

Abstract
Animals develop in the presence of complex microbial communities, and early host responses to these microbes can influence key aspects of development, such as maturation of the immune system, in ways that impact adult physiology. We previously showed that the zebrafish intestinal alkaline phosphatase (ALPI) gene alpi.1 was induced by Gram-negative bacterium-derived lipopolysaccharide (LPS), a process dependent on myeloid differentiation primary response gene 88 (MYD88), and functioned to detoxify LPS and prevent excessive host inflammatory responses to commensal microbiota in the newly colonized intestine. In the present study, we examined whether the regulation and function of ALPI were conserved in mammals. We found that among the mouse ALPI genes, Akp3 was specifically upregulated by the microbiota, but through a mechanism independent of LPS or MYD88. We showed that disruption of Akp3 did not significantly affect intestinal inflammatory responses to commensal microbiota or animal susceptibility to Yersinia pseudotuberculosis infection. However, we found that Akp3−/− mice acquired LPS tolerance during postweaning development, suggesting that Akp3 plays an important role in immune education. Finally, we demonstrated that inhibiting LPS sensing with a mutation in CD14 abrogated the accelerated weight gain in Akp3−/− mice receiving a high-fat diet, suggesting that the weight gain is caused by excessive LPS in Akp3−/− mice.
INTRODUCTION
Mammals coexist with a consortium of microorganisms, their microbiota. The most populous microbial community is present in the gastrointestinal tract. A mutually beneficial relationship has been forged between the host and its associated gut microbiota. While the host intestine provides a nutrient-rich environment for the microbes, the gut microbiota modulate host metabolism (1), promote immune maturation (2), preserve gut epithelial barrier function (3), and prevent growth of pathogens (4). Despite these benefits, the intestinal microbiota are a continuous source of antigens and toxins, which can provoke host inflammatory responses. Regulatory mechanisms therefore must exist to prevent unlimited immune activation by microbial products.
Alkaline phosphatases (ALPs) are a superfamily of metalloenzymes that are widely found in organisms ranging from bacteria to humans (5) and catalyze the hydrolytic removal of phosphate from a variety of molecules (6). Importantly, ALPs have been shown to remove the lipid A phosphates of the endotoxin lipopolysaccharide (LPS) (7–10). LPS is a constituent of the outer membrane of Gram-negative bacteria (11) which compose a substantial proportion of the mammalian gut microbiota (12). In mammals, LPS is transferred by LPS binding protein to CD14, which then presents the molecule to the TLR4 (Toll-like receptor 4)/MD-2 receptor complex, resulting in the activation of innate immune signaling. The two phosphate groups of the LPS lipid A moiety support stable binding to the receptor complex, and dephosphorylation of lipid A greatly reduces the inflammatory activity of LPS (13). Although this mechanism of LPS sensing is not conserved in nonmammalian species (e.g., fish lack orthologs of CD14 and MD-2 and have functionally distinct TLR4 homologs [14, 15]), the dephosphorylation of LPS by ALPs ameliorates LPS-induced inflammation and reduces LPS endotoxic properties in a wide range of organisms from cephalopod to tetrapods (16–20).
The mammalian ALP family consists of several isozymes which can be classified as tissue-nonspecific (liver-bone-kidney type) and tissue-specific ALPs (intestinal, placental, and germ cell type) (6, 12). The intestinal ALP (ALPI) is abundantly present in the apical microvilli of the brush border of enterocytes (21) and actively secreted into the intestinal lumen (22, 23). Recent studies have shown that ALPI regulates neutral pH in the intestine (24), protects gut barrier function (18, 25–27), and preserves gut microbial homeostasis (28, 29).
Our studies using zebrafish larvae have demonstrated an integral role of ALPI in establishing a balanced host immune response to the microbiota during gut colonization (16, 30). We showed that intestinal colonization of Gram-negative bacteria upregulated zebrafish ALPI gene alpi.1 through an LPS-induced innate immune signaling mechanism that required Myd88. Furthermore, we showed that ALPI functioned to prevent excessive inflammatory responses to the resident gut microbiota by detoxifying LPS. The LPS-ALPI negative-feedback loop therefore promotes zebrafish immune tolerance to commensal Gram-negative bacteria. Analysis of the evolution of vertebrate ALPI genes revealed a dynamic history of gene losses and duplications and demonstrated that the extant zebrafish, mouse, and human genes are not derived from a common descendant but instead arose independently in each lineage (12). This raises the question of whether the LPS-induced regulation and anti-inflammatory function of ALPI that we described in zebrafish are conserved in mammals. To answer this, we investigated the regulation and function of the mouse ALPI gene Akp3 in host-microbe interactions.
The mouse genome contains four ALP genes: the tissue-nonspecific type Alpl (MGI:87983), the embryonic Alppl2 (MGI:108009), the intestinal Alpi (MGI:1924018), and the duodenal Akp3 (MGI:87984) (6, 12). Alppl2, Alpi, and Akp3 all contribute to ALP activity in the mouse intestine but exhibit distinct expression patterns (31). Alppl2 and Alpi are expressed after birth throughout the intestine but are enriched in the duodenum. In comparison, Akp3 expression starts around postnatal day 13 to 15 and is restricted to the duodenum. The concordant timing of Akp3 expression with weaning and establishment of the adult gut microbiota led us to test the possibility that Akp3 is induced by the microbiota.
Since the generation of Akp3−/− mice (32), several studies have probed the role of Akp3 in animal health and disease. Like other ALPs, AKP3 has LPS dephosphorylation activity, and Akp3−/− mice had significantly lower ALP enzyme and LPS dephosphorylating activities in stools and duodenal mucosa compared to wild-type littermates (18). Akp3−/− mice had increased gut permeability and hence increased LPS translocation from the intestinal lumen into the blood (27). Consequently, Akp3−/− mice displayed higher hepatic expression of major histocompatibility complex class II molecules (20) and signs of metabolic endotoxemia (27). Akp3−/− mice were found to contain dramatically fewer and also different types of aerobic and anaerobic microbes in stools compared to wild-type mice (28, 29). Together, these observations from routinely maintained animals indicate that Akp3 plays a role in maintaining gut barrier function and microbial homeostasis. The role of AKP3 as a gut mucosal defense factor is more evident when mice were challenged. Compared to wild-type mice, Akp3−/− mice suffered from more severe gut mucosal disruption and higher bacterial translocation after ischemia/reperfusion injuries (18) and were more vulnerable to dextran sulfate sodium-induced colitis (33). Interestingly, Akp3−/− mice showed immune tolerance to intraperitoneal LPS injection and resistance to Salmonella enterica serovar Typhimurium infection (20).
All of the aforementioned results were obtained from full-grown mice, but little is known about the biological function of Akp3 in developing animals. In this report, we compared intestinal inflammatory responses of wild-type and Akp3−/− mice at different ages to resident microbiota, orally administered LPS, and the Gram-negative pathogen Yersinia pseudotuberculosis. Based on our previous observations in zebrafish (16), we speculated that the partial reduction in ALPI activity due to disruption of one ALPI gene would result in subtle inflammatory reactions to the microbiota in Akp3−/− mice during development, which might lead to long-term changes in their immune sensitivity to future bacterial or LPS challenges. In addition, to test whether the reported resistance of Akp3−/− mice to colonic S. Typhimurium infection is pathogen or tissue specific, we investigated the requirement of Akp3 for defense against infection by Y. pseudotuberculosis, which infects the mouse ileum, as well as the colon (34).
Finally, we explored a possible connection between the inflammatory status of Akp3−/− mice and their accelerated weight gain when maintained on a high-fat diet (HFD) (27, 32). Long-term HFD feeding has been shown to increase plasma LPS (35–38), and continuous infusion of LPS is sufficient to induce weight gain to a similar extent as HFD feeding (36). Furthermore, loss-of-function mutation of LPS receptors TLR4 or CD14 protects mice against HFD-induced obesity (36, 39–41). We report that LPS sensing is required for the accelerated weight gain observed in Akp3−/− mice receiving HFD.
MATERIALS AND METHODS
Mice.
Animals were maintained and all procedures were performed with approval of the University of Oregon Institutional Animal Care and Use Committee. Pairs of Akp3+/− C57BL/6 mice (32) were shipped from the Millán laboratory at the Sanford-Burnham Medical Research Institute, La Jolla, CA. Genotyping of Akp3−/− mice was performed as previously established (32). Pairs of Myd88+/− and CD14+/− C57BL/6 mice were obtained from the Jackson Laboratory, and genotyping was performed according to instructions in JAX mouse database. Heterozygous mice were bred at the University of Oregon animal facility to acquire homozygous mutants and wild-type littermates. For HFD feeding, a female Akp3−/− mouse was crossed to a male CD14−/− mouse to generate Akp3+/−;CD14+/− mice, which were subsequently mated to produce progeny of desired genotypes. Mice undergoing the same experiment were mixed housed. Intestinal tissue samples from age-matched Swiss Webster isogenic germfree and “restricted flora” mice, originally from Taconic, were provided by Justin Sonnenburg (Stanford University, Stanford, CA).
ALP activity assay.
One-centimeter segments of the proximal duodenum from mice were collected and cut open longitudinally. The lumen side was gently washed with phosphate-buffered saline (PBS). Cells on the lumen side were harvested by using cell scrapers, homogenized in 1 ml of lysis buffer (50 mM Tris-HCl[pH 7.6], 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100), and centrifuged at 17,900 × g for 15 min at 4°C. The cell-free supernatant was diluted in double-distilled H2O (ddH2O), and the protein concentration was measured using a Bio-Rad protein assay kit (Bio-Rad Laboratories, Inc.). Mouse stools were collected, weighed, homogenized in 1 ml of ddH2O, and centrifuged at 17,900 × g for 15 min at 4°C to collect the supernatant. Diluted supernatants processed from duodenal samples and stools were assayed for ALP activities using a phosphate substrate kit (Thermo Fisher Scientific, Inc.). The ALP activities were normalized to the protein concentration for duodenal samples and to the mass for stools.
LPS administration.
LPS (purified from Salmonella enterica serotype Typhimurium [Sigma-Aldrich]) was dissolved in ddH2O by a 2-min sonication. Age-matched female wild-type and Akp3−/− mice were deprived of food for 3 h, and then LPS was introduced by gavage at indicated doses. Oral gavage was performed using sterile 20 G 1.5-in. feeding needles (curved, ball tipped; Popper & Sons) in 8- and 10-week-old mice and sterilized 24 G 1-in. needles (straight, ball tipped; Roboz Surgical Instrument) in 19-day-old and 4-week-old mice. Water gavage was performed on the control group. Mice were euthanized 3 h after gavage. One-centimeter segments of the proximal duodenum from mice were collected, and the transcription levels of ALP genes were examined by quantitative PCR (qPCR). Then, 2-cm segments of the ileum were collected for qPCR examination of inflammatory markers.
Yersinia pseudotuberculosis infection.
The wild-type Y. pseudotuberculosis strain (YPIII) was provided by Joan Mecsas (Tufts University, Medford, MA). The infection was performed according to the protocol described previously (34). Briefly, female wild-type and Akp3−/− mice (10 weeks old) were fasted overnight. Infection was performed by gavage at 2 × 109 CFU/animal. Uninfected animals were used as the control. Three to five mice of the infected or control group were weighed daily for 5 days. To quantify bacterial colonization, infected mice were euthanized 24 h after the oral inoculation, and Peyer's patches along the small intestine were collected, homogenized in sterile PBS, and plated on TSA plates containing 2 μg of Irgasan (Sigma-Aldrich)/ml to determine the CFU/g of tissue. Two-centimeter segments of the ileum tissue from mice were collected for qPCR examination of the inflammatory markers.
qPCR.
The total RNA was isolated from collected mouse intestinal tissues using a PureLink RNA minikit (Life Technologies, Grand Island, NY), and cDNA was synthesized using a SuperScript III first-strand synthesis system (Life Technologies). qPCR was performed on the StepOnePlus real-time PCR system (Life Technologies) using the Kapa SYBR Fast qPCR kit (Kapa Biosystems). Endogenous reference genes were tested, and 36B4, Gapdh, and Rpl13a were selected for qPCR normalization based on the GeNorm and the Normfinder algorithms using the GenEx software (Multid Analyses AB). Primers used in qPCR are listed in Table 1. Primer amplification efficiency was determined using the LinRegPCR software and incorporated to normalize the relative levels of gene expression to endogenous references using ΔΔCT analysis.
TABLE 1.
Primers used in qPCR
| Primer | Sequence (5′–3′) |
|---|---|
| 36B4fwd | TCCAGGCTTTGGGCATCA |
| 36B4rev | CTTTATCAGCTGCACATCACTCAGA |
| Gapdhfwd | GCACCACCAACTGCTTAGC |
| Gapdhrev | GGCATGGACTGTGGTCATGAG |
| Rpl13afwd | TTCTGGTATTGGATGGCCGAG |
| Rpl13arev | ATGTTGATGCCTTCACAGCG |
| Akp3fwd | ACATTGCTACACAACTCATCTCC |
| Akp3rev | TCCTGCCATCCAATCTGGTTC |
| Alppl2fwd | AATGCCTGCCTCAGCGCTACAGGA |
| Alppl2rev | TGGTCTGGTGTCCCCTTGGGAA |
| Alpifwd | GGCTACACACTTAGGGGGACCTCCA |
| Alpirev | AGCTTCGGTGACATTGGGCCGGTT |
| Tnfαfwd | AGGCTGCCCCGACTACGT |
| Tnfαrev | GACTTTCTCCTGGTATGAGATAGCAAA |
| Il1βfwd | GAAATGCCACCTTTTGACAGTG |
| Il1βrev | TGGATGCTCTCATCAGGACAG |
| Il6fwd | TGGTACTCCAGAAGACCAGAGG |
| Il6rev | AACGATGATGCACTTGCAGA |
| Lcn2fwd | TGGCCCTGAGTGTCATGTG |
| Lcn2rev | CTCTTGTAGCTCATAGATGGTGC |
HFD feeding.
Male mice of indicated genotypes (≥10 of each) from the same generation were fed a 36.0% fat diet (Bio-Serv, F3282/S3282) after weaning. HFD feeding started when mice were 3 weeks old and continued for 8 weeks. Mouse body weights were recorded weekly.
Neutrophil staining and quantification.
Mouse ileum tissue was collected, embedded in agar, and cryosectioned at a thickness of 16 μm. Slides were washed in PBDT (1% bovine serum albumin, 1% dimethyl sulfoxide, and 0.1% Triton X-100 in PBS) for 30 min, blocked in the block buffer (2% normal goat serum in PBDT), and incubated with rat anti-mouse Ly-6G primary antibody (number 550291; BD Pharmingen, San Jose, CA) at 4°C overnight. The next day, the slides were washed with PBST (0.1% Triton X-100 in PBS) and then incubated with biotin-conjugated mouse anti-rat IgG2b antibody (number 550327; BD Pharmingen) at 4°C overnight. The slides were washed with PBST, incubated with Alexa Fluor 555-streptavidin (catalog no. S21381; Life Technologies), and finally mounted with Vectashield mounting medium with DAPI (4′,6′-diamidino-2-phenylindole; Vector Laboratories, Burlingame, CA). The total number of Ly6G-positive cells was counted from five ileal cross sections from each individual mouse and was used as an indicator of neutrophil infiltration in the mouse.
Statistics.
All data were analyzed using Prism software (GraphPad). The statistical analyses used for different data sets are indicated in the figure legends.
RESULTS
Akp3 is upregulated by the microbiota.
To test whether Akp3 expression was induced by the microbiota, as is the case for zebrafish alpi.1, we measured the transcription levels of ALP genes in duodenal samples of age-matched, isogenic germfree and conventionally reared mice using qPCR. We found that germfree mice had reduced Akp3 transcription compared to conventional mice at 4 weeks of age (Fig. 1A). No significant differences in Alppl2 or Alpi transcription levels were observed between germfree and conventional mice. Consistent results were observed in two independent experiments (one shown in Fig. 1A), suggesting that the microbiota specifically upregulate Akp3.
FIG 1.

Akp3 is upregulated by the microbiota in an LPS- and MYD88-independent manner. (A) Comparison of Akp3, Alppl2, and Alpi levels between 4-week-old conventionally raised (CONR) and germfree (GF) mice. (B) Comparison of Akp3 transcription levels between control, water-treated, and LPS-treated wild-type mice (19 days, 4 weeks, and 8 weeks old). (C) Comparison of Akp3 transcription levels between 4-week-old wild-type (WT) and Myd88−/− mice. Error bars represent standard deviations. The number below the graph indicates the sample size. Asterisks indicate a significant difference (***, P < 0.001 [Student t test]).
We next investigated whether MYD88-dependent LPS signaling induced Akp3, similar to the regulation of zebrafish alpi.1. We treated wild-type mice with water or LPS by oral gavage at various time points, concurrent with and after the normal onset of Akp3 expression (at 19 days and at 4 and 8 weeks of age), and measured Akp3 transcription levels. We observed no induction of Akp3 in the mouse duodenum upon oral LPS administration at any of these time points (Fig. 1B). Furthermore, Myd88−/− mice and their wild-type littermates (4 weeks old) had comparable levels of Akp3 transcription (Fig. 1C). We conclude that, whereas the microbiota upregulate Akp3 in the mouse intestine, the mechanism is different from that in zebrafish.
Basal inflammatory responses are normal in postweaning and adult Akp3−/− mice.
We used the Akp3 loss-of-function mice to study the role of ALP in modulating intestinal inflammatory responses to resident microbiota during postweaning development. We first confirmed that ALP activities were significantly reduced in the intestines of Akp3−/− mice versus wild-type mice after the onset of Akp3 expression. We detected ALP deficiency in Akp3−/− mice, as measured by enzymatic assay on duodenal epithelial cells (Fig. 2A). ALP deficiency was also detected in stools of Akp3−/− mice (Fig. 2B), indicating that the mutant mice have reduced ALP activity throughout the intestine.
FIG 2.

Akp3−/− mice have reduced ALPI in the intestine. Comparison of ALP activities between wild-type (WT) and Akp3−/− mice in duodenal epithelia (dALP) (A) and stools (sALP) (B) at different ages, as indicated at the top of each bar graph. Error bars represent the standard deviations. The number below the graph indicates the sample size. Asterisks mark significant differences (**, P < 0.01; ***, P < 0.001 [Student t test]).
We expected that ALP deficiency in the intestine of postweaning Akp3−/− mice would result in enhanced intestinal basal inflammatory responses to excess microbiota-derived LPS. To assess inflammatory responses, we measured the transcription levels of proinflammatory cytokines (tumor necrosis factor alpha [TNF-α], interleukin-1β [IL-1β], and IL-6) and the innate immune protein LCN2 using qPCR and compared wild-type mice to Akp3−/− mice. Although the duodenum is the site of Akp3 expression, it is sparsely colonized by bacteria (42), and we detected only very low levels of cytokine transcripts in duodenal samples of both wild-type and Akp3−/− mice (data not shown). We hypothesized that the LPS-detoxifying function of AKP3 would be more important in the distal intestine, which is more densely colonized and exposed to secreted ALP. We therefore examined basal inflammatory responses in the ileum. We detected comparable levels of the four inflammatory markers in the ilea of postweaning wild-type and Akp3−/− mice (19 days, 4 weeks, 8 weeks, or 10 weeks old) (Fig. 3). We also measured neutrophil infiltration in the ileum by counting Ly6G-positive cells and observed no difference between wild-type and Akp3−/− mouse ileal tissues (data not shown). We therefore conclude that loss of AKP3 does not result in significant intestinal inflammation.
FIG 3.

Akp3−/− mice show normal basal intestinal inflammatory responses. Comparison of ileal transcriptional levels of Tnf-α, Il-1β, Il-6, and Lcn2 between wild-type (white columns) and Akp3−/− (gray columns) mice at different ages. Error bars represent the standard deviations. The number below the graph indicates the sample size.
Akp3 is not required for defense against Yersinia pseudotuberculosis infection.
Although disruption of Akp3 has no obvious effect on basal intestinal inflammatory responses in healthy animals, we reasoned that the functional requirement for Akp3 might be more pronounced when the equilibrium between host and the gut microbiota is severely disturbed, as in the case of pathogen infection. We speculated that Akp3−/− mice would be more susceptible to Gram-negative pathogen infection due to the reduced capability to dephosphorylate and detoxify the pathogen-derived LPS.
Y. pseudotuberculosis is a well-studied Gram-negative pathogen that colonizes the mouse small intestine (34, 43). We infected 10-week-old wild-type and Akp3−/− female mice with the pathogen by oral gavage and monitored animal weight losses over 5 days. We observed similar weight losses in wild-type and Akp3−/− mice (Fig. 4A), indicating equal susceptibilities to infection in both genotypes. To investigate the infection dynamics, we examined dissemination of Y. pseudotuberculosis into Peyer's patches along the mouse small intestine 24 h after the initial inoculation. We found that Y. pseudotuberculosis colonized at similar levels in Peyer's patches of wild-type and Akp3−/− mice (Fig. 4B), indicating that Akp3 does not prevent translocation of the pathogen. We then assessed the ileal inflammatory responses of wild-type and Akp3−/− mice at this time point, when they were equally colonized (Fig. 4C). We found that Tnf-α and Il-1β transcription levels were significantly elevated in wild-type and Akp3−/− mice. Il-6 also seemed to be induced, although not significantly, from the basal level in wild-type and Akp3−/− mice. Interestingly, Lcn2 appeared only upregulated in Akp3−/− mice, which might suggest increased sensitivity of Akp3−/− mice to Y. pseudotuberculosis infection in this specific innate immune response. Taken together, our data demonstrated no clear requirement for Akp3 in animal defense against Y. pseudotuberculosis infection.
FIG 4.

Akp3−/− and wild-type mice show similar susceptibilities to Y. pseudotuberculosis infection. (A) Body weight changes of wild-type (WT) and Akp3−/− mice, uninfected or infected with Y. pseudotuberculosis (Yp). Error bars indicate the standard errors of the mean (SEM). (B) Colonization of Y. pseudotuberculosis in Peyer's patches. Each dot represents a mouse. (C) qPCR quantification of transcription levels of Tnf-α, Il-1β, Il-6, and Lcn2 in the small intestines of uninfected or infected wild-type (WT) and Akp3−/− mice (10 weeks old). Error bars represent the standard deviations. The numbers below the graphs indicate the sample sizes. Asterisks mark the significant differences (*, P < 0.05; **, P < 0.01; determined by one-way analysis of variance [ANOVA], followed by the Bonferroni test).
Akp3−/− mice acquire LPS tolerance in the small intestine during development.
We speculated that oral administration of Y. pseudotuberculosis might lead to influx of large amounts of LPS, which greatly surpassed the dephosphorylation capacity of the mouse intestine, thus masking a possible requirement for AKP3 to detoxify LPS in a less extreme situation. To experimentally test this, we challenged mice with different doses of LPS.
We first challenged 10-week-old mice with a low dose of LPS (100 mg/kg of body weight) by a one-time oral gavage and compared intestinal inflammatory responses between wild-type and Akp3−/− mice. Comparable LPS dosing was shown not to induce antimicrobial factors such as RegIIIγ, CRP-ductin, and RELMβ in germfree mice (44). We reasoned that the small amount of LPS might be effectively dephosphorylated only in wild-type mice, resulting in stronger inflammatory responses in Akp3−/− mutants. On the contrary, we found that all four inflammatory markers (TNF-α, IL-1β, IL-6, and LCN2) were significantly upregulated in wild-type but not Akp3−/− mice after the low-dose LPS gavage (Fig. 5A). Moreover, after LPS gavage wild-type mice showed significantly higher transcription levels of Tnf-α, Il-1β, Il-6, and Lcn2 in the small intestine than did Akp3−/− mice (Fig. 5A). However, when challenged with a higher dose of LPS (200 mg/kg of body weight), wild-type and Akp3−/− mice exhibited similar intestinal inflammatory responses (Fig. 5B).
FIG 5.

Adult Akp3−/− mice show intestinal immune tolerance to low-dose LPS. qPCR quantification of transcription levels of Tnf-α, Il-1β, Il-6, and Lcn2 in the small intestines of water- or LPS-treated 10-week-old wild-type (WT) and Akp3−/− mice (LPS at 100 mg/kg of body weight [A]; LPS at 200 mg/kg of body weight [B]). Error bars represent the standard deviations. The numbers below the graphs indicate the sample sizes. Asterisks mark significant differences (*, P < 0.05; **, P < 0.01; ***, P < 0.001 [one-way ANOVA, followed by the Bonferroni test]).
The blunted response of the intestinal innate immune system to the low-dose LPS in adult Akp3−/− mice reminded us of a well-recognized phenomenon, endotoxin tolerance. Endotoxin/LPS tolerance describes a situation where animals or innate immune cells become refractory to endotoxin challenge after a prior exposure to small amounts of LPS (45). We reasoned that reduced ALP activity in the Akp3−/− mouse intestine could result in higher luminal concentrations of phosphorylated LPS and continuous exposure to more immunostimulatory LPS during development would prime the intestinal innate immune system, in the long term making it less responsive to further LPS challenge. To test this theory, we examined intestinal inflammatory responses to low doses of LPS in younger mice, expecting to see no LPS tolerance at early stages of development. Indeed, we found that the low-dose LPS challenge caused similar inflammatory responses in the small intestines of wild-type and Akp3−/− mice at 19 days and 4 weeks (Fig. 6A and B) old, as demonstrated by the transcription levels of Tnf-α, Il-1β, Il-6, and Lcn2. Interestingly, 8-week-old Akp3−/− mice exhibited “partial” tolerance to the low-dose LPS (Fig. 6C): whereas Tnf-α and Il-1β were significantly induced, Il-6 and Lcn2 transcription remained at low levels. Together, the data demonstrated that Akp3−/− mice acquired tolerance to low doses of LPS through a long period of postweaning development, a finding consistent with these mice being exposed to higher concentrations of microbiota-derived LPS due to deficiency in intestinal ALP LPS-detoxification activity.
FIG 6.

Akp3−/− mice acquire endotoxin tolerance during postweaning development. qPCR quantification of transcription levels of Tnf-α, Il-1β, Il-6, and Lcn2 in the small intestines of wild-type (WT) and Akp3−/− mice, treated with water or LPS (100 mg/kg of body weight) at 19 days (A), 4 weeks (B), or 8 weeks (C) of age. Error bars represent the standard deviations. The numbers below the graphs indicate the sample sizes. Asterisks mark the significant difference (*, P < 0.05; **, P < 0.01 [one-way ANOVA, followed by the Bonferroni test]).
LPS-induced innate immune signaling is required for HFD-induced accelerated weight gain of Akp3−/− mice.
Akp3−/− mice exhibit accelerated weight gain when maintained on HFD (32). Obesity is associated with elevated serum LPS and chronic low-grade inflammation of adipose tissue through mechanisms that are not fully understood but require LPS sensing (46). We hypothesized that reduced LPS detoxification in the Akp3−/− mouse intestine and hence excessive LPS accumulation could underlie the accelerated weight gain observed in these animals when maintained on HFD. To test the hypothesis, we sought to determine whether the HFD-induced accelerated weight gain in Akp3−/− mice was dependent on their ability to sense LPS. We reasoned that if we blocked LPS-sensing in Akp3−/− mice by mutating the LPS coreceptor CD14, they would no longer be affected by excessive microbiota-derived LPS and therefore should be resistant to the HFD-induced faster weight gain. We thus bred wild-type, Akp3−/−, CD14−/−, and Akp3−/−;CD14−/− mice and kept them on an HFD, starting from 3 weeks of age. Animal body weight gains were monitored and compared (Fig. 7). As previously reported (32), Akp3−/− mice showed faster weight gain compared to wild-type mice. The weight gain curve of Akp3−/−;CD14−/− double mutants, however, was statistically indistinguishable from that of wild-type or CD14−/− mice. The results demonstrate that LPS-sensing is required for the HFD-induced accelerated weight gain of Akp3−/− mice, suggesting that their accelerated weight gain is caused by the accumulation of excessive microbiota-derived LPS.
FIG 7.
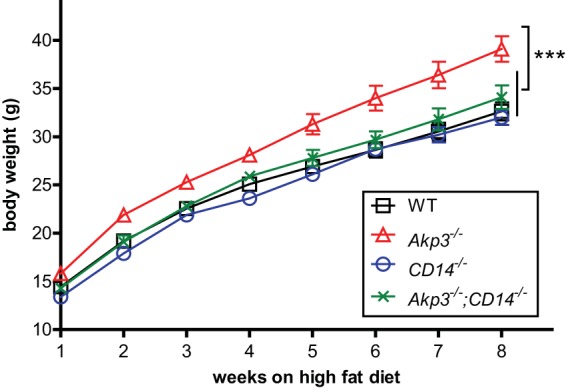
LPS sensing is required for HFD-induced obesity in Akp3−/− mice. Body weight changes of wild-type (WT), Akp3−/−, CD14−/−, and Akp3−/−;CD14−/− mice fed an HFD were determined. Error bars represent the SEM. Asterisks indicate a significant difference (***, P < 0.001 [two-way ANOVA]).
DISCUSSION
The intestinal microbiota are an important source of signals that influence gut development and impact animal physiology. We show here that the microbiota upregulate the mouse ALPI gene Akp3. Although we observed no overt signs of intestinal inflammation or increased susceptibility to challenge with an intestinal pathogen in Akp3-deficient animals, we uncovered a role for AKP3's LPS detoxification activity in the finding that Akp3−/− mice become desensitized to low doses of LPS during the first few weeks after weaning. We further showed that this early subtle defect in LPS detoxification was related to the susceptibility of adult Akp3−/− mice to HFD-induced obesity, which was blocked with a mutation in the LPS coreceptor CD14. These findings demonstrate how subtle alterations in host-microbiota interactions early in development can have long-lasting and systemic effects on animal physiology.
Akp3 is upregulated by the microbiota through a nonconserved mechanism.
The intestinal microbiota are an important source of signals that regulate gene transcription in animal intestines (47–50). Consistent with the upregulation of Akp3 at the time of weaning and establishment of the adult gut microbiota, we show that Akp3 expression is reduced in germfree mouse intestines. In contrast with zebrafish alpi.1, we found that mouse Akp3 is upregulated by the microbiota independent of MYD88. Furthermore, we showed that LPS did not induce Akp3, even when administered at 19 days when several cytokine genes showed robust LPS induction. The discrepancy is not entirely surprising considering the evolution history of the ALPI genes (12). Akp3, along with Alppl2, arose from recent tandem duplications of Alpi in the mouse lineage, long after the divergence of zebrafish alpi.1 and mouse Alpi, and following the divergence between rodents and primates. Since levels of Alpi and Alppl2 are not significantly reduced in germfree mice, the most parsimonious explanation for Akp3 induction by microbiota is through a newly acquired, rodent-specific mechanism. In addition, mammals and teleosts have evolved different strategies for LPS reception, with divergent TLR4 sensing functions (14, 15).
We speculate that the microbiota upregulate Akp3 through metabolic interactions with the host. Many dietary components such as fat (51–53), as well as micronutrients such as calcium (54), have been reported to modulate ALPI expression or activity. The gut microbiota perform essential metabolic functions that are not encoded in the host genome, including the processing of complex proteins and indigestible dietary fibers, and the synthesis of vitamins such as vitamin K (55). Interestingly, vitamin K has been shown to elevate ALPI activity in rodents (56), and oral administration of vitamin K specifically induced Akp3 and Alpi expression in the mouse intestine, with a more dramatic effect on Akp3 than Alpi (56, 57). Vitamin K production, which is dependent on the gut microbiota but independent of immune responses, may be one mechanism by which the microbiota upregulate Akp3.
A requirement for Akp3 in host-microbiota equilibrium or enteric infection is not detectable.
We demonstrate that Akp3 is not required for maintaining mouse immune tolerance to resident microbiota in the intestine, as shown by comparable levels of inflammatory markers in wild-type and Akp3−/− mice. We measured considerable ALP activity retained in the Akp3−/− mouse intestine, presumably from Alppl2 and Alpi, which is likely sufficient to prevent excessive inflammatory responses to the gut microbiota. Furthermore, Alppl2 and Alpi expression was reported to be increased in Akp3−/− mice (31). The ALPI gene redundancy and compensatory gene regulation likely combine to keep the inflammatory responses in check in Akp3−/− mice. The unaffected basal inflammatory responses in the small intestine after disruption of Akp3 suggest a robust equilibrium between host immunity and the gut microbiota.
Exogenous ALPI treatment has been reported to preserve the normal gut barrier function (26, 27, 33) and prevent gut bacterial translocation after experimental injury in rodent models (25). Consistent with the role of ALPI in promoting gut barrier function, disruption of Akp3 caused increased gut permeability (27) and more gut bacterial translocation into mesenteric tissues after intestinal ischemia/reperfusion injury (18). We observed here that Akp3 provides no protection against Y. pseudotuberculosis invasion into Peyer's patches. We think that these results are not contradictive but could represent fundamental differences between commensal bacteria and virulent pathogens. ALPI's role in promoting intestinal epithelial integrity was sufficient to block the passive translocation of commensal bacteria in the injury models. However, pathogens such as Y. pseudotuberculosis have developed sophisticated strategies to breach the gut barrier, overcoming the protective activity of ALPI.
Our results also showed that Akp3 confers no benefits with respect to intestinal inflammatory responses to Y. pseudotuberculosis infection. We speculated that the large amount of LPS introduced by Y. pseudotuberculosis inoculation exceeded the dephosphorylation and detoxification capacity of the mouse intestine. We tested this experimentally and found that Akp3 does not affect mouse intestinal inflammatory responses to high doses of LPS, which could explain why exogenous supplementation of ALPI is usually necessary to elicit its beneficial effects in various injury and disease models (8–10, 16, 25, 26, 29, 33, 58–69).
Akp3 is required for normal LPS sensitivity.
Although Akp3 does not appear to be required for regulating the basal level of inflammatory responses to the gut microbiota or for defending against Y. pseudotuberculosis infection, we demonstrated an important role of Akp3 in establishing LPS sensitivity during postweaning development. By characterizing the intestinal inflammatory responses to low doses of LPS in developing animals, we showed that Akp3−/− mice acquired intestinal immune tolerance to LPS during postweaning development, with LPS desensitization occurring around 8 to 10 weeks of age. We hypothesize that reduced ALPI activity in Akp3−/− mice causes accumulation of phosphorylated LPS in the gut lumen, which induces a slightly more active innate immune response and in the long term trains the intestinal immune system to be less responsive to further LPS challenge.
Endotoxin tolerance profoundly influences animal responses to infections. Mice with induced endotoxin tolerance were reported to be more resistant to S. Typhimurium and Cryptococcus infections (70, 71) but more susceptible to E. coli (72). Consistent with our observation of increased LPS tolerance in Akp3−/− mice, they have also been shown to be more resistant to S. Typhimurium infection, exhibiting less dramatic weight loss and reduced bacterial translocation compared to wild-type mice (20). Our observation that wild-type and Akp3−/− mice have similar susceptibilities to Y. pseudotuberculosis infection emphasizes the complexity of host responses to LPS during infection with different pathogens. Notably, Yersinia species are known to modify their LPS structure upon host colonization to adopt a less immunostimulatory form, suggesting that these bacteria's pathogenesis strategy may be better adapted to an LPS-tolerant host than S. Typhimurium (73).
Subtle dysregulation of LPS-induced innate immune signaling due to Akp3 deficiency contributes to HFD-induced obesity.
ALPI activities are inversely related to the obesity proneness in rats receiving HFD (74, 75). Analogously, mice lacking Akp3 exhibited accelerated weight gain when maintained on HFD compared to wild-type mice (32). A direct role of the mouse gene Alpi in promoting fatty acid intake was proposed, and upregulation of Alpi in Akp3−/− mice could therefore contribute to the HFD-induced faster weight gain (31, 76, 77). Akp3−/− mice have also been shown to have elevated serum LPS levels, presumably due to their impaired LPS dephosphorylation in the intestine and increased gut permeability (27). We hypothesized that elevated basal levels of LPS in Akp3−/− mice result in a physiological state in which the innate immune system no longer responses acutely to low-level LPS challenge but rather the tissues exist in a state of chronic, low-grade inflammation that promotes weight gain (46). Here, we tested the specific contribution of LPS-induced innate immune signaling to HFD-induced accelerated weight gain in Akp3−/− mice and found that by blocking LPS sensing with CD14 deficiency we abrogated the faster weight gain phenotype in these mice. This provides evidence that LPS-mediated innate immune signaling is required for the HFD-induced phenotype of Akp3−/− mice. Elevated, LPS-dependent, chronic, low-grade inflammation signaling might also underlie the inverse relationship between ALPI activities and the obesity proneness of rats receiving HFD. It would be interesting to evaluate the contribution of LPS sensing in these models.
In conclusion, we have discovered that the mouse ALPI gene Akp3 is specifically upregulated by the microbiota in an LPS- and MYD88-independent manner. We have showed that disruption of Akp3 leads to immune desensitization to LPS during postweaning development, demonstrating a requirement for Akp3's LPS detoxifying activity in immune education. Finally, we have linked these earlier subtle defects in LPS detoxification to later onset of HFD-induced obesity in Akp3−/− mice by showing that the LPS-sensing machinery is required for the accelerated weight gain in Akp3−/− mice maintained on HFD. Collectively, these studies show that subtle dysregulation of host-microbiota interactions in the gut can have long-term systemic effects on host immunity and physiology.
ACKNOWLEDGMENTS
We thank Jennifer Bates for her contributions to this project in its early stages, Lora Hooper for valuable early assistance, and Justin Sonnenberg for providing germfree mouse tissue samples. We especially thank the staff of the University of Oregon Mouse Facility for animal management and Poh Kheng Loi and the staff of the University of Oregon histology facility for histology services.
The project described was supported by awards R01DK075549 from the National Institute of Diabetes and Digestive and Kidney Diseases and award P50GM098911 from the National Institute of General Medical Sciences of the National Institutes of Health.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We have no conflicting financial interests.
REFERENCES
- 1.Tremaroli V, Backhed F. 2012. Functional interactions between the gut microbiota and host metabolism. Nature 489:242–249. doi: 10.1038/nature11552. [DOI] [PubMed] [Google Scholar]
- 2.Belkaid Y, Hand TW. 2014. Role of the microbiota in immunity and inflammation. Cell 157:121–141. doi: 10.1016/j.cell.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goto Y, Kiyono H. 2012. Epithelial barrier: an interface for the cross-communication between gut flora and immune system. Immunol Rev 245:147–163. doi: 10.1111/j.1600-065X.2011.01078.x. [DOI] [PubMed] [Google Scholar]
- 4.Kamada N, Chen GY, Inohara N, Nunez G. 2013. Control of pathogens and pathobionts by the gut microbiota. Nat Immunol 14:685–690. doi: 10.1038/ni.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McComb RB, Bowers GN Jr, Posen S. 1979. Alkaline phosphatase. Plenum Press, Inc, New York, NY. [Google Scholar]
- 6.Millán JL. 2006. Mammalian alkaline phosphatases: from biology to applications in medicine and biotechnology. Wiley-VCH, Weinheim, Germany. [Google Scholar]
- 7.Bentala H, Verweij WR, Huizinga-Van der Vlag A, van Loenen-Weemaes AM, Meijer DK, Poelstra K. 2002. Removal of phosphate from lipid A as a strategy to detoxify lipopolysaccharide. Shock 18:561–566. doi: 10.1097/00024382-200212000-00013. [DOI] [PubMed] [Google Scholar]
- 8.Riggle KM, Rentea RM, Welak SR, Pritchard KA Jr, Oldham KT, Gourlay DM. 2013. Intestinal alkaline phosphatase prevents the systemic inflammatory response associated with necrotizing enterocolitis. J Surg Res 180:21–26. doi: 10.1016/j.jss.2012.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tuin A, Poelstra K, de Jager-Krikken A, Bok L, Raaben W, Velders MP, Dijkstra G. 2009. Role of alkaline phosphatase in colitis in man and rats. Gut 58:379–387. doi: 10.1136/gut.2007.128868. [DOI] [PubMed] [Google Scholar]
- 10.van Veen SQ, Dinant S, van Vliet AK, van Gulik TM. 2006. Alkaline phosphatase reduces hepatic and pulmonary injury in liver ischaemia: reperfusion combined with partial resection. Br J Surg 93:448–456. doi: 10.1002/bjs.5275. [DOI] [PubMed] [Google Scholar]
- 11.Elin RJ, Wolff SM. 1976. Biology of endotoxin. Annu Rev Med 27:127–141. doi: 10.1146/annurev.me.27.020176.001015. [DOI] [PubMed] [Google Scholar]
- 12.Yang Y, Wandler AM, Postlethwait JH, Guillemin K. 2012. Dynamic evolution of the LPS-detoxifying enzyme intestinal alkaline phosphatase in zebrafish and other vertebrates. Front Immunol 3:314. doi: 10.3389/fimmu.2012.00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park BS, Lee JO. 2013. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp Mol Med 45:e66. doi: 10.1038/emm.2013.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sullivan C, Charette J, Catchen J, Lage CR, Giasson G, Postlethwait JH, Millard PJ, Kim CH. 2009. The gene history of zebrafish tlr4a and tlr4b is predictive of their divergent functions. J Immunol 183:5896–5908. doi: 10.4049/jimmunol.0803285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sepulcre MP, Alcaraz-Perez F, Lopez-Munoz A, Roca FJ, Meseguer J, Cayuela ML, Mulero V. 2009. Evolution of lipopolysaccharide (LPS) recognition and signaling: fish TLR4 does not recognize LPS and negatively regulates NF-κB activation. J Immunol 182:1836–1845. doi: 10.4049/jimmunol.0801755. [DOI] [PubMed] [Google Scholar]
- 16.Bates JM, Akerlund J, Mittge E, Guillemin K. 2007. Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe 2:371–382. doi: 10.1016/j.chom.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rader BA, Kremer N, Apicella MA, Goldman WE, McFall-Ngai MJ. 2012. Modulation of symbiont lipid A signaling by host alkaline phosphatases in the squid-vibrio symbiosis. mBio 3:e00093-12. doi: 10.1128/mBio.00093-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldberg RF, Austen WG Jr, Zhang X, Munene G, Mostafa G, Biswas S, McCormack M, Eberlin KR, Nguyen JT, Tatlidede HS, Warren HS, Narisawa S, Millan JL, Hodin RA. 2008. Intestinal alkaline phosphatase is a gut mucosal defense factor maintained by enteral nutrition. Proc Natl Acad Sci U S A 105:3551–3556. doi: 10.1073/pnas.0712140105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen KT, Malo MS, Moss AK, Zeller S, Johnson P, Ebrahimi F, Mostafa G, Alam SN, Ramasamy S, Warren HS, Hohmann EL, Hodin RA. 2010. Identification of specific targets for the gut mucosal defense factor intestinal alkaline phosphatase. Am J Physiol Gastrointest Liver Physiol 299:G467–G475. doi: 10.1152/ajpgi.00364.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen KT, Malo MS, Beasley-Topliffe LK, Poelstra K, Millan JL, Mostafa G, Alam SN, Ramasamy S, Warren HS, Hohmann EL, Hodin RA. 2011. A role for intestinal alkaline phosphatase in the maintenance of local gut immunity. Dig Dis Sci 56:1020–1027. doi: 10.1007/s10620-010-1396-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanna SD, Mircheff AK, Wright EM. 1979. Alkaline phosphatase of basal lateral and brush border plasma membranes from intestinal epithelium. J Supramol Struct 11:451–466. doi: 10.1002/jss.400110404. [DOI] [PubMed] [Google Scholar]
- 22.McConnell RE, Higginbotham JN, Shifrin DA Jr, Tabb DL, Coffey RJ, Tyska MJ. 2009. The enterocyte microvillus is a vesicle-generating organelle. J Cell Biol 185:1285–1298. doi: 10.1083/jcb.200902147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shifrin DA Jr, McConnell RE, Nambiar R, Higginbotham JN, Coffey RJ, Tyska MJ. 2012. Enterocyte microvillus-derived vesicles detoxify bacterial products and regulate epithelial-microbial interactions. Curr Biol 22:627–631. doi: 10.1016/j.cub.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizumori M, Ham M, Guth PH, Engel E, Kaunitz JD, Akiba Y. 2009. Intestinal alkaline phosphatase regulates protective surface microclimate pH in rat duodenum. J Physiol 587:3651–3663. doi: 10.1113/jphysiol.2009.172270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez-Moya P, Ortega-Gonzalez M, Gonzalez R, Anzola A, Ocon B, Hernandez-Chirlaque C, Lopez-Posadas R, Suarez MD, Zarzuelo A, Martinez-Augustin O, Sanchez de Medina F. 2012. Exogenous alkaline phosphatase treatment complements endogenous enzyme protection in colonic inflammation and reduces bacterial translocation in rats. Pharmacol Res 66:144–153. doi: 10.1016/j.phrs.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 26.Rentea RM, Liedel JL, Welak SR, Cassidy LD, Mayer AN, Pritchard KA Jr, Oldham KT, Gourlay DM. 2012. Intestinal alkaline phosphatase administration in newborns is protective of gut barrier function in a neonatal necrotizing enterocolitis rat model. J Pediatr Surg 47:1135–1142. doi: 10.1016/j.jpedsurg.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 27.Kaliannan K, Hamarneh SR, Economopoulos KP, Nasrin Alam S, Moaven O, Patel P, Malo NS, Ray M, Abtahi SM, Muhammad N, Raychowdhury A, Teshager A, Mohamed MM, Moss AK, Ahmed R, Hakimian S, Narisawa S, Millan JL, Hohmann E, Warren HS, Bhan AK, Malo MS, Hodin RA. 2013. Intestinal alkaline phosphatase prevents metabolic syndrome in mice. Proc Natl Acad Sci U S A 110:7003–7008. doi: 10.1073/pnas.1220180110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malo MS, Alam SN, Mostafa G, Zeller SJ, Johnson PV, Mohammad N, Chen KT, Moss AK, Ramasamy S, Faruqui A, Hodin S, Malo PS, Ebrahimi F, Biswas B, Narisawa S, Millan JL, Warren HS, Kaplan JB, Kitts CL, Hohmann EL, Hodin RA. 2010. Intestinal alkaline phosphatase preserves the normal homeostasis of gut microbiota. Gut 59:1476–1484. doi: 10.1136/gut.2010.211706. [DOI] [PubMed] [Google Scholar]
- 29.Malo MS, Moaven O, Muhammad N, Biswas B, Alam SN, Economopoulos KP, Gul SS, Hamarneh SR, Malo NS, Teshager A, Mohamed MM, Tao Q, Narisawa S, Millan JL, Hohmann EL, Warren HS, Robson SC, Hodin RA. 2014. Intestinal alkaline phosphatase promotes gut bacterial growth by reducing the concentration of luminal nucleotide triphosphates. Am J Physiol Gastrointest Liver Physiol 306:G826–G838. doi: 10.1152/ajpgi.00357.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bates JM, Mittge E, Kuhlman J, Baden KN, Cheesman SE, Guillemin K. 2006. Distinct signals from the microbiota promote different aspects of zebrafish gut differentiation. Dev Biol 297:374–386. doi: 10.1016/j.ydbio.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 31.Narisawa S, Hoylaerts MF, Doctor KS, Fukuda MN, Alpers DH, Millan JL. 2007. A novel phosphatase upregulated in Akp3 knockout mice. Am J Physiol Gastrointest Liver Physiol 293:G1068–G1077. doi: 10.1152/ajpgi.00073.2007. [DOI] [PubMed] [Google Scholar]
- 32.Narisawa S, Huang L, Iwasaki A, Hasegawa H, Alpers DH, Millan JL. 2003. Accelerated fat absorption in intestinal alkaline phosphatase knockout mice. Mol Cell Biol 23:7525–7530. doi: 10.1128/MCB.23.21.7525-7530.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramasamy S, Nguyen DD, Eston MA, Alam SN, Moss AK, Ebrahimi F, Biswas B, Mostafa G, Chen KT, Kaliannan K, Yammine H, Narisawa S, Millan JL, Warren HS, Hohmann EL, Mizoguchi E, Reinecker HC, Bhan AK, Snapper SB, Malo MS, Hodin RA. 2011. Intestinal alkaline phosphatase has beneficial effects in mouse models of chronic colitis. Inflamm Bowel Dis 17:532–542. doi: 10.1002/ibd.21377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Logsdon LK, Mecsas J. 2003. Requirement of the Yersinia pseudotuberculosis effectors YopH and YopE in colonization and persistence in intestinal and lymph tissues. Infect Immun 71:4595–4607. doi: 10.1128/IAI.71.8.4595-4607.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Amar J, Burcelin R, Ruidavets JB, Cani PD, Fauvel J, Alessi MC, Chamontin B, Ferrieres J. 2008. Energy intake is associated with endotoxemia in apparently healthy men. Am J Clin Nutr 87:1219–1223. [DOI] [PubMed] [Google Scholar]
- 36.Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmee E, Cousin B, Sulpice T, Chamontin B, Ferrieres J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R. 2007. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56:1761–1772. doi: 10.2337/db06-1491. [DOI] [PubMed] [Google Scholar]
- 37.Erridge C, Attina T, Spickett CM, Webb DJ. 2007. A high-fat meal induces low-grade endotoxemia: evidence of a novel mechanism of postprandial inflammation. Am J Clin Nutr 86:1286–1292. [DOI] [PubMed] [Google Scholar]
- 38.Ghanim H, Abuaysheh S, Sia CL, Korzeniewski K, Chaudhuri A, Fernandez-Real JM, Dandona P. 2009. Increase in plasma endotoxin concentrations and the expression of Toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: implications for insulin resistance. Diabetes Care 32:2281–2287. doi: 10.2337/dc09-0979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis JE, Gabler NK, Walker-Daniels J, Spurlock ME. 2008. Tlr-4 deficiency selectively protects against obesity induced by diets high in saturated fat. Obesity 16:1248–1255. doi: 10.1038/oby.2008.210. [DOI] [PubMed] [Google Scholar]
- 40.Radin MS, Sinha S, Bhatt BA, Dedousis N, O'Doherty RM. 2008. Inhibition or deletion of the lipopolysaccharide receptor Toll-like receptor-4 confers partial protection against lipid-induced insulin resistance in rodent skeletal muscle. Diabetologia 51:336–346. doi: 10.1007/s00125-007-0861-3. [DOI] [PubMed] [Google Scholar]
- 41.Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, Prada PO, Hirabara SM, Schenka AA, Araujo EP, Vassallo J, Curi R, Velloso LA, Saad MJ. 2007. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes 56:1986–1998. doi: 10.2337/db06-1595. [DOI] [PubMed] [Google Scholar]
- 42.Uhlig HH, Powrie F. 2003. Dendritic cells and the intestinal bacterial flora: a role for localized mucosal immune responses. J Clin Invest 112:648–651. doi: 10.1172/JCI200319545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Autenrieth IB, Firsching R. 1996. Penetration of M cells and destruction of Peyer's patches by Yersinia enterocolitica: an ultrastructural and histological study. J Med Microbiol 44:285–294. doi: 10.1099/00222615-44-4-285. [DOI] [PubMed] [Google Scholar]
- 44.Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, Hooper LV. 2008. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc Natl Acad Sci U S A 105:20858–20863. doi: 10.1073/pnas.0808723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fan H, Cook JA. 2004. Molecular mechanisms of endotoxin tolerance. J Endotoxin Res 10:71–84. doi: 10.1177/09680519040100020301. [DOI] [PubMed] [Google Scholar]
- 46.Cani PD, Osto M, Geurts L, Everard A. 2012. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes 3:279–288. doi: 10.4161/gmic.19625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Larsson E, Tremaroli V, Lee YS, Koren O, Nookaew I, Fricker A, Nielsen J, Ley RE, Backhed F. 2012. Analysis of gut microbial regulation of host gene expression along the length of the gut and regulation of gut microbial ecology through MyD88. Gut 61:1124–1131. doi: 10.1136/gutjnl-2011-301104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mutch DM, Simmering R, Donnicola D, Fotopoulos G, Holzwarth JA, Williamson G, Corthesy-Theulaz I. 2004. Impact of commensal microbiota on murine gastrointestinal tract gene ontologies. Physiol Genomics 19:22–31. doi: 10.1152/physiolgenomics.00105.2004. [DOI] [PubMed] [Google Scholar]
- 49.Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. 2001. Molecular analysis of commensal host-microbial relationships in the intestine. Science 291:881–884. doi: 10.1126/science.291.5505.881. [DOI] [PubMed] [Google Scholar]
- 50.Kanther M, Sun X, Muhlbauer M, Mackey LC, Flynn EJ III, Bagnat M, Jobin C, Rawls JF. 2011. Microbial colonization induces dynamic temporal and spatial patterns of NF-κB activation in the zebrafish digestive tract. Gastroenterology 141:197–207. doi: 10.1053/j.gastro.2011.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eliakim R, Mahmood A, Alpers DH. 1991. Rat intestinal alkaline phosphatase secretion into lumen and serum is coordinately regulated. Biochim Biophys Acta 1091:1–8. doi: 10.1016/0167-4889(91)90213-H. [DOI] [PubMed] [Google Scholar]
- 52.Kaur J, Madan S, Hamid A, Singla A, Mahmood A. 2007. Intestinal alkaline phosphatase secretion in oil-fed rats. Dig Dis Sci 52:665–670. doi: 10.1007/s10620-006-9384-x. [DOI] [PubMed] [Google Scholar]
- 53.Vazquez CM, Zanetti R, Santa-Maria C, Ruiz-Gutierrez V. 2000. Effects of two highly monounsaturated oils on lipid composition and enzyme activities in rat jejunum. Biosci Reports 20:355–368. doi: 10.1023/A:1010377900745. [DOI] [PubMed] [Google Scholar]
- 54.Brun LR, Brance ML, Rigalli A. 2012. Luminal calcium concentration controls intestinal calcium absorption by modification of intestinal alkaline phosphatase activity. Br J Nutr 108:229–233. doi: 10.1017/S0007114511005617. [DOI] [PubMed] [Google Scholar]
- 55.Resta SC. 2009. Effects of probiotics and commensals on intestinal epithelial physiology: implications for nutrient handling. J Physiol 587:4169–4174. doi: 10.1113/jphysiol.2009.176370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sogabe N, Maruyama R, Hosori T, Goseki-Sone M. 2007. Enhancement effects of vitamin K1 (phylloquinone) or vitamin K2 (menaquinone-4) on intestinal alkaline phosphatase activity in rats. J Nutr Sci Vitaminol 53:219–224. doi: 10.3177/jnsv.53.219. [DOI] [PubMed] [Google Scholar]
- 57.Haraikawa M, Sogabe N, Tanabe R, Hosoi T, Goseki-Sone M. 2011. Vitamin K1 (phylloquinone) or vitamin K2 (menaquinone-4) induces intestinal alkaline phosphatase gene expression. J Nutr Sci Vitaminol 57:274–279. doi: 10.3177/jnsv.57.274. [DOI] [PubMed] [Google Scholar]
- 58.Bol-Schoenmakers M, Fiechter D, Raaben W, Hassing I, Bleumink R, Kruijswijk D, Maijoor K, Tersteeg-Zijderveld M, Brands R, Pieters R. 2010. Intestinal alkaline phosphatase contributes to the reduction of severe intestinal epithelial damage. Eur J Pharmacol 633:71–77. doi: 10.1016/j.ejphar.2010.01.023. [DOI] [PubMed] [Google Scholar]
- 59.Heemskerk S, Masereeuw R, Moesker O, Bouw MP, van der Hoeven JG, Peters WH, Russel FG, Pickkers P. 2009. Alkaline phosphatase treatment improves renal function in severe sepsis or septic shock patients. Crit Care Med 37:417–423. doi: 10.1097/CCM.0b013e31819598af. [DOI] [PubMed] [Google Scholar]
- 60.Heinzerling NP, Liedel JL, Welak SR, Fredrich K, Biesterveld BE, Pritchard KA Jr, Gourlay DM. 2014. Intestinal alkaline phosphatase is protective to the preterm rat pup intestine. J Pediatr Surg 49:954–960. doi: 10.1016/j.jpedsurg.2014.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kats S, Brands R, Seinen W, de Jager W, Bekker MW, Hamad MA, Tan ME, Schonberger JP. 2009. Anti-inflammatory effects of alkaline phosphatase in coronary artery bypass surgery with cardiopulmonary bypass. Recent Patents Inflamm Allergy Drug Discov 3:214–220. doi: 10.2174/187221309789257388. [DOI] [PubMed] [Google Scholar]
- 62.Lukas M, Drastich P, Konecny M, Gionchetti P, Urban O, Cantoni F, Bortlik M, Duricova D, Bulitta M. 2010. Exogenous alkaline phosphatase for the treatment of patients with moderate to severe ulcerative colitis. Inflamm Bowel Dis 16:1180–1186. doi: 10.1002/ibd.21161. [DOI] [PubMed] [Google Scholar]
- 63.Pickkers P, Heemskerk S, Schouten J, Laterre PF, Vincent JL, Beishuizen A, Jorens PG, Spapen H, Bulitta M, Peters WH, van der Hoeven JG. 2012. Alkaline phosphatase for treatment of sepsis-induced acute kidney injury: a prospective randomized double-blind placebo-controlled trial. Crit Care 16:R14. doi: 10.1186/cc11159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rentea RM, Liedel JL, Fredrich K, Pritchard K Jr, Oldham KT, Simpson PM, Gourlay DM. 2013. Enteral intestinal alkaline phosphatase administration in newborns decreases iNOS expression in a neonatal necrotizing enterocolitis rat model. J Pediatr Surg 48:124–128. doi: 10.1016/j.jpedsurg.2012.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rentea RM, Liedel JL, Fredrich K, Welak SR, Pritchard KA Jr, Oldham KT, Simpson PM, Gourlay DM. 2012. Intestinal alkaline phosphatase administration in newborns decreases systemic inflammatory cytokine expression in a neonatal necrotizing enterocolitis rat model. J Surg Res 177:228–234. doi: 10.1016/j.jss.2012.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Veen SQ, van Vliet AK, Wulferink M, Brands R, Boermeester MA, van Gulik TM. 2005. Bovine intestinal alkaline phosphatase attenuates the inflammatory response in secondary peritonitis in mice. Infect Immun 73:4309–4314. doi: 10.1128/IAI.73.7.4309-4314.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Verweij WR, Bentala H, Huizinga-van der Vlag A, Miek van Loenen-Weemaes A, Kooi K, Meijer DK, Poelstra K. 2004. Protection against an Escherichia coli-induced sepsis by alkaline phosphatase in mice. Shock 22:174–179. doi: 10.1097/01.shk.0000132485.05049.8a. [DOI] [PubMed] [Google Scholar]
- 68.Whitehouse JS, Riggle KM, Purpi DP, Mayer AN, Pritchard KA Jr, Oldham KT, Gourlay DM. 2010. The protective role of intestinal alkaline phosphatase in necrotizing enterocolitis. J Surg Res 163:79–85. doi: 10.1016/j.jss.2010.04.048. [DOI] [PubMed] [Google Scholar]
- 69.Alam SN, Yammine H, Moaven O, Ahmed R, Moss AK, Biswas B, Muhammad N, Biswas R, Raychowdhury A, Kaliannan K, Ghosh S, Ray M, Hamarneh SR, Barua S, Malo NS, Bhan AK, Malo MS, Hodin RA. 2014. Intestinal alkaline phosphatase prevents antibiotic-induced susceptibility to enteric pathogens. Ann Surg 259:715–722. doi: 10.1097/SLA.0b013e31828fae14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lehner MD, Ittner J, Bundschuh DS, van Rooijen N, Wendel A, Hartung T. 2001. Improved innate immunity of endotoxin-tolerant mice increases resistance to Salmonella enterica serovar Typhimurium infection despite attenuated cytokine response. Infect Immun 69:463–471. doi: 10.1128/IAI.69.1.463-471.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rayhane N, Fitting C, Lortholary O, Dromer F, Cavaillon JM. 2000. Administration of endotoxin associated with lipopolysaccharide tolerance protects mice against fungal infection. Infect Immun 68:3748–3753. doi: 10.1128/IAI.68.6.3748-3753.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lu M, Varley AW, Ohta S, Hardwick J, Munford RS. 2008. Host inactivation of bacterial lipopolysaccharide prevents prolonged tolerance following gram-negative bacterial infection. Cell Host Microbe 4:293–302. doi: 10.1016/j.chom.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rebeil R, Ernst RK, Gowen BB, Miller SI, Hinnebusch BJ. 2004. Variation in lipid A structure in the pathogenic yersiniae. Mol Microbiol 52:1363–1373. doi: 10.1111/j.1365-2958.2004.04059.x. [DOI] [PubMed] [Google Scholar]
- 74.de La Serre CB, Ellis CL, Lee J, Hartman AL, Rutledge JC, Raybould HE. 2010. Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am J Physiol Gastrointest Liver Physiol 299:G440–G448. doi: 10.1152/ajpgi.00098.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sefcikova Z, Hajek T, Lenhardt L, Racek L, Mozes S. 2008. Different functional responsibility of the small intestine to high-fat/high-energy diet determined the expression of obesity-prone and obesity-resistant phenotypes in rats. Physiol Res 57:467–474. [DOI] [PubMed] [Google Scholar]
- 76.Hansen GH, Niels-Christiansen LL, Immerdal L, Nystrom BT, Danielsen EM. 2007. Intestinal alkaline phosphatase: selective endocytosis from the enterocyte brush border during fat absorption. Am J Physiol Gastrointest Liver Physiol 293:G1325–G1332. doi: 10.1152/ajpgi.00379.2007. [DOI] [PubMed] [Google Scholar]
- 77.Lynes M, Narisawa S, Millan JL, Widmaier EP. 2011. Interactions between CD36 and global intestinal alkaline phosphatase in mouse small intestine and effects of high-fat diet. Am J Physiol Regul Integr Comp Physiol 301:R1738–R1747. doi: 10.1152/ajpregu.00235.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]