

Abstract
Genetically engineered mouse (GEM) models of cancer currently comprise the most accurate way to experimentally recapitulate the human disease in the laboratory. Given recent advances in genomics and genetic screens, however, as well as an increasing urgency for the translation of effective preclinical treatments into the clinic, there is a pressing need to make these models easier and more efficient to work with. Accordingly, we have developed a versatile lentivirus-based approach to induce tumours from somatic cells of GEMs, add or subtract gene expression and render the tumours imageable from a simple breeding stock. The vectors deliver a tamoxifen-inducible and self-inactivating Cre recombinase, conditional bioluminescent and fluorescent proteins and an shRNA component. Following the transduction of somatic cells, tumours are initiated by Cre-mediated recombination of the inherited floxed alleles. Self-inactivation of Cre expression switches on the expression of luciferase, thereby rendering the recombined cells and resulting tumours bioluminescent. We demonstrate proof of concept of this approach by inducing bioluminescent lung tumours in conditional Kras and p53 mice. We also show that a variant vector expressing shRNA alters tumour growth dynamics and the histological grade associated with the inherited genotype. This approach comprises a versatile means to induce imageable and spontaneous tumour burden in mice. The vectors can be readily customized at the bench to modify reporter readout or tumour phenotype without additional transgenic strain development or breeding. They should also be useful for inducing imageable tumours in organs other than the lung, provided that the inherited conditional genotype is sufficiently penetrant. © 2013 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Keywords: preclinical model, tumour, imaging, lentivirus
Introduction
Genetically engineered mouse (GEM) models of cancer currently comprise the most accurate way to experimentally recapitulate the human disease in the laboratory 1. On a practical level, however, these models are difficult to work with, for several reasons. First, the genetic modifications underpinning these models are typically brought together through the germline, which is a time-consuming, inflexible and inefficient process. Second, tumour development in GEM models is stochastic and not readily visible at many locations in the body until a late stage. This limits the ability of these models to evaluate novel cancer treatments, as it is difficult to know when to initiate treatment or how to assess therapeutic response. An alternative embryonic stem (ES) cell chimera-based approach has been described recently that can broadly address this first limitation 2,3; however, it is highly technical and relatively expensive, and consequently is not available to all researchers. Bioluminescence imaging (BLI) could efficiently obviate the second issue, as it has been shown to be useful in determining viable tumour burden non-invasively in mouse models in vivo 4–6. When used optimally, BLI is the most sensitive preclinical imaging modality available and its relatively high-throughput nature makes it well suited for the analysis of small experimental cohorts. BLI imaging of tumours in mice relies upon the introduction of tumour-specific luciferase expression, however 7–10, which re-emphasises the first limitation and the technical considerations associated with the inflexible nature of GEM models.
We have developed an alternative and broadly accessible approach to address these limitations and to practically enhance GEM tumour models. It utilizes lentiviral vectors to stably deliver multiple transgenic elements to somatic cells of adult GEMs that have a simple conditional (floxed) oncogene and/or tumour suppressor genotype. The vectors contain inducible Cre recombinase (for tumour induction) and luciferase transgenes (for imaging), so resultant Cre-induced tumours in these mice stably express luciferase and are rendered visible by non-invasive BLI.
To demonstrate proof of concept, we generated a bioluminescent model of non-small cell lung carcinoma (NSCLC) by administering our vectors to the lungs of conditional KrasG12D only (K) and KrasG12D +p53 fl/fl (KP) mice by intranasal inhalation 11. We show that imaging is sensitive, even from a relatively deep body location such as the lung, and highly specific, as only transduced cells emit light, a situation that cannot be easily achieved with existing approaches. In terms of breeding our approach is efficient, as the inherited genotype of the mice that develop bioluminescent tumours is greatly simplified. Our approach also has the capacity to add or subtract specific gene expression from the base inherited conditional genotype. Accordingly, we describe a variant vector with an additional shRNA component that alters tumour phenotype relative to the inherited genotype via the knock-down of endogenous p53 expression.
Materials and methods
Vectors and lentivirus production, purification and concentration
Constructs were cloned into the third-generation pBOBI lentiviral system (from Inder Verma, Salk Institute) and VSV-G-pseudotyped lentiviral particles were produced as described previously 12,13. The relevant plasmid maps are included in the supplementary material (Figures S1–3). All viral supernatants were concentrated approximately 100-fold by ultracentrifugation at 25 000 × g for 2 h on a 20% sucrose cushion. Lentiviral titres were established by infection of HEK 293T cells with serially diluted virus and flow cytometry for red fluorescence. We routinely produced 1–5 × 108 infectious units/ml.
In vitro validation of lentiviral constructs
HEK 293T cells were grown to 50% confluence on six-well dishes in phenol red-free Dulbecco's modified Eagle's medium (DMEM) with 10% charcoal-stripped fetal bovine serum (FBS; Thermo Scientific HyClone, HYC-001-325B) and 2 mm l-glutamine. The cells were infected with approximately 2 × 106 lentiviral particles and 500 ng/ml 4-hydroxytamoxifen (4-OHT; 70% Z-isoform, Sigma-Aldrich H6258) was added immediately to half of the wells. Media (with or without 4-OHT) was refreshed 24 h post-infection and reporter functionality was clearly evident 72 h post-transduction and induction. Fluorescence was assessed by flow cytometry (FACScalibur, BD Biosciences) and microscopy (Leica TCS SP5) by standard methods. In vitro bioluminescence was measured with an IVIS 200 series camera (Perkin-Elmer), approximately 5 min after supplementing the tissue culture medium with 150 µg/ml d-luciferin (Perkin-Elmer).
Bacterial promoter prediction
Plasmid vector DNA sequences were analysed for predicted consensus bacterial promoters, using BPROM 14 and BDGP Neural Network Promoter Prediction 15. Both algorithms predicted a consensus bacterial promoter at the lox71 site of LV–indLS1, with scores of 80 and 0.98, respectively.
Mouse genotyping
Genomic DNA was prepared from earclip material, as described previously 16. The genotype of mice with the LSL-KrasG12D allele 17 was determined by PCR, as described in 18, using primers 1 (5′-gtctttccccagcacagtgc-3′), 2 (5′-ctcttgcctacgccaccagctc-3′) and 3 (5′-agctagccaccatggcttgagtaagtctgca-3′), cycled at 95°C for 2 min, followed by 34 cycles at 95°C for 30 s, 61°C for 30 s and 72°C for 45 s, then 72°C for 5 min. The LSL-KrasG12D allele was 500 bp and the wild-type Kras allele 622 bp. The p53 floxed allele was genotyped by PCR, as described previously 19.
In vivo experiments
All mouse experiments were performed under the terms of a UK Government Home Office licence and were subject to internal institutional ethical review. All the mice used in this study were on a pure-bred C57/BL6 genetic background. Our mouse colony was established from breeding stock that had previously inherited a mutant tyrosinase allele. This resulted in infrequent albinism amongst our experimental cohorts but did not affect imaging sensitivity, as hair was removed prior to imaging. We found that maintaining mice on an alfalfa-free diet at all times (Testdiet, AIN93G) reduced non-specific background from the abdomen, which was otherwise more pronounced with our standard imaging settings at early time points, when light emission from the lungs was close to background.
Induction of lung tumours
K or KP mice, 8–12 weeks old, were anaesthetized with isofluorane and 1–5 × 106 lentiviral particles, made up to a total volume of 70 µl in phosphate-buffered saline (PBS; 2 × 35 µl administrations), were administered to the lungs via intranasal inhalation 11. One week after infection, the mice were imaged for bioluminescence to establish the day 0 baseline, then immediately injected intraperitoneally (i.p.) with 100 µl tamoxifen (T5648, Sigma-Aldrich; 10 mg/ml in sunflower oil). Imaging and tamoxifen administration were repeated 48 h later.
In vivo bioluminescence imaging
Mice were anaesthetized with isofluorane and injected i.p. with 10 µl/g d-luciferin (15 mg/ml; Perkin-Elmer). Sequential bioluminescence images, each typically 3 min, large binning, f/stop 1, were then taken 10–20 min post-luciferin injection, using an IVIS 200 series imaging system (Perkin-Elmer). Light output was quantified at peak signal intensity, typically 14 or 18 min post-luciferin injection, using Living Image software (Perkin-Elmer).
Histology and immunohistochemistry
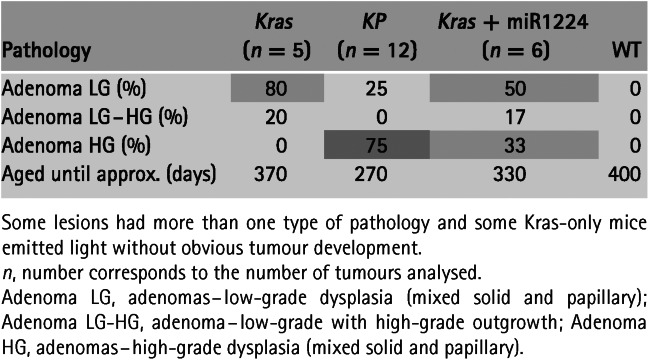
At the experimental end-point and immediately after a routine in vivo bioluminescence image, tumour-bearing or control animals were euthanized; the lungs were then quickly removed, inflated with PBS and imaged ex vivo with an IVIS 200 series camera. The lungs were then fixed in 4% PFA for 24 h, embedded in paraffin, and five 4 µm-thick sections were cut at 100 µm steps, with one slide stained with haematoxylin and eosin (H&E) at each step, resulting in 8–12 step sections covering the whole three dimensions for each pair of lungs. These step sections were analysed for tumour formation, with counting of tumours (which were often present in several adjacent step sections). Tumours were assessed for the presence of invasion to distinguish adenocarcinomas from adenomas, and the adenomas were graded into those showing low-grade or high-grade dysplasia, using established criteria 20.
Immunohistochemistry (IHC) was performed on unstained sections following microwave-based antigen retrieval in citrate buffer and incubation with an anti-RFP primary antibody (ab34771; Abcam) at 1:400 dilution at 4°C overnight. The primary antibody was then detected by incubation with a biotinylated secondary antibody (diluted 1:200) and horseradish peroxidase (Vectastain ABC Kit, PK4005, Vector Laboratories). The slides were counterstained with haematoxylin.
Results
In vitro validation
To induce conditional and bioluminescent tumours in floxed mice, we built a lentivirus (LV–indLS1) that contained CreERT2 (tamoxifen-inducible Cre recombinase), as well as firefly luciferase and mStrawberry expression cassettes. To couple expression of the transgenic reporters with tumour initiation and to limit the total size of the vector, we employed a single CAG promoter 21, flanked by inverted mutant loxP sites 22, to drive transgene expression. As depicted in Figure 1, only CreERT2 is expressed from the CAG promoter upon cell transduction. Following the functional activation of Cre recombinase by administration of tamoxifen, the promoter inverts and locks in the opposite orientation, resulting in expression of the reporter transgenes and Cre inactivation. Should other conditional tumour-related alleles also be present in the cell, they too should be activated or deleted by Cre recombinase. Consequently, tumours initiated in this way will be amenable to in vivo visualization by BLI.
Figure 1.

(a) Schematic of lentiviral vector constructs: (1) LV–LS (control vector); (2) LV–indLS1; (3) LV–indLS2; and (4) LV–indLS2miR1224; CAG; robust and ubiquitous promoter; Lox66 and lox71, mutant loxP sites; Luc_Strawberry, luciferase and mStrawberry co-expression cassette; CreERT2, tamoxifen-inducible Cre recombinase; i, synthetic intron in CreERT2; miR, miR30 cassette containing p53 (1224) hairpin. (b) Outline of the transgenic strategy to induce conditional and bioluminescent tumours in GEMs following transduction with LV–indLS2 and induction with tamoxifen: loxP and loxDM, wild-type and double-mutant lox P sites, respectively. (c) Fluorescence images of culture plates showing tamoxifen inducibility of mStrawberry expression in HEK293T cells (top panels) and bioluminescence images showing tamoxifen inducibility of luciferase expression in HEK293T cells (lower panels). (d) Flow-cytometric analysis of HEK293T cells transduced with LV–indLS2, showing transition from low- to high-level expression of mStrawberry following tamoxifen induction
We first validated the functionality of the LV–indLS1 lentivirus in vitro by transducing HEK 293T cells, inducing with 4-hydroxy-tamoxifen (4-OHT) and measuring fluorescent and bioluminescent reporter transgene function by microscopy, flow cytometry and bioluminescence measurement. We initially observed high levels of background bioluminescence arising from transduced but non-induced cells (data not shown). Analysis of plasmid DNA showed that the lox71 site positioned immediately upstream of CreERT2 comprised a strong consensus bacterial promoter (see Materials and methods) and was recombining the vector prepackaging. To prevent this, we cloned a synthetic intron within the coding sequence of CreERT2 23. This intron disrupted the expression of functional Cre from the lox71 site in bacteria, but splicing permitted functional CreERT2 expression from the CAG promoter in mammalian cells. Transduction of this variant lentiviral vector (LV–indLS2) into HEK 293T cells showed much lower levels of non-induced reporter expression and an approximate six-fold increase in bioluminescence following the administration of 4-OHT to the tissue culture medium.
Flow-cytometric analysis indicated that the cells that switch on reporter expression in response to 4-OHT originate from a population of cells that already exhibit very low levels of reporter expression. Fluorescence from these cells was barely visible by fluorescence microscopy pre-4-OHT addition, but increased by two orders of magnitude post-induction. As the induction of reporter expression is binary (off or on), this observation implies that transduced and non-induced cells possess very low levels of reporter expression, possibly originating from either weak reverse activity of the CAG promoter or from the lox site (as observed in Escherichia coli). We exploited this, as the low intensity fluorescence from non-induced cells facilitated rapid titration of lentiviral packaging by flow cytometry. The robust and practically useful levels of reporter activity induced by 4-OHT, however, functionally validated all the transgenic components of LV–indLS2.
In vivo application
To induce enhanced spontaneous lung tumours in mice, we administered LV–indLS2 to the lungs of K and KP mice by intranasal inhalation (see Materials and methods). One week later, we imaged these mice to establish the non-induced baseline levels of bioluminescence, then induced Cre function with tamoxifen and imaged according to the schedule shown in Figure 2. Increased levels of bioluminescence were evident 48 h after the first dose of tamoxifen, indicating that the construct was switching in vivo. Levels of bioluminescence continued to increase until day 17–20 post-tamoxifen, when levels returned to near baseline. The levels of bioluminescence remained low for several weeks before starting to rise again. Unambiguous bioluminescent foci then became evident in both cohorts, with levels of light increasing faster in KP than K mice, consistent with the fact that KP mice additionally lack p53 expression 24,25. The histological grade of lesions from these mouse cohorts (as summarized in Table1) was broadly consistent with the imaging data, with KP mice developing higher-grade lesions than the K mice. All of the tumours present were classified as adenomas and most were 1–2 mm in maximum dimension, with no evidence of invasion into surrounding structures. Heterogeneous regions of high-grade dysplasia, as evidenced by marked nuclear polymorphism and readily identifiable mitotic figures (indicative of low- to high-grade progression), were evident in the tumours from KP mice. A mild lymphoid hyperplasia within the lung parenchyma of some mice was also noted, but we have not determined whether this was due to tumour growth alone or whether lentiviral infection contributed to it.
Figure 2.
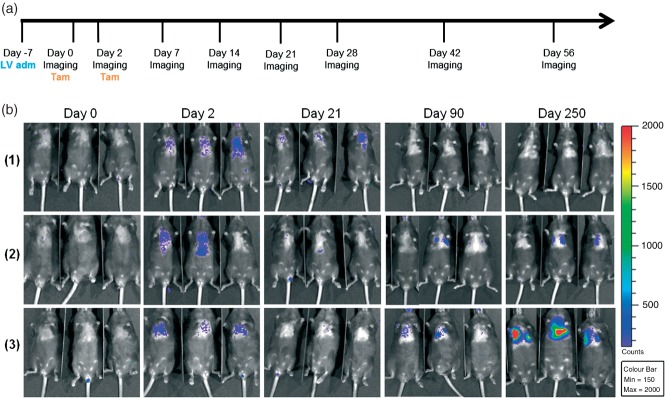
Spontaneous and bioluminescent lung tumour induction in mice. (a) Lentivirus administration (LV adm), induction (Tam) and imaging schedule. (b) Longitudinal BLI of the different experimental cohorts of mice: (1) wild-type; (2) Kras only; and (3) KP
Table 1.
Histological analysis of the lungs from the different experimental groups
We next asked whether we could convert the phenotype of a K mouse into a KP mouse by introducing an shRNA into the lentiviral vector to knock down endogenous p53 expression. We used a miR30 cassette that contained the 1224 p53 hairpin, as it had previously been shown to robustly knock down murine p53 when expressed from a RNA pol II promoter 26,27. By positioning the miR30-1224 cassette immediately after the stop codon of the optical reporters 28, we were able to add this functionality to our base LV–indLS2 vector with only a 350 bp increase in total packaging size. Bioluminescence from cells transduced with this variant vector also indicates knock-down of endogenous gene expression.
The measured lung bioluminescence from K mice transduced with LV-indLSmiR1224 developed in a manner intermediate to that of to its inherited genotype and KP mice (Figure 3). Unambiguous lung signal appeared approximately 1 month earlier in this cohort than in K or KP mice, however; once bioluminescence was established (after ˜100 days), the tumours did not appear to develop as quickly as those from the KP cohort. Histological analysis of lung tumours from these mice (Figures 4, S4, S5 and Table1) at 7 months post-Cre induction showed that the tumour phenotype was broadly similar, but intermediate in terms of grade, to mice with inherited K and KP genotypes. In addition to developing tumours faster than K mice, K+mir1224 mice developed more frequent high-grade adenomas, but not as frequently as KP mice.
Figure 3.

(a) Lentiviral miR variant construct (LV–indLS2miR1224) before and after tamoxifen administration. (b) Longitudinal analysis of tumour development in K, KP, K+miR and wild-typemice after infection with LV–indLS2 or LV–indLS2miR1224. Each symbol corresponds to an individual animal. Bioluminescence emission (photons/s/cm2/sr) on y axis and time (days) on x axis. (c) Longitudinal BLI of Kras mice following infection with LV–indLS2miR1224
Figure 4.

(a) Representative end stage bioluminescence images of mice (in vivo) and lungs (ex vivo) of: (1) K; (2) KP; (3) K+miR; and (4) wild-type mice, following infection with LV–indLS2 (3 min; large bin acquisition in all cases). (b) End-stage histology of lungs from a KP mouse at day 270 after LV–indLS2 infection, showing H&E staining and α-RFP immunohistochemistry
Total tumour burden in all luciferase-positive mice, irrespective of mouse genotype or lentiviral vector, ranged from between two and seven lesions/pair of lungs. This level of tumour burden should be useful for applied research purposes, as the tumours can develop for a longer period of time before total burden becomes problematic. Based on the routine titres of our lentiviral productions, 10-fold more lentivirus could be administered intranasally in an equivalent volume. This should initiate more tumours within the lung and, as tumour progression is stochastic, should result in a greater number of higher-grade lesions and a reduction in tumour latency.
Discussion
In an effort to enhance GEM models of cancer and render them more practicable and versatile, we have developed a lentiviral transduction-based approach to induce spontaneous, bioluminescent and genetically variant tumours in mice. A lentiviral Cre vector has been used in the past by others to induce tumours in the lungs of conditional Kras and p53 mice 11. We therefore chose to employ mice with this inherited genotype to validate the functionality and unique attributes of our vectors by inducing genetically enhanced and variant lung tumours.
To achieve this by conventional means would require combining four, possibly five, individually segregating alleles together by breeding; a conditional oncogenic Kras allele, two copies of a floxed p53 allele, a luciferase reporter and potentially also a lung-specific Cre recombinase allele (or viral Cre) 8,24,29. The introduction or removal of further gene expression in a compound GEM is not trivial, as additional GEM mice need to be sourced or generated, then bred to generate the variant compound genotype. Should any genetic component subsequently prove suboptimal (eg unpredicted expression of reporter), it is not possible to modify without sourcing and introducing a variant GEM into the breeding stock.
An ES cell chimera approach was described recently which improves upon the conventional process 2,3. It relies upon the in vitro manipulation and genetic modification of ES cells derived from GEM tumour models. Once derived, these ES cells can be maintained and precisely modified in vitro (eg targeted knock-in or knock-out) and so is a significantly faster and more versatile way to generate variant compound GEM lines than conventional breeding. Not all researchers have access to the facilities or the expertise needed to generate ES cells or ES cell chimeric mice, however; so, although powerful, this approach may not be available to all. From an imaging perspective, too, depending upon how luciferase transgene expression is regulated and the overall percentage contribution of modified ES cells in the resulting chimeric mouse, high background levels of bioluminescence from non-transformed cells may significantly compromise the ability to detect small developing tumours.
Although not capable of introducing the same spectrum of genetic manipulations that the ES cell chimera approach can, the approach described here is a useful and broadly accessible method to generate variant and enhanced GEM tumours. Lentiviral vectors were needed for this purpose, as they have useful packaging limits (approximately 9 kb) and can stably transduce non-replicating cells. This latter attribute was essential in order to induce tumours from normal cells in adult mice and express transgenic components many cell divisions after the initial gene delivery event. Following lentiviral transduction, activation of Cre function initiates tumourigenesis by recombining the inherited conditional alleles of the GEM. Functional Cre also self-inactivates, a process that switches on bioluminescence from the conditional luciferase reporter cassette and limits the potential for Cre-induced toxicity or off-target/non-specific recombination 30,31. Bioluminescence therefore indicates that cells are stably transduced and that they or a cellular ancestor have undergone Cre-mediated recombination. Not all floxed alleles will necessarily recombine in every transduced cell prior to Cre self-inactivation, and not all partially or fully recombined cells will necessarily develop into tumours. Fully recombined cells will be more likely to develop into tumours, however, and our approach better ensures that background bioluminescence arising from nearby transduced but non-transformed cells is minimized. The lung is a relatively deep organ in the mouse, so our ability to detect light (recombined cells) at nearly all stages post-Cre-activation exemplifies the sensitivity and specificity conferred by this approach. In comparison, computed tomography (CT) imaging of mouse lung tumours cannot detect tumours until approximately 1 mm3 in size 32. Non-invasive BLI of transduced mice did not resolve individual lung lesions, but rather was indicative of overall tumour burden within the lungs. This is sufficient to facilitate the selection of animals for therapeutic intervention scheduling or for more advanced, high-resolution and low-throughput imaging techniques, such as positron emission tomography (PET), single-photon emission computed tomography (SPECT), magnetic resonance imaging (MRI) and CT.
We administered 1–5 × 106 lentiviral particles by intranasal inhalation to induce tumours in the lung but, in comparison to previously described adenoviral Cre studies, this generated fewer tumours, with a longer latency and proportionately fewer high-grade lesions. As spontaneous tumour development is a stochastic process, we speculate that increasing the initial number of transduced and recombined cells will result in more frequent, higher-grade lesions with a shorter latency. Based on our routine packaging efficiency, as much as 10-fold more lentivirus could be administered by inhalation without increasing the administered volume. Also, we use VSV-G envelope to pseudotype our lentivirus, which confers broad infectivity to many mammalian cell types but is not optimal for infecting lung epithelium, unless administered with lysophosphatidylcholine 33. There may also be a difference attributable to the way that Cre is expressed. Adenoviral Cre expresses functional Cre recombinase for the duration of time that it transiently persists in a transduced cell. In comparison, given the stable nature of lentiviral-mediated transgenesis, our tamoxifen-inducible and self-inactivating Cre limits functional recombination to a short period of time. It should be noted, however, that the induction of too many tumours in the lung could compromise normal lung function prior to sufficient tumour maturation, and so limit certain applied research applications.
Additional functionality was introduced with a variant construct (LV–indLS2miR1224) that co-expressed a well-validated shRNA component 26,27 with the luciferase reporter. Although not equivalent to the inherited p53 fl/fl genotype, we show that the addition of a miR specific to murine p53 could accelerate the rate and histological grade of lung tumours developing in mice with only an inherited conditional Kras genotype. Subject to possessing a suitable shRNA, variant vectors can be generated rapidly at the bench and used to investigate the contribution made by specific genes to either tumour aetiology or response to intervention.
A significant advantage of our approach is versatility. The lentiviral vectors are designed to be modular, such that the promoter, reporter and miR30 cassette can all be customized with a minimum of additional cloning steps. Varying these parameters should permit tailoring of a GEM to confer cell-type specific tumour induction within a transduced organ (promoter), imaging of variant tumour-related parameters (reporter) or the rapid generation of variant model phenotypes (shRNA or oncogenes, subject to virus packaging constraints) from a simple and common breeding stock. These vectors can also be used in the context of other tumour-related floxed alleles, eg c-myc 34 and PTEN 35,36, so, depending upon where the vectors are administered and the penetrance of the genetics, they should be able to induce imageable tumours in a variety of organs. We have observed reporter function following direct injection of a control reporter-expressing lentivirus (LV–LS) into smooth muscle, skin, pancreas and breast (data not shown). Therefore, subject to the recipient GEM possessing the relevant conditional alleles, our approach should be able to induce enhanced tumours at those body locations.
Taken together, the vectors described here add to the range of useful options available to researchers to address the pressing experimental need for accurate, but more versatile and informative, tumour modelling with GEMs.
Acknowledgments
The authors would like to thank Kris Frese and Dave Tuveson for providing the mice to start our LSL-KrasG12D and p53 fl/fl breeding colony, Masashi Narita for advice about miR30, Inder Verma for the use of the pBOBI lentiviral system, Carla Martins for advice about the KP lung tumour model, Jim Metcalfe, Doug Winton and Ken Olive for critical reading of the manuscript, Alex Schriener and Richard Grenfell for assistance with confocal microscopy and flow cytometry, respectively, and all support staff in our animal and histopathology core facilities, in particular Mark Pryor, Ellie Pryor, Matt Clayton and Mike Mitchell. This work was supported by a Cancer Research UK (CRUK) Programme Grant to KMB and is a contribution from the Cambridge–Manchester Cancer Imaging Centre.
Author contributions
ER carried out experiments, analysed and interpreted data and contributed to the writing of the manuscript; LM and PD carried out experiments; MG and MJA provided histopathological analysis of tissue samples; KMB provided funding support and contributed to both the conception of this study and the writing of the manuscript; and SKL conceived this study, analysed and interpreted data and wrote the manuscript. All authors approved the submitted manuscript.
SUPPLEMENTARY MATERIAL
The following supplementary material may be found in the online version of this article:
Figure S1. Plasmid map of the lentiviral construct LV–LS1
Figure S2. Plasmid map of the lentiviral construct LV–indLS2
Figure S3. Plasmid map of the lentiviral construct LV–indLS2miR1224
Figure S4. Further representative examples of end-stage bioluminescence images (lungs ex vivo) and histology
Figure S5. Higher-magnification image of the tumours depicted in Figure S4
Supporting Information
Figure S1. Plasmid map of the lentiviral construct LV–LS1. All single and two-cutter 6+ nucleotide restriction enzymes indicated, plus additional enzymes relevant for future vector customization
Figure S2. Plasmid map of the lentiviral construct LV–indLS2. All single and two-cutter 6+ nucleotide restriction enzymes indicated, plus additional enzymes relevant for future vector customization
Figure S3. Plasmid map of the lentiviral construct LV–indLS2miR1224. All single and two-cutter 6+ nucleotide restriction enzymes indicated, plus additional enzymes relevant for future vector customization
Figure S4. Further representative examples of end-stage bioluminescence images (lungs ex vivo) and histology. Mouse genotypes and vectors as indicated; LG, low-grade; HG, high-grade
Figure S5. Higher-magnification image of the tumours depicted in Figure S4, to better depict nuclear morphology
References
- Frese KK, Tuveson DA. Maximizing mouse cancer models. Nat Rev Cancer. 2007;7:645–658. doi: 10.1038/nrc2192. [DOI] [PubMed] [Google Scholar]
- Heyer J, Kwong LN, Lowe SW. Non-germline genetically engineered mouse models for translational cancer research. Nat Rev Cancer. 2010;10:470–480. doi: 10.1038/nrc2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huijbers IJ, Krimpenfort P, Berns A. Rapid validation of cancer genes in chimeras derived from established genetically engineered mouse models. BioEssays. 2011;33:701–710. doi: 10.1002/bies.201100018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross S, Piwnica-Worms D. Spying on cancer: molecular imaging in vivo with genetically encoded reporters. Cancer Cell. 2005;7:5–15. doi: 10.1016/j.ccr.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Lyons SK. Advances in imaging mouse tumour models in vivo. J Pathol. 2005;205:194–205. doi: 10.1002/path.1697. [DOI] [PubMed] [Google Scholar]
- Kocher B, Piwnica-Worms D. Illuminating cancer systems with genetically engineered mouse models and coupled luciferase reporters in vivo. Cancer Discov. 2013;3:616–629. doi: 10.1158/2159-8290.CD-12-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vooijs M, Jonkers J, Lyons S. Noninvasive imaging of spontaneous retinoblastoma pathway-dependent tumors in mice. Cancer Res. 2002;62:1862–1867. [PubMed] [Google Scholar]
- Lyons SK, Meuwissen R, Krimpenfort P. The generation of a conditional reporter that enables bioluminescence imaging of Cre/loxP-dependent tumorigenesis in mice. Cancer Res. 2003;63:7042–7046. [PubMed] [Google Scholar]
- Lyons SK, Lim E, Clermont AO. Noninvasive bioluminescence imaging of normal and spontaneously transformed prostate tissue in mice. Cancer Res. 2006;66:4701–4707. doi: 10.1158/0008-5472.CAN-05-3598. [DOI] [PubMed] [Google Scholar]
- Zhang N, Lyons S, Lim E. A spontaneous acinar cell carcinoma model for monitoring progression of pancreatic lesions and response to treatment through noninvasive bioluminescence imaging. Clin Cancer Res. 2009;15:4915–4924. doi: 10.1158/1078-0432.CCR-08-2256. [DOI] [PubMed] [Google Scholar]
- DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc. 2009;4:1064–1072. doi: 10.1038/nprot.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nat Protoc. 2006;1:241–245. doi: 10.1038/nprot.2006.37. [DOI] [PubMed] [Google Scholar]
- Kutner RH, Zhang XY, Reiser J. Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat Protoc. 2009;4:495–505. doi: 10.1038/nprot.2009.22. [DOI] [PubMed] [Google Scholar]
- BPROM. http://linux1.softberry.com/berry.phtml?topic=bprom&group=programs&subgroup=gfindb.
- BDGP neural network promoter prediction. http://www.fruitfly.org/seq_tools/promoter.html.
- Laird PW, Zijderveld A, Linders K. Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 1991;19:4293. doi: 10.1093/nar/19.15.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks Lab website: http://web.mit.edu/jacks-lab/protocols_table.html.
- Jonkers J, Meuwissen R, van der Gulden H. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29:418–425. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- Nikitin AY, Alcaraz A, Anver MR. Classification of proliferative pulmonary lesions of the mouse: recommendations of the mouse models of human cancers consortium. Cancer Res. 2004;64:2307–2316. doi: 10.1158/0008-5472.can-03-3376. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Lutz B. Cre recombinase-mediated inversion using lox66 and lox71: method to introduce conditional point mutations into the CREB-binding protein. Nucleic Acids Res. 2002;30:e90. doi: 10.1093/nar/gnf089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahonen AJ, Airenne KJ, Lind MM. Optimized self-excising Cre-expression cassette for mammalian cells. Biochem Biophys Res Commun. 2004;320:366–371. doi: 10.1016/j.bbrc.2004.05.175. [DOI] [PubMed] [Google Scholar]
- Jackson EL, Olive KP, Tuveson DA. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65:10280–10288. doi: 10.1158/0008-5472.CAN-05-2193. [DOI] [PubMed] [Google Scholar]
- Junttila MR, Karnezis AN, Garcia D. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature. 2010;468:567–571. doi: 10.1038/nature09526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickins RA, Hemann MT, Zilfou JT. Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet. 2005;37:1289–1295. doi: 10.1038/ng1651. [DOI] [PubMed] [Google Scholar]
- Fellmann C, Zuber J, McJunkin K. Functional identification of optimized RNAi triggers using a massively parallel sensor assay. Mol Cell. 2011;41:733–746. doi: 10.1016/j.molcel.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premsrirut PK, Dow LE, Kim SY. A rapid and scalable system for studying gene function in mice using conditional RNA interference. Cell. 2011;145:145–158. doi: 10.1016/j.cell.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland KD, Proost N, Brouns I. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell. 2011;19:754–764. doi: 10.1016/j.ccr.2011.04.019. [DOI] [PubMed] [Google Scholar]
- Hameyer D, Loonstra A, Eshkind L. Toxicity of ligand-dependent Cre recombinases and generation of a conditional Cre deleter mouse allowing mosaic recombination in peripheral tissues. Physiol Genom. 2007;31:32–41. doi: 10.1152/physiolgenomics.00019.2007. [DOI] [PubMed] [Google Scholar]
- Bersell K, Choudhury S, Mollova M. Moderate and high amounts of tamoxifen in α-MHC-MerCreMer mice induce a DNA damage response, leading to heart failure and death. Dis Models Mechan. 2013;6:1459–1469. doi: 10.1242/dmm.010447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch DG, Grimm J, Guimaraes AR. Imaging primary lung cancers in mice to study radiation biology. Int J Radiat Oncol Biol Phys. 2010;76:973–977. doi: 10.1016/j.ijrobp.2009.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer KL, Dunning KR, Parsons DW. Gene delivery to airway epithelial cells in vivo: a direct comparison of apical and basolateral transduction strategies using pseudotyped lentivirus vectors. J Gene Med. 2007;9:362–368. doi: 10.1002/jgm.1025. [DOI] [PubMed] [Google Scholar]
- Wang X, Cunningham M, Zhang X. Phosphorylation regulates c-Myc's oncogenic activity in the mammary gland. Cancer Res. 2011;71:925–936. doi: 10.1158/0008-5472.CAN-10-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesche R, Groszer M, Gao J. Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis. 2002;32:148–149. doi: 10.1002/gene.10036. [DOI] [PubMed] [Google Scholar]
- Ma X. der Made AC, Autar B. Targeted biallelic inactivation of Pten in the mouse prostate leads to prostate cancer accompanied by increased epithelial cell proliferation but not by reduced apoptosis. Cancer Res. 2005;65:5730–5739. doi: 10.1158/0008-5472.CAN-04-4519. Ziel-van. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Plasmid map of the lentiviral construct LV–LS1. All single and two-cutter 6+ nucleotide restriction enzymes indicated, plus additional enzymes relevant for future vector customization
Figure S2. Plasmid map of the lentiviral construct LV–indLS2. All single and two-cutter 6+ nucleotide restriction enzymes indicated, plus additional enzymes relevant for future vector customization
Figure S3. Plasmid map of the lentiviral construct LV–indLS2miR1224. All single and two-cutter 6+ nucleotide restriction enzymes indicated, plus additional enzymes relevant for future vector customization
Figure S4. Further representative examples of end-stage bioluminescence images (lungs ex vivo) and histology. Mouse genotypes and vectors as indicated; LG, low-grade; HG, high-grade
Figure S5. Higher-magnification image of the tumours depicted in Figure S4, to better depict nuclear morphology