

Abstract
Biofilm formation is the primary virulence factor of Staphylococcus epidermidis. S. epidermidis biofilms preferentially form on abiotic surfaces and may contain multiple matrix components, including proteins such as accumulation-associated protein (Aap). Following proteolytic cleavage of the A domain, which has been shown to enhance binding to host cells, B domain homotypic interactions support cell accumulation and biofilm formation. To further define the contribution of Aap to biofilm formation and infection, we constructed an aap allelic replacement mutant and an icaADBC aap double mutant. When subjected to fluid shear, strains deficient in Aap production produced significantly less biofilm than Aap-positive strains. To examine the in vivo relevance of our findings, we modified our previously described rat jugular catheter model and validated the importance of immunosuppression and the presence of a foreign body to the establishment of infection. The use of our allelic replacement mutants in the model revealed a significant decrease in bacterial recovery from the catheter and the blood in the absence of Aap, regardless of the production of polysaccharide intercellular adhesin (PIA), a well-characterized, robust matrix molecule. Complementation of the aap mutant with full-length Aap (containing the A domain), but not the B domain alone, increased initial attachment to microtiter plates, as did in trans expression of the A domain in adhesion-deficient Staphylococcus carnosus. These results demonstrate Aap contributes to S. epidermidis infection, which may in part be due to A domain-mediated attachment to abiotic surfaces.
INTRODUCTION
Staphylococcus epidermidis is a normal inhabitant of human skin (1). While generally innocuous (2), the species is an opportunistic pathogen with a propensity for forming biofilms on abiotic surfaces. Accordingly, S. epidermidis is responsible for 50 to 70% of catheter-related infections and more than half of central nervous system (CNS) shunt infections, and it is the number one cause of orthopedic implant infections (3–5). Cells within a biofilm are held together via a self-produced matrix and have altered physiology due to nutrient and oxygen gradients, leading to differential gene expression (6, 7). Thus, in addition to the rise in antibiotic resistance in multiple bacterial species worldwide, biofilms confer antimicrobial recalcitrance irrespective of genetic determinants, thereby decreasing treatment options (8, 9).
Biofilm formation requires the initial attachment of cells to a surface and subsequent cell proliferation and accumulation mediated by cell-cell interactions. S. epidermidis biofilm matrix composition varies between strains and may be composed of a combination of polysaccharide (polysaccharide intercellular adhesin; PIA), proteins (e.g., accumulation-associated protein [Aap] and extracellular matrix-binding protein [Embp]), and/or extracellular DNA (eDNA) (2, 10–14). Although these components have unique structural characteristics, PIA-, Embp-, and Aap-dependent biofilms all are resistant to phagocytosis due to their ability to inhibit NF-κB activation and interleukin-1β (IL-1β) production (15, 16). Recently, it also was demonstrated that S. aureus biofilms skew the immune response, generating alternatively activated macrophages (17, 18) that promote a fibrotic rather than bactericidal response, thereby facilitating bacterial persistence. All of these factors, i.e., ineffectiveness of antimicrobial therapy, alteration of host immune responses, and underlying patient health status, complicate treatment, often leading to chronic infections (19, 20). Most often the only treatment option is device removal. In addition to the increase in patient morbidity and medical expense, implant revisions in previously infected individuals are twice as likely to become infected (21–24). Thus, a more thorough understanding of biofilm-mediated infections is critical to improve medical practice and patient outcomes.
While much has been made of PIA-dependent biofilm and the relationship between icaADBC carriage and disease, it is well established that at least one-third of disease-causing isolates lack the icaADBC operon (12, 25). Clearly, additional molecules are capable of supporting biofilms in vivo. Among these, Aap is the most well studied and frequently identified component of PIA-independent, protein-mediated S. epidermidis biofilms (12, 26–43). Aap is a cell wall-anchored protein (41) of approximately 220 kDa; however, the size varies between strains due to differences in the number of B domain repeats (Fig. 1). Several studies have demonstrated that Aap is a rod-like fibril extending from the cell surface (26, 31, 44). Bioinformatics analysis suggests Aap consists of two distinct domains, the A domain and B domain (28, 45). Within the A domain, most strains possess a variable number of 16-amino-acid (aa) repeats located N terminally to a conserved 222-aa L-type lectin domain (38, 45, 46). The C-terminal B domain contains between 3 and 17 repeating units (12). Each of these B repeats is composed of 2 subdomains, G5 and E. The G5 unit is the functional component of the B repeat due to its propensity for self association; thus, it coordinates Aap-dependent intercellular interactions (27, 30). E spacer regions are located between G5 units and likely prevent misfolding of the protein, a potential consequence of its extended fibrillar nature and repetitive domain architecture (44). Following extracellular export directed by the N-terminal signal sequence, Aap is anchored to the cell wall at the C-terminally located LPDTG sortase recognition motif (28, 47).
FIG 1.

Domain architecture of accumulation-associated protein (Aap) varies among S. epidermidis strains. Published Aap sequences were used to create diagrams for strains NCTC 11047 (GenBank accession no. HM587132.1), RP62A (AJ249487.1), 5179 (AY359815.1), and ATCC 12228 (NP_763730.1). Draft genomic sequencing results from S. epidermidis 1457 were used to construct the 1457 Aap diagram (GenBank accession no. KJ920749). Chevrons (><) indicate regions amplified for the construction of 1457 Δaap.
Aap and its S. aureus orthologue, SasG, have been shown to mediate biofilm formation in the absence of PIA. To do so, the A domain must be cleaved, enabling dimerization of B repeats on adjacent bacteria, thereby facilitating bacterial accumulation (27, 30, 31). While the overall processing dynamics of Aap and SasG differ, inhibiting proteolytic cleavage of the A domain prevents biofilm formation in both species (39, 46, 48). Aap and SasG both are able to bind human epithelial cells via their A domains, specifically the conserved globular L-type lectin domain, which has 59% amino acid identity between the two species (35, 38, 45). While no specific ligand for the domain has been identified in either species, antibodies directed against the globular region of the A domain blocked attachment to epithelial cells, suggesting this domain recognizes a cellular receptor. In support of this, ligand binding of other staphylococcal microbial surface components recognizing adhesive molecules (MSCRAMMs) also occurs at domains within their A regions (45). Together, these studies suggest Aap and SasG have dual functions. They also present a scenario where coordination of staphylococcal or host proteases dictate protein function.
Recent structural analysis has enabled the development of models explaining how B domain interactions facilitate cell accumulation. There is strong evidence that Aap G5 units dimerize in a zinc-dependent manner that Conrady et al. refer to as a zinc zipper (30, 31). While there has been substantial progress defining the biology of Aap, an allelic replacement knockout has not been generated to examine the contribution of Aap to biofilm dynamics and its relevance during infection. We constructed an Aap allelic replacement mutant in S. epidermidis strain 1457 (49) and assessed the importance of Aap during in vitro adherence and biofilm formation, as well as in vivo using a rat model of catheter-related infection.
MATERIALS AND METHODS
Aap sequence comparison and analysis.
Aap domain schematics were constructed based on the publically available aap sequences HM587132.1, AJ249487.1, AY359815.1, and NP_763730.1, from strains NCTC 11047 (ATCC 14990), RP62A (ATCC 36984), 5179, and ATCC 12228, respectively (Fig. 1). The aap sequence of strain 1457 (GenBank accession number KJ920749) was determined by draft genome analysis via PacBio DNA sequence generation. DNA sequencing was performed at UC Irvine in the Genomic High-Throughput Facility. Sequence was assembled into one SMRT cell using the RS_HGAP_Assembly.2 (HGAP2) protocol (50) of PacBio's SMRT analysis software, v2.2.0. Default assembly parameters were used with an estimated genome length of 2,560,000 bp. Mean DNA sequence coverage depth across the genome was 105×.
Bacterial strains and culture conditions.
The bacterial strains and plasmids used are listed in Table 1. Escherichia coli was propagated in lysogeny broth (LB) or agar (Becton Dickinson, Franklin Lakes, NJ). Staphylococcal strains were cultured in tryptic soy broth (TSB; Becton Dickinson). If not specified, all chemicals and reagents were purchased from Sigma (St. Louis, MO). When appropriate, antibiotics were used at the following concentrations for E. coli: 50 μg/ml ampicillin (Amp), 500 μg/ml erythromycin (Erm), 10 μg/ml chloramphenicol (Cam), 10 μg/ml tetracycline (Tet), and 10 μg/ml trimethoprim (Tmp). For staphylococci, all antibiotics were used at a concentration of 10 μg/ml. Unless otherwise stated, cultures were grown aerobically (1:10 medium-to-flask ratio; 250 rpm) at 37°C. For Aap Western blot analysis, biofilms were grown in 6-well polystyrene plates (Corning Inc., Corning, NY). Fifty μl of overnight cultures was inoculated into 5 ml of TSB with or without 250 μl α2-macroglobulin (2.5 U/ml; Roche, Mannheim, Germany) and grown statically for 24 h at 37°C.
TABLE 1.
Bacterial strains, bacteriophage, and plasmids used in this study
| Bacterial strain, bacteriophage, or plasmid | Characteristicsa | Source or reference |
|---|---|---|
| Escherichia coli DH5α | Cloning host | Invitrogen |
| Staphylococcus aureus strains | ||
| RN4220 | NCTC 8325-4 derivative, restriction negative, modification positive | 51 |
| RN4220/pGO1-pC221 | RN450 (NCTC 8325-4) containing conjugative (pGO1) and mobilizeable (pC221) plasmids for conjugative mobilization | 52 |
| Staphylococcus epidermidis strains | ||
| 1457 | CVC infection isolate, PIA-positive, Aap-positive | 49 |
| 1457NR | 1457 derivative used for selection following conjugative mobilization, Novr Rifr | This work |
| 1457 Δica | icaADBC allelic replacement mutant, PIA negative, Aap positive, Tmpr | 53 |
| 1457 Δaap | aap allelic replacement mutant, PIA positive, Aap negative, Tetr | This work |
| 1457 Δica Δaap | icaADBC and aap allelic replacement mutant, PIA negative, Aap negative, Tmpr Tetr | This work |
| 1457 Δ ica Δaap pRBaap | PIA negative, Aap negative, contains plasmid carrying full-length aap | This work |
| 1457 Δ ica Δaap pNF110 | PIA negative, Aap negative, contains plasmid carrying icaADBC operon | This work |
| 1457 Δaap pRBaap | PIA negative, Aap negative, contains plasmid carrying full-length aap | This work |
| 1457 Δaap pRBaapDomB | PIA negative, Aap negative, contains plasmid carrying the B domain of aap | This work |
| Staphylococcus carnosus strains | ||
| TM300 | Adhesion negative | 54 |
| TM300 pCNaapDomA | Contains plasmid encoding the A domain of aap | This work |
| TM300 pRBaapDomB | Contains plasmid encoding the B domain of aap | This work |
| Bacteriophage 71 | S. epidermidis transducing phage | 55 |
| Plasmids | ||
| pGEM-T Easy | Cloning vector | Invitrogen |
| pUC19 | Cloning vector containing MCS, Ampr | 56 |
| pCR4 | Cloning vector | Invitrogen |
| pROJ6448 | pE194(ts) with pC221 nick site cloned into the unique ClaI site, Ermr | 52 |
| pGO1 | Conjugative plasmid, Genr Tmpr | 57 |
| pC221 | Mobilizable plasmid, Camr | 52 |
| pCN57 | Staphylococcal expression vector | 58 |
| pRBaap | aap complementation plasmid, Camr | 39 |
| pCRExDomA | pCR4 containing A domain of aap | This work |
| pCRDomA_LPXTG | pCRExDomA with N-terminal region of aap inserted between the AvaI and AscI sites | This work |
| pCNaapDomA | Complementation plasmid containing A domain of aap, Camr | This work |
| pRBaapDomB | Complementation plasmid containing B domain of aap, Camr | 39 |
| pNF110 | icaADBC complementation plasmid, Ermr | 53 |
| pNF137 | Used for amplification of tetM | 59 |
| pNF208 | pGEM with 3′ aap fragment inserted between the SalI and PstI sites | This work |
| pNF210 | pBT2 with 5′ aap fragment inserted between the EcoRI and BamHI sites | This work |
| pNF211 | pUC19 with 3′ aap fragment inserted between the SalI and PstI sites | This work |
| pNF211+ | pNF211 with pROJ6448 inserted at the PstI site | This work |
| pNF219 | pNF211+ with 5′ aap (digested from pNF210) inserted between the EcoRI and BamHI sites | This work |
| pNF220 | pNF219 with tetM inserted at the BamHI site | This work |
Amp, ampicillin; Cam, chloramphenicol; Erm, erythromycin; Gen, gentamicin; Nov, novobiocin; Rif, rifampin; Tmp, trimethoprim; MCS, multiple cloning site.
Construction of allelic replacement mutants and complementation plasmids.
PCRs were performed with Bullseye Taq DNA polymerase (MIDSCI, St. Louis, MO) and chromosomal DNA from S. epidermidis strain 1457 as the template. Primers used are listed in Table 2. Plasmids were transformed into MAX efficiency DH5α competent cells (Invitrogen, Carlsbad, CA). Plasmid DNA was isolated from E. coli using alkaline lysis or a Zyppy plasmid miniprep kit (Zymo Research, Irvine, CA) according to the manufacturer's recommendations. Conjugative mobilization and generation of 1457NR were performed as previously described (60).
TABLE 2.
Primers used in this study
| Primer | Sequencea | Gene |
|---|---|---|
| 1240 | GGG AAA TCG ATC GAT TGT CAG GCT TAA TGG | dhfr |
| 1241 | GGG AAA TCG ATG CAA CTT AGG GAA TGT TTA TGG | dhfr |
| 1249 | ACA GGA ATA ACC TCT TTT CAG TGGG | tetM |
| 1250 | GGA CCT CGA TGT GTT GAT GAA TAAA | tetM |
| 1638 | ATCCTA GGATCC GGT ACA TGA TTA CAG ATAC | tetM |
| 1639 | ATCCTA GGATCC CGA TCT CCT CCT TTC CAC | tetM |
| 1855 | CGGC GAATTC GTA AAA GAA GGT AAC ATT AC | aap |
| 1856 | CGGC GGATCC GTT GTA GGT TCT ATT TTA CTG TC | aap |
| 1857 | CGGC GTCGAC CCA ACA AAA GCA GAA CCA GG | aap |
| 1858 | CGGC CTGCAG CTA ACA CTC TAT CGAC | aap |
| 1866 | CGGC CATATG GGC AAA CGT AGA CAA GGT CC | aap |
| DomA_for | ATT TAA AAT TAA ATA CAT GGG AGGT | aap |
| DomA_rev | GGCGCGCC GAA TTC CCCGGG GAC TGC TTT AGG AGT GTA TGT CA | aap |
| DomA_for2 | AAGCTTC GGTACC ATT TAA AAT TAA ATA CAT GGG AGGT | aap |
| DomA_rev2 | AAGCTTC GGCGCGCC AGT TTT TAT ATG AAA TTA TTT TTC ATT AC | aap |
| Col_for | CCCGGG GGT CCA ACA AAA GCA GAAC | aap |
| LPXTG_rev | GGCGCGCC AGT TTT TAT ATG AAA TTA TTT TTC ATT AC | aap |
Underlined sequences indicate restriction sites.
To construct the aap allelic replacement plasmid, a 728-bp fragment, containing the 3′ region of aap and downstream sequence, was amplified from 1457 genomic DNA using primers 1857 and 1858. The fragment was inserted into pUC19 between the SalI and PstI sites, creating plasmid pNF211. pROJ6448, containing a temperature-sensitive origin of replication and conferring erythromycin resistance (52), was linearized with PstI and then ligated into the PstI site of pNF211, generating pNF211+. A 1,040-bp fragment, containing the 5′ region of aap and upstream sequence, was amplified from 1457 genomic DNA using primers 1855 and 1856. The fragment was inserted into pNF211+ between the EcoRI and BamHI sites, creating pNF219. A 2,383-bp fragment containing the tetM gene and its promoter was amplified from pNF137 (59) with BamHI ends using primers 1638 and 1639. The fragment was inserted in the BamHI site of pNF219, creating the complete allelic replacement plasmid pNF220. The plasmid subsequently was transformed into chemically competent E. coli DH5α and then sequentially electroporated into S. aureus RN4220 (51) competent cells and S. aureus RN4220/pGO1-pC221 (52) competent cells. pNF220 was transferred by conjugative mobilization to S. epidermidis 1457NR and finally transduced into S. epidermidis 1457 (49) using phage 71 (61).
To generate single recombinants, TSB plus Erm was inoculated with a single colony of 1457/pNF220 and incubated at 30°C until exponential phase. The culture was diluted 1:1,000 in fresh TSB without antibiotics and incubated at 43°C for approximately 24 h. After the fourth such dilution, culture broth was streaked for isolation onto tryptic soy agar (TSA) plus Erm and incubated at 45°C. Resultant colonies were patched onto TSA plus Tet and TSA plus Erm and grown at 45°C. Isolates able to grow on both antibiotics were screened by pulsed-field gel electrophoresis (PFGE) and Southern blotting using probes for aap (primers 1856 and 1866) and tetM (primers 1249 and 1250).
To produce double recombinants, sterile saline was inoculated to a McFarland standard of 3.0 from a single recombinant culture. The inoculum was diluted 1:1,000 into TSB without antibiotics and grown overnight at 43°C. The culture was diluted and plated onto TSA plus Tet and incubated at 45°C. Colonies were patched onto TSA plus Tet and TSA plus Erm and incubated at 45°C. Colonies able to grow on TSA plus Tet but not TSA plus Erm were streaked onto TSA plus Tet, TSA plus Erm, and modified mannitol salt agar (53).
Following allelic replacement, the aap mutation was backcrossed into strain 1457 using the transducing phage 71 as described previously (61). Mutant construction was confirmed by PCR, PFGE, and immunoblotting with antibodies to both the A and B domains of Aap (12, 46). Chromosomal integration and plasmid curing were verified by pulsed-field gel electrophoresis and Southern blotting using a previously described methodology (62, 63).
The 1457 icaADBC aap double mutant (1457 Δica Δaap) was generated by transducing the aap mutation into 1457 icaADBC::dhfr (53) using phage 71 and confirmed by PFGE and Southern blot analysis using a dhfr probe. Construction of the plasmids pRBaap and pRBaapDomB, encoding full-length aap and the B domain only, respectively, was described previously (39). For in trans expression of the Aap A domain, the export signal and A domain (nucleotides [nt] 1 to 1827) were amplified using primers DomA_for and DomA_rev. The resulting fragment was cloned into pCR4 (Invitrogen, Carlsbad, CA), producing plasmid pCRExDomA. Nucleotides encoding the collagen triple helix motifs and the LPXTG motif (nt 3973 to 4539) were amplified using primers Col_for and LPXTG_rev. The resulting amplicon was digested using AvaI and AscI and cloned into pCRExDomA. The resulting plasmid, pCRDomA_LPXTG, was used as the template for PCR using primers DomA_for2 and DomA_rev2. The resulting amplicon was digested with KpnI and AscI and ligated into pCN57 (58), producing pCNaapDomA. pCNaapDomA was introduced into S. aureus RN4220 and then into S. epidermidis 1457 Δaap and Staphylococcus carnosus TM300 as previously described (64). All cloning products were verified by sequence analysis. Protein production from Aap complementation plasmids was confirmed by Western blotting (see Fig. S1 in the supplemental material). Strains 1457 Δica Δaap pRBaap and 1457 Δica Δaap pRBaapDomB were generated by phage 71 transduction of pRBaap and pRBaapDomB, respectively, into 1457 Δica Δaap.
Static biofilm assay.
Microtiter plate assays were based on the procedure of Christensen et al., with modifications (65, 66). Overnight planktonic cultures were diluted 1:100 in fresh TSB. Each well of a 96-well plate was inoculated with 200 μl with samples run in triplicate. Plates were incubated statically at 37°C for 24 h. The supernatant was removed, and wells were washed three times with phosphate-buffered saline (PBS). Cells were fixed by drying plates at 45°C for 1 h. Adherent cells were stained with 0.4% crystal violet for 15 min, and excess stain was removed by washing in distilled water. To quantify biofilm formation, absorbance was measured at 595 nm using a Wallac 1420 multilabel counter (PerkinElmer, Waltham, MA). Results presented are from at least 4 independent experiments.
Flow cell biofilms.
S. epidermidis overnight cultures were grown in 5 ml TSB with shaking (200 rpm) at 37°C. Measurements of the optical density at 600 nm (OD600) were taken to ensure cultures grew to similar ODs, and the cultures were diluted 1:100 in sterile PBS. One ml of each diluted culture was injected into flow cell chambers prepared with acid-etched glass coverslips as previously described (67). Bacteria were allowed to attach for 1 h at room temperature before laminar flow was initiated at a rate of 3.75 rpm. Flow cells were grown in 2% TSB and incubated at 37°C for 2 days for PIA-producing strains (1457 and 1457 Δaap) and 4 days for PIA-negative strains (1457 Δica and 1457 Δica Δaap). For imaging, live/dead poststaining was performed using the fluorescent nucleic acid stains Syto9 and ToPro3 (Invitrogen) at the manufacturer's recommended concentrations. Confocal laser scanning microscopy (CLSM) was performed on a Nikon Eclipse E600 microscope using the Radiance 2100 image capturing system (Bio-Rad, Hercules, CA). Images were acquired with Laser Sharp 2000 software (Zeiss, Oberkochen, Germany) and processed using the Volocity program (PerkinElmer, Waltham, MA). Quantitative analysis of biofilms was performed in Matlab with the COMSTAT script (68).
Biofilm harvest, protein preparation, and Western blotting.
Protein isolation procedures are based on previously published methods (38, 69), with modifications. Supernatant was removed from wells, and 2 ml of PBS was added to each well. Cells were removed from wells using cell scrapers (Nalge Nunc, Rochester, NY) and pipetted into 2-ml tubes. Cells were pelleted and washed in PBS twice and then resuspended in 1 ml lysis buffer (50 mM Tris-HCl, 20 mM MgCl2, pH 7.5, with 30% [wt/vol] raffinose, containing 1 mM EDTA, 20 U DNase I [Roche], 4 mM phenylmethylsulfonyl fluoride, 1 mM N-ethylmaleimide, 25 mM aminocaproic acid, lysostaphin [100 μg/ml; AMBI Products, Lawrence, NY], and lysozyme [100 μg/ml]) and incubated at 37°C with shaking for 30 min. Cell walls were separated from protoplasts by centrifugation at 6,000 × g for 20 min. The supernatant (cell wall fraction) was removed and concentrated using 100-kDa-molecular-size cutoff columns (Millipore, Billerica, MA) according to the manufacturer's instructions. Concentrated samples were stored at −20°C.
Proteins were separated on 5% SDS-PAGE gels before transfer to polyvinylidene difluoride (PVDF) membranes. Membranes were blocked with 5% nonfat milk in Tris-buffered saline-Tween. Aap B domain antiserum (12) was diluted 1:100,000 in 5% nonfat milk. Alkaline phosphatase (AP)-conjugated mouse anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA) was diluted 1:25,000. Blots were developed with enhanced chemifluorescence (ECF) substrate (GE Healthcare Life Sciences; Piscataway, NJ) and visualized on a Typhoon FLA 7000 (GE Healthcare Life Sciences).
Attachment assays.
Initial attachment was measured by enzyme-linked immunosorbent assay (ELISA) as described by Mack et al. (70). Briefly, bacteria were grown overnight in TSB with antibiotics as appropriate. Overnight cultures were diluted 1:100 in fresh, prewarmed TSB without antibiotics and grown for 6 h at 37°C, 200 rpm. Cells were harvested and resuspended in PBS at a final concentration of 4 × 109 CFU/ml. Serial dilutions were added to wells of a 96-well NunclonΔ polystyrene plate (Nunc, Roskilde, Denmark) and incubated at room temperature for 1 h. Wells then were washed four times with 200 μl PBS plus Tween 20 (0.1%, vol/vol), and adherent cells were fixed with 3.7% (vol/vol) formaldehyde. Wells again were washed with PBS and then blocked with protein-free blocking reagent (Thermo Fisher Scientific, Bonn, Germany). Bacteria were detected using polyclonal rabbit anti-S. epidermidis antiserum (39) and AP-coupled anti-rabbit IgG. After the addition of AP substrate and incubation at 37°C for 45 min, absorbance was measured in an ELISA plate reader (Tecan Infinite) at 405 nm using 492 nm as a reference wavelength. Results presented are from at least 2 independent experiments, each with at least 3 technical replicates.
In vivo rat jugular catheter model.
Animal experimentation was performed as previously described (71) under a University of Nebraska Medical Center-approved Institutional Animal Care and Use Committee (IACUC) protocol for P. D. Fey. The University of Nebraska Medical Center is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AALAC). All animals at the University of Nebraska Medical Center are maintained in accordance with the Animal Welfare Act and the DHHS Guide for the Care and Use of Laboratory Animals (72). Briefly, a catheter segment was inserted into the jugular vein of anesthetized rats. After recovery from surgery, rats were immunosuppressed by intraperitoneal injection of 100 mg/kg of body weight cyclophosphamide (CP), followed by a second injection of 50 mg/ml CP. Twenty-four hours later, rats were inoculated via the tail vein with 109 CFU of S. epidermidis. Three days after infection, rats were sacrificed and catheters, blood, and spleens were harvested and processed to determine bacterial burden. Leukopenia was confirmed by white blood cell count and determination of lymphocyte differentials.
For model validation studies, 28 male Sprague-Dawley rats (Charles River, Portage, MI), weighing approximately 300 g, were divided into four groups: (i) catheter and CP treatment, (ii) catheter only, (iii) CP treatment only, and (iv) no catheter or CP treatment. For virulence studies, 66 male Sprague-Dawley rats (Charles River), weighing approximately 300 g, were inoculated with either wild-type 1457, 1457 Δica, 1457 Δaap, or 1457 Δica Δaap.
Statistical analysis.
A Shapiro-Wilk's test (P > 0.05) verified that absorbance values were approximately normally distributed; therefore, the overall difference in biofilm formation in the static biofilm assay was assessed using one-way analysis of variance (ANOVA) with Dunnett's T3 to correct for multiple comparisons. Comparisons between individual strains were performed by independent-samples t test with equal variances not assumed (P < 0.05 on the basis of the parametric Levene's test). The significance of COMSTAT results was determined by t tests (1457 versus 1457 Δaap and 1457 Δica versus 1457 Δica Δaap), assuming equal variance (Microsoft Excel). Initial attachment binding curves were calculated by regression analysis of the data sets.
In vivo analysis was done on the log10 scale with 1 added (i.e., log10 + 1) prior to taking the logarithm due to 0 values and to make the data more normally distributed. For model validation studies, the unpaired t test was used to compare bacterial load on catheters between CP-treated and untreated groups. The comparison of bacterial burdens in the blood and spleen between all 4 treatment groups was performed by ANOVA with the Tukey-Kramer adjustment. One-way ANOVA also was used to compare outcomes (blood, catheter, and spleen) between bacterial strains. Bartlett's test was used to test for equal variance. If the Bartlett's test indicated different variances among groups, ANOVA with heterogeneous variance was used. If results were significant, pairwise comparisons were made between groups, adjusting for multiple comparisons using the Bonferroni method. Except where specified, PASW Statistics was used for statistical determinations. For all analyses, P values less than 0.05 were considered statistically significant.
Nucleotide sequence accession number.
The aap sequence of strain 1457 was submitted to GenBank under accession number KJ920749.
RESULTS
S. epidermidis strain 1457 produces and processes Aap in vitro.
Previous investigations have demonstrated that Aap domain architecture and subsequent processing is variable among clinical and laboratory strains (12, 26, 33, 35, 39). Draft genomic sequencing revealed that Aap of 1457 has an A domain with 11 short, 16-aa repeats, followed by the conserved L-type lectin domain, while the B domain contains 6 full and 1 partial B repeat followed by 2 putative collagen-like repeat motifs. The protein is 1,639 residues long with an estimated in silico size of 174 kDa and a predicted pI of 4.75. Comparison of Aap from 1457 to that of the 4 publically available Aap sequences revealed 92 to 95% protein identity (Fig. 1).
To confirm strain 1457 was appropriate for studying the function of Aap in biofilm-mediated infections, Western blot analysis was performed using B domain antiserum (39) on the cell wall fraction (Fig. 2). This assay not only confirmed that Aap was translated but also demonstrated that the resulting protein was cleaved in vitro. While cells grown in TSB without the protease inhibitor α2-macroglobulin (α2m) produced a short, approximately 150-kDa Aap isoform (most likely representing the B domain), cells grown in the presence of α2m predominantly displayed a band of approximately 250 kDa, corresponding to full-length Aap (Fig. 2). While Aap runs at a higher apparent molecular mass (∼250 kDa) than predicted from its sequence (Fig. 2), this is common of cell wall-associated proteins (38) and may in part be explained by the protein preparation method. Although cell wall-specific proteins are separated from the rest of the cell, cell wall fragments remain covalently bound to the proteins, thereby retarding their migration through the gel.
FIG 2.

S. epidermidis strain 1457 produces and processes Aap in vitro. Antiserum raised against the recombinant B domain of Aap specifically binds to an approximately 150-kDa band present on the cell wall of bacteria grown in standard medium (left lane). The ∼250-kDa band corresponding to full-length Aap is apparent when bacteria are cultured in medium containing the protease inhibitor α2-macroglobulin (α2m; right lane). While Aap runs at a higher apparent molecular mass (∼250 kDa) than that predicted from its sequence (174 kDa), this is common of cell wall-associated proteins and may be a consequence of cell wall fragments remaining covalently bound to the protein.
Construction and preliminary characterization of an isogenic aap allelic replacement mutant.
Various studies have demonstrated that Aap functions to mediate biofilm accumulation of S. epidermidis strains in a PIA-independent manner (12, 32, 39). However, none of these studies were performed in isogenic allelic replacement mutants. Therefore, to determine the function of Aap in biofilm accumulation, an aap allelic replacement mutant was constructed in S. epidermidis 1457 (1457 Δaap). However, no significant difference was noted between 1457 and 1457 Δaap in the Christensen static biofilm assay (Fig. 3). This result was not necessarily surprising, as our experience and that of others (53, 66, 73) suggests in vitro static biofilm assays essentially measure PIA synthesis and a hallmark of 1457 is constitutive production of PIA (9, 49, 53). Therefore, to determine the contribution of Aap in biofilm accumulation in the absence of PIA, bacteriophage 71 was used to transduce the aap::tetM marker from 1457 Δaap into 1457 Δica (53), generating 1457 Δica Δaap. Initial quantification of static biofilm formation after 24 h was in agreement with previous findings (53), demonstrating that 1457 Δica produced significantly less biofilm than PIA-positive 1457 (P < 0.0001) (Fig. 3). However, it also was noted that 1457 Δica Δaap formed significantly less biofilm than 1457 Δica, highlighting the contribution of Aap in biofilm accumulation and/or maturation in the absence of PIA (P < 0.01). Complementation of 1457 Δica Δaap with icaADBC (pNF110) restored biofilm formation to the level of 1457 Δaap (P < 0.001).
FIG 3.
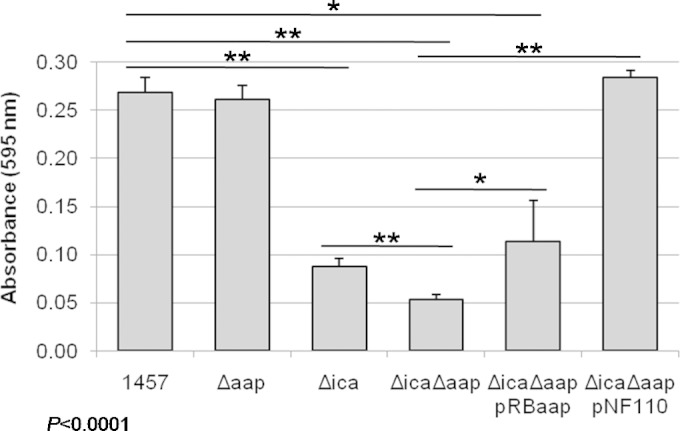
Deletion of aap decreases static biofilm formation in a PIA-negative background. Isogenic ica and aap allelic replacement mutants were constructed in the S. epidermidis 1457 background. Bacteria were grown statically in TSB for 24 h in a 96-well plate. Adherent cells were stained with crystal violet, and the absorbance was measured at 595 nm on a multiplate reader. The deletion of icaADBC (Δica) significantly impaired biofilm formation compared to that for 1457 (P < 0.0001), while no statistical difference was noted between 1457 and 1457 Δaap with regard to biofilm formation. However, a significant difference was detected between 1457 Δica and 1457 Δica Δaap (P < 0.01), suggesting that the presence of PIA masks the accumulation function of Aap in this assay. The biofilm phenotype was complementable in 1457 Δica Δaap with an icaADBC plasmid (pNF110). ***, P < 0.001; **, P < 0.01; *, P < 0.05 (n ≥ 4 biological replicates).
To further examine the function of Aap in biofilm development under dynamic conditions, we grew 1457, 1457 Δica, 1457 Δaap, and 1457 Δica Δaap in a flow cell system with a continual supply of fresh media. Based on the results of the static biofilm assay, we first examined the biofilm-forming ability of both PIA-deficient strains (1457 Δica and 1457 Δica Δaap). After 4 days, live/dead staining showed that both strains produced biofilm mats containing live and dead cells; however, the spatial organization of the 1457 Δica biofilm was similar to that of other reports, with dead cells localized at the biofilm-substrate interface (9). In contrast, the 1457 Δica Δaap biofilm was much less structured, and dead cells were visible throughout the media within the flow cell chamber, suggesting the cells were more loosely adherent than those of the Aap-positive 1457 Δica mutant (Fig. 4A and B). In addition to the decreased biofilm structure, in the absence of Aap the biomass of 1457 Δica Δaap was noticeably decreased compared to that of 1457 Δica (P < 0.001) (Fig. 4C). After confirming Aap synthesis affected biofilm formation in a flow cell, we next sought to determine the effect of Aap in the PIA-positive strains 1457 and 1457 Δaap. After 4 days of growth, PIA-mediated biofilms were too thick to be imaged using standard CLSM methodology; thus, biofilm production was analyzed after 2 days rather than 4. In contrast to the results observed in the static biofilm assay (Fig. 3), 1457 Δaap had significantly less biomass after 2 days of growth than 1457 (P < 0.05) (Fig. 5A and B). Note that in contrast to confocal analysis following 4 days of growth (Fig. 4A and B), dead cells were not apparent in the 2-day biofilms of 1457 or 1457 Δaap, which most likely is a consequence of the decreased duration of the experiment. Collectively, these data suggest Aap enhances biofilm maturation in both a PIA-dependent and independent biofilm; however, the presence of PIA may significantly mask the contribution of Aap in many in vitro biofilm accumulation assays.
FIG 4.
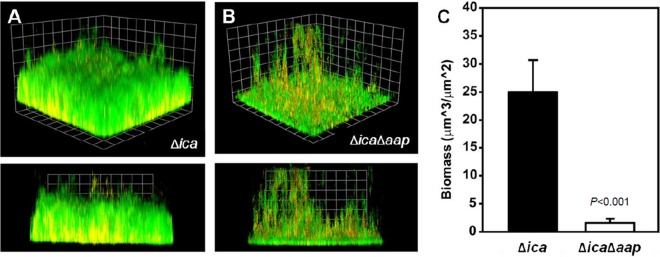
Flow cell biomass is decreased in 1457 Δica Δaap compared to that in 1457 Δica. S. epidermidis 1457 Δica (A) and 1457 Δica Δaap (B) were grown for 4 days in 2% TSB at a flow rate of 3.75 rpm. Biofilms were poststained for live (Syto9; green) and dead (ToPro3; red) cells and then visualized by confocal light microscopy. Magnification, ×60. The close proximity of live and dead cells causes regions containing dead cells to appear yellow. (C) COMSTAT analysis of live cell biomass from three-dimensional biofilm reconstructions demonstrating decreased biomass in 1457 Δica Δaap compared to that in 1457 Δica (P < 0.001) (n ≥ 3).
FIG 5.

Flow cell biomass is decreased in 1457 Δaap compared to that in 1457. S. epidermidis 1457 (A) and 1457 Δaap (B) were grown for 2 days in 2% TSB at a flow rate of 3.75 rpm. Biofilms were poststained for live (Syto9; green) and dead (ToPro3; red) cells and then visualized by confocal light microscopy. Magnification, ×20. (C) COMSTAT analysis of three-dimensional biofilm reconstructions demonstrating decreased biomass in 1457 Δaap compared to that in 1457 (P < 0.05) (n = 5).
Development and validation of an in vivo rat model of intravenous catheter infection.
A modification of our previously described rat model of intravenous catheter-related infection (74) was developed to assess the effect of Aap on S. epidermidis virulence and/or catheter colonization (71). The adapted model contains three major changes: catheter type, inoculation method, and animal immune status. First, after a Tygon catheter, sealed at the proximal end, was inserted into the jugular vein, the extravascular portion of the catheter was positioned in the soft tissue of the neck and was not exposed outside the animal as previously described (74, 75); accordingly, rodent restraint jackets were not necessary. Second, rats were inoculated with 1 × 109 CFU of S. epidermidis via the tail vein rather than through the catheter. Third, as it is well documented that immunocompetent rats are able to clear high inocula of S. epidermidis from the bloodstream in the absence of a foreign body (76, 77), to increase the percentage of catheters seeded following S. epidermidis challenge, rats were given intraperitoneal injections of CP to produce leukopenia. To validate our model and confirm that the presence of a catheter and immunosuppression are critical for S. epidermidis infection, rats were divided into four groups prior to bacterial inoculation: (i) catheter plus CP, (ii) catheter only, (iii) CP only, and (iv) no catheter or CP (no treatment). After recovering from surgery, all groups were inoculated with S. epidermidis 1457 via the lateral tail vein. Three days postinfection, rats were sacrificed and the catheter, blood, and spleen were harvested and plated to determine bacterial load. As anticipated, bacterial burden on the catheters of CP-treated rats was significantly greater than that of immunocompetent rats (P < 0.05); bacteria were recovered from only 1 of 7 catheters in the untreated group (Fig. 6A). Bacterial counts from the blood and spleen also were significantly decreased in the absence of a catheter or CP treatment, and counts were the lowest in the uncatheterized, untreated group (Fig. 6B and C). Collectively, similar to S. epidermidis infections in humans, these data suggest that both a foreign body and immunosuppression are required for successful S. epidermidis infection in this model.
FIG 6.

A foreign body and immunosuppression are required for S. epidermidis infection. Prior to bacterial inoculation, rats were divided into four groups: catheter plus cyclophosphamide (CP), catheter only, CP only, and no catheter or CP (no treatment). Three days after infection, rats were sacrificed and the catheter (A), blood (B), and spleen (C) were harvested and plated for bacterial load. Bacterial burdens from the catheter plus CP rats were significantly higher than those in the other three groups (P < 0.05). Bacterial counts were normalized (log10 + 1) due to complete clearance of bacteria in some animals. ***, P < 0.001; **, P < 0.01; *, P < 0.05. Cath, catheter; tx, treatment.
Loss of Aap decreases virulence in a rat catheter model of S. epidermidis infection.
Based on the high prevalence of strains encoding aap (12, 28, 42, 78–82) and the ability of ica-negative strains to cause infection (12, 32, 39, 81, 83–86), we hypothesized that Aap contributed to S. epidermidis virulence. To address this question, we compared the infectivity of our isogenic strain set in our validated model of rat central venous catheter (CVC) infection. Rats were inoculated with equal amounts (109 CFU) of either 1457, 1457 Δica, 1457 Δaap, or 1457 Δica Δaap. Based on multiple animal experiments demonstrating that PIA synthesis enhanced S. epidermidis virulence (74, 87), we were surprised to find that both 1457 and 1457 Δica adhered to catheters with similar efficiency, suggesting catheter adherence in this model is PIA independent. However, we did find that catheter adherence of both 1457 Δaap and 1457 Δica Δaap were significantly impaired (P < 0.001); 13 of 15 and 15 of 17 catheters of 1457 Δaap and 1457 Δica Δaap, respectively, were sterile (Fig. 7A). Consistent with these data, there was also a significant decrease in bacterial burden in the blood, compared to the level in 1457, in both 1457 Δaap and 1457 Δica Δaap (P < 0.01 and P < 0.05, respectively) but not in 1457 Δica (Fig. 7B). Furthermore, the finding that 1457 Δica Δaap phenocopied 1457 Δaap rather than 1457 Δica supports the physiological importance of Aap during S. epidermidis infection, even in PIA-producing strains. Of note, similar bacterial counts were recovered from the spleen in all four strains (P > 0.05) (Fig. 7C), suggesting differences from the catheter and blood were not due to differential fitness of the mutants.
FIG 7.

Aap is required for bacterial adherence in a rat model of intravenous catheter infection. Rats with implanted jugular catheters were immunosuppressed with cyclophosphamide and then inoculated via the tail vein with 109 CFU of S. epidermidis 1457, 1457 Δica, 1457 Δaap, or 1457 Δica Δaap. Three days postinfection, the catheter (A), blood (B), and spleen (C) were harvested and plated to assess bacterial burden. Both Aap-deficient strains (1457 Δaap and 1457 Δica Δaap) had significantly lower bacterial burdens on the catheter and in the blood than strains with Aap (1457 and 1457 Δica). Similar titers were obtained from the spleen for all four strains, suggesting the differences in catheter and blood counts were not due to decreased fitness of the mutants. ***, P < 0.001; **, P < 0.01; *, P < 0.05.
The A domain of Aap mediates initial attachment.
Previous studies have established that the A domain of Aap and its S. aureus orthologue, SasG, functions to mediate initial attachment to mammalian cells (35, 38, 45). Therefore, we hypothesized that initial binding to an abiotic surface (e.g., rat catheter) was Aap A domain dependent. To test this hypothesis, initial attachment to a plastic surface was compared between 1457 Δaap and 1457 Δaap complemented in trans with full-length aap (pRBaap) or with the B domain only (pRBaapDomB) (Fig. 8A and B). As shown in Fig. 8A, complementation with full-length aap (containing both the A and B domains) significantly increased initial adherence to plastic compared to that for 1457 Δaap. However, complementation of 1457 Δaap with the Aap B domain only (pRBaapDomB) did not alter initial adherence of 1457 aap (Fig. 8B), suggesting the Aap A domain was responsible for initial adherence to plastic. To further demonstrate the ability of the A domain to facilitate attachment to abiotic surfaces, a plasmid containing the Aap A domain linked to the LPDTG cell wall anchor motif (pCNaapDomA) was introduced into the adhesion-negative strain S. carnosus TM300. As predicted from the results of the S. epidermidis Aap complementation studies (Fig. 8A), in trans expression of the Aap A domain in a surrogate host substantially increased initial attachment (Fig. 8C), thereby confirming that the A domain of Aap facilitates binding to artificial surfaces. Additionally, the expression of the B domain in S. carnosus did not noticeably enhance attachment, reiterating the specificity of the A domain in this process.
FIG 8.

The A domain of Aap mediates initial attachment to plastic surfaces. Complementation of 1457 Δaap (dark gray circles) with full-length aap (pRBaap; black squares) (A) but not the B domain alone (pRBaapDomB; black squares) (B) enhances initial attachment. (C) Expression of the A domain (pCNaapDomA; black diamonds) in S. carnosus TM300 (dark gray circles) mediates attachment to a plastic surface, while in trans expression of the B domain (pRBaapDomB; light gray squares) does not.
DISCUSSION
S. epidermidis is the most common cause of nosocomial infections, specifically those related to implanted materials, due to its propensity to form biofilms. Biofilm-mediated infections are especially difficult to treat; therefore, over the past few decades research has focused on better understanding these structures. The biofilm matrix may be composed of numerous factors, including PIA, various proteins (e.g., Aap), and eDNA. Due to the diversity and functional redundancy of biofilm matrix molecules, we generated isogenic mutants to allow the investigation of the specific contribution of Aap during biofilm formation and infection. Although no difference was noted between 1457 and 1457 Δaap using a static microtiter biofilm assay, a striking difference in biomass was detected when strains were grown in a flow cell system, suggesting Aap contributes to biofilm accumulation during maturation (Fig. 5). An even greater difference in biomass was observed when 1457 Δica Δaap was compared to 1457 Δica (Fig. 4). These data suggest the lack of Aap, as well as PIA, severely inhibits the ability of S. epidermidis to form a mature biofilm.
Dynamics of host interactions are a key component of infections; therefore, to correlate our in vitro findings with S. epidermidis virulence, we modified our previously developed rat catheter model (74, 75). In validating our model, we found that rats required immunosuppression and a high inoculum, consistent with previous reports (76, 77, 88, 89). Following validation, we used the model to assess virulence of the mutant S. epidermidis strains 3 days postinfection. Ultimately, we found that in the absence of Aap, regardless of PIA production, there were significantly lower bacterial burdens on catheters and in the blood. Additionally, bacterial burdens of 1457 Δica were the same as those for wild-type 1457. Previous reports regarding the importance of PIA in other animal models of infection conflict with our observations. Investigations using a mouse foreign body and rat catheter-related models found that PIA-negative strains were less virulent (74, 87, 90). However, three separate studies employing guinea pig tissue cage models of infection showed no difference in virulence with non-PIA producing strains (91–93). The type of infection generated in each model can explain in part these differences. Both the guinea pig tissue cage model and the mouse foreign body model are more replicative of localized, subcutaneous infections (87) than the systemic infections modeled in the rat catheter studies. In all cases, experiments utilizing direct application of bacteria into the implant material presents a different scenario than indirect inoculation requiring bacterial seeding of the device, complicating comparison between earlier results and those we observed. Together, these data suggest PIA has various levels of importance in different niches.
Multiples studies also have demonstrated a fitness defect resulting from decreased resistance to the host immune response in PIA-negative strains (15, 94); however, this difference is negated in our model as a consequence of CP treatment. Additionally, as S. epidermidis produces few virulence factors, the tissue damage observed in the PIA-producing strain most likely was a consequence of the host immune response that again would not be expected in our model due to the severe CP-induced immunosuppression (76) (see Fig. S1 in the supplemental material). Regarding the previously described rat catheter model (74), a large bolus of bacteria was delivered directly into the catheter lumen, followed by daily heparin flushes to maintain catheter patency. The force from the fluid flushes may have detached non-PIA-producing bacteria; as demonstrated during our flow cell experiments, the ica mutant strain biofilms were less robust. Further support for the importance of PIA during high shear stress is demonstrated by increased ica expression and PIA synthesis upon exposure to fluid flow (95, 96). Given the experimental differences, the disparity in PIA virulence is not surprising; rather, it emphasizes the importance of utilizing multiple animal models to ascertain the function of proposed virulence factors.
As detailed above, the model presented here differs from the aforementioned models by examining an alternative step in the infection process of adhesion and initial attachment. By inoculating via the tail vein, bacteria subsequently isolated from the catheter are the result of hematogenous seeding and attachment (88). Another important component of our model is the interaction between host proteins and bacterial adhesins, or MSCRAMMs. Foreign materials placed in the body are rapidly coated with host proteins that have been demonstrated to enhance bacterial colonization (88, 89, 97). By allowing at least 8 days between catheter introduction and bacterial inoculation, this essential aspect of bacterial attachment and subsequent infection is an integral facet of our model.
Based on the finding that adherence is essential for bacterial colonization in our model, we next sought to determine if the observed requirement of Aap for catheter infection resulted from Aap-mediated attachment. Although a specific target has not been identified, both Aap and SasG bind specifically to epithelial cells via their A domains (35, 38, 45); however, it was previously unclear if this also was true for abiotic surfaces. To answer this question, we performed initial attachment ELISAs with an aap mutant and complement strains. In trans complementation of 1457 Δaap with full-length aap (Fig. 8A), but not the B domain alone (Fig. 8B), enhanced S. epidermidis attachment. Furthermore, introduction of the A domain, but not the B domain, into adhesion-deficient S. carnosus also markedly increased its adherence (Fig. 8C). These data demonstrate that the A domain of Aap indeed binds to artificial surfaces.
The cleavage of Aap is accomplished by bacterial and host-derived proteases (39). Among S. epidermidis isolates, the degree of cleavage varies, including strains that do not appear to process the protein (26, 39). If the same holds true during infection, that would support A domain-mediated attachment as the primary function of Aap rather than B domain-dependent intercellular accumulation. Based on our results and previously published data, we developed the following hypothetical model regarding the bifunctional activity of Aap (Fig. 9). The expression of full-length Aap facilitates initial attachment of bacterial cells via interactions between the A domain and a surface ligand (e.g., abiotic surface or host protein). Once bound, protease cleavage removes the A domain, enabling interactions between B domain repeats, resulting in intercellular accumulation and biofilm formation. In S. epidermidis and S. aureus, the production of secreted proteases is controlled by the agr quorum-sensing (QS) system (98–100). The proposed model is further supported by previous findings of QS-dependent control of MSCRAMM cleavage and subsequent function in S. aureus, Streptococcus pyogenes, and Enterococcus faecalis (101).
FIG 9.

Model of Aap dual functionality. S. epidermidis expressing full-length Aap attaches to a foreign body through unknown interactions within the A domain. Following binding, Aap is cleaved by staphylococcal or host-derived proteases, enabling intercellular accumulation via the B domain.
Our proposal that the importance of Aap in our model is at least partially due to A domain-mediated attachment highlights the importance of in vivo modeling. Prior to this study, the A domain was mentioned in biofilm literature only to state that its cleavage was required for Aap-mediated biofilm formation. Collectively, our data suggest Aap is a bifunctional biofilm matrix protein, with the A domain mediating initial attachment and the B domain facilitating cell accumulation. This is not unprecedented, as dual-function proteins have been identified in S. epidermidis and other species (14, 102–105). Additional studies are needed to define the cleavage dynamics of Aap and the corresponding contribution to biofilm formation and virulence of S. epidermidis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Timothy Foster for the α-Aap A domain antibody, Roxanne Alter for assistance with hematological analysis, Fang Yu for help with statistical analysis, and Jeremy Yethon, Leah E. Cole, and Hubert Lam for helpful discussions.
This work was supported in part by Public Health Service grant P01AI083211 (P.D.F. and A.R.H.) from the National Institute of Allergy and Infectious Diseases and an investigator-initiated grant from Sanofi-Pasteur (P.D.F.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02177-14.
REFERENCES
- 1.Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, Program NCS, Bouffard GG, Blakesley RW, Murray PR, Green ED, Turner ML, Segre JA. 2009. Topographical and temporal diversity of the human skin microbiome. Science 324:1190–1192. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Otto M. 2009. Staphylococcus epidermidis–the “accidental” pathogen. Nat Rev Microbiol 7:555–567. doi: 10.1038/nrmicro2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rogers KL, Fey PD, Rupp ME. 2009. Coagulase-negative staphylococcal infections. Infect Dis Clin N Am 23:73–98. doi: 10.1016/j.idc.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Rupp ME, Fey PD. 2010. Staphylococcus epidermidis and other coagulase-negative staphylococci, p 2579–2589. In Mandell GL, Bennett JE, Dolin R (ed), Principles and practice of infectious disease, vol 2 Churchill Livingstone, Philadelphia, PA. [Google Scholar]
- 5.Rupp ME. 2014. Clinical characteristics of infections in humans due to Staphylococcus epidermidis. Methods Mol Biol 1106:1–16. doi: 10.1007/978-1-62703-736-5_1. [DOI] [PubMed] [Google Scholar]
- 6.Moormeier DE, Endres JL, Mann EE, Sadykov MR, Horswill AR, Rice KC, Fey PD, Bayles KW. 2013. Use of microfluidic technology to analyze gene expression during Staphylococcus aureus biofilm formation reveals distinct physiological niches. Appl Environ Microbiol 79:3413–3424. doi: 10.1128/AEM.00395-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stewart PS, Franklin MJ. 2008. Physiological heterogeneity in biofilms. Nat Rev Microbiol 6:199–210. doi: 10.1038/nrmicro1838. [DOI] [PubMed] [Google Scholar]
- 8.Anderl JN, Zahller J, Roe F, Stewart PS. 2003. Role of nutrient limitation and stationary-phase existence in Klebsiella pneumoniae biofilm resistance to ampicillin and ciprofloxacin. Antimicrob Agents Chemother 47:1251–1256. doi: 10.1128/AAC.47.4.1251-1256.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang SJ, Nast CC, Mishra NN, Yeaman MR, Fey PD, Bayer AS. 2010. Cell wall thickening is not a universal accompaniment of the daptomycin nonsusceptibility phenotype in Staphylococcus aureus: evidence for multiple resistance mechanisms. Antimicrob Agents Chemother 54:3079–3085. doi: 10.1128/AAC.00122-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mack D, Fischer W, Krokotsch A, Leopold K, Hartmann R, Egge H, Laufs R. 1996. The intercellular adhesin involved in biofilm accumulation of Staphylococcus epidermidis is a linear beta-1,6-linked glucosaminoglycan: purification and structural analysis. J Bacteriol 178:175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rice KC, Mann EE, Endres JL, Weiss EC, Cassat JE, Smeltzer MS, Bayles KW. 2007. The CidA murein hydrolase regulator contributes to DNA release and biofilm development in Staphylococcus aureus. Proc Natl Acad Sci U S A 104:8113–8118. doi: 10.1073/pnas.0610226104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rohde H, Burandt EC, Siemssen N, Frommelt L, Burdelski C, Wurster S, Scherpe S, Davies AP, Harris LG, Horstkotte MA, Knobloch JK, Ragunath C, Kaplan JB, Mack D. 2007. Polysaccharide intercellular adhesin or protein factors in biofilm accumulation of Staphylococcus epidermidis and Staphylococcus aureus isolated from prosthetic hip and knee joint infections. Biomaterials 28:1711–1720. doi: 10.1016/j.biomaterials.2006.11.046. [DOI] [PubMed] [Google Scholar]
- 13.Williams RJ, Henderson B, Sharp LJ, Nair SP. 2002. Identification of a fibronectin-binding protein from Staphylococcus epidermidis. Infect Immun 70:6805–6810. doi: 10.1128/IAI.70.12.6805-6810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christner M, Franke GC, Schommer NN, Wendt U, Wegert K, Pehle P, Kroll G, Schulze C, Buck F, Mack D, Aepfelbacher M, Rohde H. 2010. The giant extracellular matrix-binding protein of Staphylococcus epidermidis mediates biofilm accumulation and attachment to fibronectin. Mol Microbiol 75:187–207. doi: 10.1111/j.1365-2958.2009.06981.x. [DOI] [PubMed] [Google Scholar]
- 15.Schommer NN, Christner M, Hentschke M, Ruckdeschel K, Aepfelbacher M, Rohde H. 2011. Staphylococcus epidermidis uses distinct mechanisms of biofilm formation to interfere with phagocytosis and activation of mouse macrophage-like cells 774A. 1. Infect Immun 79:2267–2276. doi: 10.1128/IAI.01142-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vuong C, Durr M, Carmody AB, Peschel A, Klebanoff SJ, Otto M. 2004. Regulated expression of pathogen-associated molecular pattern molecules in Staphylococcus epidermidis: quorum-sensing determines pro-inflammatory capacity and production of phenol-soluble modulins. Cell Microbiol 6:753–759. doi: 10.1111/j.1462-5822.2004.00401.x. [DOI] [PubMed] [Google Scholar]
- 17.Hanke ML, Kielian T. 2012. Deciphering mechanisms of staphylococcal biofilm evasion of host immunity. Front Cell Infect Microbiol 2:62. doi: 10.3389/fcimb.2012.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thurlow LR, Hanke ML, Fritz T, Angle A, Aldrich A, Williams SH, Engebretsen IL, Bayles KW, Horswill AR, Kielian T. 2011. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J Immunol 186:6585–6596. doi: 10.4049/jimmunol.1002794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Costerton JW. 1999. Introduction to biofilm. Int J Antimicrob Agents 11:217–221. doi: 10.1016/S0924-8579(99)00018-7. [DOI] [PubMed] [Google Scholar]
- 20.Fey PD. 2010. Modality of bacterial growth presents unique targets: how do we treat biofilm-mediated infections? Curr Opin Microbiol 13:610–615. doi: 10.1016/j.mib.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bozic KJ, Kurtz SM, Lau E, Ong K, Chiu V, Vail TP, Rubash HE, Berry DJ. 2010. The epidemiology of revision total knee arthroplasty in the United States. Clin Ortho Related Res 468:45–51. doi: 10.1007/s11999-009-0945-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bozic KJ, Kurtz SM, Lau E, Ong K, Vail TP, Berry DJ. 2009. The epidemiology of revision total hip arthroplasty in the United States. J Bone Joint Surg 91:128–133. doi: 10.2106/JBJS.H.00155. [DOI] [PubMed] [Google Scholar]
- 23.Kurtz SM, Lau E, Schmier J, Ong KL, Zhao K, Parvizi J. 2008. Infection burden for hip and knee arthroplasty in the United States. J Arthroplasty 23:984–991. doi: 10.1016/j.arth.2007.10.017. [DOI] [PubMed] [Google Scholar]
- 24.Urquhart DM, Hanna FS, Brennan SL, Wluka AE, Leder K, Cameron PA, Graves SE, Cicuttini FM. 2010. Incidence and risk factors for deep surgical site infection after primary total hip arthroplasty: a systematic review. J Arthroplasty 25:1216–1222 e1211–1213. doi: 10.1016/j.arth.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 25.Spiliopoulou AI, Krevvata MI, Kolonitsiou F, Harris LG, Wilkinson TS, Davies AP, Dimitracopoulos GO, Karamanos NK, Mack D, Anastassiou ED. 2012. An extracellular Staphylococcus epidermidis polysaccharide: relation to polysaccharide intercellular adhesin and its implication in phagocytosis. BMC Microbiol 12:76. doi: 10.1186/1471-2180-12-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Banner MA, Cunniffe JG, Macintosh RL, Foster TJ, Rohde H, Mack D, Hoyes E, Derrick J, Upton M, Handley PS. 2007. Localized tufts of fibrils on Staphylococcus epidermidis NCTC 11047 are comprised of the accumulation-associated protein. J Bacteriol 189:2793–2804. doi: 10.1128/JB.00952-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bateman A, Holden MT, Yeats C. 2005. The G5 domain: a potential N-acetylglucosamine recognition domain involved in biofilm formation. Bioinformatics 21:1301–1303. doi: 10.1093/bioinformatics/bti206. [DOI] [PubMed] [Google Scholar]
- 28.Bowden MG, Chen W, Singvall J, Xu Y, Peacock SJ, Valtulina V, Speziale P, Hook M. 2005. Identification and preliminary characterization of cell-wall-anchored proteins of Staphylococcus epidermidis. Microbiology 151:1453–1464. doi: 10.1099/mic.0.27534-0. [DOI] [PubMed] [Google Scholar]
- 29.Broekhuizen CA, de Boer L, Schipper K, Jones CD, Quadir S, Feldman RG, Vandenbroucke-Grauls CM, Zaat SA. 2009. The influence of antibodies on Staphylococcus epidermidis adherence to polyvinylpyrrolidone-coated silicone elastomer in experimental biomaterial-associated infection in mice. Biomaterials 30:6444–6450. doi: 10.1016/j.biomaterials.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 30.Conrady DG, Brescia CC, Horii K, Weiss AA, Hassett DJ, Herr AB. 2008. A zinc-dependent adhesion module is responsible for intercellular adhesion in staphylococcal biofilms. Proc Natl Acad Sci U S A 105:19456–19461. doi: 10.1073/pnas.0807717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conrady DG, Wilson JJ, Herr AB. 2013. Structural basis for Zn2+-dependent intercellular adhesion in staphylococcal biofilms. Proc Natl Acad Sci U S A 110:E202–211. doi: 10.1073/pnas.1208134110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hennig S, Nyunt Wai S, Ziebuhr W. 2007. Spontaneous switch to PIA-independent biofilm formation in an ica-positive Staphylococcus epidermidis isolate. Int J Med Microbiol 297:117–122. doi: 10.1016/j.ijmm.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 33.Hu J, Xu T, Zhu T, Lou Q, Wang X, Wu Y, Huang R, Liu J, Liu H, Yu F, Ding B, Huang Y, Tong W, Qu D. 2011. Monoclonal antibodies against accumulation-associated protein affect EPS biosynthesis and enhance bacterial accumulation of Staphylococcus epidermidis. PLoS One 6:e20918. doi: 10.1371/journal.pone.0020918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hussain M, Herrmann M, von Eiff C, Perdreau-Remington F, Peters G. 1997. A 140-kilodalton extracellular protein is essential for the accumulation of Staphylococcus epidermidis strains on surfaces. Infect Immun 65:519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Macintosh RL, Brittan JL, Bhattacharya R, Jenkinson HF, Derrick J, Upton M, Handley PS. 2009. The terminal A domain of the fibrillar accumulation-associated protein (Aap) of Staphylococcus epidermidis mediates adhesion to human corneocytes. J Bacteriol 191:7007–7016. doi: 10.1128/JB.00764-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Monk AB, Boundy S, Chu VH, Bettinger JC, Robles JR, Fowler VG Jr, Archer GL. 2008. Analysis of the genotype and virulence of Staphylococcus epidermidis isolates from patients with infective endocarditis. Infect Immun 76:5127–5132. doi: 10.1128/IAI.00606-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pintens V, Massonet C, Merckx R, Vandecasteele S, Peetermans WE, Knobloch JK, Van Eldere J. 2008. The role of sigmaB in persistence of Staphylococcus epidermidis foreign body infection. Microbiology 154:2827–2836. doi: 10.1099/mic.0.2007/015768-0. [DOI] [PubMed] [Google Scholar]
- 38.Roche FM, Meehan M, Foster TJ. 2003. The Staphylococcus aureus surface protein SasG and its homologues promote bacterial adherence to human desquamated nasal epithelial cells. Microbiology 149:2759–2767. doi: 10.1099/mic.0.26412-0. [DOI] [PubMed] [Google Scholar]
- 39.Rohde H, Burdelski C, Bartscht K, Hussain M, Buck F, Horstkotte MA, Knobloch JK, Heilmann C, Herrmann M, Mack D. 2005. Induction of Staphylococcus epidermidis biofilm formation via proteolytic processing of the accumulation-associated protein by staphylococcal and host proteases. Mol Microbiol 55:1883–1895. doi: 10.1111/j.1365-2958.2005.04515.x. [DOI] [PubMed] [Google Scholar]
- 40.Schumacher-Perdreau F, Heilmann C, Peters G, Gotz F, Pulverer G. 1994. Comparative analysis of a biofilm-forming Staphylococcus epidermidis strain and its adhesion-positive, accumulation-negative mutant M7. FEMS Microbiol Lett 117:71–78. doi: 10.1111/j.1574-6968.1994.tb06744.x. [DOI] [PubMed] [Google Scholar]
- 41.Sun D, Accavitti MA, Bryers JD. 2005. Inhibition of biofilm formation by monoclonal antibodies against Staphylococcus epidermidis RP62A accumulation-associated protein. Clin Diagn Lab Immunol 12:93–100. doi: 10.1128/CDLI.12.1.93-100.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vandecasteele SJ, Peetermans WE, Merckx R, Van Eldere J. 2003. Expression of biofilm-associated genes in Staphylococcus epidermidis during in vitro and in vivo foreign body infections. J Infect Dis 188:730–737. doi: 10.1086/377452. [DOI] [PubMed] [Google Scholar]
- 43.Yang XM, Li N, Chen JM, Ou YZ, Jin H, Lu HJ, Zhu YL, Qin ZQ, Qu D, Yang PY. 2006. Comparative proteomic analysis between the invasive and commensal strains of Staphylococcus epidermidis. FEMS Microbiol Lett 261:32–40. doi: 10.1111/j.1574-6968.2006.00327.x. [DOI] [PubMed] [Google Scholar]
- 44.Gruszka DT, Wojdyla JA, Bingham RJ, Turkenburg JP, Manfield IW, Steward A, Leech AP, Geoghegan JA, Foster TJ, Clarke J, Potts JR. 2012. Staphylococcal biofilm-forming protein has a contiguous rod-like structure. Proc Natl Acad Sci U S A 109:E1011–E1018. doi: 10.1073/pnas.1119456109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roche FM, Massey R, Peacock SJ, Day NP, Visai L, Speziale P, Lam A, Pallen M, Foster TJ. 2003. Characterization of novel LPXTG-containing proteins of Staphylococcus aureus identified from genome sequences. Microbiology 149:643–654. doi: 10.1099/mic.0.25996-0. [DOI] [PubMed] [Google Scholar]
- 46.Corrigan RM, Rigby D, Handley P, Foster TJ. 2007. The role of Staphylococcus aureus surface protein SasG in adherence and biofilm formation. Microbiology 153:2435–2446. doi: 10.1099/mic.0.2007/006676-0. [DOI] [PubMed] [Google Scholar]
- 47.Schneewind O, Mihaylova-Petkov D, Model P. 1993. Cell wall sorting signals in surface proteins of gram-positive bacteria. EMBO J 12:4803–4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Geoghegan JA, Corrigan RM, Gruszka DT, Speziale P, O'Gara JP, Potts JR, Foster TJ. 2010. Role of surface protein SasG in biofilm formation by Staphylococcus aureus. J Bacteriol 192:5663–5673. doi: 10.1128/JB.00628-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mack D, Siemssen N, Laufs R. 1992. Parallel induction by glucose of adherence and a polysaccharide antigen specific for plastic-adherent Staphylococcus epidermidis: evidence for functional relation to intercellular adhesion. Infect Immun 60:2048–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, Turner SW, Korlach J. 2013. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10:563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 51.Kreiswirth BN, Lofdahl S, Betley MJ, O'Reilly M, Schlievert PM, Bergdoll MS, Novick RP. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709–712. doi: 10.1038/305709a0. [DOI] [PubMed] [Google Scholar]
- 52.Projan SJ, Archer GL. 1989. Mobilization of the relaxable Staphylococcus aureus plasmid pC221 by the conjugative plasmid pGO1 involves three pC221 loci. J Bacteriol 171:1841–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Handke LD, Slater SR, Conlon KM, O'Donnell ST, Olson ME, Bryant KA, Rupp ME, O'Gara JP, Fey PD. 2007. σB and SarA independently regulate polysaccharide intercellular adhesin production in Staphylococcus epidermidis. Can J Microbiol 53:82–91. doi: 10.1139/w06-108. [DOI] [PubMed] [Google Scholar]
- 54.Rosenstein R, Nerz C, Biswas L, Resch A, Raddatz G, Schuster SC, Gotz F. 2009. Genome analysis of the meat starter culture bacterium Staphylococcus carnosus TM300. Appl Environ Microbiol 75:811–822. doi: 10.1128/AEM.01982-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dean BA, Williams RE, Hall F, Corse J. 1973. Phage typing of coagulase-negative staphylococci and micrococci. J Hygiene 71:261–270. doi: 10.1017/S0022172400022737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 57.Archer GL, Johnston JL, Vazquez GJ, Haywood HB III. 1983. Efficacy of antibiotic combinations including rifampin against methicillin-resistant Staphylococcus epidermidis: in vitro and in vivo studies. Rev Infect Dis 5(Suppl 3):S538–S542. doi: 10.1093/clinids/5.Supplement_3.S538. [DOI] [PubMed] [Google Scholar]
- 58.Charpentier E, Anton AI, Barry P, Alfonso B, Fang Y, Novick RP. 2004. Novel cassette-based shuttle vector system for gram-positive bacteria. Appl Environ Microbiol 70:6076–6085. doi: 10.1128/AEM.70.10.6076-6085.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lindgren JK, Thomas VC, Olson ME, Chaudhari SS, Nuxoll AS, Schaeffer CR, Lindgren KE, Jones J, Zimmerman MC, Dunman PM, Bayles KW, Fey PD. 2014. Arginine deiminase in Staphylococcus epidermidis functions to augment biofilm maturation through pH homeostasis. J Bacteriol 196:2277–2289. doi: 10.1128/JB.00051-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maliszewski KL, Nuxoll AS. 2014. Use of electroporation and conjugative mobilization for genetic manipulation of Staphylococcus epidermidis. Methods Mol Biol 1106:125–134. doi: 10.1007/978-1-62703-736-5_11. [DOI] [PubMed] [Google Scholar]
- 61.Nedelmann M, Sabottke A, Laufs R, Mack D. 1998. Generalized transduction for genetic linkage analysis and transfer of transposon insertions in different Staphylococcus epidermidis strains. Zentralbl Bakteriol 287:85–92. doi: 10.1016/S0934-8840(98)80151-5. [DOI] [PubMed] [Google Scholar]
- 62.McDougal LK, Steward CD, Killgore GE, Chaitram JM, McAllister SK, Tenover FC. 2003. Pulsed-field gel electrophoresis typing of oxacillin-resistant Staphylococcus aureus isolates from the United States: establishing a national database. J Clin Microbiol 41:5113–5120. doi: 10.1128/JCM.41.11.5113-5120.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Southern EM. 1975. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol 98:503–517. doi: 10.1016/S0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
- 64.Christner M, Heinze C, Busch M, Franke G, Hentschke M, Bayard Duhring S, Buttner H, Kotasinska M, Wischnewski V, Kroll G, Buck F, Molin S, Otto M, Rohde H. 2012. sarA negatively regulates Staphylococcus epidermidis biofilm formation by modulating expression of 1 MDa extracellular matrix binding protein and autolysis-dependent release of eDNA. Mol Microbiol 86:394–410. doi: 10.1111/j.1365-2958.2012.08203.x. [DOI] [PubMed] [Google Scholar]
- 65.Christensen GD, Simpson WA, Bisno AL, Beachey EH. 1982. Adherence of slime-producing strains of Staphylococcus epidermidis to smooth surfaces. Infect Immun 37:318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Los R, Sawicki R, Juda M, Stankevic M, Rybojad P, Sawicki M, Malm A, Ginalska G. 2010. A comparative analysis of phenotypic and genotypic methods for the determination of the biofilm-forming abilities of Staphylococcus epidermidis. FEMS Microbiol Lett 310:97–103. doi: 10.1111/j.1574-6968.2010.02050.x. [DOI] [PubMed] [Google Scholar]
- 67.Boles BR, Horswill AR. 2008. Agr-mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathol 4:e1000052. doi: 10.1371/journal.ppat.1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Heydorn A, Nielsen AT, Hentzer M, Sternberg C, Givskov M, Ersboll BK, Molin S. 2000. Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology 146(Part 10):2395–2407. [DOI] [PubMed] [Google Scholar]
- 69.McCrea KW, Hartford O, Davis S, Eidhin DN, Lina G, Speziale P, Foster TJ, Hook M. 2000. The serine-aspartate repeat (Sdr) protein family in Staphylococcus epidermidis. Microbiology 146(Part 7):1535–1546. [DOI] [PubMed] [Google Scholar]
- 70.Mack D, Bartscht K, Fischer C, Rohde H, de Grahl C, Dobinsky S, Horstkotte MA, Kiel K, Knobloch JK. 2001. Genetic and biochemical analysis of Staphylococcus epidermidis biofilm accumulation. Methods Enzymol 336:215–239. doi: 10.1016/S0076-6879(01)36592-8. [DOI] [PubMed] [Google Scholar]
- 71.Schaeffer CR, Woods KM, Longo GM. 2014. Rat jugular catheter model of biofilm-mediated infection. Methods Mol Biol 1106:199–206. doi: 10.1007/978-1-62703-736-5_19. [DOI] [PubMed] [Google Scholar]
- 72.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed. National Academies Press, Washington, DC. [Google Scholar]
- 73.Wang L, Li M, Dong D, Bach TH, Sturdevant DE, Vuong C, Otto M, Gao Q. 2008. SarZ is a key regulator of biofilm formation and virulence in Staphylococcus epidermidis. J Infect Dis 197:1254–1262. doi: 10.1086/586714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rupp ME, Ulphani JS, Fey PD, Mack D. 1999. Characterization of Staphylococcus epidermidis polysaccharide intercellular adhesin/hemagglutinin in the pathogenesis of intravascular catheter-associated infection in a rat model. Infect Immun 67:2656–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ulphani JS, Rupp ME. 1999. Model of Staphylococcus aureus central venous catheter-associated infection in rats. Lab Animal Sci 49:283–287. [PubMed] [Google Scholar]
- 76.Chauhan A, Lebeaux D, Decante B, Kriegel I, Escande MC, Ghigo JM, Beloin C. 2012. A rat model of central venous catheter to study establishment of long-term bacterial biofilm and related acute and chronic infections. PLoS One 7:e37281. doi: 10.1371/journal.pone.0037281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ebert T, Smith S, Pancari G, Wu X, Zorman J, Clark D, Cook J, Burns C, Antonello JM, Cope L, Nagy E, Meinke A, McNeely T. 2011. Development of a rat central venous catheter model for evaluation of vaccines to prevent Staphylococcus epidermidis and Staphylococcus aureus early biofilms. Hum Vaccines 7:630–638. doi: 10.4161/hv.7.6.15407. [DOI] [PubMed] [Google Scholar]
- 78.de Araujo GL, Coelho LR, de Carvalho CB, Maciel RM, Coronado AZ, Rozenbaum R, Ferreira-Carvalho BT, Figueiredo AM, Teixeira LA. 2006. Commensal isolates of methicillin-resistant Staphylococcus epidermidis are also well equipped to produce biofilm on polystyrene surfaces. J Antimicrob Chemother 57:855–864. doi: 10.1093/jac/dkl071. [DOI] [PubMed] [Google Scholar]
- 79.Hellmark B, Soderquist B, Unemo M, Nilsdotter-Augustinsson A. 2013. Comparison of Staphylococcus epidermidis isolated from prosthetic joint infections and commensal isolates in regard to antibiotic susceptibility, agr type, biofilm production, and epidemiology. Int J Med Microbiol 303:32–39. doi: 10.1016/j.ijmm.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 80.Monk AB, Archer GL. 2007. Use of outer surface protein repeat regions for improved genotyping of Staphylococcus epidermidis. J Clin Microbiol 45:730–735. doi: 10.1128/JCM.02317-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Petrelli D, Zampaloni C, D'Ercole S, Prenna M, Ballarini P, Ripa S, Vitali LA. 2006. Analysis of different genetic traits and their association with biofilm formation in Staphylococcus epidermidis isolates from central venous catheter infections. Eur J Clin Microbiol Infect Dis 25:773–781. doi: 10.1007/s10096-006-0226-8. [DOI] [PubMed] [Google Scholar]
- 82.Stevens NT, Tharmabala M, Dillane T, Greene CM, O'Gara JP, Humphreys H. 2008. Biofilm and the role of the ica operon and aap in Staphylococcus epidermidis isolates causing neurosurgical meningitis. Clin Microbiol Infect 14:719–722. doi: 10.1111/j.1469-0691.2008.02012.x. [DOI] [PubMed] [Google Scholar]
- 83.Fitzpatrick F, Humphreys H, Smyth E, Kennedy CA, O'Gara JP. 2002. Environmental regulation of biofilm formation in intensive care unit isolates of Staphylococcus epidermidis. J Hosp Infect 52:212–218. doi: 10.1053/jhin.2002.1309. [DOI] [PubMed] [Google Scholar]
- 84.Klug D, Wallet F, Kacet S, Courcol RJ. 2003. Involvement of adherence and adhesion Staphylococcus epidermidis genes in pacemaker lead-associated infections. J Clin Microbiol 41:3348–3350. doi: 10.1128/JCM.41.7.3348-3350.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kogan G, Sadovskaya I, Chaignon P, Chokr A, Jabbouri S. 2006. Biofilms of clinical strains of Staphylococcus that do not contain polysaccharide intercellular adhesin. FEMS Microbiol Lett 255:11–16. doi: 10.1111/j.1574-6968.2005.00043.x. [DOI] [PubMed] [Google Scholar]
- 86.Qin Z, Yang X, Yang L, Jiang J, Ou Y, Molin S, Qu D. 2007. Formation and properties of in vitro biofilms of ica-negative Staphylococcus epidermidis clinical isolates. J Med Microbiol 56:83–93. doi: 10.1099/jmm.0.46799-0. [DOI] [PubMed] [Google Scholar]
- 87.Rupp ME, Ulphani JS, Fey PD, Bartscht K, Mack D. 1999. Characterization of the importance of polysaccharide intercellular adhesin/hemagglutinin of Staphylococcus epidermidis in the pathogenesis of biomaterial-based infection in a mouse foreign body infection model. Infect Immun 67:2627–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mehall JR, Saltzman DA, Jackson RJ, Smith SD. 2002. Fibrin sheath enhances central venous catheter infection. Crit Care Med 30:908–912. doi: 10.1097/00003246-200204000-00033. [DOI] [PubMed] [Google Scholar]
- 89.Keller JE, Hindman JW, Mehall JR, Smith SD. 2006. Enoxaparin inhibits fibrin sheath formation and decreases central venous catheter colonization following bacteremic challenge. Crit Care Med 34:1450–1455. doi: 10.1097/01.CCM.0000215832.40827.71. [DOI] [PubMed] [Google Scholar]
- 90.Li H, Xu L, Wang J, Wen Y, Vuong C, Otto M, Gao Q. 2005. Conversion of Staphylococcus epidermidis strains from commensal to invasive by expression of the ica locus encoding production of biofilm exopolysaccharide. Infect Immun 73:3188–3191. doi: 10.1128/IAI.73.5.3188-3191.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chokr A, Leterme D, Watier D, Jabbouri S. 2007. Neither the presence of ica locus, nor in vitro-biofilm formation ability is a crucial parameter for some Staphylococcus epidermidis strains to maintain an infection in a guinea pig tissue cage model. Microb Pathog 42:94–97. doi: 10.1016/j.micpath.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 92.Francois P, Tu Quoc PH, Bisognano C, Kelley WL, Lew DP, Schrenzel J, Cramton SE, Gotz F, Vaudaux P. 2003. Lack of biofilm contribution to bacterial colonisation in an experimental model of foreign body infection by Staphylococcus aureus and Staphylococcus epidermidis. FEMS Immunol Med Microbiol 35:135–140. doi: 10.1016/S0928-8244(02)00463-7. [DOI] [PubMed] [Google Scholar]
- 93.Olson ME, Slater SR, Rupp ME, Fey PD. 2010. Rifampicin enhances activity of daptomycin and vancomycin against both a polysaccharide intercellular adhesin (PIA)-dependent and -independent Staphylococcus epidermidis biofilm. J Antimicrob Chemother 65:2164–2171. doi: 10.1093/jac/dkq314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vuong C, Kocianova S, Voyich JM, Yao Y, Fischer ER, DeLeo FR, Otto M. 2004. A crucial role for exopolysaccharide modification in bacterial biofilm formation, immune evasion, and virulence. J Biol Chem 279:54881–54886. doi: 10.1074/jbc.M411374200. [DOI] [PubMed] [Google Scholar]
- 95.Foka A, Katsikogianni MG, Anastassiou ED, Spiliopoulou I, Missirlis YF. 2012. The combined effect of surface chemistry and flow conditions on Staphylococcus epidermidis adhesion and ica operon expression. Eur Cells Materials 24:386–402. [DOI] [PubMed] [Google Scholar]
- 96.Weaver WM, Milisavljevic V, Miller JF, Di Carlo D. 2012. Fluid flow induces biofilm formation in Staphylococcus epidermidis polysaccharide intracellular adhesin-positive clinical isolates. Appl Environ Microbiol 78:5890–5896. doi: 10.1128/AEM.01139-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hoshal VL Jr, Ause RG, Hoskins PA. 1971. Fibrin sleeve formation on indwelling subclavian central venous catheters. Arch Surg 102:353–358. doi: 10.1001/archsurg.1971.01350040115023. [DOI] [PubMed] [Google Scholar]
- 98.Thoendel M, Kavanaugh JS, Flack CE, Horswill AR. 2011. Peptide signaling in the staphylococci. Chem Rev 111:117–151. doi: 10.1021/cr100370n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vuong C, Gotz F, Otto M. 2000. Construction and characterization of an agr deletion mutant of Staphylococcus epidermidis. Infect Immun 68:1048–1053. doi: 10.1128/IAI.68.3.1048-1053.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yao Y, Vuong C, Kocianova S, Villaruz AE, Lai Y, Sturdevant DE, Otto M. 2006. Characterization of the Staphylococcus epidermidis accessory-gene regulator response: quorum-sensing regulation of resistance to human innate host defense. J Infect Dis 193:841–848. doi: 10.1086/500246. [DOI] [PubMed] [Google Scholar]
- 101.Pinkston KL, Gao P, Diaz-Garcia D, Sillanpaa J, Nallapareddy SR, Murray BE, Harvey BR. 2011. The Fsr quorum-sensing system of Enterococcus faecalis modulates surface display of the collagen-binding MSCRAMM Ace through regulation of gelE. J Bacteriol 193:4317–4325. doi: 10.1128/JB.05026-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Arrecubieta C, Lee MH, Macey A, Foster TJ, Lowy FD. 2007. SdrF, a Staphylococcus epidermidis surface protein, binds type I collagen. J Biol Chem 282:18767–18776. doi: 10.1074/jbc.M610940200. [DOI] [PubMed] [Google Scholar]
- 103.Arrecubieta C, Toba FA, von Bayern M, Akashi H, Deng MC, Naka Y, Lowy FD. 2009. SdrF, a Staphylococcus epidermidis surface protein, contributes to the initiation of ventricular assist device driveline-related infections. PLoS Pathog 5:e1000411. doi: 10.1371/journal.ppat.1000411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Heilmann C, Hussain M, Peters G, Gotz F. 1997. Evidence for autolysin-mediated primary attachment of Staphylococcus epidermidis to a polystyrene surface. Mol Microbiol 24:1013–1024. doi: 10.1046/j.1365-2958.1997.4101774.x. [DOI] [PubMed] [Google Scholar]
- 105.McNab R, Holmes AR, Clarke JM, Tannock GW, Jenkinson HF. 1996. Cell surface polypeptide CshA mediates binding of Streptococcus gordonii to other oral bacteria and to immobilized fibronectin. Infect Immun 64:4204–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.