

Abstract
Phage display is a biotechnique that fuses functional peptides on the outer surface of filamentous phage by inserting DNA encoding the peptides into the genes of its coat proteins. The resultant peptide-displayed phage particles have been widely used as biotemplates for the synthesis of functional hybrid nanomaterials. Here, we describe the bioengineering of M13 filamentous phage to surface-display bone mineral (hydroxyapatite (HAP))-nucleating peptides derived from dentin matrix protein-1 and using the engineered phage as a biotemplate to grow HAP nanocrystals.
Keywords: Phage display, Hydroxyapatite (HAP), Biomineralization, Bone mineral, Dentin matrix protein-1
1 Introduction
Powered by phage display technique, filamentous phage has been widely used as a biotemplate for the synthesis of virus hybrid nanomaterials, such as inorganic nanowires and nanotubes [1 – 5]. The beauty of using phage as biotemplates attributes to their genetically modifiable protein coat and unique linear morphology [6]. When surface-displaying functional peptides (e.g., material-nucleating/ binding peptides), phage particles can specifically nucleate or bind with functional inorganic materials to form virus hybrid nanowires/nanotubes. One particular interest is to use phage as a biotemplate for fabricating novel biomaterials for bone regeneration. Bone is a hierarchically structured composite made of organic collagen fibrils and inorganic hydroxyapatite (HAP) crystals [7]. If filamentous phage particles, which morphologically mimic the collagen fibrils in bone, can surface-display HAP-nucleating peptides, it is possible to nucleate HAP crystals on them to form a bone-mimetic phage–HAP hybrid nanomaterial for bone regeneration.
In this chapter, the synthesis of phage–HAP hybrid nanomaterial was used as an example to show the process of phage-templated synthesis of inorganic materials. Basically, the whole procedure includes three main steps (Fig. 1): (1) genetic fusion of HAP-nucleating peptides (pE: ESQES and pQ: QESQSEQDS) derived from dentin matrix protein-1 (DMP-1) [8] to the outer surface of M13 phage, which forms engineered E phages (displaying pE) and Q phages (displaying pQ), respectively; (2) assembly of E and Q phage particles into phage bundles; and (3) nucleation of HAP nanocrystals on phage bundles by incubating the bundles in a HAP precursor solution.
Fig. 1.
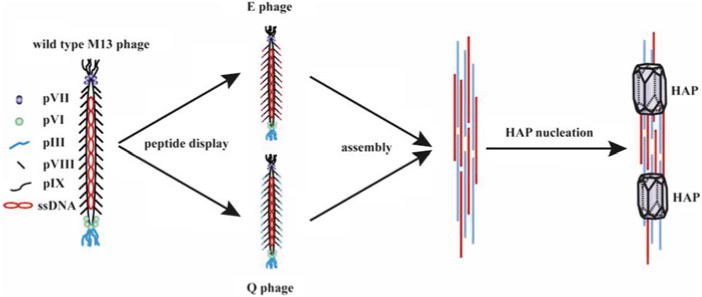
Scheme of phage surface engineering and using engineered phage to grow bone mineral nanocrystals (hydroxyapatite, HAP). Structurally, wild-type M13 phage is composed of coat proteins surrounding a circular ssDNA, which encodes its coat proteins including major coat protein (pVIII) constituting the sidewall and four other structural proteins (pIII, pVI, pVII, and pIX) at two tips (called minor coats). By taking advantage of phage display technique, engineered phage displaying HAP-nucleating peptides (pE: ESQES and pQ: QESQSEQDS) can be produced and assembled into phage bundles driven by the formation of beta-structure between pE and pQ. The nucleation of HAP nanocrystals is induced by the beta-structures formed between pE and pQ within phage bundles
M13 filamentous phage, a nanofiber-like virus (~7 nm wide and 900 nm long) that specifically infects bacteria, is an excellent candidate for phage-templated synthesis (Fig. 1). Structurally, it is composed of coat proteins surrounding a circular single-stranded (ss) DNA, which encodes coat proteins, including major coat protein (pVIII) constituting the sidewall and four other structural proteins (pIII, pVI, pVII, and pIX) at two tips (called minor coats) (Fig. 1) [6]. By inserting DNA encoding foreign peptides into the gene of pVIII, the peptides are themselves displayed on the outer surface of phage along its sidewall (called pVIII display), resulting in bioengineered phage [6]. Peptide display on phage is mostly done by inserting DNA into a phagemid, a vector that can both work as a plasmid and be packaged as ssDNA in phage. In this protocol, we cloned the engineered pVIII gene into a phage expression vector to form recombinant phage vectors. In the production of engineered phages in this method, wild-type (WT) M13 phage (helper phage) is needed to infect the bacteria containing recombinant phage vectors. Engineered pVIII coat proteins, with a foreign peptide (either pE or pQ) fused to the N-terminal end, are expressed in bacteria and assembled along replicated ssDNA to form the main body of new phage particles (pVIII display, Fig. 1). In step 2, the assembly of E and Q phage particles into phage bundles is driven by the formation of beta-structure between pE and pQ (Fig. 1). In step 3, in the presence of HAP precursor solution, the oriented nucleation of HAP nanocrystals is induced by the beta-structures formed from pE and pQ within phage bundles [2] (Fig. 1). Through the three steps, we successfully obtained E and Q phage particles, assembled them into bundle structures, and produced phage–HAP hybrid nanomaterials (Fig. 1). Our results (Fig. 2) show that HAP crystals were nucleated on phage bundles assembled from E and Q phages. The nucleated HAP crystals have preferred orientation with their c-axis along the long axis of phage bundles (Fig. 2 inset), mimicking the feature of c-axis preferred orientation of HAP along collagen fibers in bone.
Fig. 2.

TEM image of HAP crystals nucleated on phage bundles assembled from E and Q phages. The nucleated HAP crystals are preferentially oriented with their c-axis (i.e., [002] direction) along the long axis of phage bundles [2] (reprinted with permission from Biomacromolecules 12, 2193–2199, Copyright 2013 American Chemical Society)
2 Materials
Prepare all solutions and gels using ultrapure water and analytical grade reagents. All the test tubes, flasks, centrifuge tubes, and bottles are sterile or pre-autoclaved before use.
2.1 DNA Gel Electrophoresis System
10× TBE buffer (1 L): Dissolve 108 g Tris and 55 g boric acid in 900 mL of water. Adjust volume to 1 L. Store at room temperature. 10× TBE may take some time to dissolve, even with fast stirring. Dilute 10× buffer to 1× or 0.5× prior to use in electrophoresis.
1 and 1.5 % (w/v) agarose gel: Dissolve 1 g (for 1 %) or 1.5 g (1.5 %) of agarose in 100 mL of 1× TBE buffer. Mark the level of solution on a bottle or flask and heat for about 2.5 min under medium power of microwave. If the solution level has decreased, add water to the correct volume. Pour the gel and allow it to cool.
1 kb and 100 bp DNA ladders (NEB).
Gel Loading Dye, Blue (6×) (NEB) (see Note 1).
Ethidium bromide (EB) staining solution: Dissolve 0.2 g EB in 20 mL of 1× TBE buffer. Mix well, and store at 4 °C in the dark (stock solution is 10 mg/mL concentration). Add 5 μL of 10 mg/mL EB stock solution to 100 mL 1× TBE buffer to make EB staining solution (see Note 2).
Gel electrophoresis apparatus: Horizontal gel chamber with gel tray and well combs.
Power supply.
Razor blades.
2.2 DNA Purifications
2.3 Bacteria Culture, Phage Amplification, and Purification
Escherichia coli (E. coli) strains: XL1-Blue cells harboring phage vector, ER2738 cells (NEB), and TG1 cells (Lucigen).
M13 helper phage solution (NEB).
Luria broth (LB) medium: Mix 20 g of LB powder (Sigma) in 1 L of distilled water. Autoclave for 30–60 min at 121 °C.
Chloramphenicol (35 mg/mL): Dissolve 0.35 g chloramphenicol in 10 mL of 100 % ethanol, aliquot, and store at −20 °C. Use at 1:1,000 dilution in LB broth or LB agar plates.
Tetracycline (20 mg/mL): Dissolve 0.2 g tetracycline in 10 mL of 100 % ethanol, aliquot, and store at −20 °C. Use at 1:1,000 dilution in LB broth or LB agar plates.
Kanamycin (70 mg/mL): Dissolve 0.7 g kanamycin in 10 mL of H2O, filter through 0.22 μm filter, sterilize, aliquot, and store at −20 °C. Use at 1:1,000 dilution in LB broth or LB agar plates.
LB chloramphenicol plates: Mix 35 g of LB agar powder (Sigma) in 1 L of water and heat to boiling while stirring to dissolve all ingredients completely. Autoclave for 30–60 min at 121 °C. Cool the solution to about 56–58 °C in a 56 °C water bath for 1 h. Once the solution is cooled, add 1 mL of 35 mg/mL chloramphenicol. Dispense approximately 20 mL per plate into 100 × 15 mm Petri dishes. Let the plates solidify and then store at 4 °C.
LB chloramphenicol and kanamycin plates: Follow the same procedures as making LB chloramphenicol plates, except adding 1 mL of 70 mg/mL kanamycin along with chloramphenicol.
100 mM CaCl2: Dissolve 1.48 g CaCl2·2H2O in H2O, bring the volume to 100 mL, and autoclave to sterilize. Store at 4 °C.
1 M IPTG: Mix 238 mg of isopropyl-1-thio-β-D-galactopyranoside (IPTG) with 900 μl of H2O, and add H2O to adjust to a final volume of 1 mL. Store at −20 °C.
PEG stock solution (2.5 M NaCl/20 % PEG-8000): Add 146 g of NaCl and 200 g of PEG-8000 to a beaker with 500 mL H2O, stir to mix, and then bring the volume to 1 L (see Note 4).
TBS buffer (1×): Dissolve 8 g of NaCl, 0.2 g of KCl, and 3 g of Tris base in 800 mL of H2O; adjust the pH to 7.4; bring the volume to 1 L by adding H2O; and autoclave to sterilize. Store at room temperature.
Glycerol.
1.5 mL Eppendorf (EP) tubes.
50 mL Conical tubes.
Erlenmeyer flasks: 125 mL and 4 L.
Centrifuge tubes: 40 mL and 1 L.
Benchtop microcentrifuge (Thermo Scientific).
Centrifuge (Beckman Avanti J-E).
2.4 Preparation of Recombinant Vectors and Their Transformation into Competent TG1 Cells
Primers used are listed in Table 1.
10 mM dNTP.
Phusion DNA polymerase (2,000 units/mL, NEB), 5× Phusion GC buffer (NEB).
Restriction endonucleases: Nco I (10,000 units/mL), Hind III (10,000 units/mL), 10× NEB buffer 2 (NEB).
T4 DNA ligase (2,000,000 units/mL, NEB), 10× ligase buffer.
200 μL PCR tubes.
MJ Mini Personal Thermal Cycler (Bio-Rad).
Water bath.
Table 1.
Primers used for preparation of E and Q inserting fragments
2.5 Analysis of the Concentration of Phage Solution and Morphology of Phage Particles
1 % Uranyl acetate (UA) staining solution: Dissolve 0.1 g uranyl acetate powder in 10 mL water and filter through 0.45 μm filter. Store at 4 °C and avoid exposure to light.
UV photometer.
Transmission electron microscope (TEM).
TEM formvar-coated grid (Ted Pella).
2.6 Nucleation of HAP Nanocrystals on Phage Bundles Co-assembled from E and Q Phages
HAP stock solution: Dissolve HAP powder with 100 mM of HCl to reach the final concentration of 50 mM calcium. Store the solution in 4 °C (see Note 7).
HAP-supersaturated solution: Dilute HAP stock solution with 250 mM NaCl solution to reach the final concentration of 200 mM NaCl and 4 mM Ca2+. Carefully adjust the solution pH to 7.01 with 0.05 M KOH. This is the HAP-supersaturated solution. Prepare fresh HAP-supersaturated solution each time.
3 Methods
Bacteria are always cultured in a shaking incubator at 37 °C with a speed of 250 rpm unless otherwise indicated. When using commercial kits, we follow the manufacturer’s recommended protocol unless otherwise stated.
3.1 Linearization of Phage Vectors
Isolate phage vectors using a QIAGEN plasmid miniprep kit as per the manufacturer’s recommended protocol, and elute the isolated vector with 50 μL of water (see Note 8).
Double digest 5 μL of the isolated phage vectors (~200 μg/mL) in 20 μL reaction volume with 1 μL each of Nco I (10,000 units/mL) and Hind III (10,000 units/mL) in a 37 °C water bath for 2.5 h.
Run the digested vector in 1 % TBE agarose gel, purify the digested phage vectors using DNA gel purification kit following the manufacturer’s recommended protocol, and elute the DNA fragments with 20 μL of water (see Note 8).
Run both undigested and digested vectors in another 1 % TBE agarose gel to compare the supercoiled (undigested) and linear (digested) phage vector fragments by gel electrophoresis. Supercoiled vectors are expected to run faster than the linear ones.
Store the linearized phage vectors at −20 °C.
3.2 Preparation of M13 Phage Replicative Form (RF) DNA
Inoculate 5 mL of LB media with 1 μL of ER 2738 bacteria in a test tube (see Note 9).
Add 5 μL of tetracycline solution into the test tube and incubate overnight.
Transfer 20 μL of overnight culture into 5 mL of LB media in a new test tube.
Add 1 μL of M13 helper phage solution into the test tube and incubate for 30 min.
Add 5 μL of kanamycin stock solution and incubate overnight.
Isolate the phage RF DNA using a QIAGEN plasmid miniprep kit following the manufacturer’s recommended protocol and elute the phage RF DNA with 50 μL of water (see Note 8).
Analyze the phage RF DNA by gel electrophoresis (see Note 10).
Store phage RF DNA at −20 °C.
3.3 Preparation of Insert Fragments
Amplify the insert fragments by PCR using isolated RF DNA as template. The PCR reactions were performed in 50 μL volumes containing 1× Phusion GC buffer (NEB), 200 μM of dNTPs, 1 μM each of primer 1 and 2, 1 U of Phusion DNA polymerase (NEB), and 2 μL of RF DNA as template (~300 μg/mL) (see Note 11).
Carry out the amplification reactions in MJ Mini Personal Thermal Cycler (Bio-Rad) with preliminary denaturation at 98 °C for 30 s, followed by 30 cycles of 98 °C for 10 s, 56 °C for 30 s, 72 °C for 15 s, and a final extension at 72 °C for 4 min (see Note 12).
Analyze the PCR products (~200–300 bp) by gel electrophoresis.
Purify the PCR products using a QIAGEN quick PCR purification kit following the manufacturer’s recommended protocol, and elute the purified PCR products with 20 μL of water (see Note 8).
Double digest 10 μL of the purified PCR products in 40 μL reaction volume with 1.5 μL each of Nco I (10,000 units/mL) and Hind III (10,000 units/mL) in a 37 °C water bath for 2.5 h.
Run the digested PCR products in 1 % TBE agarose gel, purify the digested PCR products with DNA gel purification kit following the manufacturer’s recommended protocol, and elute the DNA fragments with 20 μL of water (see Note 8).
Analyze the digested PCR products by gel electrophoresis, and store the gel-purified PCR products at −20 °C.
3.4 Preparation of Competent E. coli TG1 Cells by CaCl2 (See Note 13)
Inoculate 5 mL of LB media with 1 μl of TG1 E. coli cells in a test tube and incubate overnight.
Transfer 20 μL of overnight culture into 20 mL of LB media in a 125-mL sterile flask and incubate until its OD 600 reaches 0.4.
Collect the TG1 bacteria cells by centrifugation at 2,500 × g for 10 min in a 50-mL sterile conical tube.
Discard the supernatant, and gently re-suspend cells in 10 mL of cold sterile CaCl2 solution (100 mM).
Incubate the suspended TG1 cells in CaCl2 solution on ice for 30 min without shaking.
Collect the TG1 cells again by centrifugation at 2,500 × g for 10 min in a 50-mL sterile conical tube.
Discard supernatant, and gently re-suspend TG1 cells in 1 mL of ice-cold sterile 100 mM CaCl2 solution.
Add 200 μL of 50 % glycerol into the 1 mL TG1 competent cell solution and mix well (see Note 14).
Aliquot 60 μL of the glycerol-containing TG1 competent cell solution into 1.5 mL Eppendorf tubes and store at −80 °C (see Note 15).
3.5 Ligation and Transformation of Ligated Recombinant Vector into Competent TG1 Cells
Ligate the digested, cleaned PCR products (3 μL) to digested and cleaned phage vector (1 μL) with T4 DNA ligase in a 20 μL reaction volume containing 1× ligation buffer at room temperature for 10 min (see Notes 16 and 17).
Add 5 μL of ligation products to 60 μL of TG1 competent cells in a 1.5 mL Eppendorf tube and incubate on ice for 1 h.
Allow the cells to be subjected to heat shock in a 42 °C water bath for 90 s.
Add 1 mL of autoclaved LB media immediately into the tube and incubate at 37 °C with shaking (150 rpm) for 1 h.
Spread 100 μL of the transformed TG1 bacteria onto an LB plate with chloramphenicol and kanamycin and incubate overnight at 37 °C without shaking.
Select colonies for screening successful recombinant phage vectors by inoculating several tubes containing 5 mL of LB media with a single TG1 colony picked from the LB plate and incubate overnight.
Use 1 mL of overnight culture for DNA sequencing to verify the successful construction of recombinant phage vector.
Mix the rest of overnight culture (~4 mL) with 800 μL glycerol thoroughly and store at −80 °C (see Note 13).
3.6 Amplification and Purification of Engineered Phage Particles
Inoculate 5 mL of LB media with 1 μL of stored TG1 bacteria harboring recombinant phage vector (from Subheading 3.5) in a test tube.
Add 5 μL of chloramphenicol solution into the tube and incubate overnight.
Transfer 20 μL of overnight culture into 20 mL of LB media in a 125-mL flask.
Add 1 μL of phage solution and incubate for 1 h.
Transfer all 25 mL of bacteria culture to 1 L of LB media in a 4-L flask.
Add 1 mL of kanamycin stock solution and 100 μl of IPTG stock solution to the flask and incubate for 20 h (see Notes 18 and 19).
Collect the supernatant by centrifugation for 40 min at 4,000 × g in a 1-L centrifuge bottle.
Add 150 mL of PEG stock solution to the 1 L of supernatant and mix well.
Precipitate phages by incubating the mixed solution overnight at 4 °C.
Collect the phages by centrifugation for 55 min at 8,200 × g in two 1-L centrifuge bottles.
Re-suspend the phage precipitates with 50 mL of water.
Remove any insoluble fraction by centrifugation for 10 min at 13,000 × g in two 40-mL centrifuge bottles.
Collect supernatant, add 15 mL of PEG stock solution for the second phage precipitation, and mix well.
Incubate the mixed solution overnight at 4 °C.
Collect the phage precipitates by centrifugation for 55 min at 11,000 × g in two 40-mL centrifuge bottles.
Re-suspend the phage precipitates with 1 mL of water (see Note 20).
Remove any insoluble fraction again by centrifugation for 30 min at 13,000 × g in a 1.5 mL Eppendorf tube, and save the supernatant that contains high concentration of engineered phage particles.
Store the phage particles at 4 °C.
3.7 Analysis of the Concentration and Morphology of Phage Particles
Determine the concentration of phage solution by UV adsorption at 269 nm. OD269 = 1 approximately equates to 1 × 1013 pfu/mL [1] (see Note 21).
Negatively stain the phage particles with 1 % UA staining solution. Drop 10 μL of phage solution onto a formvar-coated TEM grid, and wait for 3 min. During this process, phage particles are absorbed to the supporting film. Wick the rest of the phage solution from the edge of the grid by the edge of filter paper. Then apply 10 μL of 1 % UA solution to the specimen for 10 s. Wick the rest of the UA solution by filter paper.
Air-dry the TEM grid and visualize their morphologies under a TEM [9].
3.8 Nucleation of HAP Nanocrystals on Phage Bundles Co-assembled from E and Q Phages
Dilute both E and Q phage solutions to the concentration of 1 × 1013 pfu/mL with water (see Note 22).
Mix each of 750 μL of diluted E and Q phage solution in a 1.5 mL Eppendorf tube and incubate at room temperature for more than 24 h.
Drop 10 μL of the mixed phage solution on a TEM grid, negatively stain the sample with 1 % UA, and visualize the phage bundles under a TEM [9].
Mix 20 μL of the premixed phage solution with 1 mL of supersaturated HAP solution in a 1.5 mL Eppendorf tube.
Allow HAP nanocrystals to nucleate on phage bundles for different time intervals (5, 10, 15, and 20 days) at room temperature.
Collect the phage–HAP complexes by centrifugation for 1 min at 13,000 × g.
Wash the complexes with water twice (1 mL of water each time) by centrifugation and re-suspension.
Re-suspend the complexes in 20 μL of water after washing.
Apply the 20 μl of phage–HAP complex solution on a TEM grid and air-dry it in a hood (see Note 23).
Visualize the phage–HAP complexes from different time points under a TEM.
Acknowledgments
We would like to thank the financial support from National Science Foundation (DMR-0847758, CBET-0854414, CBET-0854465, CMMI-1234957, and CBET-1229309), Department of Defense Peer Reviewed Medical Research Program Discovery Award (W81XWH-12-1-0384), National Institutes of Health (5R01HL092526, 5R21EB009909, 1R21EB015190, 4R03AR056848), Oklahoma Center for the Advancement of Science and Technology (HR11-006), and Oklahoma Center for Adult Stem Cell Research (434003).
Footnotes
Gel loading dye, blue (6×), is a premixed loading buffer with one tracking dye, bromophenol blue, for agarose gel electrophoresis. This solution contains SDS, which often results in sharper bands, as some restriction enzymes are known to remain bound to DNA following cleavage. EDTA is also included to chelate magnesium (up to 10 mM) in enzymatic reactions, thereby stopping the reaction. Bromophenol blue is a standard tracking dye for electrophoresis. It migrates at approximately 300 bp on a standard 1 % TBE agarose gel.
EB solution should be kept in a sealed container in a hood to avoid possible health risks against researchers. EB is known as a mutagen that could affect biological processes of DNA.
Other kits from different manufacturers can also be used.
Incubate the mixture at 60 °C until all solids are dissolved in water and a two-layered solution is formed. Then cool down the solution to 35 °C, and gently agitate the bottle until a homogeneous transparent solution is formed.
Primer 1 should be designed for specific displayed peptide.
Primer 2 is a universal primer for phage engineering by our method.
Use a plastic bottle instead of a glass one and clean it with 1 M HCl and DI water completely before use. In the preparation of HAP-supersaturated solution, glass and any other small contaminates such as small crystals and dusts may induce the nucleation of HAP and lower the concentration of ions in the solution. Also be careful and patient to adjust the pH of HAP solution to 7.01 with a diluted (0.05 M) and filtered (220 nm) KOH solution under agitation. When the pH value is close to 7, add 10 μl of KOH solution each time and wait for 10 min for the complete diffusion of OH−. Any local high concentration of OH− may induce the unwanted nucleation of HAP crystals.
Add water to the center of the membrane in spin column, and wait for 1 min for higher DNA recovery rate.
TG1 or XL1-Blue bacteria can also be used here to prepare M13 phage RF DNA.
The size of isolated RF DNA from wild-type M13 phage is about 8.7 kb. Hence, a band corresponding to 8.7 kb is expected.
Tag DNA polymerase with its buffer can also be used for the PCR.
Increase the number of cycles to 40 if more PCR products are wanted.
XL1-Blue E. coli cells can also be used as competent cells for the production of E or Q phages. Commercially available electrotransfection TG1 competent cells (from Lucigen Inc.) can also be used, but in the transformation step (Subheading 3.6), ligated products should be electrotransfected into TG1 competent cells instead.
Pure glycerol at room temperature is sticky. We find that it is more convenient to mix cultures with 50 % glycerol at a ratio of 1:1 (v/v) for storage.
For convenience purpose, stored competent cells with glycerol at −80 °C can be used for transformation; however, freshly prepared TG1 competent cells will increase the chance of successful transformation of recombinant vectors into cells and is recommended for this protocol.
Use T4 DNA ligase with a high concentration (2,000,000 units/mL, NEB) for a quick ligation (10 min) at room temperature. Avoid a longer reaction time (like 2 h) or a higher temperature (37 °C) for the ligation when a higher concentration of T4 ligase (2,000,000 units/mL, NEB) is used.
Sometimes, transformation fails due to a bad ligation. If it happens, the ratio between linear phage vectors and insert DNA fragments can be adjusted from 1:3 to 1:10 (v/v) in ligation reaction to find the optimal condition.
If the yield of engineered phage is low, increase the incubation time from 20 to 24 h. Sometimes, engineered phage-infected bacteria grow much slower than WT phage-infected ones. If so, a longer incubation time is needed for the amplification of phage particles.
If possible, amplify phage particles in a fermenter with the parameters (pH = 7.4, agitation speed = 800 rpm, bubbled with oxygen gas) as described previously [10] to maximize the phage production yield.
Use of water instead of commonly used TBS buffer for re-suspension is to avoid introducing additional ions in the later nucleation tests.
The concentration of phage solution can also be accurately determined by titering. Prepare serial dilutions (10−2~10−8) of phage solution with TBS/gelatin buffer (0.1 g gelatin in 100 mL TBS buffer). Mix 10 μL of each dilution with 200 μL of mid-log (OD600 = 0.5) TG1 culture in 1.5 mL Eppendorf tubes and incubate for 5 min at room temperature. Spread the culture onto LB plates with kanamycin (70 μg/mL) and incubate overnight. Count the number of colonies on the plates in the following morning, and calculate the titer using the formula titer = N × 100 × dilution factor, where N is the number of the colonies on a plate.
Phage particles may aggregate when stored at 4 °C. Warm up the stocked engineered phage solutions to room temperature and vortex them for 5 min before using or dilution.
Visualize phage–HAP complexes directly under TEM without staining because the acidic UA staining solution (pH < 4) will dissolve nucleated HAP crystals on phage bundles.
References
- 1.Mao CB, Wang FK, Cao BR. Controlling nanostructures of mesoporous silica fibers by supramolecular assembly of genetically modifiable bacteriophages. Angew Chem Int Ed Engl. 2012;51:6411–6415. doi: 10.1002/anie.201107824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu H, Cao BR, George A, Mao CB. Self-assembly and mineralization of genetically modifiable biological nanofibers driven by beta-structure formation. Biomacromolecules. 2011;12:2193–2199. doi: 10.1021/bm200274r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He T, Abbineni G, Cao BR, Mao CB. Nanofibrous bio-inorganic hybrid structures formed through self-assembly and oriented mineralization of genetically engineered phage nanofibers. Small. 2010;6:2230–2235. doi: 10.1002/smll.201001108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mao CB, Flynn CE, Hayhurst A, Sweeney R, Qi JF, Georgiou G, Iverson B, Belcher AM. Viral assembly of oriented quantum dot nanowires. Proc Natl Acad Sci USA. 2003;100:6946–6951. doi: 10.1073/pnas.0832310100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mao CB, Solis DJ, Reiss BD, Kottmann ST, Sweeney RY, Hayhurst A, Georgiou G, Iverson B, Belcher AM. Virus-based toolkit for the directed synthesis of magnetic and semiconducting nanowires. Science. 2004;303:213–217. doi: 10.1126/science.1092740. [DOI] [PubMed] [Google Scholar]
- 6.Smith GP, Petrenko VA. Phage display. Chem Rev. 1997;97:391–410. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- 7.Wang FK, Cao BR, Mao CB. Bacteriophage bundles with prealigned Ca2+ initiate the oriented nucleation and growth of hydroxylapatite. Chem Mater. 2010;22:3630–3636. doi: 10.1021/cm902727s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He G, Dahl T, Veis A, George A. Nucleation of apatite crystals in vitro by self-assembled dentin matrix protein, 1. Nat Mater. 2003;2:552–558. doi: 10.1038/nmat945. [DOI] [PubMed] [Google Scholar]
- 9.Cao B, Xu H, Mao C. Transmission electron microscopy as a tool to image bioinorganic nanohybrids: the case of phage- gold nanocomposites. Microsc Res Tech. 2011;74:627–635. doi: 10.1002/jemt.21030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grieco SH, Lee S, Dunbar WS, MacGillivray RT, Curtis SB. Maximizing filamentous phage yield during computer-controlled fermentation. Bioprocess Biosyst Eng. 2009;32:773–779. doi: 10.1007/s00449-009-0303-3. [DOI] [PubMed] [Google Scholar]