

Abstract
The oral cavity harbors a diverse community of microbes that are physiologically unique. Oral microbes that exist in this polymicrobial environment can be pathogenic or beneficial to the host. Numerous oral microbes contribute to the formation of dental caries and periodontitis; however, there is little understanding of the role these microbes play in systemic infections. There is mounting evidence that suggests that oral commensal streptococci are cocolonized with Pseudomonas aeruginosa during cystic fibrosis pulmonary infections and that the presence of these oral streptococci contributes to improved lung function. The goal of this study was to examine the underlying mechanism by which Streptococcus parasanguinis antagonizes pathogenic P. aeruginosa. In this study, we discovered that oral commensal streptococci, including Streptococcus parasanguinis, Streptococcus sanguinis, and Streptococcus gordonii, inhibit the growth of P. aeruginosa and that this inhibition is mediated by the presence of nitrite and the production of hydrogen peroxide (H2O2) by oral streptococci. The requirement of both H2O2 and nitrite for the inhibition of P. aeruginosa is due to the generation of reactive nitrogenous intermediates (RNI), including peroxynitrite. Transposon mutagenesis showed that a P. aeruginosa mutant defective in a putative ABC transporter permease is resistant to both streptococcus/nitrite- and peroxynitrite-mediated killing. Furthermore, S. parasanguinis protects Drosophila melanogaster from killing by P. aeruginosa in a nitrite-dependent manner. Our findings suggest that the combination of nitrite and H2O2 may represent a unique anti-infection strategy by oral streptococci during polymicrobial infections.
INTRODUCTION
The human oral cavity provides residence to more than 700 diverse bacterial species (1). A fraction of the microbial communities that colonize the oral cavity are responsible for localized pathogenic infections, such as dental caries and aggressive periodontitis (1, 2). In contrast, other oral microbes are classified as commensal bacteria (Streptococcus parasanguinis, Streptococcus sanguinis, Streptococcus gordonii, and Streptococcus salivarius) and are involved in facilitating the attachment of pathogenic bacteria to the tooth surface (3) and modulating bacterial dysbiosis (4). Microorganisms unique to the oral cavity have been considered to be mainly involved in localized oral infections; however, there is increasing evidence that suggests that oral microbes may play a more prominent role in systemic infections (5). Oral pathogens and commensals have the ability to disseminate through the bloodstream and cause infective endocarditis (6). Moreover, periodontal pathogens have been associated with cases of atherosclerosis and respiratory tract infections (5, 7).
Current evidence suggests that some oral streptococci reside in locations of the body that are distant to the oral cavity, including the respiratory tracts of cystic fibrosis (CF) patients (8). Historically, Pseudomonas aeruginosa was considered to be the most predominant and clinically important pathogen in CF patients (9, 10). The development of a chronic P. aeruginosa infection in the lungs of CF patients typically correlated with deteriorated lung function and mortality in these patients (11). There are several reasons why P. aeruginosa has become an incredibly successful pathogen in the lungs of CF patients. First, P. aeruginosa is equipped with an assortment of both cell-associated and extracellular virulence factors (12). Second, this bacterium has the ability to alter or redirect central metabolic activities or virulence mechanisms to adapt to stressful environments or acquire nutrients (10, 13–16) and is intrinsically resistant to many antimicrobials (17). All of these adaptation strategies enable P. aeruginosa to establish persistent infections in the lungs of CF patients and thus render these infections difficult to treat. Interestingly, the oral commensal streptococci S. parasanguinis and S. salivarius have been found to be abundant in some CF patients and, remarkably, were associated with increased lung stability (8).
Previous reports have demonstrated that in the oral cavity, hydrogen peroxide (H2O2) production by oral commensal streptococci has an antagonistic effect on the pathogen Streptococcus mutans (18). Since the occurrence of oral streptococci in the lungs of CF patients has been linked to improved lung function, it is possible that the lungs of CF patients provide a unique environmental condition that allows oral streptococci to compete with P. aeruginosa in an H2O2-dependent manner. We aimed to investigate the underlying mechanism that enables oral streptococci to outcompete pathogenic P. aeruginosa under given circumstances, which may explain how the existence of oral streptococci impacts the progression of lung disease in CF patients.
We report that oral streptococci inhibit P. aeruginosa in an H2O2- and nitrite-dependent manner via the production of a reactive nitrogenous intermediate (RNI) such as peroxynitrite. In addition, S. parasanguinis/nitrite-mediated activity protects the host from killing by P. aeruginosa in the Drosophila melanogaster model of infection. Furthermore, transposon mutagenesis showed that a P. aeruginosa mutant defective in a putative ABC transporter permease is resistant to streptococcus/nitrite- and peroxynitrite-mediated killing. These data indicate that RNI production by the presence of oral streptococci and nitrite is the main antibacterial mechanism which could potentially represent an effective strategy utilized by oral commensal streptococci to inhibit the colonization or survival of P. aeruginosa in the lungs of CF patients.
MATERIALS AND METHODS
Bacterial strains, culture conditions, and reagents.
The bacteria used in this study were S. parasanguinis (FW213), S. sanguinis (SK36), S. gordonii (DL1), S. mutans (UA159), S. salivarius (K12), P. aeruginosa (PAO1 and FRD1), and Escherichia coli (TOP10). P. aeruginosa was isolated on Pseudomonas isolation agar (PIA) and subsequently cultured in Luria broth (LB) and incubated at 37°C. E. coli cells were also cultured in LB and incubated at 37°C. All oral streptococci were routinely cultured in Todd-Hewitt broth (THB) and incubated at 37°C with 5% CO2. Filtered-sterilized sodium nitrite was used at a concentration of 0.5 or 1.0 mM. Antibiotics were purchased from Sigma-Aldrich (St. Louis, MO) and used at the following concentrations: 100 μg of ampicillin ml−1 for E. coli; 125 μg of kanamycin ml−1 for S. parasanguinis; 40 and 10 μg of erythromycin ml−1 for E. coli and S. parasanguinis, respectively; and 180 μg of gentamicin ml−1 and 100 μg of carbenicillin ml−1 for P. aeruginosa. Peroxynitrite was purchased from Cayman Chemical Company (Ann Arbor, MI).
Construction of the S. parasanguinis poxL mutant.
To generate poxL mutants of S. parasanguinis, a 2.564-kb fragment containing 500 bp upstream and 500 bp downstream of the poxL coding sequence was PCR amplified from S. parasanguinis cells and ligated into the pGEM-T Easy vector. An 879-bp internal fragment of the poxL coding region was removed using inverse PCR and replaced with the aphA3 gene encoding kanamycin resistance as an XbaI fragment. The resulting plasmid was introduced into S. parasanguinis via electroporation, and potential poxL mutants were isolated as kanamycin-resistant transformants. Replacement of the wild-type poxL gene with the poxL::aphA3 allele was verified by PCR analysis.
Construction of the poxL-complemented strains.
To complement the poxL mutation, the wild-type gene was PCR amplified from S. parasanguinis cells. The resulting fragment was cloned into the SalI and KpnI sites of the streptococcus and E. coli shuttle vector pVPT-gfp. This plasmid was transformed into the poxL mutant. In cis complementation was verified by PCR analysis.
Competition assays on solid medium and in liquid medium.
To examine the interactions between the oral streptococcal species and P. aeruginosa, a 10-μl subculture culture of each streptococcal species was inoculated onto a Todd-Hewitt agar (THA) plate (supplemented with 1 mM filtered-sterilized nitrite) as the early colonizer. After incubation overnight at 37°C with 5% CO2, 10 μl of subcultured FRD1 (chronic P. aeruginosa cystic fibrosis isolate) or PAO1 (acute-phase P. aeruginosa isolate) was inoculated next to the streptococci as the late colonizer. The plate was incubated overnight at 37°C with 5% CO2. Growth inhibition of P. aeruginosa was assessed by the presence of a proximal zone of inhibition at the intersection with the early colonizer.
For competition assays in liquid media, all streptococcal species were grown in THB overnight and 5 μl of cells was subcultured in a Costar 96-well microtiter plate (Corning, Inc., Corning, NY) containing 200 μl of fresh THB (with or without 1 mM nitrite) to an optical density at 470 nm (OD470) of 0.1, followed by the addition of 5 μl of a P. aeruginosa subculture (OD600 = 0.2 to 0.3). The cells were incubated overnight at 37°C in the presence of 5% CO2. P. aeruginosa and oral streptococcal cells were dispersed by vigorous pipetting, serially diluted, and plated on PIA or THA in duplicate; CFU counts were determined the next day.
Hydrogen peroxide and peroxynitrite quantification and peroxynitrite sensitivity assay.
The production of H2O2 by S. parasanguinis was measured using the Amplex red hydrogen peroxide/peroxidase assay kit (Invitrogen, Carlsbad, CA). Briefly, exponential-phase cultures grown in THB (supplemented with sodium nitrite where indicated below) were pelleted and the supernatant was filtered sterilized prior to quantification of H2O2. To measure peroxynitrite, exponential-phase S. parasanguinis cells grown in THB were pelleted and resuspended in M9 medium. Five microliters of the cell suspension was added to 200 μl of M9 medium supplemented with 1 mM sucrose with or without 1 mM nitrite, followed by the addition of 100 μM 2′,7′-dichlorodihydrofluorescein diacetate (Sigma-Aldrich) and incubation at 37°C. Fluorescence intensity exhibited by peroxynitrite was measured using a BioTek plate reader. To test the effect of peroxynitrite on cell viability, bacterial cultures were grown to an OD600 of 0.4, exposed to 250 μM peroxynitrite for 20 min, and plated on PIA to calculate CFU.
Transposon mutagenesis of P. aeruginosa.
A transposon insertion library was constructed in the P. aeruginosa chronic strain FRD1. FRD1 was mutagenized using a Tngfp transposon carried on plasmid pAG408 (19), which was introduced into P. aeruginosa via triparental mating. The conjugation mixtures were plated onto PIA containing gentamicin to select for P. aeruginosa containing transposon insertions. Transposon mutants were isolated as gentamicin-resistant, carbenicillin-sensitive colonies and subsequently pooled and screened for resistance to killing by oral streptococci.
Construction of the P. aeruginosa PA3252 mutant and complemented strains.
To generate mutants of PA3252, a DNA sequence containing approximately 500 bp upstream and 500 bp downstream of the PA3252 coding sequence was PCR amplified from FRD1 cells and cloned into the SmaI site of pBluescript K(+). An internal 800-bp fragment of the PA3252 coding sequence was removed and replaced with the aacC1 gene encoding gentamicin resistance as a SmaI fragment (20). This was followed by introduction of an origin of transfer (moriT) of RP4 on a 230-bp HindIII fragment (21). The resulting plasmid was introduced into P. aeruginosa strain FRD1 by triparental mating, and potential PA3252 mutants were isolated as gentamicin-resistant, carbenicillin-sensitive colonies, indicating a double-crossover event. Replacement of the wild-type PA3252 gene with the PA3252::aacC1 allele was verified by PCR analysis. To complement the PA3252 mutant, full-length PA3252 was PCR purified from FRD1 cells and cloned into the SmaI site of pBluescript K(+). The resulting plasmid was converted to a mobilizable plasmid via the addition of a moriT in the HindIII site and then introduced into P. aeruginosa through triparental mating.
S. parasanguinis and P. aeruginosa oral infection of Drosophila melanogaster.
Drosophila melanogaster flies were maintained on Jazz-Mix Drosophila food (Fisher, Pittsburgh, PA). To orally infect Drosophila, wild-type S. parasanguinis and the poxL mutant were grown to an OD470 of 2.0 and 1.5 ml of the culture was centrifuged at 5,000 × g for 10 min to pellet cells. The bacterial pellet was resuspended in 100 μl of sterile 5% sucrose (with or without 0.5 mM nitrite). The resuspended cells were spotted onto a sterile 21-mm filter (Whatman) that was placed on the surface of 5 ml of solidified 5% sucrose agar in a plastic vial (FlyBase). The filters were allowed to dry at room temperature for ∼30 min before addition of Drosophila. To ensure maximum feeding on the discs containing bacteria, male Canton S flies (1 to 3 days old) were starved for 3 h before being added to vials (10 flies per vial). Flies were allowed to feed on wild-type S. parasanguinis and the poxL mutant 24 h prior to being infected with P. aeruginosa strain PAO1, which was prepared in the same manner as described above. Flies were anesthetized using CO2 throughout the sorting and transferring process. The number of live flies to start the experiment was documented, and live flies were counted at 24-h intervals.
RESULTS
Nitrite is required for the killing of P. aeruginosa by oral streptococci.
To examine the competitive interactions that exist between oral streptococci and P. aeruginosa, we employed a variety of oral streptococci, including S. parasanguinis FW213, S. sanguinis SK36, S. gordonii DL1, S. mutans UA159, and S. salivarius K12, and two P. aeruginosa strains, an acute-phase (PAO1) and chronic (FRD1) isolate. We first evaluated the relationship between these two genera using a plate inhibition assay. As shown in Fig. 1, none of the oral streptococci had an effect on either PAO1 or FRD1 growth when plated on THA. Interestingly, when THA was supplemented with 1 mM nitrite, oral streptococci, with the exception of S. salivarius and S. mutans, displayed an inhibitory effect on bacterial growth of P. aeruginosa PAO1 and FRD1 (Fig. 1). This inhibition occurred when S. parasanguinis (or other streptococci) was placed as the early colonizer. THA supplemented with other chemical agents such as nitrate, arginine, or citrate did not support the inhibition of P. aeruginosa by S. parasanguinis (see Fig. SA1 in the supplemental material); therefore, the inhibition displayed in the plate assay was nitrite specific.
FIG 1.
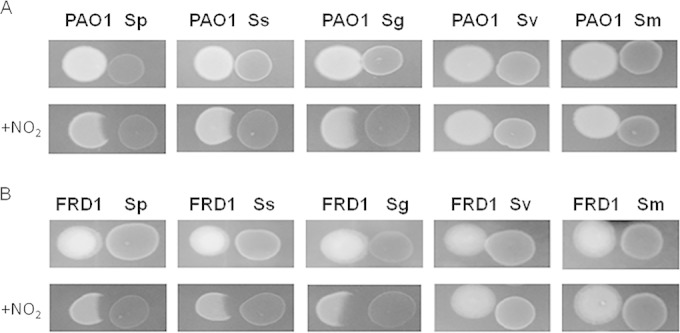
Nitrite facilitates the inhibition of P. aeruginosa by some oral streptococci. Shown is a competition assay of the oral streptococci S. parasanguinis (Sp), S. sanguinis (Ss), S. gordonii (Sg), S. salivarius (Sv), and S. mutans (Sm) with P. aeruginosa on Todd-Hewitt agar (THA) with or without 1 mM nitrite. (A) P. aeruginosa acute strain PAO1; (B) P. aeruginosa chronic strain FRD1.
H2O2 mediates the killing of P. aeruginosa by S. parasanguinis in the presence of nitrite.
S. salivarius and S. mutans had no effect on the killing of P. aeruginosa and are also non-H2O2-producing streptococci; therefore, we hypothesized that production of H2O2 is important for the killing of P. aeruginosa. To test this hypothesis, we constructed mutants that are defective in production of H2O2 and tested the mutants' ability to kill P. aeruginosa. Pyruvate oxidase is required for the optimal production of H2O2 and is a key virulence determinant in oral streptococci (22).
S. parasanguinis mutants defective in poxL, the gene that encodes pyruvate oxidase, lost the ability to produce H2O2 and concurrently failed to inhibit PAO1 and FRD1 in the presence of nitrite (Fig. 2). Complementation of poxL restored the inhibitory activity (Fig. 2). These data demonstrate that H2O2 production by oral streptococci is required for inhibition of P. aeruginosa in the presence of nitrite.
FIG 2.
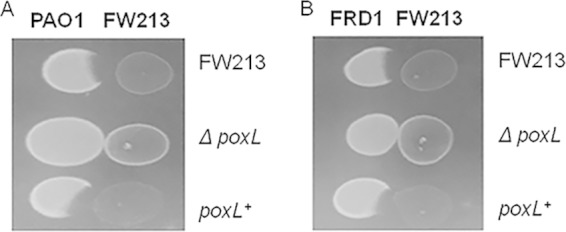
Hydrogen peroxide production is required by S. parasanguinis strain FW213 to inhibit P. aeruginosa in the presence of 1 mM nitrite. (A) Competition assay between PAO1 and S. parasanguinis FW213 (wild-type, ΔpoxL mutant, and poxL-complemented strains). (B) Competition assay between FRD1 and S. parasanguinis FW213 (wild-type, ΔpoxL mutant, and poxL-complemented strains).
To obtain a quantitative measurement of the effects of oral streptococci on P. aeruginosa, we carried out coculture assays in the broth and then determined the CFU counts of both PAO1 and FRD1 obtained from coinfection with oral streptococci in the presence and absence of nitrite. There was an ∼3-log decrease in PAO1 cells cocultured with S. parasanguinis in THB that contained 1 mM nitrite (see Fig. SA2 in the supplemental material). Coculture of PAO1 with the S. parasanguinis ΔpoxL mutant resulted in no inhibition of PAO1 in the presence of nitrite. Interestingly, coculture of PAO1 with the complemented S. parasanguinis ΔpoxL mutant resulted in no detectable PAO1. In addition, there was an ∼4-log decrease in PAO1 survival during coculture with S. sanguinis and S. gordonii. Furthermore, S. mutans and S. salivarius had no effect on PAO1 growth in cultures that contained nitrite (see Fig. SA2). There was a significant decrease in FRD1 survival in cocultures with wild-type S. parasanguinis, the S. paransanguinis poxL-complemented strain, S. sanguinis, or S. gordonii that contained 1 mM nitrite. However, there was an increase in survival of FRD1 during coculture with the S. parasanguinis ΔpoxL mutant (see Fig. SA3 in the supplemental material). These results are consistent with the data obtained from the plate inhibition assays, which further supports the conclusion that H2O2-producing streptococci have an inhibitory effect on P. aeruginosa in the presence of nitrite.
Nitrite enhances peroxynitrite production but not H2O2 in S. parasanguinis cultures.
H2O2 has been shown to have antimicrobial activity at certain concentrations. We questioned whether nitrite enhanced the production of H2O2 by oral streptococci, thereby promoting killing. To determine if 1 mM nitrite had an effect on H2O2 production by oral streptococci, we measured H2O2 production in exponential-phase cultures of S. parasanguinis and derivatives. The addition of nitrite had no significant effect on H2O2 production by oral streptococci (Fig. 3A); as a result, we hypothesized that the combination of H2O2 and nitrite generates a reactive nitrogenous intermediate (RNI), such as peroxynitrite. To test this theory, we measured peroxynitrite production by S. paransanguinis grown in the presence of nitrite. As shown in Fig. 3B, more peroxynitrite was detected by a fluorescent probe in S. parasanguinis cultures grown in nitrite than in cultures that contained no nitrite. In contrast, peroxynitrite generation was abolished in S. parasanguinis cultures defective for H2O2 production. These data suggest that in some polymicrobial infections in which nitrite is present, oral streptococci promote the generation of an RNI, which has an antagonistic effect on neighboring bacteria.
FIG 3.

Nitrite does not increase hydrogen peroxide production by S. parasanguinis FW213 but induces the generation of peroxynitrite in FW213. (A) H2O2 was measured in exponential-phase total cultures grown in THB with or without 1 mM nitrite. (B) Peroxynitrite was measured in exponential-phase cultures grown in M9 medium supplemented with 1 mM sucrose, with or without 1 mM nitrite. NS, not significant. *, P < 0.05 (Student t test).
A PA3252 FRD1 mutant is more resistant to S. parasanguinis/nitrite and peroxynitrite inhibition than wild-type FRD1.
Given the fact that nitrite increased peroxynitrite production in S. parasanguinis cultures, we hypothesized that peroxynitrite may be an intermediate responsible for P. aeruginosa inhibition. Therefore, we sought to determine the mechanism of action of peroxynitrite-mediated killing by screening the P. aeruginosa genome for mutants that confer resistance to killing by streptococcus/nitrite- and pure peroxynitrite-mediated activity. To test this, we mutagenized the FRD1 genome with a transposon and screened for mutants that were resistant to killing by S. parasanguinis. One resistant transposon mutant was isolated and mapped to PA3252, which encodes a probable permease of an ABC transporter. Next, we reconstructed a mutation in PA3252 in FRD1 to determine if a defect in PA3252 truly plays a role in resistance to both S. parasanguinis- and pure peroxynitrite-mediated killing. Indeed, the mutation in PA3252 in FRD1 background was more resistant to S. parasanguinis/nitrite-mediated (Fig. 4A) and pure peroxynitrite-mediated (Fig. 4B) inhibition than wild-type FRD1. In addition, complementation of PA3252 partially restored activity in FRD1 (Fig. 4).
FIG 4.

PA3252 is resistant to FW213 and peroxynitrite killing in the chronic isolate FRD1. (A) PA3252 mutant and FW213; (B) PA3252 resistance to peroxynitrite. Bacteria were exposed to 250 μM peroxynitrite for 20 min. *, P < 0.05 (Student t test).
Primary infection with S. parasanguinis provides protection from P. aeruginosa in the Drosophila melanogaster model of infection.
P. aeruginosa displays significant inhibition when exposed to an established oral streptococcus culture in the presence of nitrite. To determine if this is the case in vivo, we utilized an in vivo Drosophila model, as it has been extensively used in bacterium-host interaction studies and proved to be relevant to human infection (23). We infected Drosophila with wild-type S. parasanguinis and the poxL mutant 24 h prior to coinfection with P. aeruginosa strain PAO1 (0.5 mM nitrite was added in some cases). As shown in Fig. 5A, neither wild-type S. parasanguinis nor the poxL mutant had an effect on P. aeruginosa killing of Drosophila in the absence of nitrite. In contrast, when nitrite was administered, 80% of the Drosophila flies survived coinfection with wild-type S. parasanguinis and P. aeruginosa, compared to a 0% survival rate after 8 days of infection with P. aeruginosa alone (Fig. 5B). Furthermore, Drosophila coinfected with PAO1 and S. parasanguinis defective for H2O2 production had a reduction in survival rates compared to flies coinfected with PAO1 and wild-type S. parasanguinis, again suggesting that both nitrite and H2O2 are critical for the protection of Drosophila in this in vivo model of infection.
FIG 5.
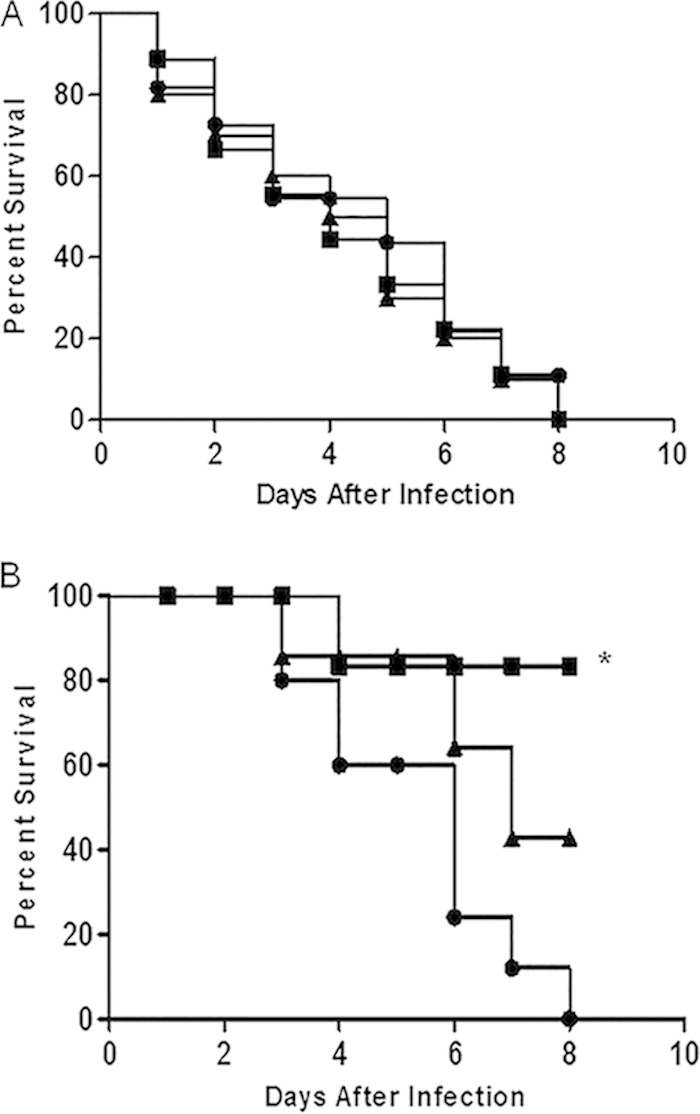
S. parasanguinis and nitrite decrease P. aeruginosa virulence in a Drosophila model of infection. Drosophila flies were infected with S. parasanguinis FW213 wild-type and ΔpoxL mutant strains 24 h prior to P. aeruginosa PAO1 infection without (A) and with (B) supplementation with 0.5 mM nitrite. Curves represent 4 biological replicates. n = 40. *, P < 0.05 (log rank test). Circles, PAO1; squares, PAO1 plus wild-type FW213; triangles, PAO1 plus the ΔpoxL mutant.
DISCUSSION
Historically, the manifestation of a chronic P. aeruginosa infection in the lungs of CF patients correlated with lung deterioration and increased mortality in these patients (11). Recent studies reveal that P. aeruginosa and oral streptococci are two types of organisms that cocolonize the polymicrobial environment constituted by the lungs of CF patients, and the presence of oral streptococci, including S. parasanguinis, offers beneficial outcomes to these patients (8). However, the underlying mechanism by which oral streptococci provide this health benefit is unknown. In this work, we report that oral streptococcal strains that produce H2O2 inhibit P. aeruginosa in a nitrite-dependent manner. More importantly, the combination of hydrogen peroxide-producing oral streptococci and nitrite was required to protect Drosophila melanogaster from killing by P. aeruginosa. These studies suggest a unique anti-infective mechanism by oral streptococci. P. aeruginosa is well known for the ability to resist antibiotics and persist during chronic infections, and our findings suggest that there may be alternative methods to combat P. aeruginosa through the use of oral commensal bacteria.
Elevated levels of nitrite have been observed in the sputum of CF patients (24, 25); therefore, exploiting the abundance of nitrite during CF may be beneficial in clearing recalcitrant pathogens that colonize the lung. The average concentrations of nitrite in sputum have been reported to be between 400 μM and 2 mM (24, 25). Although the prevalence of oral streptococci in CF patients is poorly understood, the ability of oral streptococci to exist in the lungs of these patients and utilize nitrite sources during chronic infection could potentially interfere with P. aeruginosa colonization or virulence. In support of our findings, S. sanguinis isolated from sputum has been shown to exhibit bactericidal activity against 19 nonmucoid and mucoid P. aeruginosa isolates recovered from patients with pulmonary disease, although the bactericidal agent was unknown (26). Other studies have shown that acidified nitrite inhibits P. aeruginosa cells grown under anaerobic conditions (27, 28). However, those particular studies used a very high concentration of nitrite (up to 15 mM; pH 6.5) to demonstrate killing of P. aeruginosa, which may not have physiological relevance in CF cases. In addition, S. mutans and S. salivarius produce copious amounts of lactic acid, which has been shown to permeate the outer membranes of Gram-negative cells (29, 30); however, neither of these acid-producing streptococci was able to significantly inhibit P. aeruginosa in the presence of nitrite in our study, suggesting that acidified nitrite was not the underlying killing mechanism in our study. Furthermore, the benefits conferred by production of H2O2 and the presence of nitrite may occur in the lung airways of CF patients (24). The requirement of nitrite and hydrogen peroxide for the killing suggests that combinatorial therapy may be effective in treating not only P. aeruginosa infections but also other recalcitrant infections.
The mechanism involved in P. aeruginosa inhibition by oral streptococci is not fully understood. In this study, a P. aeruginosa transposon mutant mapped to a permease of an ABC transporter (PA3252) and was resistant to the killing mediated by both S. parasanguinis/nitrite and pure peroxynitrite. Peroxynitrite has previously been shown to exhibit antibacterial activity by diffusing across bacterial cell membranes and inducing the oxidation or nitration of DNA, lipids, and proteins or promoting membrane damage (31, 32). Interestingly, Basic Local Alignment Search Tool (BLAST) analysis of the PA3252 amino acid sequence revealed that PA3252 shows significant homology to genes that encode transporters of polyamines. Polyamines have been demonstrated to decrease outer membrane permeability by inhibiting porin-mediated ion fluxes (33). Moreover, polyamines have also been shown to provide protection against nitrosative stress in Escherichia coli (34). Our working model proposes that mutation of the permease may alter membrane permeability or interfere directly with peroxynitrite transport, thereby rendering a PA3252 mutant more resistant to killing by peroxynitrite (Fig. 6). Alternatively, a mutation in PA3252 could possibly trap intracellular polyamines and enhance polyamine-mediated resistance to RNIs (Fig. 6). It is important to note that peroxynitrite may not be the only RNI produced by streptococcus/nitrite-mediated activity; therefore, we do not exclude the likelihood that other intermediates may play a role in P. aeruginosa inhibition. Furthermore, the precise function of PA3252 in the resistance to killing requires further investigation.
FIG 6.

Model of oral streptococcus- and NO2-mediated inhibition of P. aeruginosa. During polymicrobial infections when nitrite is present, H2O2-producing oral streptococci can induce the production of the antimicrobial peroxynitrite, which may be transported by a P. aeruginosa ABC transporter permease. Loss of the permease results in polyamine-mediated resistance to peroxynitrite or decreased outer membrane permeability of peroxynitrite.
The antimicrobial effects of RNIs like peroxynitrite have been established for multiple pathogens, including E. coli, Bacillus subtilis, Cryptosporidium parvum, Helicobacter pylori, and Rhodococcus equi (35–39); however, our studies reveal a bacterium-mediated production of peroxynitrite, a new avenue that has not been explored before. The current study is not the first instance in which oral streptococci have protected against P. aeruginosa. In the oral cavity, streptococcal communities prevented the integration of P. aeruginosa into salivary biofilms and inhibited growth of P. aeruginosa using a different mechanism (40). It is evident that indigenous flora can employ diverse mechanisms to interfere with the colonization or pathogenesis of incoming pathogens under different environmental conditions, which safeguards healthy homeostasis of the human body and warrants further investigation.
In summary, our study highlights a new interaction between pathogenic P. aeruginosa and oral commensal streptococci and demonstrates how the infiltration of oral commensal bacteria into polymicrobial infection sites can favorably alter the outcomes of polymicrobial diseases that harbor pathogens such as P. aeruginosa. Our working model proposes that modulation of hydrogen peroxide and nitrite within a polymicrobial environment such as the lungs of CF patients might be a valuable defense strategy against the predominant CF pathogen P. aeruginosa. Understanding the strategies these oral communities employ to compete with pathogenic bacteria will provide new insight into the development of therapeutic treatments that are effective in treating polymicrobial infections.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant R01DE017954 (to H.W.). J. A. Scoffield was supported by National Institutes of Health (National Institute of Dental and Craniofacial Research) training grant 1T90DE022736.
We thank Igor Chesnokov, Katarina Akhmetova, and Maxim Balasov for supplying the Drosophila melanogaster Canton S flies.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02396-14.
REFERENCES
- 1.Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner ACR, Yu W-H, Lakshmanan A, Wade WG. 2010. The human oral microbiome. J Bacteriol 192:5002–5017. doi: 10.1128/JB.00542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kreth J, Merritt J, Qi F. 2009. Bacterial and host interactions of oral streptococci. DNA Cell Biol 28:397–403. doi: 10.1089/dna.2009.0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nobbs AH, Lamont RJ, Jenkinson HF. 2009. Streptococcus adherence and colonization. Microbiol Mol Biol Rev 73:407–450. doi: 10.1128/MMBR.00014-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takahashi N. 2005. Microbial ecosystem in the oral cavity: metabolic diversity in an ecological niche and its relationship with oral diseases. Int Congr Ser 1284:103–112. doi: 10.1016/j.ics.2005.06.071. [DOI] [Google Scholar]
- 5.Seymour GJ, Ford PJ, Cullinan MP, Leishman S, Yamazaki K. 2007. Relationship between periodontal infections and systemic disease. Clin Microbiol Infect 4:3–10. [DOI] [PubMed] [Google Scholar]
- 6.Paik S, Senty L, Das S, Noe JC, Munro L, Kitten T, Munro CL. 2005. Identification of virulence determinants for endocarditis in Streptococcus sanguinis by signature-tagged mutagenesis. Infect Immun 73:6064–6074. doi: 10.1128/IAI.73.9.6064-6074.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gomes-Filho IS, Passos JS, Seixas da Cruz S. 2010. Respiratory disease and the role of oral bacteria. J Oral Microbiol 2:1–6. doi: 10.3402/jom.v2i0.5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Filkins LM, Hampton T, Gifford A, Gross M, Hogan D, Sogin M, Morrison H, Pastor B, O'Toole GA. 2012. The prevalence of streptococci and increased polymicrobial diversity associated with cystic fibrosis patient stability. J Bacteriol 194:4709–4717. doi: 10.1128/JB.00566-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilligan P. 1991. Microbiology of airway disease in patients with cystic fibrosis. Clin Microbiol Rev 4:35–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith E, Buckley E, Wu D, Saenphimmachak Z, Hoffman C, D'Argenio L, Miller D, Ramsey S, Speert B. 2006. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A 103:8487–8492. doi: 10.1073/pnas.0602138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bragonzi A, Paroni M, Nonis A, Cramer N, Montanari S, Rejman J, Di Serio C, Döring G, Tümmler B. 2009. Pseudomonas aeruginosa microevolution during cystic fibrosis lung infection establishes clones with adapted virulence. Am J Respir Crit Care Med 180:138–145. doi: 10.1164/rccm.200812-1943OC. [DOI] [PubMed] [Google Scholar]
- 12.Lee DG, Urbach JM, Wu G, Liberati NT, Feinbaum RL, Miyata S, Diggins LT, He J, Saucier M, Déziel E, Friedman L, Li L, Grills G, Montgomery K, Kucherlapati R, Rahme LG, Ausubel FM. 2006. Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol 7:R90. doi: 10.1186/gb-2006-7-10-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hagins JM, Scoffield JA, Suh S-J, Silo-Suh L. 2010. Influence of RpoN on isocitrate lyase activity in Pseudomonas aeruginosa. Microbiology 156:1201–1210. doi: 10.1099/mic.0.033381-0. [DOI] [PubMed] [Google Scholar]
- 14.Hagins J, Scoffield J, Suh SJ, Silo-Suh L. 2011. Malate synthase expression is deregulated in the cystic fibrosis isolate FRD1. Can J Microbiol 195:186–195. [DOI] [PubMed] [Google Scholar]
- 15.Hogardt M, Heesemann J. 2010. Adaptation of Pseudomonas aeruginosa during persistence in the cystic fibrosis lung. Int J Med Microbiol 300:557–562. doi: 10.1016/j.ijmm.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 16.Mena A, Smith EE, Burns JL, Speert DP, Moskowitz SM, Perez JL, Oliver A. 2008. Genetic adaptation of Pseudomonas aeruginosa to the airways of cystic fibrosis patients is catalyzed by hypermutation. J Bacteriol 190:7910–7917. doi: 10.1128/JB.01147-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carmeli Y, Troillet N, Eliopoulos GM, Samore MH. 1999. Emergence of antibiotic-resistant Pseudomonas aeruginosa: comparison of risks associated with different antipseudomonal agents. Antimicrob Agents Chemother 43:1379–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kreth J, Zhang Y, Herzberg MC. 2008. Streptococcal antagonism in oral biofilms: Streptococcus sanguinis and Streptococcus gordonii interference with Streptococcus mutans. J Bacteriol 190:4632–4640. doi: 10.1128/JB.00276-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suarez A, Guttler A, Stratz M, Staendner LH, Timmis KN, Guzman CA. 1997. Green fluorescent protein-based reporter systems for genetic analysis of bacteria including monocopy applications. Gene 196:69–74. doi: 10.1016/S0378-1119(97)00197-2. [DOI] [PubMed] [Google Scholar]
- 20.Schweizer HD. 1993. Small broad-host-range gentamicin resistance gene cassettes for site-specific insertion and deletion mutagenesis. Biotechniques 15:831–833. [PubMed] [Google Scholar]
- 21.Suh SJ, Silo-Suh LA, Ohman DE. 2004. Development of tools for the genetic manipulation of Pseudomonas aeruginosa. J Microbiol Methods 58:203–212. doi: 10.1016/j.mimet.2004.03.018. [DOI] [PubMed] [Google Scholar]
- 22.Chen L, Ge X, Wang X, Patel JR, Xu P. 2012. SpxA1 involved in hydrogen peroxide production, stress tolerance and endocarditis virulence in Streptococcus sanguinis. PLoS One 7:e40034. doi: 10.1371/journal.pone.0040034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Apidianakis Y, Rahme LG. 2009. Drosophila melanogaster as a model host for studying Pseudomonas aeruginosa infection. Nat Protoc 4:1285–1294. doi: 10.1038/nprot.2009.124. [DOI] [PubMed] [Google Scholar]
- 24.Grasemann H, Ioannidis I, Tomkiewicz RP, de Groot H, Rubin BK, Ratjen F. 1998. Nitric oxide metabolites in cystic fibrosis lung disease. Arch Dis Child 78:49–53. doi: 10.1136/adc.78.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Linnane SJ, Keatings VM, Costello CM, Moynihan JB, O'Connor CM, Fitzgerald MX, McLoughlin P. 1998. Total sputum nitrate plus nitrite is raised during acute pulmonary infection in cystic fibrosis. Am J Respir Crit Care Med 158:207–212. [DOI] [PubMed] [Google Scholar]
- 26.Watanabe K, Senba M, Ichinose A, Yamamoto T. 2009. Bactericidal activity in filtrated supernatant of Streptococcus sanguinis against multidrug-resistant Pseudomonas aeruginosa. Tohoku J Exp Med 219:79–84. doi: 10.1620/tjem.219.79. [DOI] [PubMed] [Google Scholar]
- 27.Major TA, Panmanee W, Mortensen JE, Gray LD, Hoglen N, Hassett DJ. 2010. Sodium nitrite-mediated killing of the major cystic fibrosis pathogens Pseudomonas aeruginosa, Staphylococcus aureus, and Burkholderia cepacia under anaerobic planktonic and biofilm conditions. Antimicrob Agents Chemother 54:4671–4677. doi: 10.1128/AAC.00379-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoon SS, Coakley R, Lau GW, Lymar SV, Gaston B, Karabulut AC, Hennigan RF, Hwang S, Buettner G, Schurr MJ, Mortensen JE, Burns JL, Speert D, Boucher RC, Hassett DJ. 2006. Anaerobic killing of mucoid Pseudomonas aeruginosa by acidified nitrite derivatives under cystic fibrosis airway conditions. J Clin Invest 116:436–446. doi: 10.1172/JCI24684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alakomi HL, Skyttä E, Saarela M, Mattila-Sandholm T, Latva-Kala K, Helander IM. 2000. Lactic acid permeabilizes gram-negative bacteria by disrupting the outer membrane. Appl Environ Microbiol 66:2001–2005. doi: 10.1128/AEM.66.5.2001-2005.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dashper SG, Reynolds EC. 1984. Lactic acid excretion by Streptococcus mutans. Microbiology 142:33–39. [DOI] [PubMed] [Google Scholar]
- 31.Pacher L, Beckman JS, Liaudet L. 2007. Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szabó C, Ischiropoulos H, Radi R. 2007. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov 6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 33.Dela Vega A, Delcour AH. 1996. Polyamines decrease Escherichia coli outer membrane permeability. J Bacteriol 178:3715–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bower JM, Mulvey MA. 2006. Polyamine-mediated resistance of uropathogenic Escherichia coli to nitrosative stress. J Bacteriol 188:928–933. doi: 10.1128/JB.188.3.928-933.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Darrah PA, Hondalus MK, Chen Q, Mosser DM, Ischiropoulos H. 2000. Cooperation between reactive oxygen and nitrogen intermediates in killing of Rhodococcus equi by activated macrophages. Infect Immun 68:3587–3593. doi: 10.1128/IAI.68.6.3587-3593.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Genest PC, Setlow B, Melly E, Setlow P. 2002. Killing of spores of Bacillus subtilis by peroxynitrite appears to be caused by membrane damage. Microbiology 148:307–314. [DOI] [PubMed] [Google Scholar]
- 37.Gookin JL, Allen J, Chiang S, Duckett L, Armstrong MU, Carolina N. 2005. Local peroxynitrite formation contributes to early control of Cryptosporidium parvum infection. Infect Immun 73:3929–3936. doi: 10.1128/IAI.73.7.3929-3936.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McLean S, Bowman LAH, Sanguinetti G, Read RC, Poole RK. 2010. Peroxynitrite toxicity in Escherichia coli K12 elicits expression of oxidative stress responses and protein nitration and nitrosylation. J Biol Chem 285:20724–20731. doi: 10.1074/jbc.M109.085506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tecder-Unal M, Can F, Demirbilek M, Karabay G, Tufan H, Arslan H. 2008. The bactericidal and morphological effects of peroxynitrite on Helicobacter pylori. Helicobacter 13:42–48. doi: 10.1111/j.1523-5378.2008.00583.x. [DOI] [PubMed] [Google Scholar]
- 40.He X, Hu W, He J, Guo L, Lux R, Shi W. 2011. Community-based interference against integration of Pseudomonas aeruginosa into human salivary microbial biofilm. Mol Oral Microbiol 26:337–352. doi: 10.1111/j.2041-1014.2011.00622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.