

Abstract
Trauma is the leading cause of death among people under the age of 44. Hemorrhage is a major contributor to deaths related to trauma in the first 48 h. Accordingly, the management of these patients is a time-sensitive and critical affair that anesthesiologists responsible for surgical resuscitation will face. Coagulopathy associated with trauma exists in one-third of all severely injured patients upon presentation to the hospital. Trauma patients presenting with coagulopathy have significantly higher mortality. This trauma-induced coagulopathy (TIC) must be managed adroitly in the resuscitation of these patients. Recent advancements in our understanding of TIC have led to new protocols and therapy guidelines. Anesthesiologists must be aware of these to effectively manage this form of shock. TIC driven by a combination of endogenous biological processes, as well as iatrogenic causes, can ultimately lead to the lethal triad of hypothermia, acidemia, and coagulopathy. Providers should understand how to promptly diagnose TIC and be aware of the early indicators of massive transfusion. The use of common laboratory studies and patient vital signs serve as our current guide, but the importance of each is still under debate. Thromboelastography is a tool used often in the diagnosis of TIC and can be used to guide blood product transfusion. Certain pharmaceutical strategies and non-transfusion strategies also exist, which aid in the management of hemorrhagic shock. Damage control surgery, rewarming, tranexamic acid, and 1:1:1 transfusion protocols are promising methods used to treat the critically wounded. Though protocols have been developed, controversies still exist on the optimal resuscitation strategy.
Keywords: Trauma, Coagulopathy, Hemorrhage, Resuscitation, Shock, Pathogenesis
Introduction
In the 1800s, surgeons John C. Warren and Samuel Gross defined shock as either a “momentary pause in the act of death” or the “rude unhinging of the machinery of life”. Both definitions have stood the test of time and depict the time-sensitive and critical nature of shock. In the United States and worldwide, trauma and associated hemorrhage is a major cause of shock and mortality. In their 2010 review of the global burden of disease, the Institute for Health Metrics and Evaluation ranks injury as the fourth leading cause of death worldwide [1]. Examples such as road injury, self harm, and falls are increasingly contributing to all-cause mortality, while non-communicable causes are decreasing. In their National Vital Statistics Reports from 2010, the Center for Disease Control shows that from ages 1–44, unintentional injuries were the leading cause of death in the United States [2]. As hemorrhage accounts for approximately 40 % of trauma-related deaths, the mechanisms and protocols needed to treat this form of shock become paramount [3]. Massive hemorrhage can be compounded by trauma-induced coagulopathy (TIC) that is present in nearly one-third of all severely injured trauma patients and carries a 50 % mortality rate [4, 5]. In fact, Cohen et al. [6] has shown that as the severity of injury or hypoperfusion increases, the degree of coagulopathy also worsens. The anesthesiologist must be familiar with current pathogenesis models or drivers of TIC as well as treatment guidelines for massive hemorrhage, as the potential for treating these patients is great. Much like Warren and Gross in the 1800s, we must come to a consensus on how we define and diagnose the coagulopathy associated with trauma. This article will review the current ideas on pathogenesis, diagnosis, medical management, and controversies surrounding TIC.
Methods
To accomplish this review, the search terms Trauma Induced Coagulopathy (TIC), Acute Coagulopathy of Trauma (ACOT), Acute Traumatic Coagulopathy (ATC), Idiopathic or Iatrogenic Coagulopathy, and Resuscitation Associated Coagulopathy (RAC) were used in PubMed. Articles from 2010 to present were preferentially cited in an attempt to have the most current information. Special attention was given to prospective studies regarding diagnosis and treatment, as well as large multi-center trials regarding blood transfusion strategies. Clinical practice guidelines from the United States Army Institute of Surgical Research that are updated yearly were used, as they pertain significantly to the patient population experiencing hemorrhagic trauma. National organizations such as the American Society of Anesthesiologists, the European Task Force for Advanced Bleeding, and the Eastern Association of Surgery Trauma were evaluated for recent consensus statements related to massive hemorrhage and trauma.
Classical Triggers
Hemodilution
For many years, the coagulopathy associated with trauma was primarily deemed the culprit of crystalloid volume resuscitation with ensuing acidemia, dilution, and hypothermia. Indeed, the hallmark of Advanced Trauma Life Support (ATLS) that many providers were taught began with the prerequisite two liters of crystalloid upon admission for hypotension. Recent advancements in our understanding have shown that this type of resuscitation is contributory, but is not the primary cause of TIC. Early in hemorrhage, the body’s small store of fibrinogen and platelets can be lost quickly. Coagulation factors can be hemodiluted further with crystalloid volume resuscitation, resulting in lower blood viscosity. Prehospital administration of crystalloid to severely injured patients has been shown to worsen coagulopathy, acidemia, and hypothermia inhibiting thrombin production, a central factor for clot formation. The PROMMT study, a large multicenter endeavor published in 2013, demonstrated that as much as 100 ml of crystalloid administered to severely injured patients was an independent risk factor for coagulopathy measured by an international normalized ratio (INR) greater than 1.3 [7•]. Pre-hospital administration of crystalloid was also shown to be detrimental by Maegele in 2007. His analysis of 8,724 patients in a German trauma registry demonstrated coagulopathy in >40 % of patients with >2,000 ml, in >50 % with >3,000 ml, and in >70 % with >4,000 ml administered [8]. Consequently, providers involved in trauma resuscitation should look to limit crystalloid resuscitation in an effort to minimize dilutional effects.
Hypothermia
Core body temperatures less than 36 °C, common in severely injured patients, is a contributor to trauma-induced coagulopathy. The immediate loss of large volumes of blood and concomitant hypoperfusion impair heat generation by the body. Again, thrombin is affected. At temperatures less than 36 °C, the production of thrombin is slowed. Stepwise reductions in temperature increase the clotting time and decrease the rate of thrombin production [9]. The effects of hypothermia appear to be exponentially greater when combined with hemodilution. Mitrophanov used computational models to show that when hemodiluted blood was analyzed at 36 °C, the prothrombin time was three times longer [9].
Acidemia and Hypoperfusion
The effects of acidemia on clot formation must also be taken into account. Engström et al. found that by artificially reducing normal human serum pH by adding hydrochloric acid, the rate of maximal clot strength on thromboelastography was slowed. At a pH of 6.8 the clot formation time was 168 % prolonged compared to a normal pH of 7.4 [10]. Serum acidemia is both an effect of crystalloid volume resuscitation and hypoperfusion, and can be difficult to delineate. In the shock state, the body has inadequate perfusion for normal cellular metabolism. In this oxygen-deprived state, cellular anaerobic metabolism produces lactate and contributes to metabolic acidemia. Normal saline’s supraphysiologic concentration of chloride can cause a hyperchloremic metabolic acidemia that can worsen a trauma patient’s existing acidemia from hypoperfusion. Hypoperfusion alone has significant effects on the coagulation system. Simmons et al. reported in a study of 777 combat trauma patients that the shock state (BD < −6 mEq/L or SBP <90 mmHg) resulted in greater INR upon admission [11]. This result strengthens the assertion that the TIC is greatest in the most severely injured.
Mechanism of Early TIC
Activated Protein C Pathway
A growing amount of evidence is pointing toward early trauma-induced coagulopathy as an endogenous biological process (Fig. 1). The endothelial cell layer lining blood vessels is damaged during trauma. This damage results in increased cellular surface expression of thrombomodulin and Endothelial Protein C Receptor (EPCR), especially in the setting of hypoperfusion or shock [12, 13•]. Thrombomodulin (TM) is a transmembrane glycoprotein on the surface of endothelial cells. Normally, thrombin is formed locally at the cite of injury, but can be released systemically due to overwhelming injury. This systemic release results in the “thrombin switch” where thrombin now acts as an anticoagulant in the following way. TM, EPCR, and thrombin (the thrombin-thrombomodulin complex) binds circulating Protein C, enhancing its activation fivefold to 20-fold [14••]. Activated Protein C (APC) is under investigation as a primary driver of acute traumatic coagulopathy [7•]. APC has profound anticoagulant activity. First, APC cleaves procoagulant Factors V and VIII. Both of these factors are essential cofactors in the production of thrombin and fibrinous clot formation. Secondly, APC inhibits Plasminogen Activator Inhibitor 1 (PAI1) resulting in increased fibrinolysis. Finally, APC acts as an anti-inflammatory molecule reducing leukocyte activation [15]. In the PROMMT study, 1,198 patients meeting the highest trauma criteria were analyzed. Patients with the highest degree of trauma evidenced by Injury Severity Score or the highest degree of hypoperfusion were found to be the most coagulopathic. In these patients, APC was a significant predictor of coagulopathy, with an adjusted odds ratio of 1.038 (p 0.03) [16].
Fig. 1.

Early changes in TIC affect multiple vital functions in the coagulation cascade. Blue normal coagulation; red endothelial glycocalyx disruption coagulopathy; orange activated protein C coagulopathy; green fibrinogen depletion coagulopathy (Color figure online)
Of significance, Cohen et al. [13•] demonstrated through a study of 203 trauma patients that higher circulating APC upon admission to the hospital correlated with increased mortality, transfusion requirements, rates of multi-organ failure and nosocomial infections, and reduced ICU ventilator free days. Cohen’s data of severely injured patients in shock (ISS >25 ± 13, BD −7 ± 1) reflects that coagulopathy after severe injury occurs nearly immediately.
Endothelial Glycocalyx Disruption
Of the current research into trauma-induced coagulopathy, injury to the endothelium is another primary focus. The endothelium can be damaged by multiple factors after trauma; tissue trauma, inflammation, hypoperfusion, and sympathoadrenal activation [17••]. Endothelial damage results in degradation of the endothelial glycocalyx (EG), a 1-μm thick, anti-adhesive and anticoagulant rich layer on the surface of blood vessels [14••]. Endothelial injury also causes degranulation of Weibel-Palade bodies located on the endothelial surface. Combined, autoheparinization and hyperfibrinolysis can occur. Autoheparinization occurs from disruption of the EG due to significant amounts of heparin-like substances contained in this surface layer [18]. Biomarkers for endothelial glycocalyx disruption such as Syndecan-1 were measured by Ostrowski and Johansson in 77 trauma patients. In their survey, 5 % of the patients presenting with coagulopathy had significant autoheparinization measured by thromboelastography. In these patients, syndecan-1 levels were fourfold higher, suggesting a possible mechanistic link between glycocalyx degradation and trauma-induced coagulopathy [19•]. Interestingly, as injury severity increased in patients with high levels of syndecan-1, there was a progressive depletion in protein C, increased soluble thrombomodulin expression, and hyperfibrinolysis [20]. Augmenting the autoheparinization caused by EG disruption is the increase in hyperfibrinolysis caused by Weibel-Palade body degranulation. Weibel-Palade bodies contain tissue plasminogen activator (TPA), Von Willebrand factor antigen, thrombomodulin, and angiopoetin-2 (Ang-2). TPA catalyzes the conversion of plasminogen to plasmin that is responsible for clot breakdown. Ang-2 is a vascular endothelial growth factor (VEGF) that modulates inflammation and increases vascular permeability [21]. Ang-2 levels were studied by Ganter et al. [22] and shown to be predictive of tissue injury severity and worse clinical outcomes. Another study by Ostrowski demonstrated that higher circulating Syndecan-1 and Ang-2 levels were associated with higher transfusion requirements, though mortality was not altered [23].
Fibrinogen Depletion
Fibrinogen is a central factor in the coagulation cascade, responsible for hemostasis as the precursor of fibrin and integral to platelet aggregation [24]. Normal values range between 2 and 4 g/L in the plasma. The effects of fibrinogen depletion early in the course of major trauma are now thought to significantly contribute to TIC. Two independent studies have shown that fibrinogen depletion is probably the first hemostatic abnormality to develop and is an early marker for massive transfusion and mortality [25•, 26]. Fibrinogen is a highly vulnerable coagulation factor owing to its limited stores in the human body. Loss and dysfunction is attributable to hemorrhage, consumption, profibrinolytic activity, acidemia, and hypothermia. In the United States, fibrinogen has not been typically replaced until levels below 0.8 g/L are reached, but the critical level during trauma has not been adequately studied. European guidelines have traditionally used 1.5 g/L as a cutoff [27••]. Rourke et al. [28•] evaluated 517 trauma patients upon arrival to hospital, with an average time from injury to admission of 90 min. They found that upon arrival, fibrinogen levels were below 1.5 g/dL in 14 %, 1 g/L in 5 % and 0.8 g/L in 3 %. Fibrinogen levels were lowest in patients with higher Injury Severity Scores, hypotension, and shock. In addition, patients administered higher volumes of isotonic crystalloid had lower fibrinogen levels. Using rotational thromboelastography (ROTEM), coagulopathy was defined as EXTEM clot amplitude of less than 35 mm at 5 min. Clot amplitude is a marker for maximal clot firmness and indicates the combined effects of fibrin and platelets. In Rourke’s study, coagulopathic patients had 33 % reduction in fibrinogen levels and 58 % reduction when defining coagulopathy as PT >1.2 [28•]. Schlimp retrospectively studied 675 trauma patients and found that in the presence of acidemia (Base Deficit <−6 mmol/L), 81 % had fibrinogen levels <2 g/L and 63 % had levels <1.5 g/L [29]. Taken together, these studies show that fibrinogen depletion is acute and results in coagulopathy, especially in the most severely injured. It should be noted that depletion of fibrinogen early in trauma is a separate process from hyperfibrinolysis, and is a result of both APC inactivation of Plasminogen Activator Inhibitor and Weibel-Palade body release of TPA.
Platelet Dysfunction
Despite its obvious and pivotal role in coagulation, the contribution of early platelet dysfunction in TIC has remained obscure. Several factors have influenced this obscurity. First, platelet dysfunction is not measured by classical plasma based clotting tests. Secondly, light transmittance aggregometry, considered a gold standard for platelet function, is both labor intensive and time consuming [30•]. Finally, the classical “cascade” teaching of the coagulation process leaves out the integral contribution of the platelet surface. In 2001, Hoffman and Monroe proposed a cell-based model of hemostasis. This model depicts coagulation in three phases: initiation, amplification, and propagation. Initiation begins after trauma when newly exposed tissue factor encounters circulating plasma FVIIa. This results in FX and FIX activation and a small amount of thrombin formation. Thrombin is potent activator of platelets. Subsequently, the surface of platelets works to facilitate thrombin generation and accumulate activated cofactors that can then amplify the procoagulant process [31]. During the amplification phase of coagulation, platelets are uniquely designed to adhere to damaged vascular sites, thereby bringing more activated cofactors into proximity of the vascular damage. Finally, in the propagation phase, the platelet now adhered to the damaged endovasculature, acts as a foundation for thrombin conversion of fibrinogen and eventual strengthening of the hemostatic clot. Kornblith et al. [32] used viscoelastic testing to determine contributions of fibrinogen and platelets to maximum clot firmness in 251 trauma patients (median ISS 9, BD −3.1, INR 1.1). In this cohort, platelets contributed 69.5 % and fibrinogen 30.5 % to clot strength.
To elucidate the unrecognized role of platelet dysfunction in TIC, Wohlauer et al. [30•] performed a prospective observational multicenter study using TEG and platelet mapping in 51 trauma patients. In this study, patients had a mean ISS of 19, BD −6.4 mEq/L, platelet count of 232 and INR of 1.14. They observed that platelets were inhibited 86.1 % in trauma patients, compared to 4.2 % in healthy volunteers using ADP as an activator [30•]. Using base deficit as a surrogate for shock and hypoperfusion, those with a greater base deficit also had worse platelet inhibition. The exact mechanism for platelet dysfunction in early trauma is not known, but extensive ADP metabolism secondary to endothelial cell injury may play in a role. Also, “platelet exhaustion” because of overwhelming immediate activation could render platelets unresponsive to ongoing hemorrhage. This prolonged refractory state would explain why platelet counts often remain in a normal range despite being dysfunctional.
The clinical importance of platelet inhibition after trauma is significant. Kutcher et al. [33•] found that 45.5 % of critically injured patients had “platelet hypofunction” upon admission and that 91.1 % would have dysfunction during their ICU stay. This is despite normal platelet counts with a mean of 257. Solomon when accounting for injury severity found similar data, augmenting the notion that platelet dysfunction is very common in early trauma [34]. Twenty-four-hour and in-hospital mortality was significantly higher in both Kutcher and Solomon’s patient group with platelet hypofunction. Kutcher’s data showed a nearly tenfold increase in 24-h mortality with platelet hypofunction (20 vs. 2.1 %) [33•]. Intensive care ventilator days and multiorgan failure rates were slightly higher in patients with platelet hypofunction.
Diagnostic Laboratory Evaluation
Functionally defining TIC based upon standard laboratory evaluation has many limitations. First, prehospital care of the trauma patient is not always consistent. Many studies will exclude patients transfused large volumes of crystalloid prior to admission based on its hemodilution effects, yet this may introduce a bias of excluding the most severely injured. Secondly, laboratory equipment used to diagnose TIC is not standard. The availability and use of Thromboelastography (TEG, Haemonetics Corporation, USA) vs. Rotational Thromboelastography (ROTEM, Pentapharm, Germany) is not consistent throughout. Next, turnaround times for certain types of laboratory evaluations often preclude usage in the setting of massive hemorrhage. In a study of 325 trauma patients, the median time for laboratory PT results to be available to clinicians in the electronic medical record was 78 min (62–103) [35•]. Finally, discrepancies between point-of-care (POC) testing and laboratory testing can introduce false negatives for TIC. Both low patient hematocrits and the temperature of the POC testing device can affect accuracy of POC testing [35•].
Classical Tests
In 2010, Frith et al. [36] performed a retrospective analysis of several major trauma centers in Europe and developed a rodent model to assess the effects of trauma and hemorrhage on coagulation. Their study revealed that prothrombin time (PT) and activated partial thromboplastin time (aPTT) significantly increased only when both shock and hemorrhage were present. To functionally define TIC based on these parameters, they suggested using a PT ratio >1.2. At this threshold, patients had a significantly higher mortality and transfusion requirement than patients with a normal PT ratio. Also, a PT ratio>1.2 captured 17 % more patients requiring greater transfusions and whom had a higher mortality versus using PT ratio>1.5. As a predictive marker, PT ratio>1.2 detected 42.9 % of patients requiring massive transfusion and had a negative predictive value of 98.4 % [35•].
Limitations
More recent evidence, however, suggests that classical tests for coagulation, such as INR, PT, and PTT, are not reliable in the setting of TIC and fail to accurately describe the complex processes taking place in the acutely traumatized patient [37•]. Because these classical tests are done on platelet-poor centrifuged plasma samples and do not actually assess the strength of the hemostatic clot, the incidence of false negatives is increased. These tests are also unable to determine the nature of the coagulopathy, whether that is depleted fibrinogen, impaired platelet function, or increased fibrinolysis. While assays for fibrinogen and platelets offer quantitative values, they do not offer qualitative or functional values.
Basics of Thromboelastography
Viscoelastic Tests
Due to the our current understanding of TIC and its global effects on the coagulation cascade, the use of viscoelastic laboratory tests have become more popular. Viscoelastic tests such as TEG or ROTEM use whole blood and provide measurements of the entire clotting mechanism. (Table 1; Fig. 2) By providing information on clot development, stabilization, and dissolution, viscoelastic tests reflect in vivo homeostasis [38]. Studies by Jeger and Doran demonstrated differences in sensitivity between classical coagulation tests and TEG [39, 40]. Though small in statistical power, Doran’s data revealed that thromboelastometry identified coagulopathy in 64 % of combat casualties versus only 10 % identification using abnormal classical tests (PT >18 s) [39]. Subsequently, Cotton et al. [40] compared rapid TEG (rTEG) results to classical coagulation tests (CCT) in a prospective study of 583 patients. Rapid TEG differs from standard thromboelastography by adding tissue factor and kaolin to accelerate the clotting mechanism. This produces clinical results approximately 10 min faster than standard TEG. In Cotton’s study, early rTEG (R and K times) results were available in 5 min and were graphically displayed on a monitor. Later rTEG results (MA) were available in 15 min. [41•] Activated clotting time (the r-time when using rTEG) and k-time had strong clinical correlation with PT and aPTT. The alpha angle and MA correlated well with PT, aPTT, and platelet count. The rTEG activated clotting time had prognostic value in determining massive transfusion needs in the first 6 h post trauma [41•].
Table 1.
Viscoelastic tests versus classical clotting tests
| Coagulation | Fibrinolysis | ||||
|---|---|---|---|---|---|
| TEG | r-time | k-time | α angle | MA | Ly30, EPL |
| ROTEM | CT | CFT | α angle | MCF | Li30 ML |
| Viscoelastic measurement | Clotting time-coagulation factors | Clot kinetics | Clot strength-platelet function | Clot stability and breakdown | |
| Coagulopathy | Indicates factor deficiency | Indicates fibrinogen deficiency | Indicates platelet and fibrinogen dysfunction | Indicates enhanced fibrinolysis | |
| Coagulation | Fibrinolysis | ||
|---|---|---|---|
| CCT | PT, INR. aPTT | Platelet count | D-dimer |
| CCT Measurement | Rate to initial clot formation | Absolute platelet number | Evidence of clot breakdown |
| Coagulopathy | Indicates factor or Fibrinogen deficiency | Unknown | Unknown |
Fig. 2.
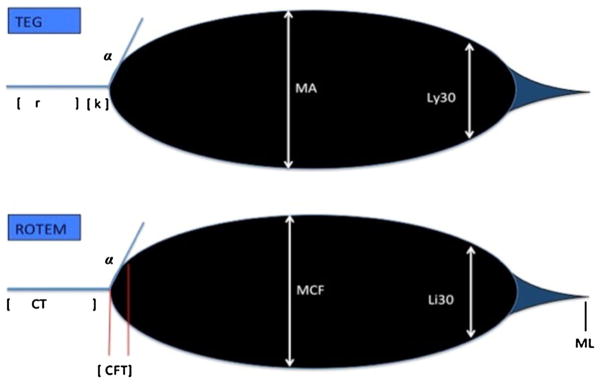
Elastometry representing TEG versus ROTEM terms
Using ROTEM, investigators have determined absolute clot strength is diminished in TIC. Clot strength was diminished by 42 % at 5 min and persisted throughout clot maturation [35•]. In Davenport’s study of 325 trauma patients, TIC produced signature ROTEM tracings showing reduction in clot strength and minimal changes in clotting time. Using clot amplitude at 5 min and a threshold of 35 mm (CA5 <35 mm), ROTEM predicted TIC in 77 % with a false negative rate of 13 % [35•]. Compared to traditional coagulation tests (PT >1.2), ROTEM identified significantly more patients with TIC (18 vs. 8 %). Davenport’s study also showed a strong correlation between CA5 <35 mm and PRBC and FFP administration. Finally, massive transfusion was predicted in 71 % using CA5 <35 mm compared to 43 % using PT based definitions. Adding to the body of evidence supporting viscoelastic testing in TIC, Tauber studied 334 trauma patients with an ISS >15. Tauber’s study demonstrated that RO-TEM accurately detects TIC and was a good predictor of mortality and transfusion. Slowed clot formation speed, reduced clot firmness, and impaired fibrin polymerization occurred at rates of 23, 22, and 30 %, respectively [42]. The body of evidence provided from viscoelastic testing has revealed much about the physical properties of the impaired clotting mechanism in TIC. Table 2 summarizes TIC characteristics. At this time, viscoelastic testing appears superior to CCT in the management of TIC due to its diagnostic sensitivity and ability to guide clinical decisions.
Table 2.
Characteristics of TIC
| Immediate effect |
| Occurs before hemodilution |
| Coagulopathy proportional to injury |
| Hypoperfusion initiates |
| Hypothermia and acidemia augment |
| Clot strength reduced |
| Clot formation minimally delayed |
| Fibrin polymerization impaired |
| Platelet dysfunction augments |
Limitations
Though viscoelastic testing is “poised to replace traditional plasma-based tests in the diagnosis of perturbations in clot formation and lysis characteristic of ATC” [32], it still has many limitations. Traditional TEG as an indicator of whole blood coagulation can be improved upon by modifications that result in faster turnaround times and more specific information as to which component of coagulation is dysfunctional or deficient. TEG assays such as Functional Fibrinogen (FF) to ascertain factor contributions to clot strength are often used in academic studies, but are not routinely available in routine clinical activities. Likewise, rTEG assays for more rapid analysis are not standard to traditional TEG. Another drawback is that analysis software for real-time viewing in the operating room is expensive and often only available in the lab. This limits its usefulness to the in-room provider needing to make decisions in a rapidly changing environment. Finally, POC TEG testing requires daily calibration by trained personnel that is not practical in some hospital settings.
Platelet Function Analysis
A promising alternative to routine platelet counts for the characterization of platelet dysfunction after trauma is impedance aggregometry (IA). This technique has been validated to existing platelet function tests [43] and exists in a point-of-care device for rapid analysis. Per Roche’s website, their Multiplate analyzer is 11 kg and uses a Microsoft Windows interface for viewing results. IA works by measuring impedance across silver-coated copper wires that are resting in whole blood. As certain platelet agonist are added to the whole blood, impedance changes depending on the functional activity of the platelets. Agonists of platelet aggregation that are often used are collagen, adenosine diphosphate (ADP), arachidonic acid (AA), and thrombin-receptor activating peptide (TRAP). Results can be obtained in approximately 10 min and require 300 microliters per test. Kutcher’s characterization of platelet dysfunction after trauma revealed that despite normal admission platelet counts, platelet dysfunction existed in 45.5 % of patients using platelet mapping [33•]. Platelet mapping or impedance aggregometry is a useful tool in the future of TIC diagnosis.
Hemostatic Resuscitation
One of the greatest on-going debates concerning TIC is protocol versus point-of-care guided therapy. The division lines are prominently geographical, with the United States favoring protocol-driven therapy and Europe using POC-guided resuscitation. The driving force behind the argument seems to be the United States experience in Iraq and Afghanistan (Operation Iraqi Freedom [OIF] and Operation Enduring Freedom [OEF]). Indeed, many of the principal investigators touting protocol-driven resuscitation are or were in the military. In fact, the current Army Institute of Surgical Research Clinical Practice Guidelines on Damage Control Resuscitation suggest using 1:1:1 (PRBC: FFP: Platelets) in acutely traumatized soldiers [44].
Protocol Guided Therapy
In the immediate resuscitation phase of trauma, traditional laboratory assessments and radiographs may not be readily available. Therefore, clinical triggers for transfusion were adapted to aide in decision-making. (Table 3) The PROMMT (Prospective Observational Multicenter Major Trauma Transfusion) study group evaluated many triggers and found that INR and base deficit derangements were the most highly predictive of needing massive transfusion [45•]. In OIF and OEF, the US military began using 1:1:1 blood component therapy in all massively transfused soldiers as part of their Damage Control Resuscitation (DCR) strategy. DCR utilizes permissive hypotension, aggressive patient rewarming, crystalloid avoidance, 1:1:1 component therapy, and short operative procedures to minimize hemodilution, acidemia, and hypothermia. Since that time, researchers have elaborated on the beneficial effects that protocol-driven therapy has on mortality and morbidity. In 2011, Holcomb et al. [46] retrospectively reviewed 2,312 patients and determined a survival benefit at 24 h and 30 days in those patients receiving near 1:1:1 therapy. Criticisms to protocol-driven therapy usually revolve around the “survivor bias” agreement. Survivor bias suggests that patients receiving 1:1:1 therapies are simply living long enough to receive 1:1:1 compared to those that did not receive the protocol, and thus have better outcomes. The PROMMT study validated this claim, recognizing that hemorrhagic death often occurred within 2.6 h and patients were not receiving platelets until 2.7 h [47]. This finding, along with the observed improved early survival of 1:1:1 therapy patients, prompted the development of the Pragmatic Randomized Optimal Platelet and Plasma Ratios (PROPPR) study. This study will yield prospective level one data comparing patients at ratios of 1:1:1 and 1:1:2. Drawbacks to protocol-driven therapy include potential waste of blood products from pre-thawed/non-used plasma and unnecessary exposure to blood antigens causing transfusion related lung injury or sepsis [48]. Furthermore, protocol-driven therapy is usually part of an overall damage control resuscitation strategy that requires system level training of personnel [49].
Table 3.
Predicting massive transfusion in early trauma
| INR | Greater than 1.2 |
| Base deficit | Less than −6 mmol/L |
| Systolic blood pressure | Less than 90 mmHg |
| Injury severity score | Greater than 15 |
| Hemoglobin | Less than 11 g/dL |
| FAST exam | Positive for hemorrhage |
| Blood pH | Less than 7.25 |
| Body temperature | Less 35.5 celsius |
| Heart rate | Greater than 120 bpm |
Point-of-Care Guided Therapy
In December 2013, Hallet et al. [50] systematically reviewed randomized control and observational studies to determine if benefit existed for high ratio Platelet: RBC transfusion (another term for 1:1:1 therapy). Their review suggested that insufficient evidence existed to support high ratio therapy in all bleeding patients, though early survival benefit did exist in massively transfused patients. Point-of-care guided therapy would then seem superior to protocol-driven therapy in that only appropriate blood products would be transfused based upon laboratory diagnosis. The European Task Force for Advanced Bleeding Care in Trauma published 2013 guidelines advocating POC therapy. This task force recognized that early intervention improves coagulation tests, reduced the overall need for transfusion of RBC, FFP and platelets, and reduced the incidence of post-traumatic multi-organ failure [27••]. This strategy relies on fast, reliable characterization of TIC using TEG or ROTEM. For each measure of the visco-elastic test, specific component therapy is given to the patient. This may include prothrombin concentrate or recombinant factor VII for prolonged R-times, fibrinogen concentrate for sup-optimal clot amplitude, platelets for reduced maximum amplitude or tranexamic acid for increased fibrinolysis [37•]. Pitfalls to this strategy are that rapid point-of-care devices such as TEG or ROTEM must be immediately available and used upon trauma admission. Rapid assays for each of these machines must be used and are not typically standard on either machine. Inappropriate interpretation of viscoelastic data could result in delayed treatment that is known to be detrimental to patient outcomes [51]. Additionally, some blood components needed for this strategy, such as fibrinogen concentrate, lyophilized plasma, and prothrombin concentrate, are not universally available in North America.
Pharmaceutical Therapy
In 2011, the CRASH-2 trial randomized 20,211 patients to receive tranexamic (TXA) acid within the first 8 h of trauma. TXA competitively inhibits the conversion of plasminogen to plasmin, thereby reducing fibrinolysis [52]. The results showed that TXA 1 gram administered over 10 min in the first 3 h post trauma lowered all-cause mortality. In this study, 1,062 patients died due to bleeding (approximately 35 %), reflecting known estimates of hemorrhagic death due to trauma [53•]. The trial also concluded that late administration of TXA (>3 h post injury) was not beneficial and could possibly cause harm. This observation could not be explained by the trial, and investigators concluded that late administration should be based upon clinical findings of on-going hemorrhage and hypotension.
Current studies are also looking at non-steroidal anti-inflammatory medications (NSAIDs) administered to trauma patients soon after injury. Recognizing inflammation as a possible trigger for TIC, the investigators analyzed the Inflammation and Host Response to Injury Large Scale Collaborative Program data. Patients deemed coagulopathic (INR >1.5 or that received at least 2 units of FFP or 1 unit of platelets within 6 h) were evaluated. Prehospital use of NSAID medications lowered the incidence of TIC by 72 % [54]. The conclusions from this data are retrospective and observational, and need further randomized control trials to validate the significance of NSAIDS as a treatment modality in TIC.
Conclusions
Trauma-induced coagulopathy is an endogenous response by the host that occurs immediately after severe trauma. The severity of trauma correlates with the degree of the coagulopathy. Severely injured patients should be aggressively managed early to reduce the incidence of multi-organ failure and death due to hemorrhage. Early management includes rapid scene to hospital transport, hemorrhage control, avoidance of crystalloid resuscitation, and early recognition of TIC. Standard coagulation tests fail to adequately describe the complex processes occurring in TIC, and thus have limited value. Activation of Protein C as well as autoheparinization following glycocalyx disruption are important instigators of TIC, but fibrinogen depletion and platelet dysfunction must also be taken into account to fully reverse the hemorrhagic process. Current literature supports early diagnosis with point-of-care testing such as TEG or ROTEM. In institutions where this is not feasible, clinical triggers are equally effective in identifying patients needing massive transfusion. Protocol guided therapy with 1:1:1 (PRBC: FFP: Platelets) is a promising strategy for treatment and seems to reduce mortality in this population. Results of the PROPPR trial as well as other on-going large multi-center trials are attempting to elucidate optimal ratios of blood components and should be available to clinicians by the end of 2014. The damage control resuscitation model has been extensively used in wartime and shown to be beneficial in reducing mortality. Existing pharmaceutical treatments such as TXA are now standard of care and should be used early in trauma. As our understanding of TIC grows, hospitals will need to develop and institute standard protocols for diagnosing and treating this early manifestation of trauma.
Footnotes
Conflict of Interest Jeffrey W. Simmons, Jean-Francois Pittet, and Bert Pierce declare that they have no conflict of interest.
Compliance with Ethics Guidelines
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
Contributor Information
Jeffrey W. Simmons, Email: jwsimmons@uabmc.edu, Trauma Section, Medical Simulation, Department of Anesthesiology, University of Alabama at Birmingham, 804 Jefferson Tower, 619 South 19th Street, Birmingham, AL 35249-6810, USA
Jean-Francois Pittet, Email: pittetj@uab.edu, Critical Care Division, Department of Anesthesiology, University of Alabama at Birmingham, 804 Jefferson Tower, 619 South 19th Street, Birmingham, AL 35249-6810, USA.
Bert Pierce, Email: bpierce@uab.edu, Department of Anesthesiology, University of Alabama at Birmingham, 804 Jefferson Tower, 619 South 19th Street, Birmingham, AL 35249-6810, USA.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Murray CJ, et al. GBD 2010: a multi-investigator collaboration for global comparative descriptive epidemiology. Lancet. 2012;380(9859):2055–8. doi: 10.1016/S0140-6736(12)62134-5. [DOI] [PubMed] [Google Scholar]
- 2.Heron M. National Vital Statistics. Deaths: leading Causes for 2010. Natl Vital Stat Syst. 2013;62(6):97. [PubMed] [Google Scholar]
- 3.Hess JR, et al. The coagulopathy of trauma: a review of mechanisms. J Trauma. 2008;65(4):748–54. doi: 10.1097/TA.0b013e3181877a9c. [DOI] [PubMed] [Google Scholar]
- 4.Maegele M. Coagulopathy after traumatic brain injury: incidence, pathogenesis, and treatment options. Transfusion. 2013;53(Suppl 1):28S–37S. doi: 10.1111/trf.12033. [DOI] [PubMed] [Google Scholar]
- 5.MacLeod JB, et al. Early coagulopathy predicts mortality in trauma. J Trauma. 2003;55(1):39–44. doi: 10.1097/01.TA.0000075338.21177.EF. [DOI] [PubMed] [Google Scholar]
- 6.Cohen MJ, West M. Acute traumatic coagulopathy: from endogenous acute coagulopathy to systemic acquired coagulopathy and back. J Trauma. 2011;70(5 Suppl):S47–9. doi: 10.1097/TA.0b013e31821a5c24. [DOI] [PubMed] [Google Scholar]
- 7•.Cohen MJ, et al. Clinical and mechanistic drivers of acute traumatic coagulopathy. J Trauma Acute Care Surg. 2013;75(1 Suppl 1):S40–7. doi: 10.1097/TA.0b013e31828fa43d. Analysis of the PROMMT trial, revealing that TIC occurs predominantly in severely injured patients in shock. Activated Protein C is identified as a mechanistic driver of TIC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maegele M, et al. Early coagulopathy in multiple injury: an analysis from the German Trauma Registry on 8724 patients. Injury. 2007;38(3):298–304. doi: 10.1016/j.injury.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 9.Mitrophanov AY, Rosendaal FR, Reifman J. Computational analysis of the effects of reduced temperature on thrombin generation: the contributions of hypothermia to coagulopathy. Anesth Analg. 2013;117(3):565–74. doi: 10.1213/ANE.0b013e31829c3b22. [DOI] [PubMed] [Google Scholar]
- 10.Engstrom M, et al. Acidosis impairs the coagulation: a thromboelastographic study. J Trauma. 2006;61(3):624–8. doi: 10.1097/01.ta.0000226739.30655.75. [DOI] [PubMed] [Google Scholar]
- 11.Simmons JW, et al. Mechanism of injury affects acute coagulopathy of trauma in combat casualties. J Trauma. 2011;71(1 Suppl):S74–7. doi: 10.1097/TA.0b013e3182218cc1. [DOI] [PubMed] [Google Scholar]
- 12.Brohi K, et al. Acute traumatic coagulopathy: initiated by hypoperfusion: modulated through the protein C pathway? Ann Surg. 2007;245(5):812–8. doi: 10.1097/01.sla.0000256862.79374.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13•.Cohen MJ, et al. Critical role of activated protein C in early coagulopathy and later organ failure, infection and death in trauma patients. Ann Surg. 2012;255(2):379–85. doi: 10.1097/SLA.0b013e318235d9e6. Trauma patients with higher APC levels upon admission had higher mortality, greater propensity towards sepsis, and higher transfusion requirements. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14••.Noel P, Cashen S, Patel B. Trauma-induced coagulopathy: from biology to therapy. Semin Hematol. 2013;50(3):259–69. doi: 10.1053/j.seminhematol.2013.06.009. The biological processes of inflammation, APC, and neurohumeral activation are described as mechanisms in TIC. [DOI] [PubMed] [Google Scholar]
- 15.Anastasiou G, et al. Thrombomodulin as a regulator of the anticoagulant pathway: implication in the development of thrombosis. Blood Coagul Fibrinolysis. 2012;23(1):1–10. doi: 10.1097/MBC.0b013e32834cb271. [DOI] [PubMed] [Google Scholar]
- 16.Rahbar MH, et al. Coordination and management of multicenter clinical studies in trauma: experience from the PRospective Observational Multicenter Major Trauma Transfusion (PROM-MTT) Study. Resuscitation. 2012;83(4):459–64. doi: 10.1016/j.resuscitation.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17••.Maegele M, Schochl H, Cohen MJ. An up-date on the coagulopathy of trauma. Shock. 2013 doi: 10.1097/SHK.0000000000000088. Weibel Palade body degranulation and glycocalyx shedding result in release of TPA and autoheparinization in TIC. [DOI] [PubMed] [Google Scholar]
- 18.Reitsma S, et al. The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. 2007;454(3):345–59. doi: 10.1007/s00424-007-0212-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19•.Ostrowski SR, Johansson PI. Endothelial glycocalyx degradation induces endogenous heparinization in patients with severe injury and early traumatic coagulopathy. J Trauma Acute Care Surg. 2012;73(1):60–6. doi: 10.1097/TA.0b013e31825b5c10. Acute endogenous autoheparinization from glycocalyx disruption contributes to TIC. [DOI] [PubMed] [Google Scholar]
- 20.Johansson PI, et al. A high admission syndecan-1 level, a marker of endothelial glycocalyx degradation, is associated with inflammation, protein C depletion, fibrinolysis, and increased mortality in trauma patients. Ann Surg. 2011;254(2):194–200. doi: 10.1097/SLA.0b013e318226113d. [DOI] [PubMed] [Google Scholar]
- 21.Whitcomb DC, et al. Angiopoietin-2, a regulator of vascular permeability in inflammation, is associated with persistent organ failure in patients with acute pancreatitis from the United States and Germany. Am J Gastroenterol. 2010;105(10):2287–92. doi: 10.1038/ajg.2010.183. [DOI] [PubMed] [Google Scholar]
- 22.Ganter MT, et al. Angiopoietin-2, marker and mediator of endothelial activation with prognostic significance early after trauma? Ann Surg. 2008;247(2):320–6. doi: 10.1097/SLA.0b013e318162d616. [DOI] [PubMed] [Google Scholar]
- 23.Ostrowski SR, et al. High levels of soluble VEGF receptor 1 early after trauma are associated with shock, sympathoadrenal activation, glycocalyx degradation and inflammation in severely injured patients: a prospective study. Scand J Trauma Resusc Emerg Med. 2012;20:27. doi: 10.1186/1757-7241-20-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weisel JW, et al. Examination of the platelet membrane glycoprotein IIb-IIIa complex and its interaction with fibrinogen and other ligands by electron microscopy. J Biol Chem. 1992;267(23):16637–43. [PubMed] [Google Scholar]
- 25•.Chambers LA, Chow SJ, Shaffer LE. Frequency and characteristics of coagulopathy in trauma patients treated with a low- or high-plasma-content massive transfusion protocol. Am J Clin Pathol. 2011;136(3):364–70. doi: 10.1309/AJCPH16YXJEFSHEO. Fibrinogen deficiency occurs very early in the process of TIC and contributes to reduced clot strength. [DOI] [PubMed] [Google Scholar]
- 26.Hiippala ST, Myllyla GJ, Vahtera EM. Hemostatic factors and replacement of major blood loss with plasma-poor red cell concentrates. Anesth Analg. 1995;81(2):360–5. doi: 10.1097/00000539-199508000-00026. [DOI] [PubMed] [Google Scholar]
- 27••.Spahn DR, et al. Management of bleeding and coagulopathy following major trauma: an updated European guideline. Crit Care. 2013;17(2):R76. doi: 10.1186/cc12685. This guideline offers comprehensive management tools for TIC. Mechanisms of TIC are discussed and protocols for management are suggested. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28•.Rourke C, et al. Fibrinogen levels during trauma hemorrhage, response to replacement therapy, and association with patient outcomes. J Thromb Haemost. 2012;10(7):1342–51. doi: 10.1111/j.1538-7836.2012.04752.x. Early fibrinogen depletion is common in TIC and can be measured effectively with ROTEM. [DOI] [PubMed] [Google Scholar]
- 29.Schlimp CJ, et al. Estimation of plasma fibrinogen levels based on hemoglobin, base excess and Injury Severity Score upon emergency room admission. Crit Care. 2013;17(4):R137. doi: 10.1186/cc12816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30•.Wohlauer MV, et al. Early platelet dysfunction: an unrecognized role in the acute coagulopathy of trauma. J Am Coll Surg. 2012;214(5):739–46. doi: 10.1016/j.jamcollsurg.2012.01.050. Platelet dysfunction occurs early in TIC despite normal platelet levels. Early platelet transfusion has a role in trauma coagulopathy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoffman M, Monroe DM., 3rd A cell-based model of hemostasis. Thromb Haemost. 2001;85(6):958–65. [PubMed] [Google Scholar]
- 32.Kornblith LZ, et al. Fibrinogen and platelet contributions to clot formation: implications for trauma resuscitation and thrombo-prophylaxis. J Trauma Acute Care Surg. 2014;76(2):255–63. doi: 10.1097/TA.0000000000000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33•.Kutcher ME, et al. Characterization of platelet dysfunction after trauma. J Trauma Acute Care Surg. 2012;73(1):13–9. doi: 10.1097/TA.0b013e318256deab. Multiple platelet aggregometry identifies platelet dysfunction in TIC despite normal platelet levels. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Solomon C, et al. Platelet function following trauma. A multiple electrode aggregometry study. Thromb Haemost. 2011;106(2):322–30. doi: 10.1160/TH11-03-0175. [DOI] [PubMed] [Google Scholar]
- 35•.Davenport R, et al. Functional definition and characterization of acute traumatic coagulopathy. Crit Care Med. 2011;39(12):2652–8. doi: 10.1097/CCM.0b013e3182281af5. Using ROTEM, TIC can be characterized by a clot amplitude of less than 35 mm at 5 minutes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frith D, et al. Definition and drivers of acute traumatic coagulopathy: clinical and experimental investigations. J Thromb Haemost. 2010;8(9):1919–25. doi: 10.1111/j.1538-7836.2010.03945.x. [DOI] [PubMed] [Google Scholar]
- 37•.Schochl H, et al. Early and individualized goal-directed therapy for trauma-induced coagulopathy. Scand J Trauma Resusc Emerg Med. 2012;20:15. doi: 10.1186/1757-7241-20-15. Viscoelastic measurements in TIC provide time-sensitive information available to guide transfusion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.da Luz LT, Nascimento B, Rizoli S. Thrombelastography (TEG(R)): practical considerations on its clinical use in trauma resuscitation. Scand J Trauma Resusc Emerg Med. 2013;21:29. doi: 10.1186/1757-7241-21-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doran CM, Woolley T, Midwinter MJ. Feasibility of using rotational thromboelastometry to assess coagulation status of combat casualties in a deployed setting. J Trauma. 2010;69(Suppl 1):S40–8. doi: 10.1097/TA.0b013e3181e4257b. [DOI] [PubMed] [Google Scholar]
- 40.Jeger V, Zimmermann H, Exadaktylos AK. Can RapidTEG accelerate the search for coagulopathies in the patient with multiple injuries? J Trauma. 2009;66(4):1253–7. doi: 10.1097/TA.0b013e31819d3caf. [DOI] [PubMed] [Google Scholar]
- 41•.Cotton BA, et al. Rapid thrombelastography delivers real-time results that predict transfusion within 1 hour of admission. J Trauma. 2011;71(2):407–14. doi: 10.1097/TA.0b013e31821e1bf0. discussion 414–7. Rapid TEG results correlate with classical coagulation tests, are rapidly available, and predict transfusion requirements. [DOI] [PubMed] [Google Scholar]
- 42.Tauber H, et al. Prevalence and impact of abnormal RO-TEM(R) assays in severe blunt trauma: results of the ‘Diagnosis and Treatment of Trauma-Induced Coagulopathy (DIA-TRE-TIC) study’. Br J Anaesth. 2011;107(3):378–87. doi: 10.1093/bja/aer158. [DOI] [PubMed] [Google Scholar]
- 43.Siller-Matula JM, et al. Cross validation of the Multiple Electrode Aggregometry. A prospective trial in healthy volunteers. Thromb Haemost. 2009;102(2):397–403. doi: 10.1160/TH08-10-0669. [DOI] [PubMed] [Google Scholar]
- 44.Eastridge B. Damage control resuscitation. San Antonio: United States Army Institute of Surgical Research; 2013. p. 32. [Google Scholar]
- 45•.Callcut RA, et al. Defining when to initiate massive transfusion: a validation study of individual massive transfusion triggers in PROMMTT patients. J Trauma Acute Care Surg. 2013;74(1):59–65. 67–8. doi: 10.1097/TA.0b013e3182788b34. discussion 66–7. Triggers for massive transfusion were identified from the PROMMT study. INR and base deficit were important early clinical triggers for massive transfusion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Holcomb JB, et al. Increased platelet: RBC ratios are associated with improved survival after massive transfusion. J Trauma. 2011;71(2 Suppl 3):S318–28. doi: 10.1097/TA.0b013e318227edbb. [DOI] [PubMed] [Google Scholar]
- 47.Holcomb JB, et al. The prospective, observational, multicenter, major trauma transfusion (PROMMTT) study: comparative effectiveness of a time-varying treatment with competing risks. JAMA Surg. 2013;148(2):127–36. doi: 10.1001/2013.jamasurg.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nascimento B, et al. Clinical review: fresh frozen plasma in massive bleedings—more questions than answers. Crit Care. 2010;14(1):202. doi: 10.1186/cc8205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cotton BA, et al. Damage control resuscitation is associated with a reduction in resuscitation volumes and improvement in survival in 390 damage control laparotomy patients. Ann Surg. 2011;254(4):598–605. doi: 10.1097/SLA.0b013e318230089e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hallet J, et al. The use of higher platelet: RBC transfusion ratio in the acute phase of trauma resuscitation: a systematic review. Crit Care Med. 2013;41(12):2800–11. doi: 10.1097/CCM.0b013e31829a6ecb. [DOI] [PubMed] [Google Scholar]
- 51.Riskin DJ, et al. Massive transfusion protocols: the role of aggressive resuscitation versus product ratio in mortality reduction. J Am Coll Surg. 2009;209(2):198–205. doi: 10.1016/j.jamcollsurg.2009.04.016. [DOI] [PubMed] [Google Scholar]
- 52.McCormack PL. Tranexamic acid: a review of its use in the treatment of hyperfibrinolysis. Drugs. 2012;72(5):585–617. doi: 10.2165/11209070-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 53•.Collaborators C, et al. The importance of early treatment with tranexamic acid in bleeding trauma patients: an exploratory analysis of the CRASH-2 randomised controlled trial. Lancet. 2011;377(9771):1096–101. 1101e1–2. doi: 10.1016/S0140-6736(11)60278-X. Early treatment with TXA (less than 3 hours from trauma) significantly reduced the risk of death due to bleeding. [DOI] [PubMed] [Google Scholar]
- 54.Neal MD, et al. Prehospital use of nonsteroidal anti-inflammatory drugs (NSAIDs) is associated with a reduced incidence of trauma-induced coagulopathy. Ann Surg. 2014 doi: 10.1097/SLA.0000000000000526. [DOI] [PMC free article] [PubMed] [Google Scholar]