

Abstract
In bacterial meningitis, excessive immune responses carry significant potential for damage to brain tissue even after successful antibiotic therapy. Bacterial meningitis is regarded primarily as the domain of innate immunity, and the role of lymphocytes remains unclear. We studied the contribution of lymphocytes to acute inflammation and neurodegeneration in experimental Toll-like receptor 2-driven meningitis, comparing wild-type mice with RAG-1-deficient mice that have no mature T and B lymphocytes. At 24 h after intrathecal challenge with the synthetic bacterial lipopeptide Pam3CysSK4, RAG-1-deficient mice displayed more pronounced clinical impairment and an increased concentration of neutrophils, reduced expression of interleukin-10 (IL-10) mRNA, and increased expression of CXCL1 mRNA in the cerebrospinal fluid. Conversely, neuronal loss in the dentate gyrus was reduced in RAG-1-deficient mice, and expression of IL-10, transforming growth factor β and CCL2 mRNA by microglia was increased compared to wild-type mice. Adoptive transfer of wild-type lymphocytes reversed the enhanced meningeal inflammation and functional impairment observed in RAG-1-deficient mice. Our findings suggest compartment-specific effects of lymphocytes during acute bacterial meningitis, including attenuation of meningeal inflammation and shifting of microglial activation toward a more neurotoxic phenotype.
INTRODUCTION
Acute bacterial meningitis continues to be associated with significant mortality and morbidity (1). Bacterial metabolic products induce damage to host tissues, but especially in the context of successful antibiotic chemotherapy, excessive host immune responses are equally responsible for the negative outcomes (2). Immunity in bacterial meningitis is regarded primarily as the domain of neutrophils and other innate immune cells, which are activated by the interaction of bacterial compounds with Toll-like and other pattern recognition receptors expressed by these cells (3). Activated leukocytes produce proinflammatory and proapoptotic cytokines, proteases, and reactive oxygen and nitrogen species, which are all capable of inducing host tissue damage. Thus, limiting the host inflammatory response is a recognized therapeutic goal in bacterial meningitis (1, 2). While strategies to reduce the life span of activated neutrophils in the cerebrospinal fluid (CSF) have proven beneficial in experimental meningitis (4, 5), the development of clinically available therapies targeting innate immunity is hampered by an incomplete understanding of the regulatory processes involved in leukocyte survival and function during meningitis.
Innate immune responses, including neutrophil and monocyte recruitment from blood, as well as activation of resident microglia in the brain, contribute to the evolution of tissue injury also in ischemic stroke (6). Here, an important role of lymphocytes in mediating brain damage was demonstrated in experiments using recombination activating gene 1 (RAG-1)-deficient mice that are devoid of functional B and T cells (7). Compared to wild-type mice, these animals displayed significant neuroprotection at 24 h after focal cerebral ischemia (8, 9).
The role of lymphocytes in bacterial meningitis, however, has not been clarified. Based on the observations in cerebral ischemia, we hypothesized that RAG-1-deficient mice show an anti-inflammatory and neuroprotective phenotype during acute meningitis. To test this hypothesis, we studied RAG-1-deficient mice in a well-characterized model of bacterial meningitis that employs a synthetic bacterial lipopeptide to induce Toll-like receptor 2 (TLR2)-driven neuroinflammation, thereby allowing us to study host immune responses without the confounding effects of bacterial metabolic products (10).
MATERIALS AND METHODS
Meningitis model.
All experimental procedures were reviewed and approved by the state authorities (Landesamt für Gesundheit und Soziales, Berlin, Germany). Experiments were conducted in 12- to 16-week-old C57BL/6 wild-type mice (n = 24) and transgenic mice with homozygous Rag-1 ablation (RAG-1−/−, n = 21) (7), using a well-established model of experimental meningitis (10). Additional experiments were performed in RAG-1-deficient mice after reconstitution with wild-type lymphocytes (n = 9). While the animals were under anesthesia with intraperitoneal ketamine (100 mg/kg; DeltaSelect) and xylazine (20 mg/kg; Bayer), a skin incision was made to expose the lumbar spine. Using a 30-gauge needle, 10 μg of Pam3CysSK4 (EMC Microcollections) dissolved in a volume of 40 μl of phosphate-buffered saline (PBS) were slowly injected into the spinal canal below vertebra L2 or L3. Controls received an equal volume of pyrogen-free PBS. After wound closure with dermal clips, the animals were allowed to wake up and were kept under standard conditions.
After 24 h, mice were assessed in the tight rope test (11). In this global test of strength and motor coordination, the time span during which a mouse is able to hold on to a horizontally suspended length of string is measured. Mice are lifted by the tail and released once they have firmly gripped the rope with both forepaws. A padded box is used to catch falling mice. The experiment is terminated after 30 s. After this test, animals were again deeply anesthetized using sodium thiopental (100 mg/kg). Under a preparation microscope, skin and suboccipital muscles were dissected. The cisterna magna was punctured with a 27-gauge butterfly cannula attached to a microliter syringe to obtain a CSF specimen. Total white blood cell concentrations in the CSF were determined microscopically by using a Fuchs-Rosenthal chamber. For manual differential leukocyte counts, CSF samples were stained with Türk's solution, and the relative proportions of granulocytes, monocytes, and lymphocytes were determined from 100 to 200 leukocytes per animal. After transcardial perfusion with cold saline, the brains were removed. The cerebellum was frozen in liquid nitrogen. The right cerebral hemisphere was snap-frozen in methylbutane on dry ice, while the left hemisphere was processed for cell isolation (see below).
Adoptive transfer of lymphocytes.
Spleens of C57BL/6 wild-type mice were excised and pushed through a 100-μm-pore-size strainer. Red blood cells were lysed in Pharm Lyse lysing buffer (BD Biosciences). T and B cells were purified by positive selection with isolation kits according to the manufacturer's protocol (CD4, CD8α, and CD19 custom-made cocktail; Miltenyi Biotec) and isolated by magnetic cell sorting (MACS; Miltenyi Biotec). RAG-1-deficient mice were intravenously injected with 1.5 × 107 wild-type lymphocytes at 24 h before the induction of meningitis. The efficiency of lymphocyte reconstitution was assessed by performing differential leukocyte counts in atrial blood 24 h after the induction of meningitis.
Histology.
Neuronal damage in mice was assessed by TUNEL (terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling) staining. First, 20-μm cryosections were thaw mounted on coated glass slides, air dried, and postfixed in methanol at −20°C. We performed TUNEL staining using a commercially available kit (Qbiogene) and a fluorescein isothiocyanate (FITC)-conjugated antibody for visualization. Sections were counterstained with Hoechst 33258 (1:10,000; Invitrogen Life Technologies). For quantification, TUNEL-positive nuclei were counted in the dentate gyrus (four sections per animal) and divided by the area of the dentate gyrus, as determined using Stereo Investigator 10 software (MicroBrightfield, Inc.). For measuring the degree of granulocyte infiltration in the tissue, sections were prepared as described above, followed by blocking in normal goat serum (3% in PBS with 0.3% Triton X-100) for 30 min and incubation with a rabbit polyclonal antibody against myeloperoxidase (MPO; 1:300 in blocking solution; Hycult Biotechnology) at 4°C overnight. A Texas Red-conjugated goat anti-rabbit secondary antibody (1:100; Jackson ImmunoResearch) was used for visualization. The density of granulocytes in the brain parenchyma was determined by counting all MPO-immunoreactive cells per area (three sections per animal). Cells in the subarachnoid space or within ∼20 μm of the brain surface were not counted. For the staining of phagocytes, 7-μm sections were prepared. After fixation in acetone for 10 min and incubation with chloroform for 7 min, the sections were blocked with species-matched serum, followed by incubation with a rat monoclonal antibody against CD11b (1:100; BD Pharmingen). A DyLight 649-conjugated goat anti-rat antibody (1:100; Jackson ImmunoResearch) was used for visualization.
Flow cytometry of brain-derived and CSF leukocytes.
In order to assess microglial activation and granulocyte invasion, as well as to obtain cell populations for subsequent PCR studies, saline-perfused brains of wild-type mice (n = 5) and RAG-1-deficient mice (n = 11) were obtained 24 h after stimulation with Pam3CysSK4. In addition, two untreated animals per genotype were included as controls. Left cerebral hemispheres of two to four animals were pooled and minced through a 70-μm mesh (BD), followed by Percoll gradient centrifugation (Amersham-Pharmacia) as described previously (12). Retrieved cells were subjected to multiple immunofluorescence staining, followed by flow cytometry. In brief, after pelleting and resuspension, the cells were blocked for unspecific binding of the Fc receptor (CD16/32 1:200; Biolegend) at 4°C for 10 min. They were then incubated for 20 min with a phycoerythrin (PE)-labeled CD11b antibody (1:400; BD Pharmingen), an allophycocyanin (APC)/Cy7-labeled CD45 antibody (1:200; BioLegend), and a Cy7/PE-labeled GR1 antibody (1:400; BioLegend). An additional sample was stained with a FITC-labeled CD4 antibody (1:200; BD Pharmingen), a Cy7/PE-labeled CD8 antibody (1:400; BioLegend), and an APC-labeled B220 antibody (1:400; BioLegend). After washing, cells were analyzed on a fluorescence-activated cell sorting (FACS) CANTO II (BD Biosciences) and evaluated with FlowJo software (Tristar). In further experiments, cerebral leukocyte populations were sorted on a FACSAria I (BD Biosciences) according to the following surface profiles: CD11b+ CD45high GR1dim for activated microglia/macrophages and CD11b+ CD45high GR1high for granulocytes. Absolute cell counts were corrected for the number of animals in each pool. Sorted cells were collected in TRIzol buffer and stored at −80°C. CSF was collected from the same animals. Due to the low sample volumes, CSF specimens were combined into two pools for each genotype. After pelleting and washing in PBS with 1% fetal calf serum, CSF cells were blocked for the Fc receptor as described above and stained for 20 min with a PE-labeled CD11b antibody (1:400; BD Pharmingen), a FITC-labeled CD3 antibody (1:200; BD Pharmingen), a PE/Cy7-labeled GR1 antibody (1:400; BioLegend), and an APC-labeled B220 antibody (1:400; BioLegend). Using FACS sorting as described above, CD11b+ GR1dim inflammatory monocytes, CD11b− GR1− CD3+ T cells, and CD11b+ GR1high CD3− granulocytes were quantified. Sorted CSF granulocytes and monocytes were stored in TRIzol at −80°C.
RNA isolation and real-time PCR.
Total RNA was obtained from different sites in order to gain information on multiple compartments. First, we studied whole cerebellar lysates to provide information on overall inflammatory changes, including the perivascular space as the presumed site of leukocyte recruitment. Furthermore, we isolated CD11b+ CD45high cells representing activated microglia and macrophages from hemispheric cerebral tissue, including the hippocampal formation and dentate gyrus. This region is characterized by pronounced neuronal damage and microglial activation in bacterial meningitis. Finally, we isolated CD11b+ cells from the CSF to provide information on cytokine production by nonlymphocytic cells in this compartment.
Total RNA was extracted using the TriFast method according to the manufacturer's instructions (peqGold TriFast; Peqlab). The concentration was determined photometrically by NanoQuant Plate (Tecan Infinite 2000). First-strand cDNA was synthesized in a volume of 20 μl using 1 μg of total RNA and TaqMan reverse transcription reagents (Applied Biosystems). To quantify target mRNA levels, transforming growth factor β (TGF-β), gamma interferon (IFN-γ), interleukin-10 (IL-10), IL-4, CXCL-1, CCL-2, and TRAIL TaqMan gene expression assays were purchased from Applied Biosystems. Hypoxanthinguanine-phosphoribosyltransferase (Hprt) was chosen as a highly stable reference gene for the analysis of frozen tissue, while β-actin (Actb) was used for FACS-sorted cells. PCR was performed in triplicates with 20 ng of cDNA using the FastStart Universal PCR master mix (Applied Biosystems) in a 7900HT real-time PCR system (Applied Biosystems) for 45 cycles. The data were processed using SDS2.4 and RQ Manager 1.2.1 software. Quantitative real-time PCR analysis was carried out using the 2−ΔΔCT method. In the corresponding figure (see Fig. 4), meningitis-induced regulation of mRNA expression was depicted as the fold change unless the level of expression in controls was below the detection limit. In this case, 1/ΔCT was used for plotting.
FIG 4.

Impact of RAG-1 deficiency on cytokine and chemokine mRNA levels 24 h after induction of meningitis in wild-type C57BL/6 mice and RAG-1-deficient mice. Quantitative real-time PCR was performed using hypoxanthinguanine-phosphoribosyltransferase (A) or beta-actin (B to G) as housekeeping genes. (A) Expression of interleukin-10 (IL-10), gamma interferon (IFN-γ), transforming growth factor β (TGF-β), forkhead box P3 (Foxp3), and tumor necrosis factor alpha-related ligand (TRAIL) mRNAs in cerebellar tissue lysates. Regulation is shown relative to nonstimulated controls of the same genotype using the ΔΔCT method. (B to D) Activated microglia/macrophages were isolated from cerebral tissue as CD11b+ CD45high cells using FACS and studied for the expression of IL-10 mRNA (B), as well as for the regulation of TGF-β mRNA (C) and CCL2 mRNA (D). (E to G) CD11b+ myeloid cells were isolated from the CSF and studied for the regulation of IL-10 mRNA (E), CCL2 mRNA (F), and CXCL1 mRNA (G) relative to controls. Graphs depict mean values and standard deviations. Statistical significance was examined using Student t tests. The detection limit was set at 40 PCR cycles. In panels B and G, negative results are marked with an “n.d.” (not detected), and expression levels are shown as 1/ΔCT.
Statistical analysis.
Outliers greater than two standard deviations above or below the mean were censored. Normal distribution was verified by using one-sample Kolmogorov-Smirnov tests. Two-group comparisons were performed by using a Student t tests for normally distributed data, whereas Mann-Whitney U tests were used in the absence of normal distribution. A log-rank test was performed to compare performances in the tight-rope test between genotypes. Multiple group comparisons were performed using Kruskal-Wallis analysis of variance (ANOVA), followed by Mann-Whitney U tests, using Holm's Bonferroni adjustment for multiple comparisons.
RESULTS
Leukocyte populations in the CSF.
At 24 h after the induction of meningitis, 4,206 ± 3,484 leukocytes per μl (range, 90 to 11,776) were present in the CSF of wild-type mice (Fig. 1A). In RAG-1-deficient mice, the CSF leukocyte counts were 9,726 ± 7,575 (mean ± the standard deviation; range, 512 to 21,248) per μl (Fig. 1A; P < 0.05, Student t test). As expected, lymphocytes were identified only in the CSF of wild-type mice but were absent in RAG-1-deficient mice (Fig. 1B; P < 0.001, Student t test). Significantly more neutrophils were found in the CSF of RAG-1-deficient mice compared to wild-type mice (Fig. 1B; P < 0.05, Student t test), while the concentration of monocytes/macrophages was similar in both genotypes (Fig. 1B; P = 0.02, Student t test). FACS analysis and cytology of pooled CSF confirmed a higher number of CD11b+ leukocytes and a strong increase in GR1high neutrophils in RAG-1-deficient mice compared to wild-type mice (Fig. 1C).
FIG 1.

Impact of RAG-1 deficiency (RAG-1−/−) on leukocyte populations in the cerebrospinal fluid 24 h after intrathecal challenge with 10 μg of Pam3CysSK4. (A) Total leukocyte concentration in C57BL/6 wild-type mice (n = 24), RAG-1-deficient mice (n = 21), and RAG-1-deficient mice reconstituted with 1.5 × 107 wild-type lymphocytes (n = 9). Graphs depict the median, minimum, and maximum values, as well as 25th and 75th percentiles. Significance was determined by Kruskal-Wallis ANOVA, followed by Mann-Whitney U tests using Holm's Bonferroni adjustment for multiple comparisons. (B) CSF concentration of lymphocytes, monocytes/macrophages, and neutrophils as determined by light microscopy of Türk-stained specimens. Graphs depict the median, minimum, and maximum values, as well as the 25th and 75th percentiles. Significance was determined using Mann-Whitney U tests. (C) FACS analysis of pooled CSF samples. Sorted CD11b+ leukocytes from wild-type mice and RAG-1-deficient mice were analyzed for Gr1 staining intensity.
Clinical disease severity.
During the observational period of 24 h, animals lost 2.3 ± 1.2 g (8.4% ± 1.2%) of body weight after challenge with Pam3CysSK4 (P < 0.001, Mann-Whitney U test). Relative weight loss was not influenced by the genotype (8.2% ± 4.5% in wild-type mice versus 8.5% ± 4.1% in RAG-1-deficient mice; P = 1.0, Mann-Whitney-U test). However, RAG-1-deficient mice showed a greater impairment of motor strength and coordination in the tight rope test (Fig. 2A; P < 0.001, log-rank test).
FIG 2.
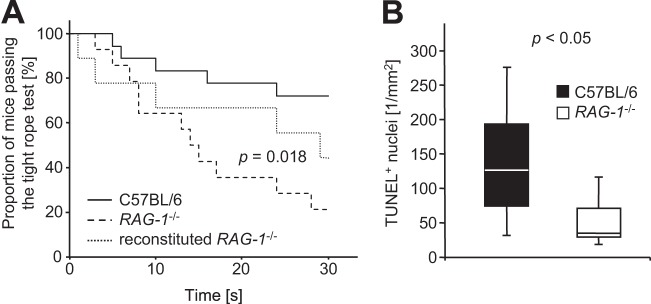
Impact of RAG-1 deficiency (RAG-1−/−) on clinical impairment (A) and neuronal damage (B) 24 h after intrathecal challenge with Pam3CysSK4. (A) Kaplan-Meyer graph depicting the proportion of mice passing the tightrope test for strength and motor coordination for the given duration of time. Significance was determined using a log-rank test. Control mice (not shown) were able to hold on to the rope for at least 30 s. (B) Density of TUNEL-positive nuclei in the dentate gyrus. Graphs depict the median, minimum and maximum values, as well as the 25th and 75th percentiles. Significance was determined using a Student t test.
Neuronal damage.
Cell damage in the dentate gyrus was assessed by TUNEL staining. In wild-type mice, 120 ± 66 TUNEL-positive cells per mm2 were detected 24 h after induction of meningitis compared to 40 ± 25 per mm2 in RAG-1-deficient mice (Fig. 2B; P < 0.05, Student t test).
Immune cells in the brain parenchyma.
Pooled brain homogenates of unstimulated RAG-1-deficient mice revealed no deficiency in CD45low resident microglia, and absence of inflammatory CD45high activated microglia/macrophages (see Fig. S1A in the supplemental material). At 24 h after the induction of meningitis, the total numbers of parenchymal leukocytes per brain retrieved from the Percoll gradient were (5.5 ± 0.5) × 105 in wild-type mice and (6.8 ± 1.9) × 105 in RAG-1-deficient mice (P = 0.26). Compared to wild-type mice, activated microglia/macrophages were significantly more prevalent in RAG-1-deficient mice (Fig. 3A to D; P < 0.01, Student t test), whereas the numbers of intraparenchymal granulocytes did not differ between genotypes (Fig. 3B; P = 0.13). In line with the FACS results, immunohistochemical analysis revealed 12 ± 11 MPO-immunoreactive granulocytes per mm2 in the brain parenchyma of wild-type mice compared to 7 ± 6 per mm2 in RAG-1-deficient mice (P = 0.53, Mann-Whitney U test). Immunostaining for CD11b revealed more activated microglia/macrophages with amoeboid morphology in the brains of RAG-1-deficient mice compared to wild-type mice after experimental meningitis (Fig. 3E and F).
FIG 3.

Characterization of myeloid cells in the brain parenchyma 24 h after induction of meningitis. (A to C) Percoll-isolated cells were subjected to FACS. (A) Activated myeloid cells from the brains of wild-type mice and RAG-1-deficient mice were gated as the CD11b+ CD45high population (left panels) and analyzed for Gr1 expression (right panels, representative examples). Activated microglia and macrophages were identified as CD11b+ CD45high Gr1dim cells, while CD11b+ CD45high Gr1high cells represent granulocytes. (B) Retrieved number of activated microglia/macrophages and granulocytes per cerebral hemisphere in wild-type and RAG-1-deficient mice, measured as outlined in panel A. Left hemispheres of two to four animals were pooled per sample, and the cell counts were corrected for the number of animals per pool. Graphs depict means and standard deviations. Statistical significance was determined using Student t tests. (C and D) Activation state of leukocytes in the parenchyma of Pam3CysSK4-challenged mice. (C) Frequency distribution of CD45 staining intensity of CD11b+ myeloid cells in the brain parenchyma of C57BL/6 mice (wild type) and RAG-1-deficient mice (RAG-1−/−). Two representative samples are shown. (D) Frequency distribution of CD45 staining intensity of CD11b+ Gr1dim microglia/macrophages in the brain parenchyma of C57BL/6 mice (wild type) and RAG-1-deficient mice (RAG-1−/−). Two representative samples are shown. (E and F) Morphology of CD11b+ microglia/macrophages in the fascia dentata region of wild-type mice (E) and RAG-1-deficient mice (F) 24 h after stimulation with Pam3CysSK4. Original magnification, ×400.
Cytokine and chemokine mRNA levels.
In the cerebellar tissues of naive controls, baseline cytokine expression was affected by the absence of RAG-1 (see Fig. S1B in the supplemental material). Although only minor effects were observed for IFN-γ and Foxp3 mRNA, baseline expression of Il-10 mRNA was increased in RAG-1-deficient mice (average ΔCT, 15.7 ± 0.1 versus 20.3 ± 0.2 in wild-type mice; P = 0.01). No significant effect of RAG-1-deficiency on the expression of TRAIL or TGF-β mRNA was detected. After stimulation with Pam3CysSK4, we observed a differential regulation of IL-10, IFN-γ, TGF-β, Foxp3, and TRAIL mRNAs when normalized to naive controls of the respective genotype (Fig. 4A). Il-4 mRNA was not detected in controls or during inflammation (data not shown). In contrast to wild-type mice, RAG-1-deficient mice displayed upregulation of IFN-γ mRNA (Fig. 4A; P < 0.01, Student t test), reduced induction of IL-10 mRNA (Fig. 4A; P < 0.05, Student t test), and failure to upregulate TGF-β and Foxp3 mRNA expression (Fig. 4A; P < 0.01, Student t test). No differences in TRAIL mRNA induction were observed between wild-type and RAG-1-deficient mice (Fig. 4A; P = 0.28, Student t test).
In brain-derived CD11b+ CD45high activated microglia/macrophages, IL-10 mRNA expression was detected at low concentrations in RAG-1-deficient mice after Pam3CysSK4 challenge but not in stimulated wild-type mice or PBS-injected controls of either genotype (Fig. 4B). Expression of monocyte chemoattractant protein 1 (CCL2) mRNA in brain-derived CD11b+ CD45high cells was upregulated in RAG-1-deficient mice and downregulated in wild-type mice (Fig. 4D; P < 0.05, Student t test). A similar pattern was observed for TGF-β mRNA, although this did not reach statistical significance (Fig. 4C; P = 0.10, Student t test).
In CD11b+ leukocytes isolated from the CSF, the induction of IL-10 mRNA at 24 h after Pam3CysSK4 challenge was less pronounced in RAG-1-deficient mice compared to wild-type mice (Fig. 4E; P < 0.01, Student t test). A trend toward increased CCL2 mRNA induction was observed for RAG-1-deficient mice (Fig. 4F; P = 0.05, Student t test). Moreover, CXCL1 mRNA expression was only induced in RAG-1-deficient mice after Pam3CysSK4 challenge but absent in control mice of either genotype and in Pam3CysSK4-stimulated wild-type mice (Fig. 4G). Using the MesoScale electrochemiluminescence-based immunoassay, we confirmed expression of IL-10 protein in brain tissue and CSF (data not shown).
Reconstitution experiments.
After adoptive transfer of 1.5 × 107 lymphocytes into RAG-1-deficient mice at 24 h prior to the induction of meningitis, partial reconstitution of circulating lymphocytes and normalization of monocytes was observed 24 h after induction of meningitis (see Fig. S2 in the supplemental material). However, the frequency of lymphocytes remained significantly reduced compared to wild-type mice (38% ± 14% versus 76% ± 15%; P < 0.01). Notably, the adoptive transfer of lymphocytes normalized the concentrations of CSF leukocytes to wild-type levels at 24 h after the induction of meningitis (Fig. 1A). Furthermore, reconstitution of lymphocytes partially reversed the increase in clinical disease severity observed in RAG-1-deficient mice during the tight-rope test (Fig. 2A). However, no consistent effect of peripheral lymphocyte reconstitution on the density of apoptotic nuclei in the dentate gyrus was observed at 24 h after meningitis induction (data not shown).
DISCUSSION
After meningitis induction with the synthetic bacterial lipopeptide, Pam3CysSK4, the absence of mature T and B lymphocytes in RAG-1-deficient mice resulted in an increased concentration of granulocytes in the CSF and more severe clinical impairment. These effects were partially reversed by the systemic reconstitution of lymphocytes using adoptive transfer of spleen-derived B and T cells. Although more activated microglia/macrophages were found in the brain parenchyma, RAG-1-deficient mice displayed a strongly reduced ratio of granulocytes versus mononuclear cells in the brain and reduced neuronal damage in the dentate gyrus of the hippocampus. These effects were paralleled by compartment-specific changes in cytokine/chemokine gene expression patterns. In particular, RAG-1-deficient mice displayed a pronounced upregulation of IL-10 mRNA in activated microglia/macrophages which was absent in wild-type mice. In CSF leukocytes, IL-10 mRNA induction was reduced in RAG-1-deficient mice, but CXCL1 mRNA expression was strongly enhanced.
The regulation of neutrophil survival by lymphocytes was previously described in vitro (13). Apoptosis and elimination of activated neutrophils was delayed in RAG-1-deficient mice following lipopolysaccharide-induced acute lung injury (14). The mechanisms by which lymphocytes promote neutrophil apoptosis have not been identified in detail. We did not detect an effect of lymphocyte deficiency on the regulation of TRAIL, a pivotal mediator of neutrophil clearance from the CSF in meningitis (4). Regulatory T cells induce anti-inflammatory phenotypes in neutrophils (15) and in monocytes/macrophages (16). Notably, IL-10 was identified both as a T cell-derived anti-inflammatory signal and as a phenotypic marker of alternative activation of the antigen-presenting cells. In a murine model of stroke, anti-inflammatory effects of CD25-positive T cells were dependent on their production of IL-10 (17). IL-10 counteracts antiapoptotic signals in activated neutrophils, thereby restoring constitutive apoptosis of neutrophils (18). Accordingly, IL-10 was previously identified as a key negative regulator of leptomeningeal infiltration of neutrophils in experimental meningitis (19). In macrophages, secretion of IL-10 and TGF-β is further stimulated by phagocytosis of apoptotic neutrophils (20, 21), suggesting the presence of a feedback effect by which reduced secretion of IL-10 may contribute to delayed apoptosis of CSF neutrophils and vice versa.
Moreover, IL-10 inhibits the production of the main granulocyte-attractant chemokine, CXCL1, in activated phagocytes (22). We consistently detected CXCL1 mRNA in RAG-1-deficient CSF leukocytes at 24 h after meningitis induction but not in wild-type mice or in unstimulated controls of either genotype. A possible interpretation of this finding is that in RAG-1-deficient mice, failure to upregulate anti-inflammatory signals, including IL-10, in the CSF could maintain a self-sustained recruitment of neutrophils, whereas in wild-type mice, CXCL1 production and influx of neutrophils have already subsided at this time point. Similar effects were described in a mouse model of neutrophil-driven skin inflammation (23). Neutrophils mediate acute complications of meningitis (24–26) and correlate with clinical disease severity (4, 5). In line with these previous results, RAG-1-deficient mice displayed more severe impairment in the tight-rope test during meningitis. Our findings suggest that lymphocytes play an immunoprotective role in the subarachnoid space by enhancing IL-10 signaling and negatively regulating the recruitment and survival of neutrophils.
In bacterial meningitis, microglial cells are regarded as important mediators of tissue damage (2). We have previously demonstrated that selective stimulation of TLR2 using Pam3CysSK4 is sufficient to induce inflammatory activation of microglia, as well as meningeal inflammation and neuronal loss in the murine dentate gyrus (10). In the present study, the absence of mature T and B lymphocytes in RAG-1-deficient mice resulted in increased numbers of activated microglia and macrophages during meningitis. However, this was associated with a neuroprotective phenotype, as shown by a lower density of TUNEL-positive nuclei in the dentate gyrus.
In recent years, a concept of variable macrophage polarization has evolved. Of the two extreme states, the so-called “M1” pattern is elicited by exposure to IFN-γ. It is characterized by the production of proinflammatory cytokines and nitric oxide. Conversely, alternative activation in response to IL-4 and IL-13, as well as acquired deactivation via IL-10 and TGF-β, result in anti-inflammatory and proregenerative phenotypes, which are broadly termed “M2” and include macrophages with regulatory functions. The concept of M1 versus M2 activation patterns has recently been expanded to include microglia (27), although the simple dichotomy cannot account for the multiplicity of functional states of microglia and brain macrophages (28). We could not verify a hypothetical contribution of IL-4-induced alternative activation (29) in our meningitis model since IL-4 mRNA concentrations were below the detection limit irrespective of the genotype and treatment group. In isolated microglia and macrophages from the brain parenchyma, increased expression of IL-10 and TGF-β mRNAs during meningeal inflammation was observed in RAG-1-deficient mice but not in wild-type mice. Moreover, the baseline expression of IL-10, TGF-β, and Foxp3 mRNAs was increased in the brains of naive RAG1-deficient mice, while their baseline expression of IFN-γ was slightly reduced. These findings support the concept of microglia/macrophage polarization toward acquired deactivation in RAG-1-deficient mice. Thus, the absence of lymphocytes in RAG-1-deficient mice appears to result in compartment-specific effects: in the subarachnoid space of RAG-1-deficient mice, a proinflammatory shift induced by IFN-γ leads to an increased ratio of CSF neutrophils versus monocytes/macrophages, whereas in the parenchyma, a more immunosuppressive cytokine environment has the opposite effect.
The increased density of activated microglia and macrophages in RAG-1-deficient mice was paralleled by a significantly higher expression of CCL2 mRNA by these cells. CCL2 signaling is critically involved in the recruitment of inflammatory monocytes from the bloodstream during meningitis (30) and promotes the migration and proliferation of microglia (31). However, CCL2 per se does not induce the production of proinflammatory cytokines in microglia (31), indicating that additional signals may be required to determine a pro- versus anti-inflammatory phenotype of microglia and monocytes.
Other than in the periphery, regulatory T cells are not thought to act as a relevant source of IL-10 and TGF-β during acute CNS inflammation. Instead, IL-10 production has been reported in microglia and astrocytes in response to bacterial compounds, suggesting paracrine or autocrine effects (32). In line with our findings, RAG-1-deficient mice were previously reported to display an increased production of IL-10 by microglia and relative neuroprotection in chronic Borrelia turicatae infection (33). Lymphocytes could therefore play an essential role in the parenchyma by shifting the balance from immunoregulatory microglia polarization and neuroprotection toward a more neurotoxic phenotype, involving the downregulation of IL-10.
Taken together, our findings suggest novel regulatory functions of lymphocytes in early TLR2-dependent meningitis, which include compartment-specific modulation of the cytokine environment, polarization of microglia and macrophages, control of neutrophil accumulation, and ultimately clinical impairment and neuronal damage. The precise role of lymphocyte subsets needs to be addressed in further studies.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grants from the DFG to J.P. (SFB/TRR43 A7, FOR1336 C4, NeuroCure) and by an unrestricted research grant from Biogen Idec to O.H. and O.R.
We thank Alexandra Döser for excellent technical assistance.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02682-14.
REFERENCES
- 1.van de Beek D, Brouwer MC, Thwaites GE, Tunkel AR. 2012. Advances in treatment of bacterial meningitis. Lancet 380:1693–1702. doi: 10.1016/S0140-6736(12)61186-6. [DOI] [PubMed] [Google Scholar]
- 2.Gerber J, Nau R. 2010. Mechanisms of injury in bacterial meningitis. Curr Opin Neurol 23:312–318. doi: 10.1097/WCO.0b013e32833950dd. [DOI] [PubMed] [Google Scholar]
- 3.Koedel U. 2009. Toll-like receptors in bacterial meningitis. Curr Top Microbiol Immunol 336:15–40. doi: 10.1007/978-3-642-00549-7_2. [DOI] [PubMed] [Google Scholar]
- 4.Hoffmann O, Priller J, Prozorovski T, Schulze-Topphoff U, Baeva N, Lünemann JD, Aktas O, Mahrhofer C, Stricker S, Zipp F, Weber JR. 2007. TRAIL limits excessive host immune responses in bacterial meningitis. J Clin Invest 117:2004–2013. doi: 10.1172/JCI30356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koedel U, Frankenberg T, Kirschnek S, Obermaier B, Häcker H, Paul R, Häcker G. 2009. Apoptosis is essential for neutrophil functional shutdown and determines tissue damage in experimental pneumococcal meningitis. PLoS Pathog 5:e1000461. doi: 10.1371/journal.ppat.1000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jin R, Yang G, Li G. 2010. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol 87:779–789. doi: 10.1189/jlb.1109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. 1992. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68:869–877. doi: 10.1016/0092-8674(92)90030-G. [DOI] [PubMed] [Google Scholar]
- 8.Yilmaz G, Arumugam TV, Stokes KY, Granger DN. 2006. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation 113:2105–2112. doi: 10.1161/circulationaha.105.593046. [DOI] [PubMed] [Google Scholar]
- 9.Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, Iwaki T, Okada Y, Iida M, Cua DJ, Iwakura Y, Yoshimura A. 2009. Pivotal role of cerebral interleukin-17-producing γδ T cells in the delayed phase of ischemic brain injury. Nat Med 15:946–950. doi: 10.1038/nm.1999. [DOI] [PubMed] [Google Scholar]
- 10.Hoffmann O, Braun JS, Becker D, Halle A, Freyer D, Dagand E, Lehnardt S, Weber JR. 2007. TLR2 mediates neuroinflammation and neuronal damage. J Immunol 178:6476–6481. doi: 10.4049/jimmunol.178.10.6476. [DOI] [PubMed] [Google Scholar]
- 11.Miquel J, Blasco M. 1978. A simple technique for evaluation of vitality loss in aging mice, by testing their muscular coordination and vigor. Exp Gerontol 13:389–396. doi: 10.1016/0531-5565(78)90049-9. [DOI] [PubMed] [Google Scholar]
- 12.Stenzel W, Soltek S, Sanchez-Ruiz M, Akira S, Miletic H, Schlüter D, Deckert M. 2008. Both TLR2 and TLR4 are required for the effective immune response in Staphylococcus aureus-induced experimental murine brain abscess. Am J Pathol 172:132–145. doi: 10.2353/ajpath.2008.070567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lewkowicz P, Lewkowicz N, Sasiak A, Tchorzewski H. 2006. Lipopolysaccharide-activated CD4+ CD25+ T regulatory cells inhibit neutrophil function and promote their apoptosis and death. J Immunol 177:7155–7163. doi: 10.4049/jimmunol.177.10.7155. [DOI] [PubMed] [Google Scholar]
- 14.D'Alessio FR, Tsushima K, Aggarwal NR, West EE, Willett MH, Britos MF, Pipeling MR, Brower RG, Tuder RM, McDyer JF, King LS. 2009. CD4+ CD25+ Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J Clin Invest 119:2898–2913. doi: 10.1172/JCI36498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lewkowicz N, Klink M, Mycko MP, Lewkowicz P. 2013. Neutrophil–CD4+ CD25+ T regulatory cell interactions: a possible new mechanism of infectious tolerance. Immunobiology 218:455–464. doi: 10.1016/j.imbio.2012.05.029. [DOI] [PubMed] [Google Scholar]
- 16.Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. 2007. CD4+ CD25+ Foxp3+ regulatory T cells induce alternative activation of human monocytes/ macrophages. Proc Natl Acad Sci U S A 104:19446–19451. doi: 10.1073/pnas.0706832104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, Giese T, Veltkamp R. 2009. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med 15:192–199. doi: 10.1038/nm.1927. [DOI] [PubMed] [Google Scholar]
- 18.Keel M, Ungethüm U, Steckholzer U, Niederer E, Hartung T, Trentz O, Ertel W. 1997. Interleukin-10 counterregulates proinflammatory cytokine-induced inhibition of neutrophil apoptosis during severe sepsis. Blood 90:3356–3363. [PubMed] [Google Scholar]
- 19.Zwijnenburg PJ, van der Poll T, Florquin S, Roord JJ, van Furth AM. 2003. Interleukin-10 negatively regulates local cytokine and chemokine production but does not influence antibacterial host defense during murine pneumococcal meningitis. Infect Immun 71:2276–2279. doi: 10.1128/IAI.71.4.2276-2279.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Byrne A, Reen DJ. 2002. Lipopolysaccharide induces rapid production of IL-10 by monocytes in the presence of apoptotic neutrophils. J Immunol 168:1968–1977. doi: 10.4049/jimmunol.168.4.1968. [DOI] [PubMed] [Google Scholar]
- 21.Savill J, Dransfield I, Gregory C, Haslett C. 2002. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol 2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 22.Olszyna DP, Pajkrt D, van Deventer SJ, van der Poll T. 2001. Effect of interleukin 10 on the release of the CXC chemokines growth related oncogene GRO-alpha and epithelial cell-derived neutrophil activating peptide (ENA)-78 during human endotoxemia. Immunol Lett 78:41–44. doi: 10.1016/S0165-2478(01)00224-3. [DOI] [PubMed] [Google Scholar]
- 23.Richards H, Williams A, Jones E, Hindley J, Godkin A, Simon AK, Gallimore A. 2010. Novel role of regulatory T cells in limiting early neutrophil responses in skin. Immunology 131:583–592. doi: 10.1111/j.1365-2567.2010.03333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Angstwurm K, Weber JR, Segert A, Bürger W, Weih M, Freyer D, Einhäupl KM, Dirnagl U. 1995. Fucoidin, a polysaccharide inhibiting leukocyte rolling, attenuates inflammatory responses in experimental pneumococcal meningitis in rats. Neurosci Lett 191:1–4. doi: 10.1016/0304-3940(95)11541-4. [DOI] [PubMed] [Google Scholar]
- 25.Weber JR, Angstwurm K, Bürger W, Einhäupl KM, Dirnagl U. 1995. Anti ICAM-1 (CD 54) monoclonal antibody reduces inflammatory changes in experimental bacterial meningitis. J Neuroimmunol 63:63–68. doi: 10.1016/0165-5728(95)00131-X. [DOI] [PubMed] [Google Scholar]
- 26.Weber JR, Angstwurm K, Rosenkranz T, Lindauer U, Freyer D, Bürger W, Busch C, Einhäupl KM, Dirnagl U. 1997. Heparin inhibits leukocyte rolling in pial vessels and attenuates inflammatory changes in a rat model of experimental bacterial meningitis. J Cereb Blood Flow Metab 17:1221–1229. [DOI] [PubMed] [Google Scholar]
- 27.Colton CA. 2009. Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol 4:399–418. doi: 10.1007/s11481-009-9164-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prinz M, Priller J. 2014. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci 15:300–312. doi: 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- 29.Stein M, Keshav S, Harris N, Gordon S. 1992. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med 176:287–292. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mildner A, Djukic M, Garbe D, Wellmer A, Kuziel WA, Mack M, Nau R, Prinz M. 2008. Ly-6G+ CCR2− myeloid cells rather than Ly-6Chigh CCR2+ monocytes are required for the control of bacterial infection in the central nervous system. J Immunol 181:2713–2722. doi: 10.4049/jimmunol.181.4.2713. [DOI] [PubMed] [Google Scholar]
- 31.Hinojosa AE, Garcia-Bueno B, Leza JC, Madrigal JL. 2011. CCL2/MCP-1 modulation of microglial activation and proliferation. J Neuroinflammation 8:77. doi: 10.1186/1742-2094-8-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ledeboer A, Brevé JJ, Wierinckx A, van der Jagt S, Bristow AF, Leysen JE, Tilders FJ, Van Dam AM. 2002. Expression and regulation of interleukin-10 and interleukin-10 receptor in rat astroglial and microglial cells. Eur J Neurosci 16:1175–1185. doi: 10.1046/j.1460-9568.2002.02200.x. [DOI] [PubMed] [Google Scholar]
- 33.Gelderblom H, Schmidt J, Londono D, Bai Y, Quandt J, Hornung R, Marques A, Martin R, Cadavid D. 2007. Role of interleukin 10 during persistent infection with the relapsing fever spirochete Borrelia turicatae. Am J Pathol 170:251–262. doi: 10.2353/ajpath.2007.060407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.