

Abstract
Vibrio cholerae causes human infection through ingestion of contaminated food and water, leading to the devastating diarrheal disease cholera. V. cholerae forms matrix-encased aggregates, known as biofilms, in the native aquatic environment. While the formation of V. cholerae biofilms has been well studied, little is known about the dispersal from biofilms, particularly upon entry into the host. In this study, we found that the exposure of mature biofilms to physiologic levels of the bile salt taurocholate, a host signal for the virulence gene induction of V. cholerae, induces an increase in the number of detached cells with a concomitant decrease in biofilm mass. Scanning electron microscopy micrographs of biofilms exposed to taurocholate revealed an altered, perhaps degraded, appearance of the biofilm matrix. The inhibition of protein synthesis did not alter rates of detachment, suggesting that V. cholerae undergoes a passive dispersal. Cell-free media from taurocholate-exposed biofilms contains a larger amount of free polysaccharide, suggesting an abiotic degradation of biofilm matrix by taurocholate. Furthermore, we found that V. cholerae is only able to induce virulence in response to taurocholate after exit from the biofilm. Thus, we propose a model in which V. cholerae ingested as a biofilm has coopted the host-derived bile salt signal to detach from the biofilm and go on to activate virulence.
INTRODUCTION
Vibrio cholerae is an aquatic bacterium that can cause the diarrheal disease cholera when ingested by humans. As the bacteria transition from an aquatic environment to the human gut, they encounter many host signals during passage to the primary site of colonization, the small intestine. Pathology is caused predominantly by the enterotoxin cholera toxin (CT) (1). A toxin-coregulated pilus (TCP) also is required for colonization (2). This type IV pilus is important for bacterium-bacterium interactions and microcolony formation in the small intestine, although its functions are not fully understood. The regulation of these two key virulence factors is tightly controlled and activated in response to cues from the host environment (reviewed in reference 3). In the small intestine, one of these host factors is bile. Bile is a heterogeneous mixture, with the main constituents being bile salts, cholesterol-derived amphipathic compounds that aid in digestion and limit bacterial growth by disrupting membranes and proteins (4). Certain bile salts, particularly those with taurine or glycine conjugates, activate the virulence program of V. cholerae, resulting in the expression of the two major virulence factors, CT and TCP, by promoting the activation of upstream regulatory factors (5).
Bile salts affect the physiology of V. cholerae and other enteric bacteria (4) and are antimicrobial due to their surfactant and detergent properties (4). In response to bile salt exposure, V. cholerae can induce active efflux, enhance motility, remodel outer membrane protein composition, and promote biofilm formation (6–9). Entrance into a biofilm state may aid in persistence in the aquatic environment and is a multistep developmental process (10–12). When V. cholerae encounters bile or other biofilm-inducing conditions, it expresses a set of vibrio polysaccharide (VPS) polysynthetic genes (vpsA-Q) that result in the production of an exopolysaccharide that encases the bacterial aggregate (13). In addition to this major biofilm matrix component, V. cholerae also produces several proteins that stabilize the matrix (14). This induction is regulated by the major regulators VpsT and VpsR (15, 16), which, in turn, are regulated by quorum sensing (17, 18). This pathway can be exogenously activated by signals such as starvation and bile (8).
Once they have entered a biofilm state of development, bacteria may employ methods to actively exit from a biofilm. Strategies may include egress by enhanced motility or repressed adherence and targeting of matrix components for degradation by bacterially produced enzymes and small molecules (19). Often, these systems involve complex genetic regulatory mechanisms that cue dispersal in response to internal and external cues, such as quorum sensing and nutrient availability, respectively. For example, Escherichia coli K-12 dispersion is controlled by the global regulator CsrA, which modulates carbon utilization and can drive the bacteria into a biofilm-repressive state (20). For Staphylococcus aureus, glucose limitation promotes the expression of the arg system, the activation of which leads to extracellular protease production and ultimately dispersion (21). The opportunistic pathogen Pseudomonas aeruginosa can promote its own dispersal through the production of surfactants that nonspecifically disrupt cell-cell and cell-biofilm interactions, as well as a more explosive seeding dispersal in which hollowed-out biofilm structures burst open to release highly motile planktonic cells (22, 23). P. aeruginosa-detached planktonic cells utilize a distinct regulatory profile compared to biofilm or free-living planktonic cells, displaying enhanced virulence induction (22), once again highlighting the notion that biofilm-resident or previously biofilm-resident cells are physiologically distinct from their free-living planktonic counterparts.
In its natural aquatic environment, V. cholerae often is associated with chitinous surfaces and believed to be in a matrix-encased biofilm (10, 12). V. cholerae may be more infectious when ingested in this natural biofilm state, as a lower infectious dose is necessary to cause disease with biofilms than with free-living planktonic cells. Additionally, the incidence of cholera can be greatly reduced by the filtration of contaminated water through a sari cloth to remove the majority of copepod and biofilm-associated bacteria (24). Evidence also suggests that biofilm-like aggregates from rice water stool persist in the environment (25). The efficacy of biofilm-initiated infection has been attributed to a concentrated infectious dose as well as protection during gastric passage (17). Even when biofilms are disrupted prior to infection, biofilm-derived V. cholerae cells outcompete their free-living planktonic counterparts (26). This observation highlights the importance of understanding the early steps of infection in a more natural, biofilm route, especially taking into consideration the physiologic state of those biofilm-resident cells during this process. In this study, we have demonstrated one mechanism by which V. cholerae has coopted host signals to sequentially detach from biofilms and induce virulence factor production.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
V. cholerae El Tor C6706 (27) was used as the wild-type strain in this study. In-frame deletions were constructed by cloning the regions flanking the target genes into suicide vector pWM91, containing a sacB counterselectable maker (28). PvpsA-lacZ transcriptional fusions were generated by cloning the vpsA promoter into a plasmid containing a lacZ reporter (29). The transcriptional lux reporter of the promoter region of tcpA has been described previously (30). Strains were propagated in LB containing appropriate antibiotics at 37°C, unless otherwise noted.
Biofilm formation assays.
Assays to quantify biofilms were performed as previously described (17). A 1:100 dilution of overnight-grown culture of V. cholerae was inoculated in LB broth into 10- by 75-mm borosilicate glass tubes and incubated for 22 to 24 h at 22°C. Subsequently, the tubes were rinsed three times with phosphate-buffered saline (PBS) and then filled with crystal violet stain. After 5 min, excess stain was rinsed off with deionized water. The biofilm-associated crystal violet was solubilized in dimethylsulfoxide (DMSO), and the optical density at 570 nm (OD570) of the resulting suspension was measured. All experiments were performed at least three independent times, and samples were performed in triplicate.
Biofilm detachment assays.
Supernatants from mature biofilms were aspirated. Biofilms on tubes were rinsed three times with PBS, and then fresh medium premixed with the indicated compound was added gently so as not to manually disrupt biofilm structure. After the indicated time, biofilm-derived planktonic cells were drawn from the supernatant, serially diluted, and plated on LB agar plates with appropriate antibiotics for enumeration. Remaining biofilms were quantified by crystal violet as described above, or biofilm-resident cells were collected after manual disruption by vortexing in the presence of PBS and glass beads.
Microscopy to examine biofilm structures.
For scanning electron microscopy (SEM), a 1:100 dilution of overnight-grown culture of wild-type V. cholerae was inoculated into 5 ml of LB in 50-ml Falcon tubes containing 22- by 22-mm sterile glass coverslips. After 24 h, coverslips were washed three times with PBS and then placed into 6-well plates with 2 ml of fresh media alone or premixed with 1 mM taurocholate. Biofilms on coverslips for SEM were fixed in 2.5% glutaraldehyde and 2.0% paraformaldehyde in 0.1 M cacodylate buffer, pH 7.4, overnight at 4°C. After several buffer washes the samples were postfixed in 2.0% osmium tetroxide for 1 h, washed again in buffer, and dehydrated in a graded ethanol series. Samples were treated with several changes of hexamethyldisilazane (HMDS) and then allowed to air dry prior to mounting and sputter coating with gold/palladium. SEM images were collected using a Philips XL20 scanning electron microscope.
Estimation of carbohydrate content.
Supernatant from biofilm detachment assays were sterilized through 0.2-μm-pore-size filters before carbohydrate content was estimated using a modified phenol-sulfuric acid method (31). Briefly, 50 μl of each sample was added to each well of a 96-well microplate. To this, we mixed in 150 μl of concentrated sulfuric acid, followed by 30 μl of 5% phenol in water. Plates were covered and incubated for 10 min in a 90°C water bath. Plates were cooled at room temperature and dried, and the resulting OD490 was measured. A standard glucose curve generated during each independent experiment was used to convert sample OD490 values to percent sugar. To reduce background noise, M9 minimal media supplemented with 0.2% glycerol was used during detachment, and biofilms were washed four times with PBS.
Beta-galactosidase assays.
Biofilms of the V. cholerae ΔlacZ strain containing the vpsA-lacZ reporter plasmid were allowed to form and were disrupted as described above. Biofilm-associated and detached cells were collected, washed, and resuspended in PBS to an OD600 of ∼0.2. Cultures were assayed for ß-galactosidase activity, which was normalized against the optical density at 600 nm and reported as Miller units as previously described (32).
Measurement of virulence gene expression.
V. cholerae strains containing virulence promoter luxCDABE transcriptional fusions were used for detachment assays. Luminescence was measured using a Bio-Tek Synergy HT spectrophotometer and normalized for growth against the OD600. Luminescence expression is reported as light units/OD600 unit. V. cholerae strains containing a constitutive Ptet-mCherry and inducible PtcpA-gfp construct (30) were used for the detachment assay. Immediately following detachment assay, data were collected on an LSR II flow cytometer (BD Biosciences), and postcollection data were analyzed using FlowJo version 9.7.5 (TreeStar).
RESULTS
The bile salt TC promotes detachment of mature biofilms in vitro.
To better understand the interplay between V. cholerae biofilms and the host signal bile salts, we incubated V. cholerae under biofilm-inducing conditions in the presence of various bile salts. Modifications to the primary bile salt, cholate, include dehydroxylation at the 7α position to form secondary bile acids, such as deoxycholate, and covalent bonding with taurine or glycine to form conjugated bile salts, such as taurocholate (TC) and glycocholate (GC), respectively. Conjugated bile acids comprise the bulk of bile salts in the gut, with all permutations of these modifications being present (Fig. 1A) (33). As previously reported, V. cholerae formed thicker biofilms in the presence of the bile salts deoxycholate (DC) and cholate (CC) (8) (Fig. 1B). Surprisingly, biofilm formation was reduced in the presence of TC (Fig. 1B). Biofilm formation did not decrease when grown in the presence of another conjugated bile salt, glycocholate (Fig. 1B). In the presence of DC and CC, V. cholerae biofilm formation is enhanced via increased production of VPS (8). Therefore, we measured vpsA expression during biofilm formation in the presence of other bile salts. At 18 h, vpsA expression was increased when grown in the presence of DC but was unaltered in the presence of TC (Fig. 1C), suggesting that decreased VPS production was not the cause of lower biofilm formation in the presence of TC.
FIG 1.
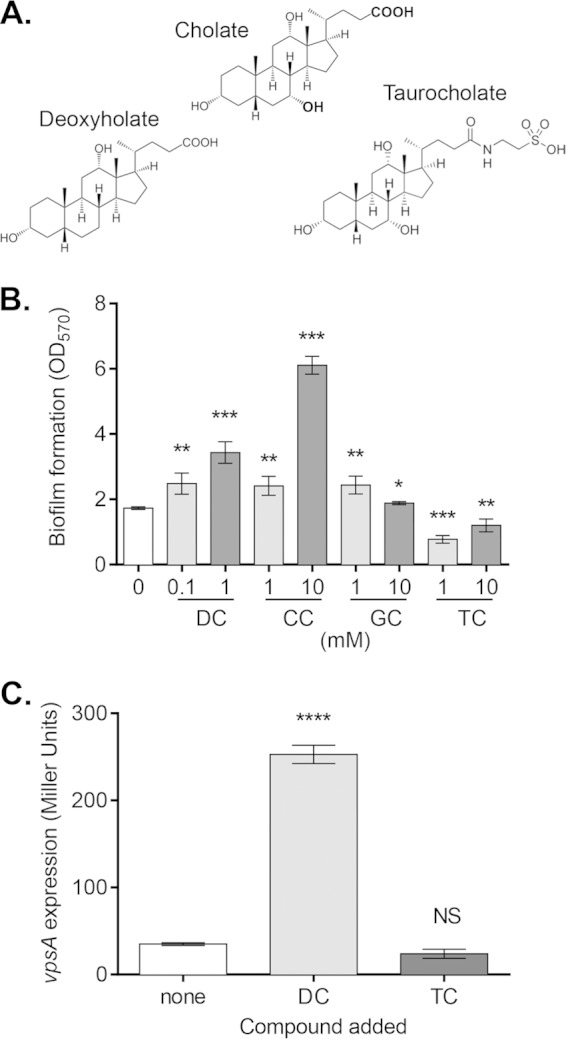
Effects of bile salts on V. cholerae biofilm formation. (A) Structures of select bile salts. Cholate can be dehyroxylated at the 7α position to form deoxycholate or conjugated with taurine to form taurocholate. (B) V. cholerae was inoculated into glass tubes and allowed to form biofilms for 24 h at 22°C. Biofilms of cultures grown in the presence of deoxycholate (DC), cholate (CC), glycocholate (GC), or taurocholate (TC) were quantified with crystal violet staining, and data are presented as OD570 values. (C) Expression of vpsA during formation of WT V. cholerae biofilms in the presence of 1 mM DC and TC for 18 h, as measured by the β-galactosidase assay. Data are means and standard deviations (SD) from three independent experiments. NS, no significance; *, P < 0.05; **, P < 0.005; ***, P < 0.0005; ****, P < 0.0001.
We next considered whether cells were less capable of residing in the biofilm, perhaps detaching at a higher rate than attaching or growing. To test this possibility, we performed detachment assays to determine whether bile salts affect the rate at which cells exit the biofilm. After 24 h of growth, V. cholerae forms robust biofilms that display a characteristic pillar-and-tunnel architecture (11, 34). After washing away free-living cells, V. cholerae biofilms were incubated with fresh media supplemented with various bile salts. When biofilms were exposed to physiologic levels of TC (35), we found that there was a decrease in biofilm mass compared to that of biofilms incubated either with DC or without bile salts (Fig. 2A). Over this same time, we also observed an increase in the number of biofilm-derived planktonic cells in the samples incubated with TC (Fig. 2B). This enhanced dispersal was not observed with CC, which shares structure with TC, but lacks a conjugated taurine group, with another conjugated bile salt, GC, or with the detergent Triton X-100 (TX) (Fig. 2C and D), indicating the specificity of the bile salt taurocholate to promote detachment. At the concentrations shown in Fig. 2, the bile salts and detergent used do not retard the growth of V. cholerae (data not shown).
FIG 2.
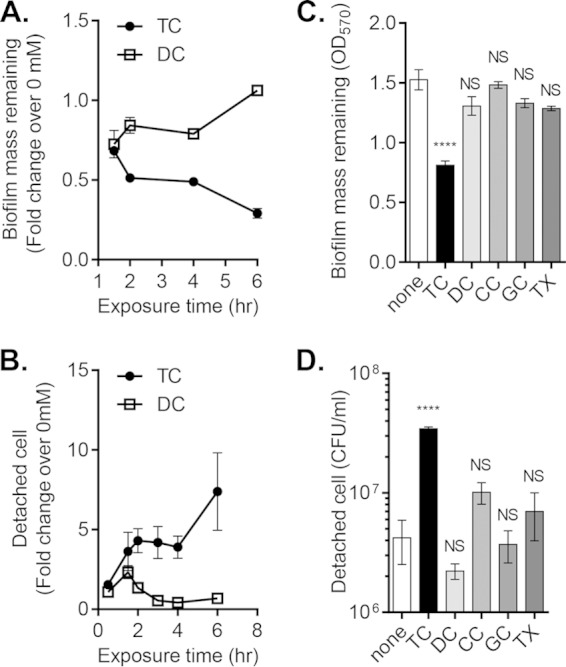
Effect of taurocholate (TC) on detachment of mature V. cholerae biofilms. (A) Remaining biofilm mass after detachment for the indicated time in the presence of 1 mM taurocholate (TC) or 1 mM deoxycholate (DC), presented as fold change of OD570 over no bile salts added for the same incubation period. (B) Biofilm-derived planktonic cells incubated in the presence of 1 mM TC or 1 mM DC, presented as fold change of CFU/ml over samples with no bile salts added for the same incubation period. (C and D) Remaining biofilm mass (OD570; C) and detached cells (CFU/ml; D) following 2 h of exposure to TC, DC, cholate (CC), glychocholate (GC), and 0.1% Triton X (TX). All bile salts are at 1 mM final concentration. Data are means and SD from three independent experiments. NS, no significance; ****, P < 0.0001.
Taurocholate alters V. cholerae biofilm matrix structure.
To further understand the nature of TC-enhanced dispersal, V. cholerae biofilms were imaged using scanning electron microscopy following incubation with or without TC for 1 h (Fig. 3). At ×2,000 and ×8,000 magnification, individual cells within the biofilm matrix are clearly visible, as is a sheet-like substance (Fig. 3C to F). Based on comparison to other previously published SEM images of biofilms, the sheets seen in the images likely are biofilm matrix (36–38). Visual analysis suggests that TC-exposed biofilms have an altered appearance compared to unexposed biofilms, in that there is less visible matrix and cells are less densely clustered. This initial assessment supports our hypothesis that TC enhances the dispersal of biofilms and prompted us to explore more quantitative measures. Thus, we explored two possibilities: one in which active egress of biofilm-resident cells damages the biofilm matrix or, conversely, one in which the biofilm matrix is physically disturbed, freeing cells in the process.
FIG 3.
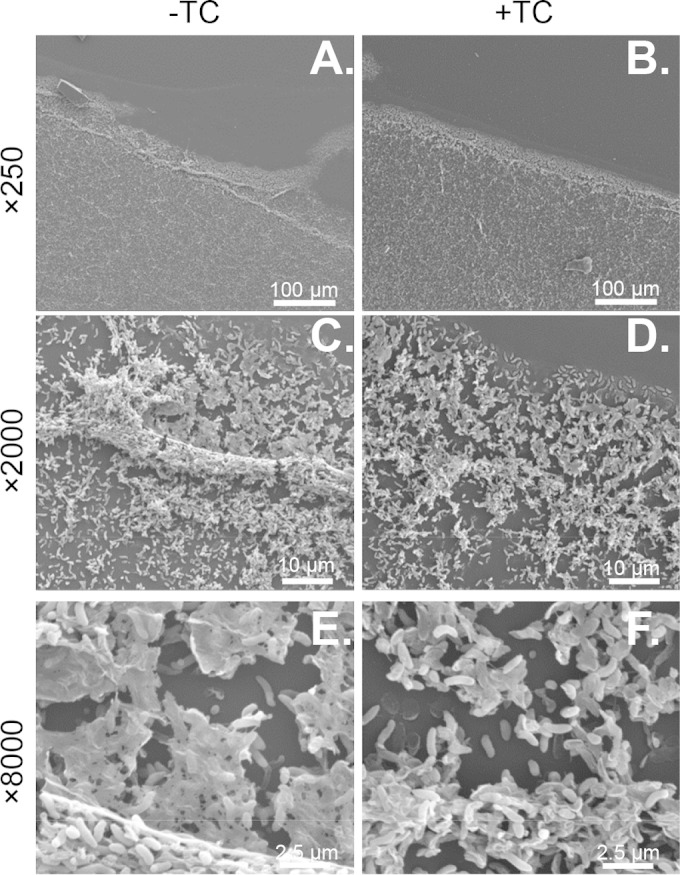
Representative images of V. cholerae biofilms imaged using scanning electron microscopy after 24 h of growth on a 55- by 55-mm glass coverslip followed by 1 h of exposure to 1 mM TC (B, D, and F) or no TC (A, C, and E). Images are at ×250 (A and B), ×2,000 (C and D), and ×8,000 (E and F) magnification.
V. cholerae TC-induced biofilm dispersal is passive.
To understand how taurocholate affects V. cholerae biofilm matrix, we first tested whether V. cholerae actively promotes its own dispersal from the biofilm in response to bile salts, possibly disrupting the biofilm matrix during this process. To examine whether TC enhances the detachment of V. cholerae biofilms through the altered production of VPS, we found that vpsA expression was not significantly altered in detached cells exposed to TC or DC compared to unexposed cells (Fig. 4A). As TC stimulates the induction of the virulence cascade in V. cholerae (5), we then asked whether the detachment and expression of the virulence program were related. We measured detachment in a ΔtoxT mutant strain of V. cholerae in which all virulence gene expression is abolished (39). Virulence gene expression is not required for detachment, as the detachment rate of the ΔtoxT mutant biofilms did not differ significantly from the wild type when exposed to TC (see Fig. S1A in the supplemental material). We then tested whether TC promotes detachment through quorum-sensing pathways involving HapR. However, the ΔhapR mutant strain displayed TC-enhanced detachment at levels similar to that of the wild type (see Fig. S1B). Likewise, the ΔhapR mutant strain grew thinner biofilms in the presence of TC (data not shown). To test whether V. cholerae induces other proteins to promote its own detachment in response to TC, we repeated detachment experiments in the presence of the protein synthesis inhibitor chloramphenicol. At low levels, chloramphenicol can inhibit growth without causing cell death. Because biofilms may display enhanced resistance to antibiotics, we confirmed that chloramphenicol is able to inhibit protein synthesis in biofilm-resident cells (40). When biofilms of wild-type V. cholerae harboring an arabinose-inducible green fluorescent protein (GFP) (pBAD-gfp) construct were treated with arabinose and chloramphenicol for 2 h, fluorescence induction was blocked compared to treatment with arabinose only (see Fig. S2). We then found that V. cholerae detachment was not significantly altered by the addition of chloramphenicol in the presence or absence of TC (Fig. 4B), suggesting that dispersal is not dependent on V. cholerae proteins induced in response to TC. Taken together, these data suggest that V. cholerae undergoes a passive dispersal.
FIG 4.
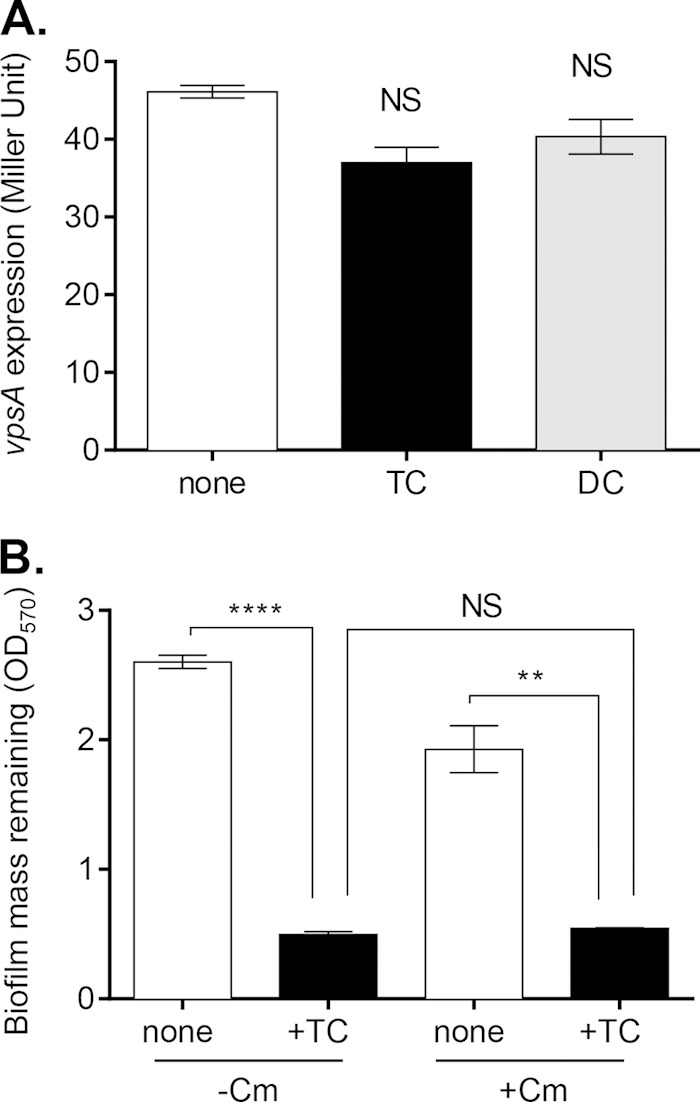
V. cholerae response to TC during detachment. (A) Level of expression of vpsA in detached cells, as measured with the β-galactosidase assay. Cells were collected following 2 h of detachment. TC and DC were supplemented to a final concentration of 1 mM. (B) Following growth for 24 h in biofilm-inducing conditions, wild-type V. cholerae biofilms were incubated with the protein synthesis inhibitor chloramphenicol (Cm) and TC for 2 h. Remaining biofilm was quantified with CV staining. Data are means and SD from three independent experiments. NS, no significance; **, P < 0.005; ****, P < 0.0001.
Polysaccharide content is released during taurocholate treatment of biofilms.
In order to better understand the nature of biofilm matrix alteration of TC-exposed biofilms, we first investigated the main component of this matrix, VPS. Using the phenol-sulfuric acid method, we estimated the polysaccharide content released during detachment. Cell-free filtered supernatants from biofilms exposed to TC contain a greater amount of carbohydrates than those exposed to DC or left unexposed (Fig. 5). Because cells may lyse during filtration, we tested whether lysed cell contents contribute to carbohydrate content by filtering increasing concentrations of cells of both wild-type and ΔvpsA mutant strains of V. cholerae, with the latter being unable to produce VPS. At all cell concentrations, filtrates had no detectable polysaccharide content compared to that of a blank medium control (see Fig. S3 in the supplemental material). We considered the possibility of abiotic degradation, either directly or indirectly resulting from TC exposure, and sought to find a possible mechanism for such degradation. Calcium and other divalent cations stabilize exopolysaccharide, and removal can cause the collapse of the biofilm (41). Because some bile salts can chelate calcium and other divalent cations, we questioned whether TC chelates Ca2+ to destabilize or degrade the biofilm matrix (42). However, supplemental calcium did not prevent TC-enhanced detachment (see Fig. S4). Due to the specificity of the detachment phenomenon to taurocholate, we looked more closely at the taurine side group of this bile salt. Taurine alone is insufficient to enhance dispersal, as no significant difference in biofilm formation and biofilm dispersal was seen between biofilms incubated with and without taurine (see Fig. S5A to C). We repeated experiments with another bile acid, taurodeoxycholate (TDC), which also contains a conjugated taurine group. Grown in the presence of TDC, V. cholerae formed thinner biofilms and detached at an enhanced rate, similar to TC (see Fig. S5A to C). Likewise, TDC-exposed biofilms have a greater polysaccharide content released from them than unexposed biofilms, while no difference is seen for those biofilms exposed to taurine (Fig. 5).
FIG 5.
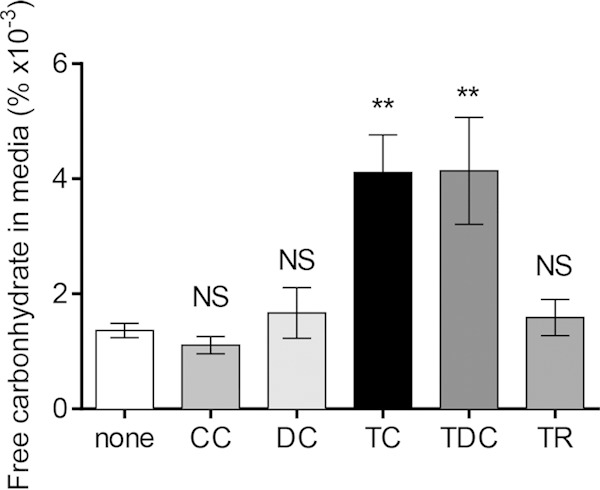
Carbohydrate content released from V. cholerae biofilms during incubation with bile salts. To reduce growth and background from rich media, biofilms were incubated in M9 minimal media with 0.2% glycerol for 2 h. Medium was filter sterilized before the carbohydrate content was estimated by the phenol-sulfuric acid method, and percent carbohydrate values were calculated by fitting to a standard glucose curve. For bile salts and taurine (TR), the final concentration is 1 mM. Data are means and SD from three independent experiments. NS, no significance; **, P < 0.005.
Detachment precedes induction of virulence program.
It is likely that biofilms or biofilm-like particles, rather than cells in a planktonic state, serve as infectious agents for human infection by V. cholerae. Presumably, cells must exit from these biofilms before they can swim to sites of infection at the intestinal epithelium, as highlighted by the importance of motility during infection (43). Because TC may be the cue for this in vivo detachment and is also a host-derived virulence inducer (5, 17), we investigated the relationship between biofilm dispersal and virulence induction upon exposure to taurocholate. We first used a luminescence reporter to test the overall population-level expression of the major virulence determinant tcpA in cells from different states. Figure 6A shows that tcpA expression was low for biofilm-resident cells and for detached cells under noninducing conditions, as was expected. Following incubation with TC, tcpA was robustly induced in cells that had detached from biofilms, but expression remained low in those cells remaining biofilm resident (Fig. 6A). We then used a PtcpA-gfp reporter paired with flow cytometry to assess individual cell levels of virulence gene expression (Fig. 6B). To track live cells, this strain also harbors a constitutive Ptet-mCherry construct. As seen in Fig. 6Bi and iii, tcpA expression is uniformly low in noninducing conditions (top right quadrant), with very few outliers. In TC-exposed samples, the majority of the detached cell population induces tcpA at least an order of magnitude above background levels (Fig. 6Biv, top right quadrant). In agreement with population-level experiments, TC-exposed biofilm-resident cells do not induce tcpA (Fig. 6Bii). Interestingly, the lack of induction in this population also was uniform; that is, robust tcpA induction was observed in very few, or perhaps none, of the biofilm-resident cells. This result suggests that although biofilm-resident cells are exposed to TC, they are unable to fully promote the expression of their virulence program until they have exited the biofilm environment. It is unclear whether detachment enhances the expression of virulence factors or merely enables it, as manually disrupted biofilms also were able to induce tcpA in response to TC (data not shown). The sequential nature of these events highlights the potential importance of detachment during natural infection with V. cholerae biofilm particles.
FIG 6.
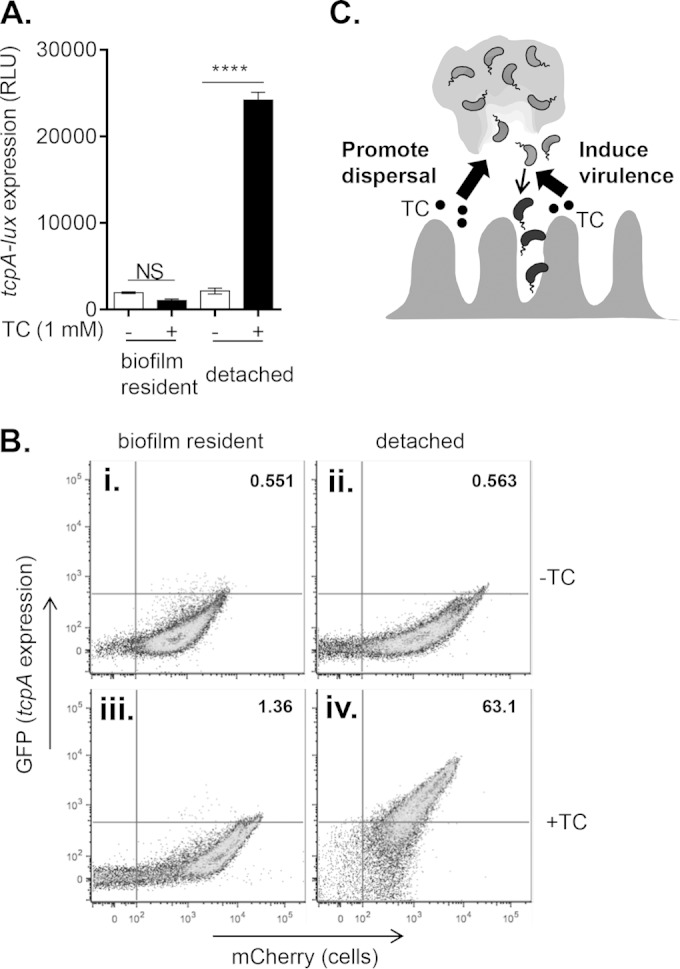
Virulence gene expression during TC-promoted biofilm detachment. (A) Expression of major virulence factor tcpA in biofilm-resident cells and detached cells as shown by a tcpA promoter-luciferase reporter fusion (PtcpA-lux). Data are means and SD from three independent experiments. ****, P < 0.0001. RLU, relative light units. (B) Flow cytometry plot of V. cholerae harboring a plasmid with constitutive mCherry (Ptet-mCherry) and PtcpA-gfp, in which mCherry indicates cells and GFP indicates tcpA expression. Average percentages of GFP-positive cells from three independent experiments are listed in each graph. (C) TC promotes both dispersal of individual cells from V. cholerae biofilms as well as induction of virulence program in the small intestine, synergistically enhancing colonization.
DISCUSSION
Using the suckling mouse model of infection, the field has learned a great deal about the physiologic and regulatory changes that V. cholerae undergoes as it transitions from aquatic-resident to enteric pathogen. However, a majority of these studies have been conducted with planktonic, free-swimming bacteria. Less is known about the changes that biofilm-resident V. cholerae undergo during this transition and while traversing through the gut. Infection dynamics differ based on biofilm residence, as biofilm-derived cells are able to colonize the small intestine more quickly and to a higher bacterial burden than planktonic-grown cultures (26). In this study, we have demonstrated that the host intestinal signal, taurocholate, promotes detachment of V. cholerae from mature biofilms in vitro. Additionally, this step precedes the induction of virulence factors, also affected by bile salts. Careful mouse animal model studies to determine the timing of virulence factor production in vivo suggest that there is an early TCP induction phase that occurs within 2 h of inoculation, while cells are in the lumen, without which CT induction does not occur. Our data are consistent with this model, in that exit from biofilm and induction of the major virulence factor, tcpA, occur within this time frame (44). Bile salts are most concentrated where they empty into the proximal small intestine and decrease in abundance in more distal portions of the gut (35). Thus, V. cholerae likely will encounter high levels of TC upon entrance into the small intestine. The idea that TC enhances detachment from V. cholerae biofilms in the proximal small intestine is consistent with previous work indicating that disrupted biofilms were able to colonize the proximal small intestine both more quickly and to a greater bacterial burden (26).
All bile salts are surface active and can act as detergents. Because only taurine-conjugated bile salts enhance the detachment of V. cholerae biofilms, it is likely that the mechanism of action is not simply detergent or surfactant in nature. The requirement for taurine conjugation hints at biochemistry at play that is unique to this class of molecules. Taurine contains a sulfonic acid, which is relatively rare among naturally occurring compounds. The most simple sulfonic acid, sulfuric acid, has long been used to degrade cellulose for industrial purposes, with recent studies describing similar properties for other sulfonic acid-containing compounds (45, 46). Work from the Wiredu group showed that out of an array of sulfonic acid-containing compounds, those with hydrophobic R groups were the most efficient at catalyzing the hydrolysis of cellulose (45). Because taurocholate orients at interfaces, with the sterol group along the interface, this hydrophobic side group may be important to enable the acidic group to come in close enough proximity with the carbohydrate and perhaps to position it properly (47). These observations could explain why taurine is insufficient to promote detachment without a hydrophobic R group and why only TC and TDC can promote detachment. Further work is needed to confirm or rule out this possibility, but we speculate that TC and TDC enhance V. cholerae detachment through directly promoting hydrolysis of the VPS component of the biofilm matrix. The role of a small molecule in disruption of biofilm matrix is not unprecedented, as P. aeruginosa produces rhamnolipid, a surfactant that, when highly expressed, can disrupt cell-matrix interactions.
For many bacteria, dispersal is a means of accessing new niches during different stages of their life cycle or in response to various cues (19). P. aeruginosa active dispersal is orchestrated and achieved through an explosive release of bacteria from a hollowed-out shell in the biofilm. Chua et al. found not only that these dispersed cells have a distinct gene expression signature compared to both planktonic and biofilm cells but also that dispersion can specifically induce a hyperinfectious state that primes the detached bacteria for further infection (22). Other studies have highlighted the importance of hyperinfectivity gained during host passage and biofilm residence for V. cholerae (48). For example, V. cholerae strains that produce excess VPS exhibited lower stochastic shedding of bacteria from biofilms, likely because cells were more deeply embedded in the matrix. These trapped cells were deficient in their ability to establish colonies in new locations (49). Our findings suggest that the converse occurs as well: reduction of biofilm matrix, perhaps due to TC-mediated degradation, frees cells and enables them to induce new genes and colonize locations inaccessible to biofilm aggregates. Thus, we propose a model (Fig. 6C) in which ingested biofilms encounter bile salts in the proximal small intestine that degrade the biofilm matrix, perhaps directly through hydrolysis. As they are freed, individual cells are able to induce a virulence program in response to TC and go on to their sites of infection. We are currently investigating whether other phenotypes relevant to colonization are affected during this detachment step.
Supplementary Material
ACKNOWLEDGMENTS
We thank Zenyu Liu and Alan Copenhaver for technical assistance, as well as Mark Goulian for helpful discussion.
This study was supported by NIH/NIAID R01 (AI080654) (to J.Z.) and NSF GRFP fellowship DGE-1321851 (to A.J.H.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.02617-14.
REFERENCES
- 1.Sack DA, Sack RB, Nair GB, Siddique AK. 2004. Cholera. Lancet 363:223–233. doi: 10.1016/S0140-6736(03)15328-7. [DOI] [PubMed] [Google Scholar]
- 2.Krukonis ES, DiRita VJ. 2003. From motility to virulence: sensing and responding to environmental signals in Vibrio cholerae. Curr Opin Microbiol 6:186–190. doi: 10.1016/S1369-5274(03)00032-8. [DOI] [PubMed] [Google Scholar]
- 3.Matson JS, Withey JH, DiRita VJ. 2007. Regulatory networks controlling Vibrio cholerae virulence gene expression. Infect Immun 75:5542–5549. doi: 10.1128/IAI.01094-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Begley M, Gahan CGM, Hill C. 2005. The interaction between bacteria and bile. FEMS Microbiol Rev 29:625–651. doi: 10.1016/j.femsre.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 5.Yang M, Liu Z, Hughes C, Stern AM, Wang H, Zhong Z, Kan B, Fenical W, Zhu J. 2013. Bile salt-induced intermolecular disulfide bond formation activates Vibrio cholerae virulence. Proc Natl Acad Sci U S A 110:2348–2353. doi: 10.1073/pnas.1218039110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bina JE, Provenzano D, Wang C, Bina XR, Mekalanos JJ. 2006. Characterization of the Vibrio cholerae vexAB and vexCD efflux systems. Arch Microbiol 186:171–181. doi: 10.1007/s00203-006-0133-5. [DOI] [PubMed] [Google Scholar]
- 7.Gupta S, Chowdhury R. 1997. Bile affects production of virulence factors and motility of Vibrio cholerae. Infect Immun 65:1131–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hung DT, Zhu J, Sturtevant D, Mekalanos JJ. 2006. Bile acids stimulate biofilm formation in Vibrio cholerae. Mol Microbiol 59:193–201. doi: 10.1111/j.1365-2958.2005.04846.x. [DOI] [PubMed] [Google Scholar]
- 9.Wibbenmeyer JA, Provenzano D, Candice F, Klose KE, Delcour AH, Landry CF. 2002. Vibrio cholerae OmpU and OmpT porins are differentially affected by bile Vibrio cholerae OmpU and OmpT porins are differentially affected by bile. Infect Immun 70:121–126. doi: 10.1128/IAI.70.1.121-126.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lutz C, Erken M, Noorian P, Sun S, McDougald D. 2013. Environmental reservoirs and mechanisms of persistence of Vibrio cholerae. Front Microbiol 4:375–375. doi: 10.3389/fmicb.2013.00375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watnick PI, Kolter R. 1999. Steps in the development of a Vibrio cholerae El Tor biofilm. Mol Microbiol 34:586–595. doi: 10.1046/j.1365-2958.1999.01624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yildiz FH, Visick KL. 2009. Vibrio biofilms: so much the same yet so different. Trends Microbiol 17:109–118. doi: 10.1016/j.tim.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fong JCN, Syed Ka Klose KE, Yildiz FH. 2010. Role of Vibrio polysaccharide (vps) genes in VPS production, biofilm formation and Vibrio cholerae pathogenesis. Microbiology 156:2757–2769. doi: 10.1099/mic.0.040196-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Absalon C, Van Dellen K, Watnick PI. 2011. A communal bacterial adhesin anchors biofilm and bystander cells to surfaces. PLoS Pathog 7:e1002210–e1002210. doi: 10.1371/journal.ppat.1002210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Casper-Lindley C, Yildiz FH. 2004. VpsT is a transcriptional regulator required for expression of vps biosynthesis genes and the development of rugose colonial morphology in Vibrio cholerae O1 El Tor. J Bacteriol 186:1574–1578. doi: 10.1128/JB.186.5.1574-1578.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yildiz FH, Dolganov NA, Schoolnik GK. 2001. VpsR, a member of the response regulators of the two-component regulatory systems, is required for expression of vps biosynthesis genes and EPS(ETr)-associated phenotypes in Vibrio cholerae O1 El Tor. J Bacteriol 183:1716–1726. doi: 10.1128/JB.183.5.1716-1726.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu J, Mekalanos JJ. 2003. Quorum sensing-dependent biofilms enhance colonization in Vibrio cholerae. Dev Cell 5:647–656. doi: 10.1016/S1534-5807(03)00295-8. [DOI] [PubMed] [Google Scholar]
- 18.Hammer BK, Bassler BL. 2003. Quorum sensing controls biofilm formation in Vibrio cholerae. Mol Microbiol 50:101–104. doi: 10.1046/j.1365-2958.2003.03688.x. [DOI] [PubMed] [Google Scholar]
- 19.Kaplan JB. 2010. Biofilm dispersal: mechanisms, clinical implications, and potential therapeutic uses. J Dent Res 89:205–218. doi: 10.1177/0022034509359403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jackson DW, Suzuki K, Oakford L, Simecka JW, Hart ME, Romeo T. 2002. Biofilm formation and dispersal under the influence of the global regulator CsrA of Escherichia coli biofilm formation and dispersal under the influence of the global regulator CsrA of Escherichia coli. J Bacteriol 184:290–301. doi: 10.1128/JB.184.1.290-301.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boles BR, Horswill AR. 2008. Agr-mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathog 4:e1000052. doi: 10.1371/journal.ppat.1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chua SL, Liu Y, Yam JKH, Chen Y, Vejborg RM, Tan BGC, Kjelleberg S, Tolker-Nielsen T, Givskov M, Yang L. 2014. Dispersed cells represent a distinct stage in the transition from bacterial biofilm to planktonic lifestyles. Nat Commun 5:4462–4462. doi: 10.1038/ncomms5462. [DOI] [PubMed] [Google Scholar]
- 23.Davey ME, Caiazza NC, O'Toole GA. 2003. Rhamnolipid surfactant production affects biofilm architecture in Pseudomonas rhamnolipid surfactant production affects biofilm architecture in Pseudomonas aeruginosa PAO1. J Bacteriol 185:1027–1036. doi: 10.1128/JB.185.3.1027-1036.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colwell RR, Huq A, Islam MS, Aziz KMa, Yunus M, Khan NH, Mahmud A, Sack RB, Nair GB, Chakraborty J, Sack Da, Russek-Cohen E. 2003. Reduction of cholera in Bangladeshi villages by simple filtration. Proc Natl Acad Sci U S A 100:1051–1055. doi: 10.1073/pnas.0237386100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nelson EJ, Chowdhury A, Harris JB, Begum Ya Chowdhury F, Khan AI, Larocque RC, Bishop AL, Ryan ET, Camilli A, Qadri F, Calderwood SB. 2007. Complexity of rice-water stool from patients with Vibrio cholerae plays a role in the transmission of infectious diarrhea. Proc Natl Acad Sci U S A 104:19091–19096. doi: 10.1073/pnas.0706352104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tamayo R, Patimalla B, Camilli A. 2010. Growth in a biofilm induces a hyperinfectious phenotype in Vibrio cholerae. Infect Immun 78:3560–3569. doi: 10.1128/IAI.00048-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joelsson A, Liu Z, Zhu J. 2006. Genetic and phenotypic diversity of quorum-sensing systems in clinical and environmental isolates of Vibrio cholerae. Infect Immun 74:1141–1147. doi: 10.1128/IAI.74.2.1141-1147.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Metcalf WW, Jiang W, Daniels LL, Kim SK, Haldimann A, Wanner BL. 1996. Conditionally replicative and conjugative plasmids carrying lacZ alpha for cloning, mutagenesis, and allele replacement in bacteria. Plasmid 35:1–13. doi: 10.1006/plas.1996.0001. [DOI] [PubMed] [Google Scholar]
- 29.Akakura R, Winans SC. 2002. Mutations in the occQ operator that decrease OccR-induced DNA bending do not cause constitutive promoter activity. J Biol Chem 277:15773–15780. doi: 10.1074/jbc.M200109200. [DOI] [PubMed] [Google Scholar]
- 30.Liu Z, Yang M, Peterfreund GL, Tsou AM, Selamoglu N, Daldal F, Zhong Z, Kan B, Zhu J. 2011. Vibrio cholerae anaerobic induction of virulence gene expression is controlled by thiol-based switches of virulence regulator AphB. Proc Natl Acad Sci U S A 108:810–815. doi: 10.1073/pnas.1014640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Masuko T, Minami A, Iwasaki N, Majima T, Nishimura S-I, Lee YC. 2005. Carbohydrate analysis by a phenol-sulfuric acid method in microplate format. Anal Biochem 339:69–72. doi: 10.1016/j.ab.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 32.Miller J. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 33.Hofmann AF. 1999. Bile acids: the good, the bad, and the ugly. News Physiol Sci 14:24–29. [DOI] [PubMed] [Google Scholar]
- 34.Kierek-Pearson K, Karatan E. 2005. Biofilm development in bacteria. Adv Appl Microbiol 57:79–111. doi: 10.1016/S0065-2164(05)57003-5. [DOI] [PubMed] [Google Scholar]
- 35.Eastwood MA, Boyd GS. 1967. The distribution of bile salts along the small intestine of rats. Biochim Biophys Acta 137:393–396. doi: 10.1016/0005-2760(67)90116-6. [DOI] [PubMed] [Google Scholar]
- 36.Reguera G, Kolter R. 2005. Virulence and the environment: a novel role for Vibrio cholerae toxin-coregulated pili in biofilm formation on chitin. J Bacteriol 187:3551–3555. doi: 10.1128/JB.187.10.3551-3555.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marks LR, Parameswaran GI, Hakansson AP. 2012. Pneumococcal interactions with epithelial cells are crucial for optimal biofilm formation and colonization in vitro and in vivo. Infect Immun 80:2744–2760. doi: 10.1128/IAI.00488-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bomchil N, Watnick P, Kolter R. 2003. Identification and characterization of a Vibrio cholerae gene, mbaA, involved in maintenance of biofilm architecture. J Bacteriol 185:1384–1390. doi: 10.1128/JB.185.4.1384-1390.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Higgins DE, DiRita VJ. 1994. Transcriptional control of toxT, a regulatory gene in the ToxR regulon of Vibrio cholerae. Mol Microbiol 14:17–29. doi: 10.1111/j.1365-2958.1994.tb01263.x. [DOI] [PubMed] [Google Scholar]
- 40.Karatan E, Watnick P. 2009. Signals, regulatory networks, and materials that build and break bacterial biofilms. Microbiol Mol Biol Rev 73:310–347. doi: 10.1128/MMBR.00041-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kostakioti M, Hadjifrangiskou M, Hultgren SJ. 2013. Bacterial biofilms: development, dispersal, and therapeutic strategies in the dawn of the postantibiotic era. Cold Spring Harb Perspect Med 3:a010306. doi: 10.1101/cshperspect.a010306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rajagopalan N, Linderbaum S. 1982. The binding of Ca2+ to taurine and glycine-conjugated bile salt micelles. Biochim Biophys Acta 15:66–74. [DOI] [PubMed] [Google Scholar]
- 43.Butler SM, Camilli A. 2005. Going against the grain: chemotaxis and infection in Vibrio cholerae. Nat Rev Microbiol 3:611–620. doi: 10.1038/nrmicro1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee SH, Hava DL, Waldor MK, Camilli A. 1999. Regulation and temporal expression patterns of Vibrio cholerae virulence genes during infection. Cell 99:625–634. doi: 10.1016/S0092-8674(00)81551-2. [DOI] [PubMed] [Google Scholar]
- 45.Amarasekara AS, Wiredu B. 2012. Aryl sulfonic acid catalyzed hydrolysis of cellulose in water. Appl Catal A Gen 417-418:259–262. doi: 10.1016/j.apcata.2011.12.048. [DOI] [Google Scholar]
- 46.Amarasekara AS, Wiredu B. 2014. Sulfonic acid group functionalized ionic liquid catalyzed hydrolysis of cellulose in water: structure activity relationships. Sustainable Energy 2:102–107. doi: 10.12691/rse-2-3-4. [DOI] [Google Scholar]
- 47.Hofmann AF, Small DM. 1967. Detergent properties of bile salts: correlation with physiological function. Annu Rev Med 18:333–376. doi: 10.1146/annurev.me.18.020167.002001. [DOI] [PubMed] [Google Scholar]
- 48.Merrell DS, Butler SM, Qadri F, Dolganov NA, Alam A, Cohen MB, Calderwood SB, Schoolnik GK, Camilli A. 2002. Host-induced epidemic spread of the cholera bacterium. Nature 417:642–645. doi: 10.1038/nature00778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nadell CD, Bassler BL. 2011. A fitness trade-off between local competition and dispersal in Vibrio cholerae biofilms. Proc Natl Acad Sci U S A 108:14181–14185. doi: 10.1073/pnas.1111147108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.