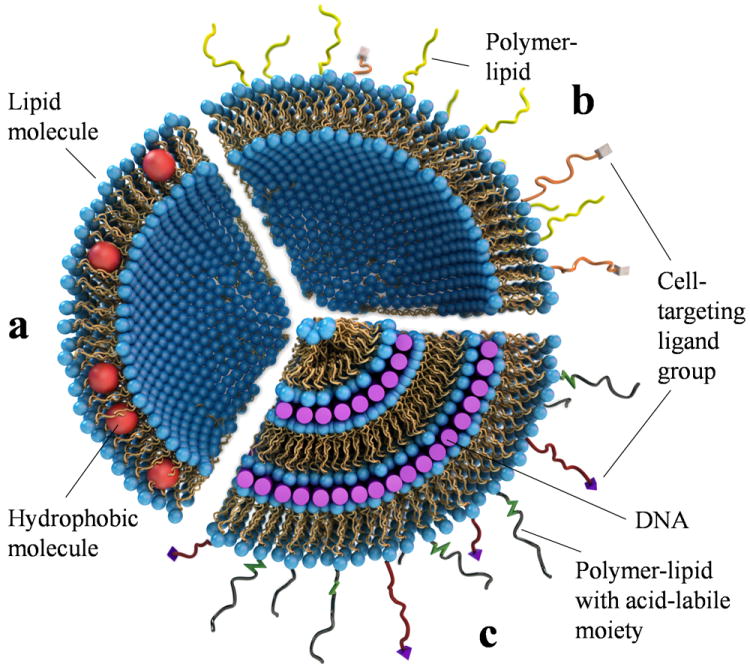
The evolution of liposomes from their discovery by A. D. Bangham and R. W. Horne in the 1960s. (a) A unilamellar liposome consisting of a self-assembly of amphiphilic lipid molecules. The liposome can trap hydrophobic molecules (red spheres) within its hydrophobic bilayer and hydrophilic molecules in its aqueous interior. (b) A “stealth” liposome, where the lipid bilayer contains a small percentage of polymer-lipids to enable the surface-modified liposome to avoid immune cells. Such liposomes may also incorporate cell-targeting ligand groups (e.g. peptides) attached to the distal end of the polymer-lipid (shown as white rectangular blocks). (c) A Cationic liposome–DNA complex consisting of an onion-like multilamellar structure with DNA (purple rods) sandwiched between cationic membranes. In addition to cell-targeting ligands attached to polymer-lipids, surface functionalization may also include polymer-lipids with acid-labile, hydrolysable groups for shedding of the polymer in late endosomes upon uptake of complexes by cells. Adapted and modified with permission from reference 2.
Within a decade of the initial discovery, researchers were investigating the potential of liposomes as carriers of drugs, peptides, proteins, and nucleic acids in therapeutic applications.3-5 A major early challenge in drug and gene delivery was the development of modified liposomes with long circulation times: bare drug-carrying liposomes are rapidly cleared by the mononuclear phagocytic system (immune cells) in in vivo settings.6,7
The initial attempt at developing long-circulating liposomes involved mimicking red blood cell (RBC) membranes, with the rationale being that immune cells do not attack RBCs under normal physiological conditions. This led to modification of the liposomal surface by addition of glycosphingolipids containing sialic acid groups.8,9 The final solution to this problem was the invention of PEGylated liposomes (so-called STEALTH® liposomes) that contain a coat of hydrophilic polymer resulting from the covalent attachment of poly(ethylene glycol) (PEG; e.g. 10 mol% of MW 2000 or 5 mol% of MW 5000) to the outer lipid headgroups (Fig. 1b).10-17 In vivo studies demonstrated long circulating times of PEGylated liposomes, suggesting that blood plasma opsonins are excluded from the immediate vicinity of the liposome surface since this is a necessary event for removal by immune cells.10-14
The PEG coat of STEALTH® liposomes induces a repulsive interaction (with a range on the scale of the size of the polymer chain18-20), which results in the steric stabilization of liposomes. This prevents adhesion of other particles and also flocculation of liposomes into loose aggregates due to van der Waals attractions.21-23 In the case of charged liposomes, the PEG coat effectively competes with and suppresses the adhesion of oppositely charged particles to the liposome.24 The development of PEGylated liposomes led naturally to the synthesis and use of ligand-containing PEG-lipids for targeted delivery applications (Fig. 1b). Currently, STEALTH® liposomes containing the chemotherapy drug doxorubicin-HCl (DOXIL®) have been approved by the FDA for applications in certain cancers.25
Structures of self-assembled cationic liposome–nucleic acid complexes
A pioneering new approach for gene delivery was introduced by P. Felgner and co-workers, who complexed cationic liposomes (CLs) with long strands of gene-containing DNA.26 The rationale for replacing the neutral or negative liposomes of earlier studies with CLs was the expectation that overall positively charged CL–DNA complexes would electrostatically adsorb to the sulfated, anionic proteoglycans coating mammalian cells, thus leading to more efficient complex uptake. The work by Felgner et al. was soon followed by numerous other groups, demonstrating gene expression in vivo in targeted organs27 and in human clinical trials.28
Gene carriers based on cationic lipids or polymers or a combination of these—rather than on engineered viruses—are now among the most promising technologies for transferring genes into cells for gene therapy and therapeutics.29-39 There are currently 113 ongoing gene therapy clinical trials worldwide using cationic liposomes (commonly referred to as lipofection) including those with added surface functionalities of steric stabilization and targeting ligands.40
Felgner et al. hypothesized a “bead-on-string” structure for CL–DNA complexes in their seminal paper,26,41 picturing the DNA strand decorated with distinctly attached cationic liposomes. This original description of a highly disordered complex has turned out to be an oversimplification. Quantitative structural studies of CL–DNA complexes using synchrotron x-ray scattering have revealed that mixing of long strands of DNA with cationic liposomes leads to a topological transition from liposomes into collapsed condensates in the form of distinct liquid crystalline (LC) self-assemblies.42-49 The most common structure of CL–DNA complexes corresponds to DNA monolayers sandwiched between cationic membranes, thus forming the multilamellar LαC phase.42 Fig. 1c depicts a simplified onion-like LαC phase with multiple layers of DNA encapsulated within each carrier. In addition to cell-targeting ligands, other functional units on the surface of CL–DNA complexes may include PEG-lipids with acid-labile hydrolysable units (Fig. 1c) for shedding of the PEG coat in the low-pH environment of late endosomes, in order for the complex to escape the endosome (discussed later).
Fig. 2a depicts the local, nanometer-scale structure of the LαC phase. We note that LαC complexes may contain focal conic type II defects commonly observed in multilamellar phases50-53 (see cryo-TEM images in Fig. 5b). Other self-assembled CL–DNA structures are the inverted hexagonal HIIC phase,43 with DNA encapsulated within cationic lipid monolayer tubes (Fig. 2b), and the HIC phase, with hexagonally ordered DNA rods surrounded by cylindrical micelles (Fig. 2c) that are formed from custom-synthesized lipids with highly charged (+16 e) dendritic multivalent headgroups (Fig. 2d).45
Fig. 2.
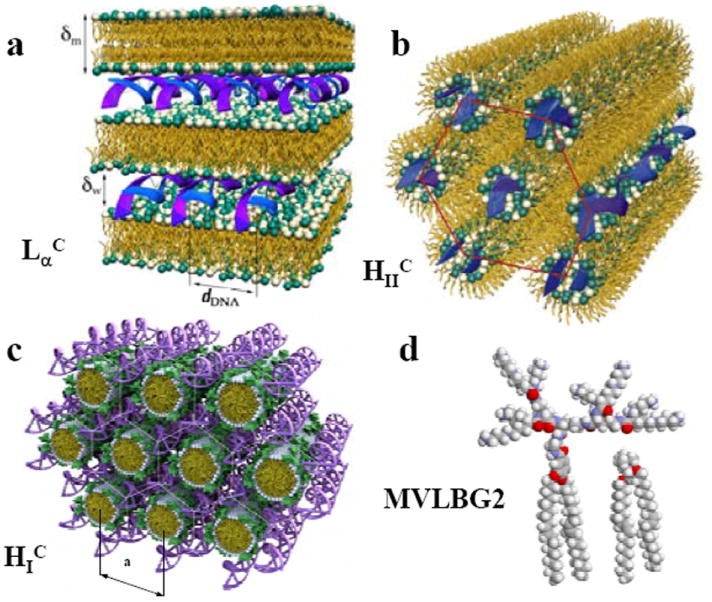
Mixing DNA and cationic liposomes (CLs) results in the spontaneous formation of CL–DNA complexes with equilibrium self-assembled structures. The schematics show the local structure of the interior of CL–DNA complexes on the nanometer scale as derived from synchrotron x-ray diffraction. (a) The lamellar LαC phase of CL–DNA complexes with alternating lipid bilayers and DNA monolayers. (b) The inverted hexagonal HIIC phase of CL–DNA complexes, composed of DNA inserted within inverse lipid tubules which are arranged on a hexagonal lattice. (c) The hexagonal HIC phase of MVLBG2/DOPC–DNA complexes, where the large lipid headgroup of the multivalent lipid MVLBG2 leads to the formation of rod-like lipid micelles arranged on a hexagonal lattice with DNA inserted within the interstices in honeycomb symmetry. (d) Molecular models of dendritic hexadecavalent MVLBG2 (headgroup charge +16 e) and univalent DOTAP (+1 e). Parts (a) and (b) reprinted with permission from references 42 and 43, respectively. Parts (c) and (d) adapted and reprinted with permission from reference 45; copyright 2006 American Chemical Society.
Fig. 5.
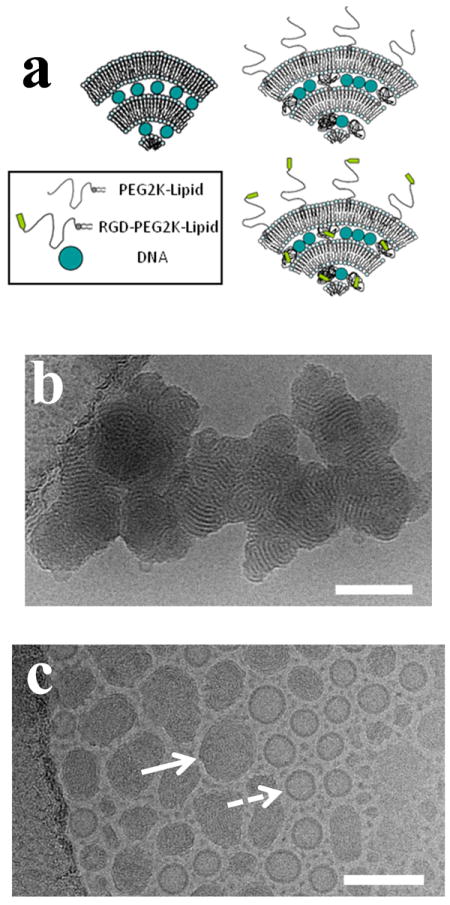
(a) Sketches of cationic liposome–DNA lamellar LαC complexes containing either no surface modification (top left), PEG-lipid (top right), or RGD-PEG-lipid (bottom right). Hydrodynamic diameter measurements via dynamic light scattering show that DOTAP/DOPC–DNA complexes with PEG2K-lipid (5 mol% or 10 mol% PEG2K-lipid) form stable ≈100 nm nanoparticles both in 150 mM NaCl and cell culture medium (DMEM). (b) Cryogenic TEM micrograph of DOTAP/DOPC–DNA complexes with no PEG2K coat, showing the multilamellar structure of the complex (80 mol% DOTAP; at cationic lipid to DNA ratio of 10, in 50 mM NaCl). (c) Cryogenic TEM micrograph showing PEGylated CL–DNA complexes forming ≈100 nm size nanoparticles (DOTAP/DOPC/PEG-lipid at 80/15/5 mol/mol/mol with cationic lipid to DNA ratio of 10, in 50 mM NaCl). Although the preparation in (c) (showing coexistence of nanometer scale complexes (solid arrow) with unilamellar cationic liposomes (dashed arrow)) has been extensively centrifuged, steric stabilization due to the PEG2K coat prevents aggregation. Scale bars correspond to 100 nm. Reprinted from reference 81, with permission from Elsevier.
The formation of CL–DNA structures with different membrane shapes is consistent with the predictions of the curvature elastic theory of membranes originally described by W. Helfrich,54 after accounting for the differences in the shapes of the lipid molecules, which influence the spontaneous curvature (C0) of membranes.55-57 For example, lipids possessing a cylindrical shape such as the univalent cationic lipid DOTAP (1,2-dioleoyl-3-trimethylammonium-propane) or the zwitterionic lipid DOPC (1,2-dioleoyl-sn-glycerophosphatidylcholine)—with a headgroup area approximately equal to the hydrophobic tail area—tend to self-assemble into lamellar structures with C0 = 0. Lipids with a head group area smaller than their tail area, such as DOPE (1,2-dioleoyl-sn-glycerophosphatidylethanolamine), have an inverse cone shape and give rise to a negative spontaneous curvature C0 < 0 and thus inverse hexagonal phases. Alternatively, lipids with a head group larger than their tail area (including custom-synthesized multivalent lipids with large headgroups45) have a cone shape, resulting in hexagonal phases with membranes with a spontaneous curvature C0 > 0. Synchrotron x-ray scattering shows that in most cases, the liquid crystalline structures of CL–DNA complexes is determined by the preferred curvature of the lipids constituting the cationic membranes.42,43,45 Further studies are needed to characterize the structures and structural transitions between lamellar and non-lamellar phases in PEGylated CL–DNA complexes.
Transfection efficiency of distinct phases of cationic liposome–DNA complexes in vitro
Intensive worldwide research over the recent past including, in particular, studies involving custom synthesis of novel multivalent lipids (MVLs), has resulted in the development of promising lipid vectors with transfection efficiencies which under optimal conditions are competitive with viral vectors for in vitro studies.29-33,58-62 Fig. 3a shows transfection efficiency (TE; a measure of expression of an exogenous gene that is transferred into the cell by the lipid carrier) as a function of mol% cationic lipid for complexes transfecting mouse fibroblast cells at various MVL/DOPC ratios with the headgroup charge of the MVLs varied between +2 e and +5 e. TE data for monovalent DOTAP mixed with DOPC is also shown for comparison. Only the amount of neutral lipid was changed between data points, while the amount of DNA and the cationic lipid/DNA charge ratio of 2.8 were kept constant.61 As the valence of the MVLs increases, TE tends to peak at a cationic lipid mol% lower than the 100% which early investigators had assumed would be the case.
Fig. 3.
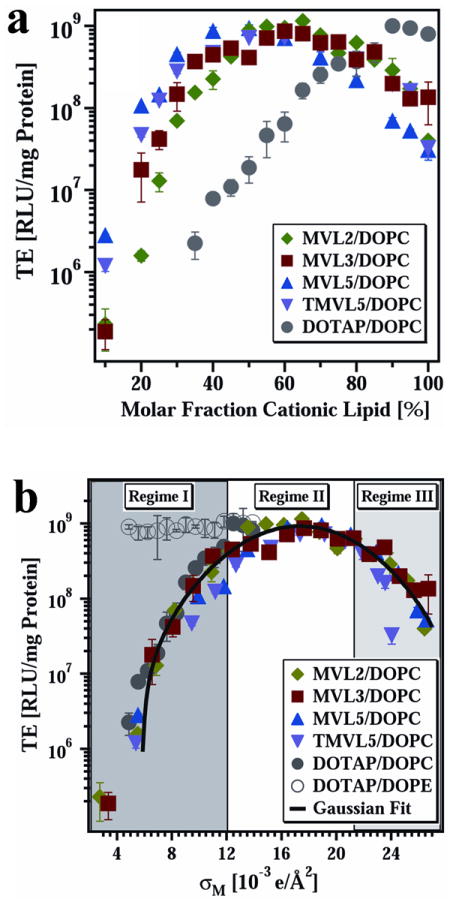
(a) Transfection efficiency (TE) as a function of mol% DOPC for DNA complexes prepared with the multivalent lipids MVL2 (green diamonds, valence Z = 2), MVL3 (red squares, valence Z = 3), MVL5 (blue triangles, valence Z = 5), TMVL5 (purple inverted triangles, valence Z = 5), and univalent DOTAP (gray circles, valence Z = 1). All data were taken at cationic lipid/DNA charge ratio ρchg = 2.8. (b) The same TE data as in (a) plotted versus the membrane charge density, σM (defined in the text). The data show that TE of the lamellar LαC complexes describes a universal, bell-shaped curve as a function of σM (the solid line is a Gaussian fit to the data). Significantly, data for DOTAP/DOPE–DNA complexes (gray open circles, HIIC phase) deviate from the universal curve, indicative of a distinctly different transfection mechanism for the inverted hexagonal phase. Three regimes of transfection efficiency are identified as discussed in the text. Reprinted with permission from reference 61. Copyright 2005, John Wiley & Sons.
The origin of this optimal mol fraction for TE of lamellar complexes is made clear if one plots the TE data as a function of the membrane charge density (σM) of the complexes.
The membrane charge density is defined as the total charge of the complex due to the MVLs divided by the total membrane area of the complex (consisting of a mixture of MVL and neutral lipids).63,61 (As described in reference 61, σM = [1-Φnl/(Φnl + rΦcl)] σcl, where r = Acl/Anl is the ratio of the headgroup areas of the cationic and the neutral lipid; σcl = eZ/Acl is the charge density of the cationic lipid with valence Z; and Φnl and Φcl are the mol fractions of the neutral and cationic lipids, respectively.) Fig. 3b depicts a plot of the same TE data shown in Fig. 3a but now plotted versus σM.61 Remarkably all the data points merge onto a single Gaussian curve with an optimal σM* = 17.0 ± 0.1 ×10−3 e/Å2. Synchrotron x-ray scattering shows that MVL- and DOTAP-based complexes used in these TE studies are in the lamellar LαC phase. The collapse of the data on a single curve implies that σM is a predictor of TE and a universal parameter for transfection by lamellar LαC CL–DNA complexes.
A simple model of transfection by LαC complexes61,63 can be used to explain the data of Fig. 3. This model hypothesizes that the TE of complexes of low σM is limited mostly because they are trapped in the endosome. Increases in σM, leading to enhanced fusion of the cationic membrane of the complex with the anionic endosomal membrane, facilitate nucleic acid delivery to the cytosol and thus enhance TE (Fig. 3b, labeled Regime I). At very high σM (Regime III), complexes escape the endosome but a fraction of the DNA remains trapped in complexes in the cytoplasm due to the strong electrostatic interactions between cationic membranes and DNA. Thus, increases in σM lead to decreases in TE in this regime III. Regime II is the regime of compromise, where the optimal σM is large enough for many complexes to escape the endosome and yet small enough to allow a larger fraction of DNA to be released from complexes (compared to Regime III).
Transfection efficiency of non-lamellar CL–DNA complexes is very different from the universal behavior found for lamellar complexes. The high TE of DOTAP/DOPE HIIC complexes (Fig. 3b, open circles), which is independent of σM, is likely related to the readily occurring fusion of the membranes of HIIC complexes with the cell’s plasma and endosomal membranes, observed in 3D confocal microscopy experiments.63 Complexes in the HIC phase of MVLs with very large headgroups (e.g. the cone-shaped lipid MVLBG2 (+16 e)) also show high TE that is independent of σM.45,64 Further studies will be required to clarify the mechanism of transfection of non-lamellar complexes where their TE behavior deviates from the bell-shaped curve observed for lamellar complexes.
siRNA embedded in a cationic gyroid cubic lipid matrix for gene silencing
The discovery of RNA interference (RNAi) as a post-transcriptional gene-silencing pathway has given rise to a major new branch in cationic lipid-based nucleic acid research worldwide.65-71 Upon complexation with cationic liposomes, short strands of double-stranded RNA (referred to as short interfering RNA, siRNA), evoke the RNA interference pathway leading to sequence-specific gene silencing.72-75 In addition to its widespread application in functional genomics, siRNA technology promises to revolutionize biotechnology and therapeutics.71 The current limiting step in siRNA gene silencing technology is the development of efficient carriers of siRNA, in particular for in vivo applications.74 Previous to the development of siRNA technology, researchers had in fact utilized single-strand antisense oligonucleotides in therapeutic applications.36 These technologies are currently being optimized further.
A recent synchrotron x-ray scattering study showed that a thermodynamically stable double-gyroid cubic lipid phase incorporating siRNA may be formed both with monovalent DOTAP and multivalent MVL5 when they are mixed with the neutral lipid GMO (1-monooleoyl-glycerol) (Fig. 4a).76,77 The study, which followed a rational design of the elastic properties of the carrier lipid membrane, revealed that the cubic CL–siRNA complex phase (labeled QIIG, siRNA) is remarkably efficient at cytoplasmic delivery and subsequent gene silencing. Notably, this was the first demonstration of CL–siRNA complexes showing highly efficient sequence-specific gene knockdown (KT) and low nonspecific silencing (KNS) (Fig. 4b) at low membrane charge density, which demonstrated that high silencing efficiency does not mandate high membrane charge density. (The previous studies of lamellar CL–siRNA complexes only showed high specific silencing efficiency at relatively high membrane charge density.72,73)
Fig. 4.
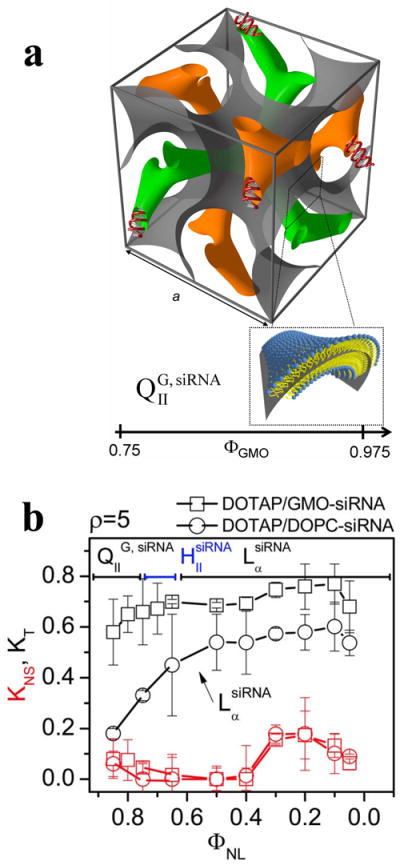
(a) The unit cell of the double-gyroid cubic phase (space group Ia3d) incorporating siRNA within its two water channels (green and orange). The structure was deduced by synchrotron x-ray scattering. For DOTAP/GMO–siRNA complexes the phase (labeled QIIG, siRNA) is observed for GMO (1-monooleoyl-glycerol) molar fractions (ΦGMO) between 0.75 and 0.975. A lipid bilayer surface separates the two intertwined but independent water channels. The bilayer is represented by a surface (grey) corresponding to a thin layer in the center of the membrane as indicated in the enlarged inset. (b) Total (KT, black lines and symbols) and nonspecific (KNS, red lines and symbols) gene knockdown for DOTAP/GMO–siRNA complexes (squares) and DOTAP/DOPC–siRNA complexes (circles) as a function of mole fraction of neutral lipid (ΦNL). DOTAP/GMO–siRNA complexes in the gyroid cubic phase (QIIG, siRNA) at low cationic lipid content (ΦGMO ≥ 0.75) show remarkably improved sequence-specific gene silencing over complexes in the lamellar phase (LαsiRNA). Reprinted with permission from reference 76. Copyright 2010, American Chemical Society.
The discovered properties of the QIIG, siRNA phase are consistent with the hypothesis that the lipids of the gyroid cubic phase favor membrane pore formation resulting from fusion of the membranes of the gyroid CL–siRNA complex and endosomal membranes. Pores, in turn, allow for cytoplasmic siRNA delivery. This inclination to pore formation arises because the surface of membrane pores and the surface of the gyroid cubic phase are both characterized by a negative Gaussian curvature.76-80
Surface-functionalized cationic liposome–DNA nanoparticles
The CL–nucleic acid complexes that we have described so far are suitable for transfection experiments in vitro (including transfection of hard-to-transfect cells), but not ideal for in vivo clinical studies because the overall positive charge of the complex results in their early clearance from circulation by immune cells through the mechanism of opsonization.10-17 As was discussed earlier in the context of unilamellar liposome delivery (see introduction), steric stabilization of complexes by addition of PEG-lipids (Fig. 5a), suppresses attachment of (overall anionic) serum opsonins, thus prolonging circulation times.18-20 Therefore, in vivo applications require the development of PEGylated CL–nucleic acid complexes with optimized TE in vitro.
Dynamic light scattering measurements show that PEGylated CL–DNA complexes containing 10 mol% PEG2K-lipid (PEG molecular weight of 2000 Da) form sterically stable nanoparticles (NPs) with an average diameter ≈ 100 nm.81 Cryogenic TEM of lamellar CL–DNA complexes in 50 mM NaCl further shows that while uncoated complexes aggregate (Fig. 5b), PEGylated CL–DNA NPs are sterically stabilized and retain their distinct size (≈ 100 nm) and morphology even after centrifugation (Fig. 5c). The TEM images are consistent with a recent synchrotron x-ray scattering study of the pathways of formation of lamellar PEGylated CL–DNA complexes, which showed that the NPs contain a significant numbers of bilayers at 50 mM and 100 mM NaCl (between 10 and 20 on average, at high membrane charge density and 10 mol% PEG2K-lipid).49 The same study showed that the NPs contain, on average, only a few bilayers if the complexation occurs at 150 mM NaCl.
PEGylated CL–DNA NPs exhibit reduced TE because the weak electrostatic adhesion of the NPs to the negative surface of cells dramatically reduces uptake of NPs by cells. TE drops between two and three orders of magnitude with the incorporation of 10 mol% PEG2K-lipid.21,81,82 Cell attachment and uptake of PEGylated NPs may be recovered by covalent attachment of a linear RGD peptide ligand (GRGDSP) to the distal end of the PEG2K-lipid. Custom synthesis again plays a crucial role in these studies, where the RGD-PEG2K-lipid was prepared by solid phase peptide synthesis methods employing a custom-synthesized carboxy-terminated PEG2K-lipid in the final coupling step to the N-terminus of the protected peptide on the resin.81 The linear RGD peptide binds to integrins (e.g. αvβ3 and αvβ5) on the cell’s surface, providing receptor-mediated endocytosis.83-85 Aside from linear RGD moieties, many current studies incorporate cyclic RGD peptides which tend to have much higher binding affinities to integrins and are more selective.86
Live-cell imaging at low membrane charge density (to fully suppress cell attachment/uptake due to electrostatic interactions), combined with quantitative particle tracking of the intracellular distribution of complexes, directly confirms the increased rate and total amount of NP uptake when PEG2K-lipid is replaced by RGD-PEG2K-lipid (Fig. 6).81 NPs containing 10 mol% PEG2K-lipid show nearly no uptake in live-cell imaging even 5 hours after addition of NPs to cells (Fig. 6a). In contrast, NPs containing linear RGD-PEG2K-lipid exhibit significantly increased surface attachment at 1 hour, followed by significant internalization at the 5 hour mark (Fig. 6b). TE data show that surface-functionalized complexes containing 10 mol% RGD-PEG2K-lipid partially recover the loss of TE induced by PEGylation. This indicates that while RGD-containing NPs have efficiently undergone receptor-mediated cell attachment and uptake (consistent with the live-cell imaging data), endosomal entrapment remains a significant barrier. Future surface functionalization is expected to ensure that the uptake of NPs with cell targeting ligands ultimately leads to endosomal release of a majority of NPs. As a first test of this concept, recent custom synthesis of PEG-lipids containing acid-labile moieties (see cartoon in Fig. 1c) has been shown to aid in shredding of the PEG coat of NPs by hydrolysis in the low-pH environment of late endosomes, thus facilitating endosomal escape and delivery of high membrane charge density NPs to the cytosol.82
Fig. 6.
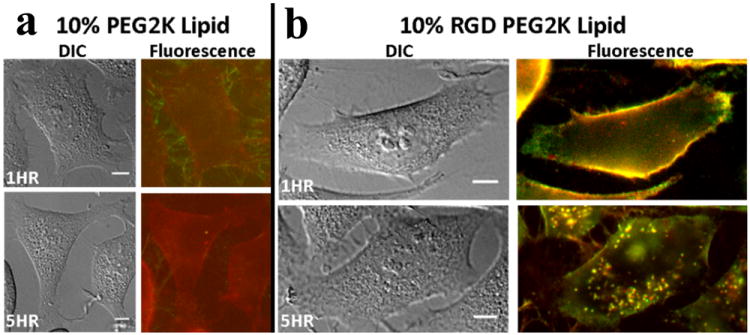
Live-cell images of PEGylated cationic liposome–DNA nanoparticles (NPs), with and without an RGD motif, at low membrane charge density (DOTAP/DOPC/PEG2K-Lipid at 30/60/10, mol/mol/mol). (a, b) Typical differential-interference-contrast and merged fluorescence micrographs (DNA: green, lipid: red). (a) The top fluorescence micrograph shows some PEGylated CL–DNA nanoparticles (rich in DNA) attached to cell filopodia, producing a small amount of staining of the plasma membrane with lipid label 1 hour after addition of NPs. The bottom fluorescence micrograph shows one or two internalized NPs after 5 hours. (b) RGD-tagged NPs (produced by replacing PEG2K-lipid with RGD-PEG2K-lipid) are seen to strongly coat the plasma membrane 1 hour after addition of NPs (top fluorescence micrograph), and many NPs are visible inside the cell at 5 hours (bottom fluorescence micrograph). Reprinted from reference 81, with permission from Elsevier.
We expect to see significant research progress in the near future in the development of a variety of functionalized lipid NPs for therapeutic purposes employing concepts along the lines of what we have described. Indeed, among the largest current efforts by numerous groups worldwide is the development of lipid NP carriers of drugs for clinical cancer chemotherapy applications.87-90
Conclusions and perspectives
An important objective in this biomedical research area is to develop a scientific understanding, which will enable the design and synthesis of optimal lipid nanoparticle carriers of DNA and siRNA for gene therapeutics and disease control. To this end, the work we have presented in this review has emphasized a rational design approach for tuning the physico-chemical and elastic properties of cationic membranes in order to overcome cellular barriers to nucleic acid delivery.
A significant finding relates to the discovery of distinct structures of CL–nucleic acid complexes by synchrotron x-ray scattering and the discovery that the structures correlate with delivery mechanisms and transfection and silencing efficiencies. The structures described in this review are the prevalent multilamellar (LαC), the inverted hexagonal (HIIC), and the hexagonal (HIC) phases for CL–DNA complexes, and the double gyroid cubic (QIIG, siRNA) phase for CL–siRNA complexes.
Custom synthesis of multivalent lipids led to the discovery that membrane charge density (σM) is a predictive chemical parameter for transfection by LαC CL–DNA complexes, while transfection efficiency of non-lamellar structures (HIIC and HIC) is independent of σM. We presented data showing that gyroid cubic CL–siRNA complexes exhibit improved silencing efficiency because of the cubic membrane’s inherent fusogenic properties which facilitate endosomal escape even at low σM. A significant success coming out of the research efforts on lipid vectors is that transfection efficiencies of multivalent lipid vectors are now competitive with viral vectors in vitro; indeed, the multivalent lipid MVL5 (originally synthesized and characterized at UC Santa Barbara) has been recently commercialized by Avanti Polar Lipids, Inc. (Alabaster, AL, USA) as an efficient transfection reagent for gene silencing with low toxicity.91
Finally, we note that current research on lipid-based vectors is heavily focused on producing nanoparticles92-95 that will be suitable for in vivo delivery.81,82,96,97 We expect that continued mechanistic studies by many laboratories worldwide of distinct strategies for cell targeting and endosomal escape, which incorporate custom synthesis for obtaining new rationally modified nanoparticle carriers, will positively impact the large number of gene therapy clinical trials which utilize lipids.
Supplementary Material
Acknowledgments
We acknowledge support by the U.S. National Institutes of Health R01-GM59288 (biological activity studies in cationic liposome–nucleic acid complexes), and the U.S. National Science Foundation DMR-1401784 (phase behavior of lipid–nucleic acid complexes). CL was funded by the Swedish Research Council (VR). The X-ray diffraction work was carried out at the Stanford Synchrotron Radiation Laboratory, a U.S. Department of Energy National Laboratory.
Biographies

Cyrus R. Safinya is a Professor at the University of California, Santa Barbara. Research in his group is centered on (1) clarifying parameters, which control the interactions between proteins derived from the neuronal cytoskeleton, with the goal of relating structure to function, and (2) developing lipid nanoparticle carriers of functional DNA and short interfering RNA for gene delivery and silencing applications.

Kai Ewert studied chemistry at the Universität Bayreuth and obtained his Ph.D. from the Universität Stuttgart in 1998 for work on polymer chemistry and structure–function relationships. He joined the group of Prof. Safinya in 1999, first as a postdoc and later as a project scientist. His work centers on the design, synthesis and characterization of lipids, polymer-lipids, and peptide-polymer-lipids.

Ramsey Majzoub earned his B.S. in Engineering Physics from the University of Colorado at Boulder in 2008 while doing research in Noel Clark’s group. He is currently a Physics Ph.D. candidate at University of California at Santa Barbara in the group of Cyrus Safinya. His Ph.D. work is centered on quantitative live cell imaging and deciphering the endosomal pathways of targeted lipid–nucleic acid nanoparticles.

Cecília Leal is an Assistant Professor of Materials Science & Engineering at the University of Illinois, Urbana-Champaign since 2012. Her research is in biomaterials structures and interactions. She graduated in Portugal before earning her Ph.D. at the University of Lund (2006) with H. Wennerström & B. Lindman. She worked at the Norwegian Radium Hospital before joining Prof. Safinya’s lab as a postdoc in late 2007.
Notes and references
- 1.Bangham AD, Horne RW. J Mol Biol. 1964;8:660–668. doi: 10.1016/s0022-2836(64)80115-7. [DOI] [PubMed] [Google Scholar]
- 2.Safinya CR, Ewert KK. Nature. 2012;489:372–374. doi: 10.1038/489372b. [DOI] [PubMed] [Google Scholar]
- 3.Gregoriadis G, Leathwood PD, Ryman BE. FEBS Lett. 1971;14:95–99. doi: 10.1016/0014-5793(71)80109-6. [DOI] [PubMed] [Google Scholar]
- 4.Gregoriadis G, Ryman BB. Eur J Biochem. 1972;24:485–491. doi: 10.1111/j.1432-1033.1972.tb19710.x. [DOI] [PubMed] [Google Scholar]
- 5.Gregoriadis G. New Engl J Med. 1976;295:704–765. doi: 10.1056/NEJM197609232951305. [DOI] [PubMed] [Google Scholar]
- 6.Lasic DD. Liposomes in Gene Delivery. CRC Press; Boca Raton: 1994. [Google Scholar]
- 7.Lasic DD, Martin FJ, editors. Stealth liposomes. CRC Press; Boca Raton: 1995. [Google Scholar]
- 8.Gabizon A, Papahadjopoulos D. Proc Natl Acad Sci U S A. 1988;85:6949–6953. doi: 10.1073/pnas.85.18.6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allen TM, Hansen C, Rutledge J. Biochim Biophys Acta. 1989;981:27–35. doi: 10.1016/0005-2736(89)90078-3. [DOI] [PubMed] [Google Scholar]
- 10.Woodle MC, Neumann M, Collins LR, Redemann C, Martin FJ. Proc Int Sym Control Release Bioact Mater. 1990;77:17–17. [Google Scholar]
- 11.Klibanov AL, Maruyama K, Torchillin VP, Huang L. FEBS Lett. 1990;268:235–237. doi: 10.1016/0014-5793(90)81016-h. [DOI] [PubMed] [Google Scholar]
- 12.Blume G, Cevc G. Biochim Biophys Acta. 1990;1029:91–97. doi: 10.1016/0005-2736(90)90440-y. [DOI] [PubMed] [Google Scholar]
- 13.Papahadjopoulos D, Allen TM, Gabizon A, Mayhew E, Matthay K, Huang S, Lee K, Woodle M, Lasic DD, Redemann C, Martin F. Proc Natl Acad Sci U S A. 1991;88:11460–11464. doi: 10.1073/pnas.88.24.11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allen TM, Martin FJ, Redemann C, Hansen C, Yau-Young A. Biochim Biophys Acta. 1991;1066:29–36. doi: 10.1016/0005-2736(91)90246-5. [DOI] [PubMed] [Google Scholar]
- 15.Woodle M, Lasic D. Biochim Biophys Acta. 1992;1113:171–199. doi: 10.1016/0304-4157(92)90038-c. [DOI] [PubMed] [Google Scholar]
- 16.Papahadjopoulos D. In: Stealth Liposomes. Lasic DD, Martin F, editors. CRC Press; Boca Raton: 1995. pp. 1–6. [Google Scholar]
- 17.Silvander M. Progr Colloid Polym Sci. 2002;120:35–40. [Google Scholar]
- 18.de Gennes PG. Scaling Concepts in Polymer Physics. Cornell University Press; New York: 1979. [Google Scholar]
- 19.Kuhl TL, Leckband DE, Lasic DD, Israelachvili JN. Biophys J. 1994;66:1479–1488. doi: 10.1016/S0006-3495(94)80938-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kenworthy AK, Hristova K, Needham D, McIntosh TJ. Biophys J. 1995;68:1921–1935. doi: 10.1016/S0006-3495(95)80369-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin-Herranz A, Ahmad A, Evans HM, Ewert K, Schulze U, Safinya CR. Biophys J. 2004;86:1160–1168. doi: 10.1016/S0006-3495(04)74190-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lasic DD. Liposomes: from Physics to Applications. Elsevier; San Diego: 1993. [Google Scholar]
- 23.Witten TA. Structured Fluids: Polymers, Colloids, Surfactants. Oxford University Press; Oxford: 2004. [Google Scholar]
- 24.Hong K, Zheng W, Baker A, Papahadjopoulos D. FEBS Lett. 1997;400:233–237. doi: 10.1016/s0014-5793(96)01397-x. [DOI] [PubMed] [Google Scholar]
- 25.Doxil: see, [July 2014]; e.g. http://www.rxlist.com/doxil-drug.htm.
- 26.Felgner PL, Gader TR, Holm M, Roman R, Chan HW, Wenz M, Northrop JP, Ringold GM, Danielsen M. Proc Natl Acad Sci U S A. 1987;84:7413–7417. doi: 10.1073/pnas.84.21.7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu N, Liggitt D, Liu Y, Debs R. Science. 1993;261:209–211. doi: 10.1126/science.7687073. [DOI] [PubMed] [Google Scholar]
- 28.Nabel GJ, Nabel EG, Yang ZY, Fox BA, Plautz GE, Gao X, Huang L, Shu S, Gordon D, Chang AE. Proc Nat Acad Sci U S A. 1993;90:11307–11311. doi: 10.1073/pnas.90.23.11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo X, Huang L. Acc Chem Res. 2012;45:971–979. doi: 10.1021/ar200151m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bielke W, Erbacher C, editors. Nucleic Acid Transfection (Topics in Current Chemistry. Vol. 296. Springer; Heidelberg: 2010. [PubMed] [Google Scholar]
- 31.Ewert KK, Zidovska A, Ahmad A, Bouxsein NF, Evans HM, McAllister CS, Samuel CE, Safinya CR. Topics Curr Chem. 2010;296:191–226. doi: 10.1007/128_2010_70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang L, Hung M-C, Wagner E, editors. Non-Viral Vectors for Gene Therapy, Part I (Advances in Genetics. 2. Vol. 53. Elsevier; San Diego: 2005. [Google Scholar]
- 33.Ewert K, Ahmad A, Evans HM, Safinya CR. Expert Opin Biol Ther. 2005;5:33–53. doi: 10.1517/14712598.5.1.33. [DOI] [PubMed] [Google Scholar]
- 34.Ewert K, Evans HM, Ahmad A, Slack NL, Lin AJ, Martin-Herranz A, Safinya CR. In: Non-viral Vectors for Gene Therapy Part I (Advances in Genetics. 2. Huang L, Hung M-C, Wagner E, editors. Vol. 53. Elsevier; San Diego: 2005. pp. 119–155. [DOI] [PubMed] [Google Scholar]
- 35.Ewert K, Slack NL, Ahmad A, Evans HM, Lin AJ, Samuel CE, Safinya CR. Curr Med Chem. 2004;11:133–149. doi: 10.2174/0929867043456160. [DOI] [PubMed] [Google Scholar]
- 36.Mahato RI, Kim SW, editors. Pharmaceutical Perspectives of Nucleic Acid-Based Therapeutics. Taylor and Francis; New York: 2002. [Google Scholar]
- 37.Safinya CR, Lin AJ, Slack NE, Koltover I. In: Pharmaceutical Perspectives of Nucleic Acid-Based Therapeutics. Mahato RI, Kim SW, editors. Taylor and Francis; New York: 2002. [Google Scholar]
- 38.Huang L, Hung M-C, Wagner E, editors. Nonviral Vectors for Gene Therapy. Academic Press; San Diego: 1999. [Google Scholar]
- 39.Safinya CR, Koltover I. In: Non-Viral Vectors for Gene Therapy. Huang L, Hung M-C, Wagner E, editors. Academic Press; San Diego: 1999. [Google Scholar]
- 40.Gene Therapy Clinical Trials Worldwide, 2014. [July, 2014];The journal of gene medicine clinical trial site. http://www.wiley.com/legacy/wileychi/genmed/clinical/
- 41.Felgner PL, Rhodes G. Nature. 1991;349:351–352. doi: 10.1038/349351a0. [DOI] [PubMed] [Google Scholar]
- 42.Rädler JO, Koltover I, Salditt T, Safinya CR. Science. 1997;275:810–814. doi: 10.1126/science.275.5301.810. [DOI] [PubMed] [Google Scholar]
- 43.Koltover I, Salditt T, Rädler JO, Safinya CR. Science. 1998;281:78–81. doi: 10.1126/science.281.5373.78. [DOI] [PubMed] [Google Scholar]
- 44.Safinya CR. Curr Opin Struct Biol. 2001;11:440–448. doi: 10.1016/s0959-440x(00)00230-x. [DOI] [PubMed] [Google Scholar]
- 45.Ewert KK, Evans HM, Zidovska A, Bouxsein NF, Ahmad A, Safinya CR. J Am Chem Soc. 2006;128:3998–4006. doi: 10.1021/ja055907h. [DOI] [PubMed] [Google Scholar]
- 46.Safinya CR, Ewert K, Ahmad A, Evans HM, Raviv U, Needleman DJ, Lin AJ, Slack NL, George CX, Samuel CE. Philosoph Trans R Soc London, Ser A. 2006;364:2573–2596. doi: 10.1098/rsta.2006.1841. [DOI] [PubMed] [Google Scholar]
- 47.Safinya CR, Ewert KK, Leal C. Liq Cryst. 2011;38:1715–1723. doi: 10.1080/02678292.2011.624364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Safinya CR, Deek J, Beck R, Jones JB, Leal C, Ewert KK, Li Y. Liq Cryst. 2013;40:1748–1758. doi: 10.1080/02678292.2013.846422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Silva BFB, Majzoub RN, Chan C-L, Li Y, Olsson U, Safinya CR. Biochim Biophys Acta – Biomembranes. 2014;1838:398–412. doi: 10.1016/j.bbamem.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Warriner HE, Idziak SHJ, Slack NL, Davidson P, Safinya CR. Science. 1996;271:969–973. doi: 10.1126/science.271.5251.969. [DOI] [PubMed] [Google Scholar]
- 51.Warriner HE, Davidson P, Slack NL, Schellhorn M, Eiselt P, Idziak SHJ, Schmidt H-W, Safinya CR. J Chem Phys. 1997;107:3707–3722. [Google Scholar]
- 52.Keller SL, Warriner HE, Safinya CR, Zasadzinski JA. Phys Rev Lett. 1997;78:4781–4784. [Google Scholar]
- 53.Warriner HE, Keller SL, Idziak SHJ, Slack NL, Davidson P, Zasadzinski JA, Safinya CR. Biophys J. 1998;75:272–293. doi: 10.1016/S0006-3495(98)77514-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Helfrich W. Z Naturforsch C. 1973;28:693–703. doi: 10.1515/znc-1973-11-1209. [DOI] [PubMed] [Google Scholar]
- 55.Safran SA. Statistical thermodynamics of surfaces, interfaces, and membranes. Addison-Wesley; Reading: 1994. [Google Scholar]
- 56.Israelachvili JN. Intermolecular and Surface Forces. 2. Academic Press; London: 1992. [Google Scholar]
- 57.Seddon JM. Biochim Biophys Acta. 1989;1031:1–69. doi: 10.1016/0304-4157(90)90002-t. [DOI] [PubMed] [Google Scholar]
- 58.Ewert K, Ahmad A, Evans HM, Schmidt H-W, Safinya CR. J Med Chem. 2002;45:5023–5029. doi: 10.1021/jm020233w. [DOI] [PubMed] [Google Scholar]
- 59.Ewert KK, Evans HM, Zidovska A, Bouxsein NF, Ahmad A, Safinya CR. Bioconjugate Chem. 2006;17:877–888. doi: 10.1021/bc050310c. [DOI] [PubMed] [Google Scholar]
- 60.Schulze U, Schmidt H-W, Safinya CR. Bioconjugate Chem. 1999;10:548–552. doi: 10.1021/bc9801068. [DOI] [PubMed] [Google Scholar]
- 61.Ahmad A, Evans HM, Ewert K, George CX, Samuel CE, Safinya CR. J Gene Med. 2005;7:739–748. doi: 10.1002/jgm.717. [DOI] [PubMed] [Google Scholar]
- 62.Chan C-L, Ewert KK, Majzoub RN, Hwu Y-K, Liang KS, Leal C, Safinya CR. J Gene Med. 2014;16:84–96. doi: 10.1002/jgm.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin AJ, Slack NL, Ahmad A, George CX, Samuel CE, Safinya CR. Biophys J. 2003;84:3307–3316. doi: 10.1016/S0006-3495(03)70055-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zidovska A, Evans HM, Ewert KK, Quispe J, Carragher B, Potter CS, Safinya CR. J Phys Chem B. 2009;113:3694–3703. doi: 10.1021/jp806863z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fire A, Xu SQ, Montgomery MK, Kostas SA, Driver SE, Mello CC. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 66.Cogoni C, Macino G. Curr Opin Genet Dev. 2000;10:638–643. doi: 10.1016/s0959-437x(00)00134-9. [DOI] [PubMed] [Google Scholar]
- 67.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 68.Caplen NJ, Parrish S, Imani F, Fire A, Morgan RA. Proc Natl Acad Sci U S A. 2001;98:9742–9747. doi: 10.1073/pnas.171251798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.See e.g., Hannon GJ, Rossi JJ. Nature. 2004;431:371–378. doi: 10.1038/nature02870..
- 70.Karagiannis TC, El-Osta A. Cancer Gene Ther. 2005;12:787–795. doi: 10.1038/sj.cgt.7700857. [DOI] [PubMed] [Google Scholar]
- 71.Sioud M. Trends Pharm Sci. 2004;25:22–28. doi: 10.1016/j.tips.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 72.Spagnou S, Miller AD, Keller M. Biochemistry. 2004;43:13348–13356. doi: 10.1021/bi048950a. [DOI] [PubMed] [Google Scholar]
- 73.Bouxsein NF, McAllister CS, Ewert KK, Samuel CE, Safinya CR. Biochemistry. 2007;46:4785–4792. doi: 10.1021/bi062138l. [DOI] [PubMed] [Google Scholar]
- 74.Gindy ME, Leone AM, Cunningham JJ. Expert Opin Drug Deliv. 2012;9:171–182. doi: 10.1517/17425247.2012.642363. [DOI] [PubMed] [Google Scholar]
- 75.Gindy ME, DiFelice K, Kumar V, Prud’homme RK, Celano R, Haas RM, Smith JS, Boardman D. Langmuir. 2014;30:4613–4622. doi: 10.1021/la500630h. [DOI] [PubMed] [Google Scholar]
- 76.Leal C, Bouxsein NF, Ewert KK, Safinya CR. J Am Chem Soc. 2010;132:16841–16847. doi: 10.1021/ja1059763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Leal C, Ewert KK, Shirazi RS, Bouxsein NF, Safinya CR. Langmuir. 2011;27:7691–7697. doi: 10.1021/la200679x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Porte G. J Phys : Condens Matter. 1992;4:8649–8670. [Google Scholar]
- 79.Siegel DP, Epand RM. Biophys J. 1997;73:3089–3111. doi: 10.1016/S0006-3495(97)78336-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang L, Huang HW. Science. 2002;297:1877–1879. doi: 10.1126/science.1074354. [DOI] [PubMed] [Google Scholar]
- 81.Majzoub RN, Chan C-L, Ewert KK, Silva BFB, Liang KS, Jacovetty EL, Carragher B, Potter CS, Safinya CR. Biomaterials. 2014;35:4996–5005. doi: 10.1016/j.biomaterials.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chan C-L, Majzoub RN, Shirazi RS, Ewert KK, Chen Y-J, Liang KS, Safinya CR. Biomaterials. 2012;33:4928–4935. doi: 10.1016/j.biomaterials.2012.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ruoslahti E, Bhatia SN, Sailor MJ. J Cell Biol. 2010;188:759–768. doi: 10.1083/jcb.200910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Temming K, Schiffelers RM, Molema G, Kok RJ. Drug Resistance Updates. 2005;8:381–402. doi: 10.1016/j.drup.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 85.Ruoslahti E. Annu Rev Cell Dev Biol. 1996;12:697–715. doi: 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
- 86.Mas-Moruno C, Rechenmacher F, Kessler H. Anticancer Agents Med Chem. 2010;10:753–768. doi: 10.2174/187152010794728639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Grossman JH, McNeil SE. Phys Today. 2012;65:38–42. [Google Scholar]
- 88.Ma P, Mumper RJ. J Nanomed Nanotechnol. 2013;4:1000164. doi: 10.4172/2157-7439.1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Feng L, Mumper RJ. Cancer Lett. 2013;334:157–175. doi: 10.1016/j.canlet.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miller AD. J Drug Deliv. 2013;2013:165981. doi: 10.1155/2013/165981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Avanti Polar Lipids Inc. [Aug 4, 2014];product page for MVL5. http://avantilipids.com/index.php?option=com_content&view=article&id=2422&catnumber=890000.
- 92.Pierrat P, Laverny G, Creusat G, Wehrung P, Strub J-M, VanDorsselaer A, Pons F, Zuber G, Lebeau L. Chemistry – A European Journal. 2013;19:2344–2355. doi: 10.1002/chem.201203071. [DOI] [PubMed] [Google Scholar]
- 93.Klein E, Leborgne C, Ciobanu M, Klein J, Frisch B, Pons F, Zuber G, Scherman D, Kichler A, Lebeau L. Biomaterials. 2010;31:4781–4788. doi: 10.1016/j.biomaterials.2010.02.047. [DOI] [PubMed] [Google Scholar]
- 94.Luvino D, Khiati S, Oumzil K, Rocchi P, Camplo M, Barthélémy P. J Control Release. 2013;172:954–961. doi: 10.1016/j.jconrel.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 95.Jin S-E, Kim C-K. J Pharm Pharm Sci. 2012;15:467–482. doi: 10.18433/j3z312. [DOI] [PubMed] [Google Scholar]
- 96.Whitehead KA, Dorkin JR, Vegas AJ, Chang PH, Veiseh O, Matthews J, Fenton OS, Zhang Y, Olejnik KT, Yesilyurt V, Chen D, Barros S, Klebanov B, Novobrantseva T, Langer R, Anderson DG. Nat Commun. 2014;5 doi: 10.1038/ncomms5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Nat Rev Genet. 2014;15:541–555. doi: 10.1038/nrg3763. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.