

Abstract
Shiga toxins (Stx) are a family of cytotoxic proteins that can cause hemolytic-uremic syndrome (HUS), a thrombotic microangiopathy, following infections by Shiga toxin-producing Escherichia coli (STEC). Renal failure is a key feature of HUS and a major cause of childhood renal failure worldwide. There are currently no specific therapies for STEC-associated HUS, and the mechanism of Stx-induced renal injury is not well understood primarily due to a lack of fully representative animal models and an inability to monitor disease progression on a molecular or cellular level in humans at early stages. Three-dimensional (3D) tissue models have been shown to be more in vivo-like in their phenotype and physiology than 2D cultures for numerous disease models, including cancer and polycystic kidney disease. It is unknown whether exposure of a 3D renal tissue model to Stx will yield a more in vivo-like response than 2D cell culture. In this study, we characterized Stx2-mediated cytotoxicity in a bioengineered 3D human renal tissue model previously shown to be a predictor of drug-induced nephrotoxicity and compared its response to Stx2 exposure in 2D cell culture. Our results demonstrate that although many mechanistic aspects of cytotoxicity were similar between 3D and 2D, treatment of the 3D tissues with Stx resulted in an elevated secretion of the kidney injury marker 1 (Kim-1) and the cytokine interleukin-8 compared to the 2D cell cultures. This study represents the first application of 3D tissues for the study of Stx-mediated kidney injury.
INTRODUCTION
Shiga toxin (Stx) is a family of AB5 protein cytotoxins expressed by Shigella dysenteriae and Shiga toxin-producing Escherichia coli (STEC) (1). Infections by Stx-producing organisms can result in the onset of a disease termed the hemolytic-uremic syndrome (HUS) that is characterized by renal failure, hemolytic anemia, and thrombocytopenia. Shiga toxins are considered the key STEC virulence factor required for the onset of HUS with Stx2 being the toxin most often associated with severe disease (2–4). In the United States, HUS is the most common cause of acute renal failure in children under 5 years of age (5–7). The majority of HUS cases are caused by STEC, and in the United States, the O157:H7 serotype is most frequently associated with this severe disease (7). Although the underreporting of cases precludes precise calculation of the incidence of STEC infection, the Centers for Disease Control and Prevention (CDC) has estimated that 73,000 cases of O157:H7 infection occur annually in the United States (http://www.cdc.gov/pulsenet/pathogens/ecoli.html) (8).
Approximately two-thirds of children who develop HUS will require dialysis (9), and many will go on to permanent renal insufficiency or renal failure. Excluding supportive therapies such as hemodialysis and volume expansion with intravenous fluids, there are no specific therapies for STEC-associated HUS. While Stxs are the primary STEC pathogenicity factors responsible for the onset of HUS by STEC, the mechanism(s) by which Stxs promote this deadly thrombotic microangiopathy in patients is still not well understood.
Shiga toxins are AB5 toxins composed of five B-subunits which bind to the host-cell surface receptor globotriaosylceramide (Gb3), and a single A-subunit with a specific N-glycosidase activity that affects protein synthesis (10–12). After binding to Gb3, Stx undergoes endocytosis and retrograde trafficking to the endoplasmic reticulum (ER), where the A-subunit separates from the B-subunits and is then transported across the ER membrane to the cytosol (13). Once in the cytosol, the A-subunit catalyzes the cleavage of an adenine from the 28S rRNA, thus inhibiting protein synthesis and activating proinflammatory signaling (11, 12). Certain types of protein synthesis inhibitors, including Stx, which interact with and/or damage the 28S rRNA have been shown to activate the mitogen activated protein kinases (MAPKs), jun-N-terminal kinases (JNKs), and p38 (14, 15). This activation of MAPKs by Stx and other protein synthesis inhibitors has been termed the ribotoxic stress response (RSR). Stx-mediated activation of the RSR has been shown to contribute to induction of cytokines such as interleukin-8 (IL-8) at both the transcriptional and the posttranscriptional level in intestinal epithelial cells (16, 17).
While renal failure is a key feature of HUS and renal samples from patients with HUS show extensive damage (2), the temporal pattern of Stx-mediated renal damage has not been determined, and it remains unclear which renal cells are the primary targets during disease. This is largely due to an inability to monitor renal damage on a molecular and cellular level during the course of disease in human patients and to access patients during the early stages of disease progression. To overcome these limitations, investigators have used either animal models (18, 19) or two-dimensional (2D) monolayer cell cultures to study Stx-mediated disease (20–22). 2D cell culture systems, while simple and low in cost, lack the architecture required to play a major role in cell-cell and cell-extracellular matrix (ECM) interactions and are thus incapable of recapitulating the complexity of the in vivo environment (23, 24), as well as inadequate at predicting cellular toxicity (25, 26). Studies using 2D human renal proximal tubule cells have demonstrated that Stx increases apoptosis in a time- and dose-dependent manner, along with the secretion of IL-1β, IL-6, and IL-8, the inhibition of protein synthesis, and water absorption (20, 21, 27, 28). Animal models are incapable of fully recapitulating human disease for numerous reasons, including differences in physiology and experimental conditions (29). Importantly, differences in Stx receptor expression patterns between mice and humans contribute to the failure of murine models of HUS to completely mimic human sequelae (30). Although nonhuman primates have been shown to recapitulate the clinical manifestation of HUS and respond to therapeutics (31), these models are expensive and require specialized staff and facilities.
The advantages of using 3D human tissues over 2D cell culture have already been demonstrated in studies of cancer progression, signal transduction, and drug toxicity (23, 32–35). These studies have revealed advantages of 3D tissue models over 2D cell cultures due to differences in cell-cell signaling, ECM interactions, ECM composition, gene expression, and epigenetic gene regulation (23, 36). Bioengineered 3D kidney tissues produced in our lab using human cells have been shown to better reflect human renal physiology than 2D cell culture (37). These tissues are composed of human telomerase reverse transcriptase (hTERT) immortalized human renal proximal tubule cells embedded within a hydrogel. After 2 weeks of culture, they form an interconnecting network of tubules (Fig. 1A). These bioengineered kidney tissues are capable of revealing toxic effects of both acute and chronic exposure to insult since they are viable for up to 8 weeks, unlike 2D cell cultures, which are viable and relevant for only days before increased cell density results in cell death.
FIG 1.

Shiga toxin-mediated cytotoxicity. (A) Representative image of the interconnecting tubules present in the 3D bioengineered human kidney tissue model. (B) Timeline of tissue formation, treatment, and sampling. (C) A [3H]leucine incorporation assay was used to determine the sensitivity of 2D and 3D cultures to overnight (24- to 26-h) treatment with Stx2 (LPS not removed). (D) H&E image of representative 3D tissues after 72 h of no treatment or treatment with noted concentrations of Stx2. Arrows indicate the hematoxylin-stained cells within the matrix. (E) IC50 values at specific time points for 2D and 3D tissues.
The goal of the present study was to adapt our 3D bioengineered human renal tissue model for the study of Stx-induced renal toxicity and to compare the responses with responses by 2D cultures. We examined cytotoxicity, activation of the RSR, renal injury biomarker expression, and inflammatory cytokine secretion. Although the overall cytotoxicity profile, protein synthesis inhibition, and RSR activation were similar between 2D and 3D cultures, there were differences in the IC50 (the dosage at which 50% toxicity was achieved) at different time points, the secretion of renal injury biomarkers, and cytokine secretion between the two systems. Importantly, treatment of the 3D tissues with Stx resulted in an elevated secretion of the kidney injury marker 1 (Kim-1) and the cytokine IL-8 compared to the 2D cell cultures. IL-8 secretion levels have been shown to significantly increase in the urine of baboons after Stx2 treatment (31) and from intestinal epithelial cells exposed to Stx2 (38). Kim-1 has been shown to be induced upon infection of mice with Stx-producing Citrobacter rodentium, a murine model for enterohemorrhagic E. coli-related disease (39); the present study is the first to show a Kim-1 renal injury biomarker response to Stx2 in vitro. This study is also the first utilization of a 3D renal tissue model for the study of Stx-mediated nephrotoxicity and reveals significant benefits of a 3D tissue model compared to 2D cell culture.
MATERIALS AND METHODS
Cell culture.
The NKi-2 cells used in all studies were derived by the serial passage in low-serum media of hTERT-immortalized human renal cortical cells. Cells were grown in high-glucose Dulbecco modified Eagle medium (DMEM)/F-12 (Invitrogen) containing 2% fetal bovine serum (Invitrogen), 20 ng/ml hEGF (Invitrogen), 72 ng/ml T3 (Sigma), 1% ITS (Invitrogen), 100 ng/ml hydrocortisone (Sigma), and 1% penicillin-streptomycin (Invitrogen). For 2D studies, cells were seeded at a density of 25,000 cells per well of a 12-well tissue culture plate 24 h prior to exposure to Stx2. Every effort was taken to ensure that matching passages were used for 2D and 3D cultures.
Shiga toxin.
Stx2 was purchased from Anne Kane of the Phoenix Laboratory at Tufts Medical Center. The toxin is purified by P1 glycoprotein affinity chromatography based on the method of Donohue-Rolfe et al. (40). Lipopolysaccharide (LPS) was removed by using Pierce high-capacity endotoxin removal resin (Thermo) on a column with a 4-h incubation. LPS removal was confirmed by using a ToxinSensor chromogenic LAL endotoxin assay kit (GenScript). As a control, Stx2 was inactivated by boiling for 8 h. For dosing, Stx2 was reconstituted in sterile phosphate-buffered saline (PBS) at 1 mg/ml and then diluted in cell culture medium in concentrations ranging from 1 fg/ml to 1 μg/ml. For 2D cultures, 2 ml of Stx2-containing medium was applied to each well, and 350 μl was removed at each time point. For 3D cultures, 1.5 ml of Stx2 culture medium was applied below the tissue, and 0.5 ml was applied on top of the tissue. Then, 350 μl of medium was removed from the bottom at each time point. The cellular supernatants were stored at −80°C and used for lactate dehydrogenase (LDH), Kim-1, neutrophil gelatinase-associated lipocalin (NGAL), and cytokine assays.
3D tissue constructs.
3D tissue constructs were formed as previously describes (37). Briefly, 100,000 NKi-2 cells were combined with a 50:50 mix of Matrigel and rat tail collagen I at a final concentration of 1 mg/ml in a 12-well transwell insert. The tissues were allowed to polymerize at 37°C for 1 h, after which NKi-2 cell culture medium was added to both the bottom well of the transwell and inside the insert. Tissues were grown at 37°C in 5% CO2, with medium changes every 2 days.
Cytotoxicity.
LDH secretion was assayed in 100 μl of cell culture supernatant from both 2D NKi-2 cells and 3D tissues using an LDH cytotoxicity assay from Clontech and according to the manufacturer's instructions. We used 2% Triton as a positive control for cell death. The percent cytotoxicity was calculated as follows: % cytotoxicity = (Stx2 treated − untreated)/(2% Triton treated − untreated) × 100. Samples from three independent experiments were analyzed and graphed using GraphPad Prism 5 software. The data are expressed as averages ± the standard errors of the mean (SEM).
IC50 calculations.
The IC50, i.e., the dosage at which 50% toxicity was achieved, was calculated using LDH data after 48 and 72 h of treatment for 2D cells and 3D tissues. Values were calculated with GraphPad Prism 5 software by using nonlinear regression analysis.
Protein synthesis inhibition.
Protein synthesis inhibition was determined by measuring the decrease in [3H]leucine incorporation. For 2D cultures, cells were plated in 96-well plates at a density of 0.6 × 105 to 1 × 105 cells/well. The following day, the medium was removed, fresh medium without toxin or with Stx2 at concentrations ranging from 1 μg/ml to 1 pg/ml was added, and the cells were incubated overnight. The next day (i.e., at 24 to 26 h after intoxication), the medium was removed, and the cells were gently washed twice with prewarmed PBS. To the cells, medium made 1:1 with RPMI with 25 mM HEPES without leucine (Chemicon International, catalog no. R-078-B) and low-glucose DMEM without Leu, Lys, and Arg (Sigma, catalog no. SLBH1080) and containing 5 μl/ml [3H]leucine (leucine [3,4,5-3H(N)], Perkin-Elmer, catalog no. NGT460001MC [1 mCi, 106.2 Ci/mmol]) was added, and the cells were incubated at 37°C in 5% CO2 for 1 h. After this incubation, the medium was removed, and the cells were washed twice with 200 μl of PBS. Then, 50 μl of 0.2 M KOH was added to each well containing cells, followed by incubation at room temperature for 15 min, followed by the addition of 150 μl of 10% trichloroacetic acid (TCA). The plates were then incubated at room temperature for 30 min, followed by transfer of the well contents to a 96-well vacuum manifold-coupled filter plate (Millipore, catalog no. MAHVN4550). Wells were washed twice with 200 μl of 5% TCA, transferring each wash to the appropriate filter well on the vacuum manifold. The filters were washed once with 5% TCA (200 μl/filter), followed by a wash with 200 μl of 1% acetic acid. The filters were allowed to dry and then punched into vials for scintillation counting.
The 3D tissues in 12-well plates were treated similarly to the 2D cultures, with the following four exceptions. (i) After the removal of toxin-containing media from the wells, the tissues were washed twice in prewarmed PBS and then incubated in leucine-free medium for 90 min to dilute out the leucine-containing medium from the matrix prior to adding the [3H]leucine-containing medium. (ii) The [3H]leucine-containing medium was incubated for 7.5 h to allow for sufficient detectable incorporation of radioactivity. The tissues were then washed twice with PBS. (iii) A total of 300 μl of Dispase (BD Scientific, catalog no. 354235) containing 100 μg of collagenase type I/ml was added to the upper chamber of the tissue-containing transwell to break down the tissue-containing matrix. This sample was incubated for 1.5 h at 37°C in 5% CO2. (iv) Lastly, the protein in the upper chamber was precipitated by adding 100% TCA to a final concentration of 25%, incubating the sample at 4°C for 30 min, and spinning the sample in a microcentrifuge for 5 min at maximum speed. One milliliter of ice-cold acetone was added to the pellet, and the tubes were centrifuged at maximum speed for 5 min. The resulting pellet was allowed to dry and suspended in scintillation fluid prior to being transferred to a scintillation vial for counting.
Immunohistochemistry.
After 72 h of Stx2 exposure, tissues were fixed with 10% buffered formalin for 48 h at 4°C and embedded in paraffin. Hematoxylin-eosin (H&E) staining was performed on 8-μm tissue sections, and images were captured with Leica Application Suite v4, using a Leica DMIL microscope equipped with a Leica DFC340FX camera for fluorescent images and a Leica DFC295FX camera for bright-field images.
Kidney injury biomarkers.
Kim-1 and NGAL secretion was assayed in 50-μl portions of the cell culture supernatant from both NKi-2 cells and 3D tissues by performing enzyme-linked immunosorbent assays (R&D Systems) according to the manufacturer's instructions. All measurements were performed with Softmax Pro 5.4 software on a SpectraMax M2 plate reader (Molecular Devices). Experiments were analyzed and graphed using GraphPad Prism 5 software and are expressed as the mean concentration ± the SEM. The statistical significance was assessed at P < 0.05 by two-way analysis of variance (ANOVA) with a Bonferroni posttest.
Cytokine secretion.
Cytokine secretion was measured in 25 μl of cell culture supernatant using a human cytokine/chemokine Milliplex MAP kit from Millipore customized to measure IL-1β, IL-6, IL-8, tumor necrosis factor alpha (TNF-α), and monocyte chemotactic protein 1 (MCP-1) and performed according to the manufacturer's instructions. Experiments were analyzed and graphed using GraphPad Prism 5 software and are expressed as the mean concentration ± the SEM. Statistical significance was assessed at P < 0.05 by two-way ANOVA with a Bonferroni posttest.
MAPK assays.
Phospho-JNKs and phospho-p38 levels were determined by using immunoprecipitation-based assays (Cell Signaling Technology, catalog numbers 8794 and 9820 for phospho-JNKs and phospho-p38, respectively) according to the manufacturer's instructions. To 200 μg of protein-containing cell lysates, were added phospho-specific JNKs or p38 antibody-conjugated beads, followed by incubation overnight. The beads were then washed twice in lysis buffer and twice in kinase reaction buffer, followed by resuspension of the beads in kinase reaction buffer containing 200 μM ATP and 1 μg of kinase substrate (c-Jun fusion protein for phospho-JNKs and ATF-2 fusion protein for phospho-p38).
MTT assay.
Cells were plated in 96-well tissue culture plates at a density of 0.6 × 105 cells/well. The next day, Stx2 was added at concentrations from 1 pg/ml to 1 μg/ml. After 24 and 48 h of incubation at 37°C in 5% CO2, the medium was removed, and 100 μl of a 0.5-mg/ml concentration of MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] in PBS was added to each well. After incubation at 37°C and 5% CO2 for 1 h, the MTT solution was removed, and 50 μl of isopropanol containing 4 mM HCl and 0.1% Nonidet P-40 was added, followed by gentle shaking at room temperature for 15 min. Optical density measurements were then made at 550 and 620 nm.
RESULTS
Shiga toxin-induced nephrotoxicity.
In order to validate our bioengineered 3D human renal model for the study of Stx-induced nephrotoxicity, we began by comparing cytotoxicity in 3D to cytotoxicity in the same cells grown in 2D (Fig. 1). 3D tissues containing immortalized renal proximal tubule cells (NKi-2) were formed and allowed to grow for 2 weeks prior to treatment with Stx2, whereas 2D cultures of the NKi-2 cells were seeded 24 h prior to Stx2 treatment. For virtually all experiments (Fig. 1C), Stx2 was purified free of LPS, which has been used as a costimulus in a murine model of HUS (41, 42) and may potentiate an Stx response by increasing the inflammatory response or increasing Gb3 receptor on the cell surface (43–46). Both the cells and the tissues were treated with a range of Stx2 concentrations (1 fg to 1 μg/ml) for up to 72 h with supernatant sampling occurring at 6, 24, 48, and 72 h posttreatment (Fig. 1B).
To first test whether Stx2 had the predicted inhibitory effect on protein synthesis, we added increasing concentrations of the toxin to either 2D or 3D cultures. Protein synthesis of both cultures, measured by determining the [3H]leucine incorporation (see Materials and Methods), was inhibited in a similar dose-dependent manner (Fig. 1C). The predicted Stx2-induced cytotoxicity was demonstrated for 3D cultures by H&E staining, which revealed the loss of nuclei at 72 h postexposure (Fig. 1D), and for 2D cultures by an MTT cell viability assay (see Fig. S1 in the supplemental material). Importantly, cell viability at 24 h after Stx2 exposure was >50% for all Stx2 concentrations tested, indicating that the inhibition of protein synthesis observed at 24 h (Fig. 1C) was not simply a reflection of cell death. Heat treatment of purified Stx2 abrogated the cytotoxic effect (data not shown), suggesting that cytotoxicity was not a reflection of possible contamination by LPS. To discern possible dramatic differences in the kinetics or dose dependence of cytotoxicity between 2D and 3D cultures, we measured the IC50 for LDH release at various times after Stx2 exposure. We found that at 48 h postexposure, 3D cultures were somewhat (7-fold) less sensitive than 2D cultures; at 72 h, 3D cultures were somewhat (3-fold) more sensitive (Fig. 1E). We conclude that although the cytotoxic responses of the two cultures may differ somewhat in kinetics and relative sensitivity, Stx2 inhibits protein synthesis and triggers cytotoxicity similarly in both 2D and 3D cultures.
Finally, since Stx-mediated damage to actively translating ribosomes results in activation of the RSR, we investigated whether a RSR was induced in 2D and 3D renal cultures. Whole-cell lysates were made from 2D and 3D cultures that were untreated or treated with heat-inactivated Stx2, Stx2 without LPS removed, and Stx2 after LPS removal for 6 h. Phospho-JNKs were immunoprecipitated from whole-cell lysates, the immunoprecipitates were incubated with c-Jun substrate, and the levels of phosphorylated c-Jun were determined by Western blotting. The activation of JNKs (Fig. 2A) was increased in Stx2-treated 2D and 3D samples but not in heat-inactivated samples. Similarly, immunoprecipitation of phosphorylated p38 (Fig. 2B) demonstrates that p38 activation was also increased by Stx2 in both 2D and 3D cultures. Taken together, these data indicate that there are similarities in the general cytotoxic response to Stx2 between 2D and 3D cultures in terms of dose response, protein synthesis inhibition, and activation of the RSR.
FIG 2.

Ribotoxic stress response. Activation of JNKs (A) or p38 (B) was determined by Western blotting, comparing in vitro phosphorylation of c-Jun (JNKs) or ATF-2 (p38) after precipitation of phosphorylated JNKs or p38 from whole-cell lysates. Bar graphs represent blot band intensity of P-c-Jun or P-ATF-2 as normalized against loading controls.
Kidney injury marker secretion in response to Stx2 treatment.
A number of biomarkers have been identified to monitor kidney damage. These include both Kim-1 and NGAL. Kim-1 secretion in the urine of patients or by kidney epithelial cells in response to Stx2 has not been examined, and only two studies have examined the secretion of NGAL in patient urine and serum in response to Stx (47, 48). NGAL has been examined for its ability to predict renal injury in HUS (48) and the need for renal replacement therapy after Stx exposure (47). We have previously shown its utility as a marker of drug-induced nephrotoxicity in 3D tissues and that it was predictive of renal proximal tubule cytotoxicity for highly toxic agents at high doses but incapable of distinguishing more subtle differences in toxicity in vitro due to high expression from untreated controls (37). None of the Stx2 doses resulted in NGAL secretion in excess of untreated controls in either 3D or 2D cultures (Fig. 3A). Interestingly, the highest doses of Stx2, 1 μg/ml and 1 ng/ml, which resulted in almost 100% cytotoxicity in 3D cultures by LDH secretion (Fig. 1C), resulted in a significant decrease in NGAL secretion (P < 0.001) by 72 h posttreatment. This may be due to the almost complete inhibition in protein synthesis seen at these doses by 24 h posttreatment. These data raise the possibility that low levels of NGAL, rather than high levels, may be predictive of Stx-induced cytotoxicity in vitro.
FIG 3.
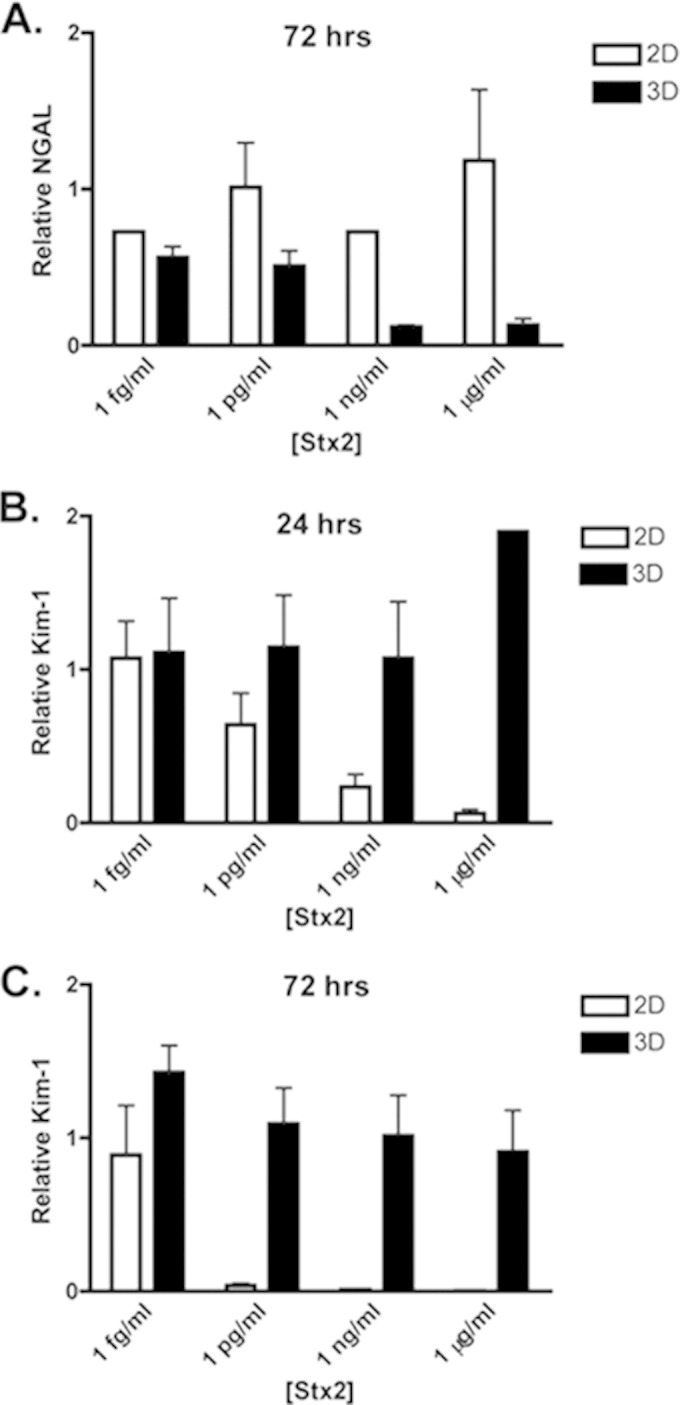
Kidney injury markers. (A) NGAL production in response to Stx2 at 72 h after toxin treatment. The Kim-1 production in response to Stx2 is shown at 24 h (B) and 72 h (C) after toxin treatment.
Kim-1 has been shown to be a very sensitive biomarker of kidney injury and we have previously shown its utility in measuring cytotoxicity in 3D with highly nephrotoxic compounds (37). However, we have also shown that secreted Kim-1 increases over time in untreated control cultures (37) making it only useful at extremely toxic concentrations and not sensitive or precise enough to reveal subtle changes in cytotoxicity in vitro. Consistent with these previous findings, Kim-1 increased in expression in both the 2D and 3D untreated controls over time (data not shown). When assayed at 72 h after exposure to 1 pg/ml Stx2 or greater, Kim-1 production by 2D cultures was inhibited, perhaps due to the protein synthesis inhibition induced by the toxin, whereas 3D cultures were largely unaffected by Stx2 exposure (Fig. 3B). When assayed at an earlier time point, i.e., 24 h after exposure, 1 μg/ml Stx2 induced Kim-1 production increased almost 2-fold above the untreated controls (Fig. 3C). In contrast, at 24 h postexposure no induction of Kim-1 was observed for 2D cultures at any Stx2 concentration (Fig. 3B). The Stx2-induced Kim-1 secretion response by 3D but not 2D cultures suggests that 3D tissues may provide a better experimental model for examining renal damage using this marker.
Stx2-induced cytokine secretion.
Since cytokines have been shown to play a role in Stx2-induced renal damage (for a review, see reference 49), are secreted by a number of different cell types in response to Stx2 (17, 20, 50–52), and are differentially secreted based upon the toxin sensitivity of the cell (20, 53), we examined whether 3D culturing would elicit a different response compared to 2D in the secretion of a set of cytokines previously shown to be secreted by cells in response to Stx, IL-1β, IL-6, IL-8, MCP-1, and TNF-α (Fig. 4) (20, 21, 31, 54–56). All cytokines were measured at 24 and 48 h after treatment with increasing concentrations of Stx2. IL-1β was not detectable in either 2D or 3D cultures (data not shown), and untreated controls had baseline secretions of IL-6, IL-8, TNF-α, and MCP-1 that increased between 24 and 48 h for both 2D and 3D cultures (Fig. 4, see open and filled bars, “untreated”).
FIG 4.

Cytokine expression. MCP-1 (A), IL-6 (B), TNF-α (C), and IL-8 (D) secretion into the cell culture supernatant after 24 and 48 h of Stx2 treatment of 3D and 2D cultures. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Upon intoxication of either model with increasing concentrations of Stx2, MCP-1 secretion was not induced. However, MCP-1 production exhibited a dramatic difference between the two models. MCP-1 was almost undetectable in 2D cultures, and while the secreted levels did increase over time, the levels never exceeded the levels of the untreated controls (Fig. 4A). In striking contrast, MCP-1 was produced at levels 20- to 40-fold higher in 3D cultures, a result that may reflect the observation that increased MCP-1 is detected in the urine of children with HUS (57).
For both 3D and 2D cultures, the secretion of IL-6 (Fig. 4B), TNF-α (Fig. 4C), and IL-8 (Fig. 4D) were each induced 48 h after exposure to Stx2 at 1 pg/ml. Cytokines were not induced by 1 ng/ml or 1 μg/ml Stx2. The fact that a significant change in expression was only seen at 1 pg/ml may be due to the fact that at the higher concentrations protein synthesis inhibition prevented cytokine production immediately. In contrast, 1 pg/ml is most similar to the IC50s determined in Fig. 1B and may not intoxicate cells rapidly enough to prevent the production and secretion of inflammatory factors. That Stx2 induces cytokine production despite promoting protein synthesis inhibition is consistent with the ability of Stx to induce gene expression and protein production via signaling pathways not directly activated by protein synthesis inhibition (16, 58, 59).
The 3D tissue IL-8 responses to Stx2 deserves particular note. First, unlike IL-6 and TNF-α, but like MCP-1, the basal production of IL-8 in the absence of Stx2 was 4- to 8-fold higher in 3D tissues than in 2D cells (Fig. 4D). Second, this already high level of secretion of IL-8 was further induced ∼1.8-fold by 1 pg/ml Stx2, resulting in IL-8 levels higher than that induced in 2D cultures by Stx2. This increase in 3D versus 2D could indicate a property of the 3D culture system that upregulates both basal and induced IL-8 levels, or it could reflect a difference in cell numbers able to secrete IL-8 at this time point. Importantly, while the exact role of IL-8 in HUS is unknown, increases in concentration have been noted in the urine of children with HUS, the urine of Stx2-treated baboons, and in human intestinal epithelial cells after Stx2 exposure (31, 38, 57).
DISCUSSION
In this study, we have shown the utility of a 3D bioengineered human renal tissue model for the examination of the mechanisms of Stx-induced renal cytotoxicity. Although many responses were similar between the 2D and 3D cultures, including overall cytotoxicity, the IC50, protein synthesis inhibition, RSR, and IL-6 and TNF-α secretion, there were also differences indicating that a 3D system may provide a more physiologically relevant model than a 2D system for examination of cellular responses to Stx2. Two distinct and notable differences were observed between the 2D and 3D systems. First, higher basal levels of Kim-1, NGAL, MCP-1, and IL-8 were observed from the 3D tissues compared to the 2D cells, indicating that these inflammatory or renal injury markers may be better expressed in the 3D tissues. Second, higher induction levels of Kim-1 and IL-8 were observed from the 3D tissues compared to the 2D cells, indicating that the 3D tissues may be a better system for monitoring Stx2-mediated renal injury in vitro through renal biomarker secretion (Kim-1) and characteristic cytokine secretion (IL-8). Taken together, these data suggest differential cellular mechanisms functioning between 2D and 3D cultures in response to Stx2.
Initially, the general features of cytotoxicity of Stx2 appeared to be very similar between the 2D and 3D cultures. For example, several studies have demonstrated that intoxication with Stx activates proinflammatory and proapoptotic signaling pathways (60, 61), in part through the activation of the MAPK pathway (62–64). This activation of the RSR contributes to both proinflammatory and proapoptotic signaling in cultured cells (62, 63, 65, 66). We showed that human renal 2D and 3D tissue both underwent an RSR after Stx2 treatment, suggesting that this signaling cascade may participate in Stx-associated inflammation and cell death in the human kidney. Whether the kinetics of the RSR in 2D and 3D models differs will be a subject of future studies, as will defining the relative contribution of the RSR to cytokine production and cell death.
A second similarity was the general trend of inflammatory cytokine expression in the cell culture supernatant. Inflammatory signaling is associated with HUS, since patients show increased urinary levels of proinflammatory cytokines, and renal biopsy specimens have revealed infiltration of neutrophils and monocytes into the kidney during disease (57, 67–69). In 2D cell culture models, Stx treatment of human proximal tubular cells has been shown to induce the secretion of IL-1β, IL-6, and IL-8 (27), and evidence suggests that IL-1, IL-8, TNF-α, and IL-6 may be elevated in the serum of D+HUS patients (55, 56). Also, IL-1 and TNF have been shown to upregulate the Stx cell surface receptor globotriaosylceramide (Gb3) and increase cellular sensitivity to Stx cytotoxicity in endothelial cells (70, 71). In nonhuman primates treated with Stx, kidney tissue had upregulated levels of IL-8, MCP-1, and TNF-α mRNA, along with increased urinary levels of IL-6, IL-8, and MCP-1 (31). Our studies revealed an increase in IL-6, IL-8, and TNF-α over time after Stx2 treatment in both 2D and 3D models. The three detectable cytokines all showed an increase above untreated controls at 48 h with 1 pg/ml Stx2, while the higher concentrations did not secrete any cytokines above the level of the untreated control. This was most likely due to the relatively high rate of cytotoxicity and protein synthesis inhibition seen with the higher doses; in contrast, the 1-pg/ml dose was close to the calculated IC50 and therefore did not result in complete cytotoxicity of the cultures. Studies designed to examine cytokine secretion at earlier time points after Stx2 treatment may reveal increased secretion at higher doses. However, studies of cytokine secretion from HK-2 cells at early time points (<4 h) did not reveal an increase in IL-1β, TNF-α, or IL-8 protein levels, although increases in mRNA transcripts were observed (20).
Examination of the IC50 at both 48 and 72 h revealed a difference between the 2D and 3D culture systems. In particular, there was a difference in the response rate to Stx2 between the 2D and 3D cultures. The 2D cultures were 7-fold more sensitive to Stx2 than the 3D cultures at 48 h posttreatment, but by 72 h the two cultures exhibited roughly similar Stx2 sensitivity. This difference does not simply reflect delayed diffusion of Stx2 into the 3D culture because, at 24 h posttreatment, Stx2 induced a similar degree of protein synthesis inhibition in both the 2D and the 3D cultures. Thus, it may be that the difference in cellular organization and structure has an impact upon Stx2-mediated cytotoxicity. Interestingly, HK-2 cells, an HPV-immortalized human renal proximal tubule cell line, were shown to have an IC50 of 20 pg/ml at 72 h posttreatment with Stx2, compared to 2.2 pg/ml and 0.7 pg/ml at the same time point for 2D and 3D NKi-2 cells, respectively, in the present study. HK-2 cells have also been shown to lack sensitivity to drug-induced toxicity (26), indicating that they may not be the best cell line for studying renal cytotoxicity.
A second significant difference between the 2D and 3D cultures was the inability to detect MCP-1 in the 2D cultures, and while it was detectable in the 3D cultures and increased with time, after Stx2 treatment it was secreted at levels significantly lower than the untreated controls. Interestingly, while MCP-1 mRNA has been shown to be elevated in the kidneys of nonhuman primates exposed to Stx2 and the protein has been shown to be elevated in the urine, its mRNA was barely detectable in cultured renal endothelial cells (31). This may indicate that the secreted MCP-1 is originating from a different cell type or the specifically high basal levels of MCP-1 might also reflect a physiologic response to the accumulation of metabolic waste products. In vivo, tubules exist adjacent to both a vasculature and a lumen through which metabolites can be either taken up into the bloodstream or discarded through the urine. Since our current 3D tubule system is not perfused, it is reasonable to hypothesize that the increase in MCP-1 over the course of 48 h reflects a normal physiological response to the accumulation of metabolic waste products. In this scenario, the tubular epithelium is signaling for resident tissue phagocytes to aid in the removal of these waste products in an effort to maintain a homeostatic environment. Furthermore, the high basal MCP-1 level does not appear to be part of an inflammatory response since neither TNF-α nor IL-6 appear to be upregulated in our model in the absence of Stx. This hypothesis is supported by the observation that the kidney maintains distinct populations of resident mononuclear phagocytes that produce anti-inflammatory and homeostatic factors (72), and the high basal levels of MCP-1 might therefore represent a homeostatic response not present in 2D cell culture.
A third significant difference between the 2D and 3D cultures was the secretion of IL-8. Although the secretion of IL-8 increased from 24 to 48 h for both the 3D tissues and the 2D cells, the amounts were ∼4-fold higher from the 3D tissues compared to the 2D cells. Importantly, this induction occurred in response to 1 pg/ml Stx2, which may be present in sera of STEC-infected patients at very low levels, i.e., often lower than the 6-pg/ml limit of detection (73). IL-8 has been shown in the urine of nonhuman primates 48 h after exposure to Stx2 at ∼600 pg/ml (31). Our cells in 2D culture did not reach that amount at any time or dose, whereas the 3D tissues more than doubled that amount at 48 h with a 1-pg/ml dose. Thus, the 3D tissue system may be more representative of the in vivo response than the cells in 2D.
Biomarkers of renal injury are extremely important for the prediction of drug-induced nephrotoxicity and the early assay of acute renal injury. A number of potential markers have been identified and are in different stages of validation and use. Kim-1 and NGAL are two such markers. Although they have been shown to be predictive of drug-induced nephrotoxicity in vivo and have been examined as biomarkers for HUS (47, 48, 74), we have previously shown that they may only be minimally useful to assess toxicity in cultured renal cells in vitro primarily due to the high levels secreted by untreated controls (37). What was notable in our studies was an increase in Kim-1 secretion at 1 μg/ml Stx2 compared to the untreated controls at 24 h that then later decreased. This spike was only evident in the 3D cultures. The 2D cultures never reached a secretion level above the untreated controls. This may indicate an initial early spike in secretion in relation to Stx2-mediated cytotoxicity that then decreases due to the inhibition of protein synthesis that we show occurred by 24 h posttreatment. Further studies at early time points need to be conducted. This spike would never have been noticed if we had only been examining the cells in 2D culture. Our 3D studies indicate that Kim-1 has the potential to act as an early biomarker of renal injury for infected patients and in vivo studies need to be performed to confirm this. If it is confirmed in vivo, then our studies also indicate a larger relevance of the 3D system over 2D cell culture in mimicking Stx2-mediated renal injury.
In conclusion, we have presented the first use of a 3D bioengineered human kidney tissue model to study renal damage upon Stx2 exposure. We have shown that the 3D tissue responds to Stx2 with a dose- and time-dependent cytotoxicity that is related to the inhibition of protein synthesis. We have also shown changes in renal injury biomarkers in a dose-dependent manner along with changes in the secretion of cytokines and activation of the RSR. Key differences between the 3D tissue over 2D cell culture included differences in the IC50 and the ability to measure significantly high levels of kidney injury biomarkers and cytokines, particularly IL-8, MCP-1, and Kim-1. Further use of this model may save on the use of inaccurate animal models while making in vitro studies more relevant to in vivo conditions.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the National Institutes of Health (grants P41 EB002520, R01 AI46454, R37 DK51050, and R01 DK099532), along with the “Tufts Collaborates!” seed grant program.
We thank Anne Kane of the Phoenix Laboratory for assistance with the Stx2 and Alexander Histed for insight on MCP-1. We also thank Milva Ricci, Carmen Preda, and Darin Goodwin for their assistance.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02143-14.
REFERENCES
- 1.Strockbine NA, Marques LR, Newland J W, Smith HW, Holmes RK, O'Brien AD. 1986. Two toxin-converting phages from Escherichia coli O157:H7 strain 933 encode antigenically distinct toxins with similar biologic activities. Infect Immun 53:135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Obrig TG, Karpman D. 2012. Shiga toxin pathogenesis: kidney complications and renal failure. Curr Top Microbiol Immunol 357:105–136. doi: 10.1007/82_2011_172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karmali MA, Petric M, Lim C, Fleming PC, Arbus GS, Lior H. 1985. The association between idiopathic hemolytic-uremic syndrome and infection by verotoxin-producing Escherichia coli. J Infect Dis 151:775–782. doi: 10.1093/infdis/151.5.775. [DOI] [PubMed] [Google Scholar]
- 4.Mora A, Leon SL, Blanco M, Blanco J E, Lopez C, Dahbi G, Echeita A, Gonzalez EA, Blanco J. 2007. Phage types, virulence genes and PFGE profiles of Shiga toxin-producing Escherichia coli O157:H7 isolated from raw beef, soft cheese and vegetables in Lima (Peru). Int J Food Microbiol 114:204–210. doi: 10.1016/j.ijfoodmicro.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 5.Ong KL, Apostal M, Comstock N, Hurd S, Webb TH, Mickelson S, Scheftel J, Smith G, Shiferaw B, Boothe E, Gould LH. 2012. Strategies for surveillance of pediatric hemolytic-uremic syndrome: Foodborne Diseases Active Surveillance Network (FoodNet), 2000-2007. Clin Infect Dis 54(Suppl 5):S424–S431. doi: 10.1093/cid/cis208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gould LH, Demma L, Jones TF, Hurd S, Vugia DJ, Smith K, Shiferaw B, Segler S, Palmer A, Zansky S, Griffin PM. 2009. Hemolytic uremic syndrome and death in persons with Escherichia coli O157:H7 infection, foodborne diseases active surveillance network sites, 2000-2006. Clin Infect Dis 49:1480–1485. doi: 10.1086/644621. [DOI] [PubMed] [Google Scholar]
- 7.Tarr PI, Gordon CA, Chandler WL. 2005. Shiga-toxin-producing Escherichia coli and hemolytic uraemic syndrome. Lancet 365:1073–1086. doi: 10.1016/S0140-6736(05)71144-2. [DOI] [PubMed] [Google Scholar]
- 8.Mead PS, Slutsker L, Dietz V, McCaig LF, Bresee J S, Shapiro C, Griffin PM, Tauxe RV. 1999. Food-related illness and death in the United States. Emerg Infect Dis 5:607–625. doi: 10.3201/eid0505.990502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheiring J, Andreoli SP, Zimmerhackl LB. 2008. Treatment and outcome of Shiga-toxin-associated hemolytic-uremic syndrome (HUS). Pediatr Nephrol 23:1749–1760. doi: 10.1007/s00467-008-0935-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fraser ME, Fujinaga M, Cherney MM, Melton-Celsa AR, Twiddy EM, O'Brien AD, James MN. 2004. Structure of Shiga toxin type 2 (Stx2) from Escherichia coli O157:H7. J Biol Chem 279:27511–27517. doi: 10.1074/jbc.M401939200. [DOI] [PubMed] [Google Scholar]
- 11.Endo Y, Tsurugi K, Yutsudo T, Takeda Y, Ogasawara T, Igarashi K. 1988. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur J Biochem 171:45–50. [DOI] [PubMed] [Google Scholar]
- 12.Fraser ME, Cherney MM, Marcato P, Mulvey GL, Armstrong GD, James MN. 2006. Binding of adenine to Stx2, the protein toxin from Escherichia coli O157:H7. Acta Crystallogr Sect F Struct Biol Crystallogr Commun 62:627–630. doi: 10.1107/S1744309106021968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sandvig K, van Deurs B. 2002. Transport of protein toxins into cells: pathways used by ricin, cholera toxin and Shiga toxin. FEBS Lett 529:49–53. doi: 10.1016/S0014-5793(02)03182-4. [DOI] [PubMed] [Google Scholar]
- 14.Iordanov MS, Pribnow D, Magun J L, Dinh TH, Pearson J A, Chen SL, Magun BE. 1997. Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol Cell Biol 17:3373–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith WE, Kane AV, Campbell ST, Acheson DW, Cochran BH, Thorpe CM. 2003. Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and JNK activation and induction of apoptosis in intestinal epithelial cells. Infect Immun 71:1497–1504. doi: 10.1128/IAI.71.3.1497-1504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thorpe CM, Smith WE, Hurley BP, Acheson DW. 2001. Shiga toxins induce, superinduce, and stabilize a variety of C-X-C chemokine mRNAs in intestinal epithelial cells, resulting in increased chemokine expression. Infect Immun 69:6140–6147. doi: 10.1128/IAI.69.10.6140-6147.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thorpe CM, Hurley BP, Lincicome LL, Jacewicz MS, Keusch GT, Acheson DW. 1999. Shiga toxins stimulate secretion of interleukin-8 from intestinal epithelial cells. Infect Immun 67:5985–5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mohawk KL, O'Brien AD. 2011. Mouse models of Escherichia coli O157:H7 infection and Shiga toxin injection. J Biomed Biotechnol 2011:258185. doi: 10.1155/2011/258185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ochoa F, Lago NR, Gerhardt E, Ibarra C, Zotta E. 2010. Characterization of stx2 tubular response in a rat experimental model of hemolytic-uremic syndrome. Am J Nephrol 32:340–346. doi: 10.1159/000319444. [DOI] [PubMed] [Google Scholar]
- 20.Lentz EK, Leyva-Illades D, Lee MS, Cherla RP, Tesh VL. 2011. Differential response of the human renal proximal tubular epithelial cell line HK-2 to Shiga toxin types 1 and 2. Infect Immun 79:3527–3540. doi: 10.1128/IAI.05139-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Creydt VP, Silberstein C, Zotta E, Ibarra C. 2006. Cytotoxic effect of Shiga toxin-2 holotoxin and its B subunit on human renal tubular epithelial cells. Microbes Infect 8:410–419. doi: 10.1016/j.micinf.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 22.Nestoridi E, Kushak RI, Duguerre D, Grabowski EF, Ingelfinger JR. 2005. Upregulation of tissue factor activity on human proximal tubular epithelial cells in response to Shiga toxin. Kidney Int 67:2254–2266. doi: 10.1111/j.1523-1755.2005.00329.x. [DOI] [PubMed] [Google Scholar]
- 23.Bissell MJ, Radisky DC, Rizki A, Weaver VM, Petersen OW. 2002. The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation 70:537–546. doi: 10.1046/j.1432-0436.2002.700907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo Q, Xia B, Moshiach S, Xu C, Jiang Y, Chen Y, Sun Y, Lahti J M, Zhang XA. 2008. The microenvironmental determinants for kidney epithelial cyst morphogenesis. Eur J Cell Biol 87:251–266. doi: 10.1016/j.ejcb.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.El MM, Laurent G, Mingeot-Leclercq MP, Tulkens PM. 2000. Gentamicin-induced apoptosis in renal cell lines and embryonic rat fibroblasts. Toxicol Sci 56:229–239. doi: 10.1093/toxsci/56.1.229. [DOI] [PubMed] [Google Scholar]
- 26.Wu Y, Connors D, Barber L, Jayachandra S, Hanumegowda UM, Adams SP. 2009. Multiplexed assay panel of cytotoxicity in HK-2 cells for detection of renal proximal tubule injury potential of compounds. Toxicol In Vitro 23:1170–1178. doi: 10.1016/j.tiv.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 27.Hughes AK, Stricklett PK, Kohan DE. 1998. Shiga toxin-1 regulation of cytokine production by human proximal tubule cells. Kidney Int 54:1093–1106. doi: 10.1046/j.1523-1755.1998.00118.x. [DOI] [PubMed] [Google Scholar]
- 28.Silberstein C, Pistone C, Gerhardt VE, Nunez P, Ibarra C. 2008. Inhibition of water absorption in human proximal tubular epithelial cells in response to Shiga toxin-2. Pediatr Nephrol 23:1981–1990. doi: 10.1007/s00467-008-0896-9. [DOI] [PubMed] [Google Scholar]
- 29.Knight A. 2008. Systematic reviews of animal experiments demonstrate poor contributions toward human healthcare. Rev Recent Clin Trials 3:89–96. doi: 10.2174/157488708784223844. [DOI] [PubMed] [Google Scholar]
- 30.Psotka MA, Obata F, Kolling GL, Gross LK, Saleem MA, Satchell SC, Mathieson PW, Obrig TG. 2009. Shiga toxin 2 targets the murine renal collecting duct epithelium. Infect Immun 77:959–969. doi: 10.1128/IAI.00679-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stearns-Kurosawa DJ, Oh SY, Cherla RP, Lee MS, Tesh VL, Papin J, Henderson J, Kurosawa S. 2013. Distinct renal pathology and a chemotactic phenotype after enterohemorrhagic Escherichia coli shiga toxins in non-human primate models of hemolytic-uremic syndrome. Am J Pathol 182:1227–1238. doi: 10.1016/j.ajpath.2012.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Andriani F, Margulis A, Lin N, Griffey S, Garlick JA. 2003. Analysis of microenvironmental factors contributing to basement membrane assembly and normalized epidermal phenotype. J Investig Dermatol 120:923–931. doi: 10.1046/j.1523-1747.2003.12235.x. [DOI] [PubMed] [Google Scholar]
- 33.Segal N, Andriani F, Pfeiffer L, Kamath P, Lin N, Satyamurthy K, Egles C, Garlick JA. 2008. The basement membrane microenvironment directs the normalization and survival of bioengineered human skin equivalents. Matrix Biol 27:163–170. doi: 10.1016/j.matbio.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mammoto T, Ingber DE. 2010. Mechanical control of tissue and organ development. Development 137:1407–1420. doi: 10.1242/dev.024166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lan SF, Starly B. 2011. Alginate based 3D hydrogels as an in vitro coculture model platform for the toxicity screening of new chemical entities. Toxicol Appl Pharmacol 256:62–72. doi: 10.1016/j.taap.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 36.Desrochers TM, Shamis Y, Alt-Holland Kudo Y, Takata T, Wang G, Jackson-Grusby L, Garlick JA. 2012. The 3D tissue microenvironment modulates DNA methylation and E-cadherin expression in squamous cell carcinoma. Epigenetics 7:34–46. doi: 10.4161/epi.7.1.18546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Desrochers TM, Suter L, Roth A, Kaplan DL. 2013. Bioengineered 3D human kidney tissue, a platform for the determination of nephrotoxicity. PLoS One 8:e59219. doi: 10.1371/journal.pone.0059219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jandhyala DM, Rogers TJ, Kane A, Paton AW, Paton J C, Thorpe CM. 2010. Shiga toxin 2 and flagellin from Shiga-toxigenic Escherichia coli superinduce interleukin-8 through synergistic effects on host stress-activated protein kinase activation. Infect Immun 78:2984–2994. doi: 10.1128/IAI.00383-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mallick EM, McBee ME, Vanguri VK, Melton-Celsa AR, Schlieper K, Karalius BJ, O'Brien AD, Butterton J R, Leong J M, Schauer DB. 2012. A novel murine infection model for Shiga toxin-producing Escherichia coli. J Clin Invest 122:4012–4024. doi: 10.1172/JCI62746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Donohue-Rolfe A, Acheson DW, Kane AV, Keusch GT. 1989. Purification of Shiga toxin and Shiga-like toxins I and II by receptor analog affinity chromatography with immobilized P1 glycoprotein and production of cross-reactive monoclonal antibodies. Infect Immun 57:3888–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keepers TR, Psotka MA, Gross LK, Obrig TG. 2006. A murine model of HUS: Shiga toxin with lipopolysaccharide mimics the renal damage and physiologic response of human disease. J Am Soc Nephrol 17:3404–3414. doi: 10.1681/ASN.2006050419. [DOI] [PubMed] [Google Scholar]
- 42.Stahl AL, Svensson M, Morgelin M, Svanborg C, Tarr PI, Mooney J C, Watkins SL, Johnson R, Karpman D. 2006. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic-uremic syndrome. Blood 108:167–176. doi: 10.1182/blood-2005-08-3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clayton F, Pysher TJ, Lou R, Kohan DE, Denkers ND, Tesh VL, Taylor FB Jr, Siegler RL. 2005. Lipopolysaccharide upregulates renal shiga toxin receptors in a primate model of hemolytic-uremic syndrome. Am J Nephrol 25:536–540. doi: 10.1159/000088523. [DOI] [PubMed] [Google Scholar]
- 44.Louise CB, Obrig TG. 1991. Shiga toxin-associated hemolytic-uremic syndrome: combined cytotoxic effects of Shiga toxin, interleukin-1 beta, and tumor necrosis factor alpha on human vascular endothelial cells in vitro. Infect Immun 59:4173–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hughes AK, Stricklett PK, Kohan DE. 1998. Cytotoxic effect of Shiga toxin-1 on human proximal tubule cells. Kidney Int 54:426–437. doi: 10.1046/j.1523-1755.1998.00015.x. [DOI] [PubMed] [Google Scholar]
- 46.Hughes AK, Stricklett PK, Schmid D, Kohan DE. 2000. Cytotoxic effect of Shiga toxin-1 on human glomerular epithelial cells. Kidney Int 57:2350–2359. doi: 10.1046/j.1523-1755.2000.00095.x. [DOI] [PubMed] [Google Scholar]
- 47.Lukasz A, Beneke J, Menne J, Vetter F, Schmidt BM, Schiffer M, Haller H, Kumpers P, Kielstein JT. 2014. Serum neutrophil gelatinase-associated lipocalin (NGAL) in patients with Shiga toxin mediated hemolytic uraemic syndrome (STEC-HUS). Thromb Haemost 111:365–372. doi: 10.1160/TH13-05-0387. [DOI] [PubMed] [Google Scholar]
- 48.Trachtman H, Christen E, Cnaan A, Patrick J, Mai V, Mishra J, Jain A, Bullington N, Devarajan P. 2006. Urinary neutrophil gelatinase-associated lipocalcin in D+HUS: a novel marker of renal injury. Pediatr Nephrol 21:989–994. doi: 10.1007/s00467-006-0146-y. [DOI] [PubMed] [Google Scholar]
- 49.Ramegowda B, Samuel JE, Tesh VL. 1999. Interaction of Shiga toxins with human brain microvascular endothelial cells: cytokines as sensitizing agents. J Infect Dis 180:1205–1213. doi: 10.1086/314982. [DOI] [PubMed] [Google Scholar]
- 50.Ramegowda B, Tesh VL. 1996. Differentiation-associated toxin receptor modulation, cytokine production, and sensitivity to Shiga-like toxins in human monocytes and monocytic cell lines. Infect Immun 64:1173–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harrison LM, van Haaften WC, Tesh VL. 2004. Regulation of proinflammatory cytokine expression by Shiga toxin 1 and/or lipopolysaccharides in the human monocytic cell line THP-1. Infect Immun 72:2618–2627. doi: 10.1128/IAI.72.5.2618-2627.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tesh VL, Ramegowda B, Samuel JE. 1994. Purified Shiga-like toxins induce expression of proinflammatory cytokines from murine peritoneal macrophages. Infect Immun 62:5085–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fujii J, Matsui T, Heatherly DP, Schlegel KH, Lobo PI, Yutsudo T, Ciraolo GM, Morris RE, Obrig T. 2003. Rapid apoptosis induced by Shiga toxin in HeLa cells. Infect Immun 71:2724–2735. doi: 10.1128/IAI.71.5.2724-2735.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hughes AK, Stricklett PK, Kohan DE. 2001. Shiga toxin-1 regulation of cytokine production by human glomerular epithelial cells. Nephron 88:14–23. doi: 10.1159/000045953. [DOI] [PubMed] [Google Scholar]
- 55.Tesh VL. 1998. Virulence of enterohemorrhagic Escherichia coli: role of molecular crosstalk. Trends Microbiol 6:228–233. doi: 10.1016/S0966-842X(98)01282-7. [DOI] [PubMed] [Google Scholar]
- 56.Karpman D, Andreasson A, Thysell H, Kaplan BS, Svanborg C. 1995. Cytokines in childhood hemolytic-uremic syndrome and thrombotic thrombocytopenic purpura. Pediatr Nephrol 9:694–699. doi: 10.1007/BF00868714. [DOI] [PubMed] [Google Scholar]
- 57.Van Setten PA, Van Hinsbergh VWM, Van Den Heuvel LPWJ, Preyers F, Dijkman HBPM, Assmann KJM, Van Der Velden TJAM, Monnens LAH. 1998. Monocyte chemoattractant protein-1 and interleukin-8 levels in urine and serum of patents with hemolytic-uremic syndrome. Pediatr Res 43:759–767. doi: 10.1203/00006450-199806000-00008. [DOI] [PubMed] [Google Scholar]
- 58.Colpoys WE, Cochran BH, Carducci TM, Thorpe CM. 2005. Shiga toxins activate translational regulation pathways in intestinal epithelial cells. Cell Signal 17:891–899. doi: 10.1016/j.cellsig.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 59.Petruzziello-Pellegrini TN, Yuen DA, Page AV, Patel S, Soltyk AM, Matouk CC, Wong DK, Turgeon PJ, Fish J E, Ho J J, Steer BM, Khajoee V, Tigdi J, Lee WL, Motto DG, Advani A, Gilbert RE, Karumanchi SA, Robinson LA, Tarr PI, Liles WC, Brunton J L, Marsden PA. 2012. The CXCR4/CXCR7/SDF-1 pathway contributes to the pathogenesis of Shiga toxin-associated hemolytic-uremic syndrome in humans and mice. J Clin Invest 122:759–776. doi: 10.1172/JCI57313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jandhyala DM, Thorpe CM, Magun B. 2012. Ricin and Shiga toxins: effects on host cell signal transduction. Curr Top Microbiol Immunol 357:41–65. doi: 10.1007/82_2011_181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tesh VL. 2012. The induction of apoptosis by Shiga toxins and ricin. Curr Top Microbiol Immunol 357:137–178. doi: 10.1007/82_2011_155. [DOI] [PubMed] [Google Scholar]
- 62.Obrig TG, Seaner RM, Bentz M, Lingwood CA, Boyd B, Smith A, Narrow W. 2003. Induction by sphingomyelinase of shiga toxin receptor and Shiga toxin 2 sensitivity in human microvascular endothelial cells. Infect Immun 71:845–849. doi: 10.1128/IAI.71.2.845-849.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cherla RP, Lee SY, Mees PL, Tesh VL. 2006. Shiga toxin 1-induced cytokine production is mediated by MAP kinase pathways and translation initiation factor eIF4E in the macrophage-like THP-1 cell line. J Leukoc Biol 79:397–407. doi: 10.1189/jlb.0605313. [DOI] [PubMed] [Google Scholar]
- 64.Hurley BP, Jacewicz M, Thorpe CM, Lincicome LL, King AJ, Keusch GT, Acheson DW. 1999. Shiga toxins 1 and 2 translocate differently across polarized intestinal epithelial cells. Infect Immun 67:6670–6677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jetzt AE, Cheng JS, Tumer NE, Cohick WS. 2009. Ricin A-chain requires c-Jun N-terminal kinase to induce apoptosis in nontransformed epithelial cells. Int J Biochem Cell Biol 41:2503–2510. doi: 10.1016/j.biocel.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shifrin VI, Anderson P. 1999. Trichothecene mycotoxins trigger a ribotoxic stress response that activates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase and induces apoptosis. J Biol Chem 274:13985–13992. doi: 10.1074/jbc.274.20.13985. [DOI] [PubMed] [Google Scholar]
- 67.Inward CD, Varagunam M, Adu D, Milford DV, Taylor CM. 1997. Cytokines in hemolytic-uraemic syndrome associated with verocytotoxin-producing Escherichia coli infection. Arch Dis Child 77:145–147. doi: 10.1136/adc.77.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Inward CD, Howie AJ, Fitzpatrick MM, Rafaat F, Milford DV, Taylor CM. 1997. Renal histopathology in fatal cases of diarrhoea-associated hemolytic uraemic syndrome. Pediatr Nephrol 11:556–559. [DOI] [PubMed] [Google Scholar]
- 69.Zoja C, Buelli S, Morigi M. 2010. Shiga toxin-associated hemolytic-uremic syndrome: pathophysiology of endothelial dysfunction. Pediatr Nephrol 25:2231–2240. doi: 10.1007/s00467-010-1522-1. [DOI] [PubMed] [Google Scholar]
- 70.Kaye SA, Louise CB, Boyd B, Lingwood CA, Obrig TG. 1993. Shiga toxin-associated hemolytic-uremic syndrome: interleukin-1 beta enhancement of Shiga toxin cytotoxicity toward human vascular endothelial cells in vitro. Infect Immun 61:3886–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van de Kar NC, Monnens LA, Karmali MA, van Hinsbergh VW. 1992. Tumor necrosis factor and interleukin-1 induce expression of the verocytotoxin receptor globotriaosylceramide on human endothelial cells: implications for the pathogenesis of the hemolytic-uremic syndrome. Blood 80:2755–2764. [PubMed] [Google Scholar]
- 72.Kawakami T, Lichtnekert J, Thompson LJ, Karna P, Bouabe H, Hohl TM, Heinecke J W, Ziegler SF, Nelson PJ, Duffield JS. 2013. Resident renal mononuclear phagocytes comprise five discrete populations with distinct phenotypes and functions. J Immunol 191:3358–3372. doi: 10.4049/jimmunol.1300342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lopez EL, Contrini MM, Glatstein E, Ayala SG, Santoro R, Ezcurra G, Teplitz E, Matsumoto Y, Sato H, Sakai K, Katsuura Y, Hoshide S, Morita T, Harning R, Brookman S. 2012. An epidemiologic surveillance of Shiga-like toxin-producing Escherichia coli infection in Argentinean children: risk factors and serum Shiga-like toxin 2 values. Pediatr Infect Dis J 31:20–24. doi: 10.1097/INF.0b013e31822ea6cf. [DOI] [PubMed] [Google Scholar]
- 74.Vinken P, Starckx S, Barale-Thomas E, Looszova A, Sonee M, Goeminne N, Vermissen L, Buyens K, Lampo A. 2012. Tissue Kim-1 and urinary clusterin as early indicators of cisplatin-induced acute kidney injury in rats. Toxicol Pathol 40:1049–1062. doi: 10.1177/0192623312444765. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.