

Abstract
The liver is the main organ involved in the metabolism of amino acids (AA), which are oxidized by amino acid catabolizing enzymes (AACE). Peroxisome proliferator-activated receptor-α (PPARα) stimulates fatty acid β-oxidation, and there is evidence that it can modulate hepatic AA oxidation during the transition of energy fuels. To understand the role and mechanism of PPARα’s regulation of AA catabolism, the metabolic and molecular adaptations of Ppara-null mice were studied. The role of PPARα on AA metabolism was examined by in vitro and in vivo studies. In wild-type and Ppara-null mice, fed increasing concentrations of the dietary protein/carbohydrate ratio, we measured metabolic parameters, and livers were analyzed by microarray analysis, histology and Western blot. Functional enrichment analysis, EMSA and gene reporter assays were performed. Ppara-null mice presented increased expression of AACE in liver affecting AA, lipid and carbohydrate metabolism. Ppara-null mice had increased glucagon/insulin ratio (7.2-fold), higher serum urea (73.1 %), lower body protein content (19.7 %) and decreased several serum AA in response to a high-protein/low-carbohydrate diet. A functional network of differentially expressed genes, suggested that changes in the expression of AACE were regulated by an interrelationship between PPARα and HNF4α. Our data indicated that the expression of AACE is down-regulated through PPARα by attenuating HNF4α transcriptional activity as observed in the serine dehydratase gene promoter. PPARα via HNF4α maintains body protein metabolic homeostasis by down-regulating genes involved in amino acid catabolism for preserving body nitrogen.
Electronic supplementary material
The online version of this article (doi:10.1007/s12263-014-0452-0) contains supplementary material, which is available to authorized users.
Keywords: Amino acid catabolism, Body nitrogen, Ppara-null mice, Dietary protein/carbohydrate ratio
Introduction
The catabolism of amino acids (AA) is an important energy source for mammalian organisms when other energy substrates are low, particularly circulating glucose and fatty acids (Young and Marchini 1990). Under an energy demanding state, such as prolonged fasting, the protein breakdown in skeletal muscle is augmented, releasing to the circulation increasing amounts of AA, which are taken up by the liver, and catabolized by specific amino acid catabolizing enzymes (AACE) (Harper 1984). This catabolism contributes to the generation of metabolic energy and provides substrates for gluconeogenesis to maintain blood glucose concentration, since the main source of glucose for the brain and other tissues is provided by gluconeogenic AA (Kaloyianni and Freedland 1990). On the other hand, after consumption of a high-protein diet, the AA supply into the liver also increases. It has been established that when dietary protein intake does not exceed the amino acid requirements, AA catabolism barely occurs. However, when protein intake is above the requirement, the excess of AA is catabolized also by the AACE (Soemitro et al. 1989). This is because the excess of dietary AA cannot be stored, and thus if excessive protein intake is not used efficiently by the body, this may impose a metabolic burden on the bones, kidneys and liver, and generate adverse effects, such as hypercalciuria and hyperuricosuria (Delimaris 2013). In fact, the AACE activity increases when the supply of circulating amino acids augments by fasting or after an excess of dietary protein (Harper 1984).
Protein homeostasis is one of the major nodal points that needs to be preserved in order to retain the cellular homeostatic balance (Chondrogianni and Gonos 2010). Proteins in the body have vital roles in regulating cell structure, cell signaling and the various enzymatic processes of the cell; consequently, protein breakdown during prolonged fasting will translate into loss of biological function. Thus, a metabolic priority is to preserve body proteins, which is accomplished through a shift in the type of fuel used from glucose to fatty acids, that are less functionally critical and more energy efficient. The liver plays an important role in the provision of alternative substrates during starvation, such as ketone bodies produced as a result of fatty acid breakdown, that are particularly important as an energy source in brain (Alberti et al. 1981). Nonetheless, the understanding of the molecular mechanisms that are implicated in this metabolic switch for preserving AA to maintain the body nitrogen balance, and utilizing fat as the main energy source, is limited.
The peroxisome proliferator-activated receptor-α (PPARα) is an important regulator during the transition of energy fuels in the body. PPARα plays a pivotal role in the management of energy stores through up-regulation of the expression of genes encoding enzymes involved in fatty acid oxidation (FAO) (Aoyama et al. 1998; Kersten et al. 1999; Lee et al. 1995; Motojima et al. 1998; Peters et al. 1998). Interestingly, it was found that the expression of mRNAs encoding enzymes involved in the metabolism of AA is lower in wild-type (WT) mice than PPARα knockout (Ppara-null) mice (Kersten et al. 2001). Recent evidence has shown that feeding mice diets containing Wy-14,643 decreases the abundance of mRNAs encoding some AACE (Aleman et al. 2013b). However, there is scarce evidence about the specific role of PPARα in the regulation of AACE gene expression, particularly about the mechanism to down-regulate their expression.
Our aim was to determine the role of PPARα in the expression of AACE, as part of the mechanism underlying the conservation of body nitrogen. To understand the role of PPARα on the molecular mechanism of the protein homeostasis, we challenged a mouse model harboring a genetic loss of function of PPARα, Ppara-null mice, with diets containing different percentages of dietary protein/carbohydrate (DP/DCH) ratio, 6/77 % (low-protein/high-carbohydrate), 20/63 % (adequate-protein/carbohydrate) or 50/33 % (high-protein/low-carbohydrate) to provide a graded increase in the supply of AA. Ppara-null mice displayed increased expression of AACE, independent of the amount of dietary protein consumed, affecting not only the metabolism of AA, but also that of lipids and carbohydrates. Our study through a functional network of differentially expressed genes modulated by the DP/DCH ratio showed changes in the expression of AACE associated with the interrelationship between PPARα and hepatocyte nuclear factor 4α (HNF4α). In fact, reporter assays and EMSA showed the role of PPARα in attenuating the transcriptional activity of HNF4α on the expression of AACE. Our work supports previous findings about the suppressive effect of PPARα on genes encoding enzymes involved in AA catabolism and gives further evidence to its role in maintaining metabolic homeostasis. Our results demonstrate that in addition to regulating lipid metabolism, PPARα also regulates AA catabolism in liver with the main aim of preserving body nitrogen.
Experimental procedures
Primary hepatocyte analyses
Male Wistar rats (200–300 g) were laparotomized under diethyl ether anesthesia, and hepatocytes were collected by in situ perfusion of the liver according to the method of Berry and Friend (1969), which consists of cannulating and exsanguinating the liver in vivo and perfusing the liver with collagenase in a recirculating system. The livers were isolated and placed in 10 % DMEM–FBS medium (Sigma-Aldrich, St. Louis, MO, USA) containing penicillin/streptomycin (100 U/ml and 100 mg/ml). The filtered hepatocytes were washed twice with the medium and then incubated at a density of 2–3 × 106 cells/ml. The medium was changed after the first 4 h to remove unattached cells, and the culture was then continued for 16 h. At 20 h after cell seeding, 200 µM Wy-14,643 (Biomol, Farmingdale, NY, USA) or 10 µM forskolin (final concentration) was added to the hepatocytes in DMEM, followed by incubation for 4 or 6 h to measure RNA expression.
Animals
Male Ppara-null mice (Lee et al. 1995) and C57BL/6 wild-type mice at 7–9 weeks of age (n = 5 per group, average weight 22.4 ± 0.5 g) were given free access to water and one of the experimental diets. Mice were provided with isoenergetic diets containing different percentages of DP/DCH, 6/77 % (low-protein/high-carbohydrate), 20/63 % (adequate-protein/carbohydrate) or 50/33 % (high-protein/low-carbohydrate), as previously reported (Menezes et al. 2013; Torres et al. 1999) for 8 days. The diets were formulated following the recommendations of the American Institute of Nutrition (AIN-93) (Reeves 1997) and adjusted to contain the indicated protein concentrations. The mice were adapted to consume their respective diets under a time-restricted feeding schedule within a period of 8 h, from 8 a.m. to 4 p.m., to synchronize their food consumption preventing metabolic and hormonal variability. At the end of the study, the mice fed with each diet were divided into two groups: animals fasted for 16 h (from 4 p.m. until 8 a.m. the next day) and those refed for 3 h (from 8 a.m. to 11 a.m.). The Animal Committee of the Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán, Mexico City, approved the animal protocol and experiments. All institutional and national guidelines for the care and use of laboratory animals were followed.
Analytical procedures
Serum glucose was measured using a YSI 2700 SELECT biochemistry analyzer (YSI Incorporated, Yellow Springs, OH, USA). Serum glucagon concentration was quantified in radioimmunoassays using a kit that utilizes 125I-labeled glucagon and a glucagon antiserum (Millipore, Billerica, MA, USA). Serum insulin concentrations were determined with an insulin ELISA kit (Millipore). Total serum nonesterified fatty acid concentrations were measured using an acyl-CoA oxidase-based colorimetric kit (Roche Applied Science, Penzberg, Upper Bavaria, Germany). Serum urea was determined through an enzymatic method with urease measured in a Synchron CX 5 Pro chemistry analyzer (Beckman Coulter, Brea, CA, USA). Serum amino acids were quantified using the EZ:faast kit (Phenomenex, Torrance, CA, USA) for physiological amino acids and analyzed on an Agilent 6850 gas chromatograph with a flame ionization detector (Agilent Technologies, Santa Clara, CA, USA).
Liver glycogen determination
Frozen liver samples (100 mg) were homogenized with 5 ml of 5 % trichloroacetic acid. Glycogen was precipitated using ethanol and detected with the Anthrone reagent. The optical density was determined using colorimetric tube, and the factor of 0.9 for converting the glucose value to a glycogen value was used in the calculations (Carroll et al. 1956).
RNA isolation/RT-PCR analysis
Total RNA was isolated from frozen mouse livers and fresh hepatocyte cultures using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA). RNA was reverse-transcribed, and PCR amplification was performed using TaqMan assays according the manufacturer’s protocol (Applied Biosystems, Carlsbad, CA, USA). Assays for each gene were carried out in triplicate in 96-well optical plates using the ABI Prism 7000 sequence detection system (Applied Biosystems).
Genome-wide microarray assay
Total RNA from the livers of 21 WT mice (n = 3 or 4 per group) and 24 Ppara-null mice (n = 4 per group), fed 6/77, 20/63 or 50/63 % diets and subjected to 16 h of fasting or 3 h of refeeding was isolated with the TRIzol reagent (Invitrogen). The RNA quality in each sample was assessed with a Bioanalyzer 2100 (Agilent Technologies). Affymetrix microarray assays were performed according to the manufacturer’s protocol using the GeneChip Mouse Gene 1.0 ST Array (Affymetrix, Santa Clara, CA, USA).
Expression profiling analysis
The low-level data analysis included the quantile normalization process (Bolstad et al. 2003) using the Bioconductor Project (Gentleman et al. 2004) implemented in R software (http://www.cran.r-project.org). To identify differentially expressed genes, a linear model involving an empirical Bayesian approach was performed using R and the Limma package (Smyth 2004, 2005). This approach relies on two matrices: The first is called the design matrix, which provides a representation of the different RNA targets hybridized to the arrays; and the second, or contrast matrix, allows the coefficients defined by the design matrix to be combined into contrasts of interest. Each contrast corresponds to a comparison of interest (Smyth 2004). To correct for multiple testing, the Benjamini–Hochberg algorithm correction was used (Reiner et al. 2003).
Body composition
After removal of the stomach and intestines, mice were weighed (eviscerated weight) and dried at 60 °C until constant weight was achieved (7 days). The water content was determined as the weight lost during drying. The dried carcasses were ground to obtain a fine powder. Samples of the powder were used for the extraction of fat contents in a Soxhlet apparatus and body nitrogen via the modified Kjeldahl method (Lane et al. 1991; Phinney et al. 1994).
Energy expenditure
Energy expenditure was assessed using indirect calorimetry (Oxymax, Columbus Instruments, Columbus, OH, USA). A constant airflow was drawn through and monitored by a mass-sensitive flow meter. The concentrations of oxygen and carbon dioxide were monitored at the inlet and outlet of the sealed chambers to calculate the oxygen consumption and respiratory exchange ratio. Measurements were obtained in each chamber at 22 min intervals.
Histological analysis
Samples of livers from WT and Ppara-null mice were dissected and immediately fixed with ice-cold 4 % (w/v) paraformaldehyde dissolved in phosphate buffer and subsequently dehydrated and embedded in paraffin. Two 4-µm sections per block were then stained with hematoxylin and eosin and imaged at a 20× magnification. To visualize neutral lipids, frozen liver sections (8 µm) were stained with Oil Red O (Sigma-Aldrich).
Functional enrichment analysis
A gene-annotation enrichment analysis was performed within the set of significant genes using the Database for Annotation, Visualization and Integrated Discovery bioinformatics tool (DAVID) (da Huang et al. 2009). The whole-genome gene expression data were tested using Ingenuity® (Redwood City, CA, USA), an integrative biomolecular tool for the analysis of complex genomics data. The Ingenuity Pathway Analysis (IPA) module was employed for identifying biological interaction networks of the significant regulated genes.
Western blotting analysis
Nuclear extracts from HepG2 cells overexpressing HNF4α, PPARα or both were obtained in RIPA protein lysis buffer containing 1 mM sodium fluoride, 2 mM sodium orthovanadate and complete protease inhibitor cocktail tablets (Roche Applied Science). Nuclear or total protein (20 μg) was loaded onto 8 % polyacrylamide gels, separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. The blots were blocked with nonfat dry milk (Bio-Rad, Hercules, CA, USA) and incubated overnight at 4 °C with the antibody anti-HNF4α, anti-PPARα or anti-SDS (Santa Cruz Biotechnology, Dallas, TX, USA). β-Actin (Santa Cruz Biotechnology) was used as a loading control. The obtained bands were analyzed using the NIH software ImageJ (http://rsbweb.nih.gov/ij/).
Preparation of nuclear extracts and electrophoretic mobility shift assays (EMSA)
Nuclear extracts from Human HepG2 hepatoblastoma cells previously transfected with HNF4α expression vector were prepared using a hypotonic lysis buffer (100 mM HEPES pH 7.9, 15 mM MgCl2, 100 mM KCl) and a high salt extraction buffer (20 mM HEPES pH 7.9, 1.5 mM MgCl2, 0.42 M NaCl, 0.2 mM EDTA, and 25 % v/v glycerol) following the instructions from the Sigma nuclear extract kit. Protein concentration was determined by the Lowry method. EMSA were performed using the Gel Shift Assay System (Promega, Fitchburg, WI, USA) according to manufacturer’s instructions. Briefly, specific probes were labeled with [γ-32P] ATP at the 5′ end of oligonucleotide using T4 polynucleotide kinase. Reaction mixtures were prepared with 6 μg of human HepG2 hepatoblastoma cells nuclear extracts previously transfected with HNF4α expression vector, 17.5 fmol of each labeled probe, 0.5–1 μg of poly(dI–dC) and binding buffer. The reaction mixtures were incubated for 30 min at room temperature. For competition, 100-fold excess unlabeled double-stranded probe were added to the binding reaction. For super shift analysis, 5 μg of specific antibody anti-HNF4α was preincubated for 1 h with the binding buffer and the nuclear extract. Binding reactions were terminated with the addition of gel-loading buffer. The complexes were separated on a nondenaturing 4 % (wt/vol) polyacrylamide gel and visualized by scan of the dried gels in the personal molecular imager (PMI) from Bio-Rad. All sequence probes can be consulted in Supp. Table 1.
Reporter gene assay
Transient transfection on human HepG2 hepatoblastoma cells was performed using FuGENE HD (Roche Applied Science) according to the manufacturer’s instructions. The Sds promoter constructs were inserted in the pGL3 basic vector (Promega), which contained a luciferase reporter gene. The PCR constructs were generated using the oligonucleotide primers presented in Supp. Table 1. Cells were co-transfected with the HNF4α and/or PPARα expression vectors; after co-transfection, the cells were incubated with or without Wy-14,643. Luciferase activity was measured as relative light units with a BD Monolight 3010C luminometer (BD Biosciences, San Diego, CA, USA). The promoterless vector (pGL3 basic) was also transfected into HepG2 cells as a basal control. The protein concentration of the samples was determined according to Lowry and was used to normalize the transfection efficiency among samples.
Site-directed mutagenesis
For the mutagenesis assay, the Quick Change Site-directed Mutagenesis kit (Stratagene, La Jolla, CA, USA) was used according to the manufacturer’s instructions. To obtain the mutated construct, the PCR was performed with the Sds promoter construct −875/+290 pb as template and specific oligos with the desired point mutations (Supp. Table 1). We added DpnI restriction enzyme for 1 h at 37 °C to digest the methylated, nonmutated parental DNA template. Two micrograms of digested product was transformed into E. coli XL1-Blue electrocompetent cells. The cells were grown in LB–ampicillin agar plates at 37 °C for 18 h. The mutated plasmid was purified using the PureYield™ Plasmid Miniprep System (Promega) from the grown colonies and verified by sequencing using the Beckman Coulter Sequencer. The activity of the HNF4α response element mutated construct was assessed following the same protocol of transfection and reporter gene assays described previously.
Statistical analysis
Results are expressed as mean ± SEM. Statistical significance was determined by two-tailed nonpaired t test, or two-way ANOVA (Bonferroni post hoc test) as appropriate, using statistical software (Prism 5.0; GraphPad Software, Inc., La Jolla, CA). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Results
Induced expression of the AACE for serine, Sds, is partially down-regulated by PPARα activation
Previous evidence has suggested that PPARα can repress the expression of genes involved in AA metabolism (Aleman et al. 2013a), then the effect of ligand activation of PPARα on the serine dehydratase (Sds) expression, which is the AACE for serine, was evaluated. Treatment of rat primary hepatocytes with Wy-14,643 did not reveal a repressive effect on Sds, despite an increase in the PPARα target gene carnitine palmitoyltransferase-1 (Cpt1) (Supp. Fig. 1a, b). When hepatocytes were stimulated with forskolin, the expression of Sds was increased, but preincubation of the cells with Wy-14,643 partially prevented this stimulatory effect. Forskolin did not modify the expression of Cpt1 and did not alter the stimulatory effect of Wy-14,643 on Cpt1 expression (Supp. Fig. 1c, d). These results suggested that PPARα had a partial repressive effect in preventing the induction of this AACE.
The lack of PPARα exacerbates glucagon response by increasing amount of protein intake
To study the role of PPARα in the mechanism underlying the conservation of body nitrogen in vivo, we challenged Ppara-null mice with diets containing different concentrations of DP/DCH to modify the supply of AA into the liver. Mice were under an 8-h restricted feeding schedule to synchronize their food consumption and prevent metabolic and hormonal variability. We did not observe difference in body weight between WT and Ppara-null mice fed 20/63 % diet. Ppara-null mice had a significant lower weight compared with WT mice in 6/77 and 50/33 % diets (Table 1). Interestingly, food intake was similar in all groups, (10.5 ± 0.5 kcal/day) despite the difference in the DP/DCH ratio. After 3 h of refeeding, the insulin concentration was higher in WT mice compared with Ppara-null mice in the three diets assessed, showing significant difference in 6/77 and 50/33 % diets. In contrast, Ppara-null mice had higher concentration of glucagon than WT mice in all groups. In WT mice, greater amounts of protein led to higher concentrations of glucagon; however, in Ppara-null mice, the glucagon concentration did not differ between 20/63 and 50/33 % diets (Table 1). The high glucagon concentration also was observed during fasting in all groups (data not shown). As a consequence, the glucagon/insulin ratio was higher in Ppara-null mice, particularly in the group fed 50/33 % (7.2-fold).
Table 1.
Body weight, food intake, metabolic parameters and body composition of WT and Ppara-null mice fed diets with different protein/carbohydrate ratio
| Dietary protein/carbohydrate ratio | p values | |||||
|---|---|---|---|---|---|---|
| 6/77 % | 20/63 % | 50/33 % | PPARα | Diet | PPARα-diet | |
| Body weight (g) | ||||||
| WT | 20.0 ± 0.5 | 21.3 ± 0.4 | 25.1 ± 0.4 | <0.0001 | <0.0001 | 0.0001 |
| Ppara-null | 17.4 ± 0.3## | 21.0 ± 0.5 | 20.0 ± 0.2#### | |||
| Food intake (Kcal/day) | ||||||
| WT | 10.1 ± 0.49 | 10.8 ± 0.47 | 10.4 ± 0.48 | 0.53 | 0.07 | 0.14 |
| Ppara-null | 10.6 ± 0.48 | 10.7 ± .0.65 | 10.3 ± 0.39 | |||
| Protein intake (g/day) | ||||||
| WT | 0.15 ± 0.01 | 0.58 ± 0.02 | 1.32 ± 0.06 | 0.76 | <0.0001 | 0.80 |
| Ppara-null | 0.16 ± 0.01 | 0.57 ± 0.04 | 1.30 ± 0.05 | |||
| Protein intake (Kcal/day) | ||||||
| WT | 0.61 ± 0.03 | 2.31 ± 0.10 | 5.26 ± 0.24 | 0.76 | <0.0001 | 0.80 |
| Ppara-null | 0.65 ± 0.03 | 2.28 ± 0.15 | 5.20 ± 0.20 | |||
| Carbohydrate intake (g/day) | ||||||
| WT | 1.95 ± 0.10 | 1.82 ± 0.08 | 0.87 ± 0.04 | 0.29 | <0.0001 | 0.08 |
| Ppara-null | 2.07 ± 0.09 | 1.80 ± 0.12 | 0.86 ± 0.03 | |||
| Carbohydrate intake (Kcal/day) | ||||||
| WT | 7.81 ± 0.38 | 7.29 ± 0.31 | 3.47 ± 0.16 | 0.29 | <0.0001 | 0.08 |
| Ppara-null | 8.29 ± 0.38 | 7.18 ± 0.48 | 3.43 ± 0.13 | |||
| Glucose (mg/ml) | ||||||
| WT | 183.4 ± 15.0 | 218.0 ± 21.0 | 174 ± 9.3 | 0.10 | 0.0008 | 0.005 |
| Ppara-null | 222.0 ± 9.6 | 176.5 ± 6.3 | 121.8 ± 3.1# | |||
| Glycogen (mg/g liver) | ||||||
| WT | 35.46 ± 1.9 | 28.06 ± 1.3 | 19.30 ± 2.7 | 0.001 | <0.0001 | <0.0001 |
| Ppara-null | 44.27 ± 1.0## | 22.26 ± 0.8 | 1.37 ± 0.62#### | |||
| Insulin (ng/ml) | ||||||
| WT | 8.0 ± 0.6 | 11.4 ± 0.1 | 8.7 ± 1.2 | <0.0001 | 0.0001 | 0.0455 |
| Ppara-null | 4.0 ± 0.9# | 8.8 ± 0.8 | 1.8 ± 0.2### | |||
| Glucagon (pg/ml) | ||||||
| WT | 35.5 ± 5.7 | 58.8 ± 3.7 | 71.5 ± 2.5 | <0.0001 | <0.0001 | 0.18 |
| Ppara-null | 59.3 ± 10.5 | 112.4 ± 3.7### | 107.6 ± 13.8# | |||
| Liver weight (% body weight) | ||||||
| WT | 5.1 ± 0.2 | 5.8 ± 0.1 | 5.3 ± 0.06 | 0.004 | 0.006 | 0.15 |
| Ppara-null | 5.0 ± 0.3 | 5.3 ± 0.1 | 4.4 ± 0.2# | |||
| Urea (mg/day) | ||||||
| WT | 14.2 ± 0.7 | 20.4 ± 0.5 | 26.0 ± 0.5 | <0.0001 | <0.0001 | 0.0002 |
| Ppara-null | 35.9 ± 2.3#### | 29.3 ± 0.7### | 45.4 ± 2.6#### | |||
| Body protein (%) | ||||||
| WT | 19.1 ± 0.4 | 22.2 ± 0.2 | 21.9 ± 0.3 | 0.0009 | 0.21 | 0.002 |
| Ppara-null | 20.0 ± 0.2 | 19.2 ± 0.8# | 17.5 ± 1.5### | |||
| Body fat (%) | ||||||
| WT | 7.3 ± 0.9 | 9.2 ± 1.5 | 5.0 ± 0.7 | <0.0001 | 0.004 | 0.15 |
| Ppara-null | 21.3 ± 1.8### | 29.4 ± 1.4#### | 17.6 ± 3.4## | |||
Data are means ± SEM. Glucose, glycogen, insulin, glucagon and liver weight data were obtained after 3 h of refeeding. Two-way ANOVA, with Bonferroni multiple comparisons post-test
WT wild-type mice, Ppara-null Ppara-null mice
# p < 0.05; ## p < 0.01; ### p < 0.001; ### p ≤ 0.0001 relative to WT group
The lack of PPARα alters the expression pattern of liver AACE induced by increasing amount of protein intake
It is known that the increase in the amount of dietary protein is accompanied by an increase in serum glucagon (Soemitro et al. 1989; Tovar et al. 2002). Consistent with this evidence, our results showed that, in WT mice, the greater the amount of dietary protein, the higher the concentration of glucagon. Interestingly, we observed an exacerbated glucagon response in Ppara-null mice independent of the DP/DCH ratio (Table 1). Since, it has been reported that glucagon stimulates the expression of AACE via cAMP-dependent PKA pathway (Aleman et al. 1998), we hypothesized that Ppara-null mice may have a different pattern of expression of AACE compared with WT mice. Thus, we performed a comparative microarray analysis of the livers samples, and it revealed that 137 and 53 genes were differentially expressed after refeeding 6/77 versus 50/33 % diets in WT and Ppara-null mice, respectively. Among the differentially expressed genes found in WT mice were genes encoding AACE, that showed increased expression as the DP/DCH ratio increased (Fig. 1a), while in Ppara-null mice, these genes were not differentially expressed (Fig. 1b). The results clearly showed that Ppara-null mice had increased expression of AACE in comparison with WT mice, independent of the DP/DCH ratio (Fig. 1c). The results for several of these genes were confirmed by RT-PCR (Fig. 1d–f). The protein abundance of Sds showed the same pattern observed in gene expression (Fig. 1g).
Fig. 1.
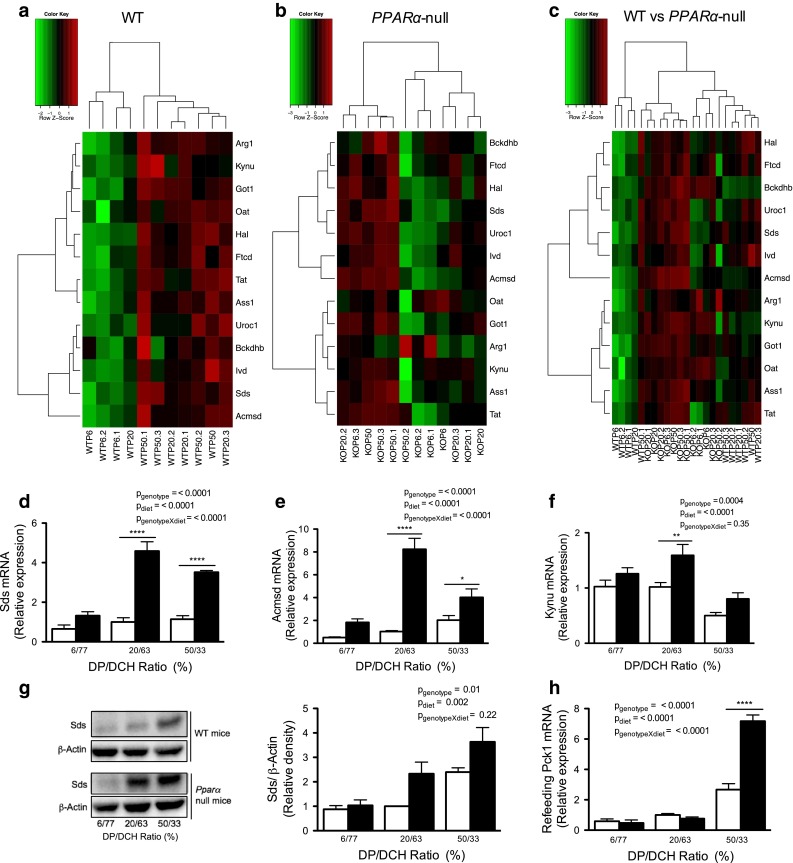
AACE gene expression in Ppara-null and WT mice fed increasing concentrations of DP/DCH. Mice were fed diets containing 6/77, 20/63 or 50/33 % DP/DCH during 8 days. a Heat maps of AACE gene expression in the livers of WT mice, b Ppara-null (KO) mice, and c WT versus Ppara-null mice (n = 3–4). d Validation of gene expression results by RT-PCR analysis of Sds, e Acmsd and f Kynu in the livers of mice. g SDS expression was analyzed in total protein liver extracts of animals; representative data are shown from one mouse per lane. h Relative expression of Pck1 in Ppara-null and WT mice by RT-PCR from liver mRNA. The expression analysis was conducted after refeeding for 3 h. White bars WT; black bars Ppara-null. Results represent mean ± SEM (n = 4–5)
Because Ppara-null mice showed increased expression of AACE, we measured serum urea and body protein contents to determine whether the changes in the expression of these genes actually modified the AA metabolism. In WT mice, serum urea increased as the DP/DCH ratio increased; interestingly, Ppara-null mice had a significantly higher serum urea concentration than WT mice regardless of the dietary protein content (Table 1). Ppara-null mice showed significantly lower body protein content compared with WT mice, particularly in the groups fed 20/63 or 50/33 % diets, which presented a 13.4 or 19.7 % reduction, respectively (Table 1). In addition, Ppara-null mice showed a decrease in serum concentration of most AA as the DP/DCH ratio increased, with the exception of the branched chain amino acids (BCAA) (Table 2). These results support previous evidence that Ppara-null mice have an enhanced AA catabolism (Kersten et al. 2001), which was associated with reduction in lean body mass as observed in the present study. In contrast, Ppara-null mice showed an increase in body fat content independent of the DP/DCH ratio (Table 1). This increase could be the consequence of an impaired fatty acid oxidation, as previously reported (Kersten et al. 1999).
Table 2.
Concentration of serum amino acids in Ppara-null and WT mice
| Dietary protein/carbohydrate ratio | p values | |||||
|---|---|---|---|---|---|---|
| 6/77 % | 20/63 % | 50/33 % | PPARα | Diet | PPARα-diet | |
| Leucine (μM) | ||||||
| WT | 327.4 ± 55.3 | 315.8 ± 82.9 | 670.3 ± 117.2 | 0.66 | 0.06 | 0.007 |
| Ppara-null | 420.8 ± 67.8 | 443.0 ± 20.5 | 374.6 ± 48.1# | |||
| Isoleucine (μM) | ||||||
| WT | 160.6 ± 24.0 | 152.8 ± 28.0 | 320.1 ± 60.0 | 0.0007 | 0.03 | 0.15 |
| Ppara-null | 287.6 ± 44.7 | 419.8 ± 62.4# | 383.6 ± 60.7 | |||
| Valine (μM) | ||||||
| WT | 371.1 ± 30.1 | 372.8 ± 80.6 | 586.3 ± 124.1 | 0.097 | 0.29 | 0.16 |
| Ppara-null | 503.9 ± 54.3 | 628.2 ± 64.4 | 531.3 ± 79.8 | |||
| Alanine (μM) | ||||||
| WT | 1,713 ± 229.9 | 1,486 ± 437.4 | 1,346 ± 295.7 | 0.74 | 0.0004 | 0.009 |
| Ppara-null | 2,941 ± 516.3# | 1,230 ± 157.7 | 634.9 ± 29.6 | |||
| Glycine (μM) | ||||||
| WT | 640.0 ± 100.8 | 363.2 ± 84.7 | 463.4 ± 76.5 | 0.43 | 0.0002 | 0.088 |
| Ppara-null | 669.9 ± 79.1 | 431.5 ± 26.5 | 218.3 ± 16.6 | |||
| Serine (μM) | ||||||
| WT | 104.0 ± 13.4 | 131.7 ± 33.1 | 83.5 ± 12.6 | 0.096 | 0.001 | 0.21 |
| Ppara-null | 112.4 ± 5.1 | 102.3 ± 9.6 | 40.0 ± 8.4 | |||
| Proline (μM) | ||||||
| WT | 75.8 ± 18.2 | 166.5 ± 83.6 | 75.0 ± 17.6 | 0.25 | 0.001 | <0.0001 |
| Ppara-null | 362.5 ± 59.8### | 40.0 ± 11.95 | 31.0 ± 8.6 | |||
| Histidine (μM) | ||||||
| WT | 430.3 ± 98.8 | 923.3 ± 143.2 | 611.9 ± 95.55 | 0.02 | 0.16 | 0.003 |
| Ppara-null | 624.4 ± 126.3 | 425.8 ± 32.6# | 325.1 ± 12.8 | |||
| Tryptophan (μM) | ||||||
| WT | 327.7 ± 84.1 | 436.0 ± 44.0 | 244.9 ± 62.0 | 0.14 | 0.002 | 0.05 |
| Ppara-null | 431.1 ± 78.4 | 226.2 ± 13.8 | 103.9 ± 10.8 | |||
| Tyrosine (μM) | ||||||
| WT | 430.3 ± 98.8 | 923.3 ± 143.2 | 611.9 ± 95.5 | 0.02 | 0.16 | 0.003 |
| Ppara-null | 624.4 ± 126.3 | 425.8 ± 32.65# | 325.1 ± 12.8 | |||
| Aspartate (μM) | ||||||
| WT | 1,073 ± 203.1 | 687.7 ± 109.9 | 995.5 ± 262.1 | 0.87 | 0.01 | 0.25 |
| Ppara-null | 1,339 ± 149.3 | 660.7 ± 46.6 | 668.3 ± 139.3 | |||
| Lysine (μM) | ||||||
| WT | 1,592 ± 463.6 | 3,119 ± 1,247 | 1,472 ± 455.8 | 0.22 | 0.01 | 0.04 |
| Ppara-null | 4,040 ± 787.6# | 3,005 ± 328.2 | 1,025 ± 105.6 | |||
Data are means ± SEM. The concentration of serum amino acids was measured after 16 h fasting. Two-way ANOVA, with Bonferroni multiple comparisons post-test
WT wild-type mice, Ppara-null Ppara-null mice
# p < 0.05; ## p < 0.01; ### p < 0.001; ### p ≤ 0.0001 relative to WT group
The lack of PPARα decreases the energy expenditure and modifies the use of energy substrates depending on the DP/DCH ratio
To study whether the increase in body fat content observed in Ppara-null mice is due to alterations in energy expenditure, we measured oxygen consumption and calculated the respiratory exchange ratio (RER). Our results indicated that Ppara-null mice showed a significant decrease in energy expenditure compared with WT mice in the three diets (Supp. Fig. 2a). We did not observe difference in RER between WT and Ppara-null mice fed 20/63 % diet; nonetheless, in 6/77 and 50/33 % diets, the RER was significantly higher in Ppara-null mice than WT mice. Interestingly, those Ppara-null mice fed 6/77 % diet showed a RER above 1.0, which is suggestive of utilization of carbohydrates for lipogenesis (Supp. Fig. 2b). These data demonstrate that the lack of PPARα decreases the energy expenditure and modifies the use of carbohydrates and lipids.
The lack of PPARα decreases serum glucose and hepatic glycogen despite increase in Pck1 expression in a high-protein/low-carbohydrate diet
Since Ppara-null mice fed 50/33 % diet increased the RER compared with WT mice, we hypothesize that the surplus of AA is channeled to synthesis of glucose by gluconeogenesis. In support to this hypothesis, most of the genes overexpressed in the Ppara-null mice, such as Sds, kynureninase (Kynu), amino carboxymuconate semialdehyde decarboxylase (Acmsd), glutamate oxaloacetate transaminase 1 (Got1), ornithine aminotransferase (Oat) and cystathionase (Cth), are AACE whose pathways generate end products that can be converted into glucose through gluconeogenesis. Therefore, we studied whether the deregulated expression of these genes was also associated with the expression of the gluconeogenic enzyme phosphoenolpyruvate carboxykinase-1 (Pck1), which is responsible for the limiting step in the synthesis of glucose. We did not observe difference of Pck1 expression between WT and Ppara-null mice fed 6/77 or 20/63 % diets. However, in WT and Ppara-null mice, there was a significant increase in Pck1 expression only after consumption of the 50/33 % diet (Fig. 1h). Interestingly, Pck1 expression was approximately 1.3-fold higher in Ppara-null mice than that in WT mice. Nonetheless, the increase in Pck1 expression was not enough to increase blood glucose and hepatic glycogen concentrations (Table 1). In our model, the high dietary protein consumption did not contribute significantly to blood glucose. These results are in agreement with previous studies in humans and rats that, despite the intrinsic differences in the metabolic response of PPARα ligands between species (Albrecht et al. 2013; Corton et al. 2014), show that the contribution of AA to glucose production via gluconeogenesis is quite modest (Fromentin et al. 2013; Stepien et al. 2011).
The lack of PPARα up-regulates lipogenesis in a greater extent in a low-protein/high-carbohydrate diet
Because Ppara-null mice showed an increase in the body fat content, we studied whether the hepatic lipid content and the expression of genes involved in fatty acid metabolism may be altered by the DP/DCH ratio. It is known that PPARα functions as a lipid sensor in the liver and recognizes and responds to the influx of fatty acids by stimulating the transcription of specific genes related to lipid metabolism (Contreras et al. 2013). Also, there is evidence that Ppara-null mice develop lipid steatosis (Costet et al. 1998) due to decreased capacity to oxidize fatty acids. In our study, hematoxylin- and eosin-stained sections did not show any morphological alterations among WT mice fed different diets. The histological analysis did not reveal differences between the Ppara-null mice and WT mice fed 20/63 % diet. Interestingly, Ppara-null mice fed 6/77 % diet showed a greater number of lipid depots than control mice (Fig. 2a). In addition, Ppara-null mice fed 6/77 or 50/33 % diets showed macro- or microvesicular steatosis, accompanied by portal inflammatory infiltrate, compared with those fed the 20/63 % diet. Furthermore, Ppara-null mice fed 50/33 % diet showed numerous hyperchromatic and condensed hepatocyte nuclei indicating apoptosis. On the other hand, in Ppara-null mice, the microarray analysis of hepatic gene expression clearly revealed a decrease in the expression of several genes involved in FAO compared with WT mice, regardless of DP/DCH ratio (Fig. 2b). These results show that the lack of PPARα in mice fed 6/77 % diet exacerbates abnormal deposition of hepatic lipids, although the DP/DCH ratio did not modify the expression of genes involved in FAO pathway.
Fig. 2.
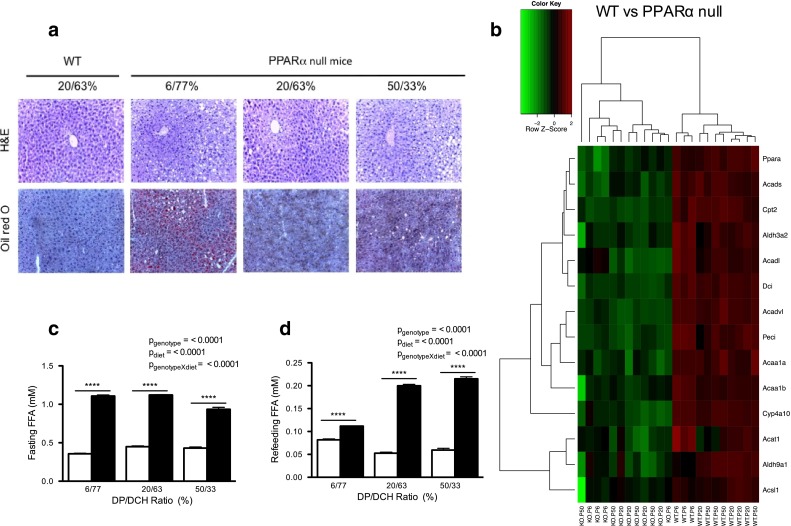
Lipid content in Ppara-null mice is altered by the DP/DCH ratio. Mice were fed diets containing 6/77, 20/63 or 50/33 % DP/DCH during 8 days and then were fasted for 16 h or refed for 3 h. a Histological analysis of liver sections stained with hematoxylin and eosin (H&E) and Oil Red O in mice after refeeding. b Heat map of genes related to FAO in the livers of WT versus Ppara-null (KO) mice after refeeding (n = 3–4). c Free fatty acids of Ppara-null and WT mice in fasted or d refeeding. White bars WT; black bars Ppara-null. Results represent mean ± SEM (n = 4–5)
To determine whether lipid depots in the liver of Ppara-null mice were the result of increased circulating fatty acids, we measured the serum concentration of free fatty acids (FFA) in all groups. Ppara-null mice showed higher concentration of circulating FFA than WT mice in the three diets (Fig. 2c, d). However, we did not observed a direct relationship of FFA concentration with the liver lipid content. Therefore, we hypothesized that high amount of dietary carbohydrate may increase lipid depots as consequence of increased hepatic lipogenesis in Ppara-null mice. A previous study showed that another peroxisome proliferator-activated receptor, PPARγ, may compensate for PPARα in liver Ppara-null mice and can up-regulate genes involved in lipogenesis (Patsouris et al. 2006). Consistent with this, our results showed increased expression of genes involved in lipid synthesis, such as fatty acid synthase (Fasn) and ELOVS family member 6, elongation of long chain fatty acids (Elovl6), in Ppara-null mice fed 6/77 % compared with those fed 50/33 %. Therefore, our data suggest that the lack of PPARα up-regulates lipogenesis in the liver of mice fed 6/77 % diet.
PPARα maintains metabolic homeostasis during increases in the DP/DCH ratio through biological pathways related to amino acid metabolism
Based on the expression patterns observed in mice fed 6/77 and 50/33 % diets, the top canonical pathways were identified. As expected, in WT mice, the main pathways that presented increased expression were those related to AA metabolism (Fig. 3a). However, for Ppara-null mice, the up-regulated expression pathways were related to growth and proliferation, inhibition of apoptosis and regulation of electrolyte and water balances through ion transport. When genes differentially expressed by the effect of DP/DCH ratio in the microarray analysis were considered, it was noted that they were associated with a functional network related to AA and lipid metabolism. Interestingly, this network established a central bidirectional interactive node between PPARα and the transcription factor HNF4α (Fig. 3b).
Fig. 3.

Biological pathways and the functional network associated with PPARα dependent on DP/DCH intake. a Top canonical pathways in Ppara-null and WT mice fed 6/77 versus 50/33 % diets. Comparisons were made using the genes that showed differential expression in the microarray analysis. b Functional network related to AA and lipid metabolism. The network was constructed taking into consideration those genes that were differentially expressed due to changes in the DP/DCH ratio. Color-coding: red, up-regulated genes; green, down-regulated genes; the intensity of the colors indicates the degree of up- or down-regulation. Shape-coding: rectangle, ligand-dependent nuclear receptor; oval, transcription regulator; rhombus, enzyme; circle, others
The interrelationship between PPARα and HNF4α influences amino acid catabolism
To understand how the interrelationship between PPARα and HNF4α affects AA catabolism, we first performed a Western blot analysis in nuclear extracts from cells overexpressing HNF4α, PPARα or both. The nuclear concentration of PPARα was enriched when cells were incubated with Wy-14,643, particularly in the group overexpressing HNF4α and PPARα, in which SDS abundance was the lowest (Fig. 4a). Bioinformatic analysis revealed the presence of putative response elements for the transcription factor HNF4α (HRE) in the promoter regions of several AACE (Supp. Table 2). EMSA analysis showed a strong binding of nuclear protein extract with the HRE-Sds sequence in the rat (Fig. 4b line-6) and mouse (Fig. 4c line-10). When HRE-Sds was mutated no binding was observed (Fig. 4b line-7; Fig. 4c line-12) and excess of cold HRE-Sds also reduced the signal (Fig. 4b line-5; Fig. 4c line-14). Preincubation with the anti-HNF4α abolished the binding or generated a supershift band, indicating that HNF4α was able to bind to the HRE in rat and mouse (Fig. 4b line-8; Fig. 4c line-15). Then, to demonstrate the ability of HNF4α to stimulate expression of AACE, luciferase reporter assays were performed. Cells co-transfected with HRE-Sds and HNF4α increased the luciferase activity, whereas mutation of the HRE-Sds abolished the transcriptional activity of HNF4α (Fig. 4d). Moreover, the addition of Wy-14,643 to cells overexpressing HNF4α and PPARα significantly decreased the activity, in contrast with those cells overexpressing only one of the two transcriptional factors (Fig. 4e). Taking together, these results showed a repressive effect of PPARα on HNF4α transcriptional activity to up-regulate Sds expression.
Fig. 4.
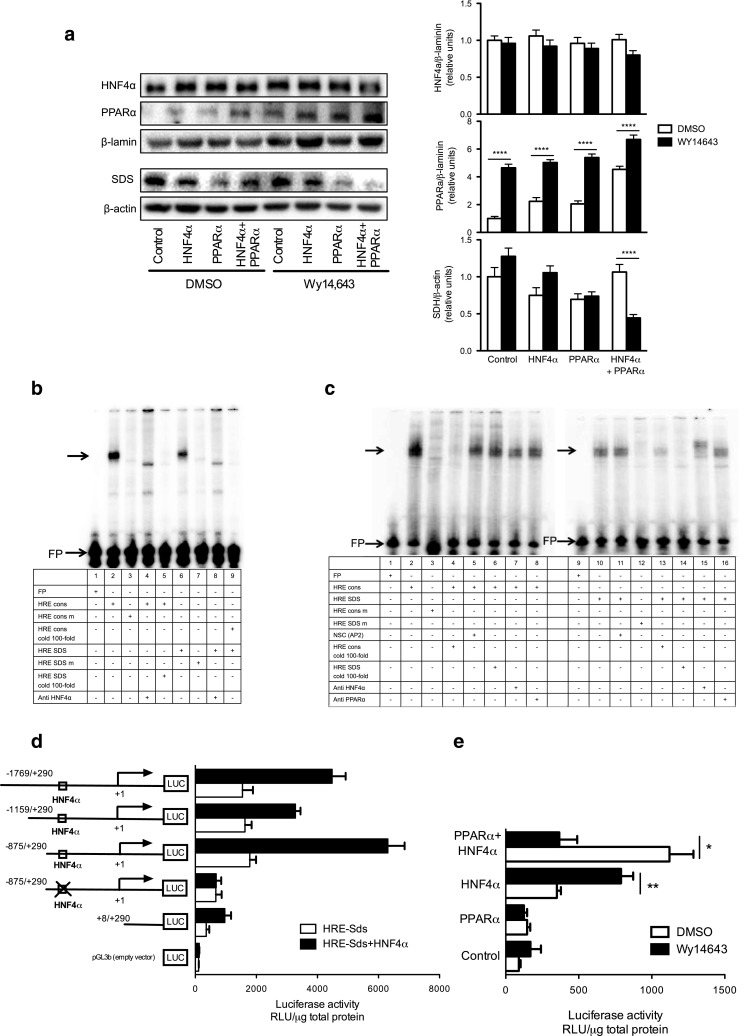
Role of HNF4α in regulating the expression of Sds. a Western blot analysis for HNF4α and PPARα in nuclear protein and SDS in total protein from HepG2 cells. Representative data are shown from one experiment per lane. b EMSA analysis of HepG2 nuclear extracts with labeled oligonucleotides containing HRE-consensus, HRE-SDS and mutated sequences for rat and c mouse. Line-1 in each panel corresponds to free probe (FP). d Gene reporter assay for the Sds gene in HepG2 cells using the HNF4α and/or e PPARα expression vectors; after co-transfection, the cells were incubated with or without Wy-14,643. NSC nonspecific competitor, m mutated sequence, SDS Serine dehydratase, cons consensus sequence, HRE HNF4α response element
Discussion
In the present study, Ppara-null mice showed a greater increase in the expression of AACE in liver, that modifies the metabolic homeostasis of carbohydrates, lipids and proteins, demonstrating the role of PPARα in preserving body nitrogen. The liver may oxidize AA, especially under two conditions: during fasting and as a result of a surplus of these substrates due to the lack of a natural storage reservoir. The feeding of diets containing increase in DP/DCH ratio induces glucagon secretion and modifies AA metabolism by inducing the expression of AACE in liver, through stimulating cAMP-dependent PKA pathway (Soemitro et al. 1989; Torres et al. 1999), and leading to the phosphorylation of cAMP response element-binding protein (CREB) (Aleman et al. 1998; Haas and Pitot 1999; Merrill and Pitot 1985; Su et al. 1990). Conversely, when protein intake is inadequate, most AACE show low activities, which should contribute to the conservation of AA for protein synthesis and the maintenance of body protein (Soemitro et al. 1989).
Higher DP/DCH ratio led to higher hepatic expression of AACE in WT mice. Intriguingly, in the liver of Ppara-null mice, increased expression of these enzymes was observed, and higher serum glucagon concentration was noticed, independent of the amount of DP/DCH received, thus indicating higher AA catabolism. This was confirmed by the increase in the serum urea concentration and decreased body protein content observed in the Ppara-null mice. Furthermore, serum concentration of most AA was lower in the Ppara-null mice, except for that of BCAA, suggesting that PPARα does not regulate the catabolism of these AA. In addition, a genome-wide scan reveled that WT mice fed 50/33 % diet elevated expression of enzymes involved in AA metabolism, including AACE, except for those related to BCAA catabolism, in agreement with previous studies (Soemitro et al. 1989; Torres et al. 1998). This suggests that in WT mice fed a low or normal protein diet, PPARα may prevent the induction of AACE to preserve body protein.
Significant metabolic adaptations are necessary to adjust the use of energy substrates in Ppara-null mice. In these mice, there was an increase in circulating FFA, low glucose and a large increase in the ratio glucagon/insulin. This phenotype implies that extensive lipolysis from adipose tissue occurs. However, these FFA cannot be oxidized due to lack of PPARα (Costet et al. 1998), resulting in a large amount of lipid deposition in liver. The consumption of a 50/33 % diet attenuated this phenotype. Interestingly, when the mice were fed a 6/77 % diet, they showed a further increase in hepatic lipid droplets, suggesting that the excess of carbohydrates increases the lipogenic capacity in the liver. The lack of PPARα not only increased the amount of hepatic lipids, but also significantly increased the total amount of body fat, by 10–20 % compared with WT mice.
The availability of intermediates for gluconeogenesis partly depends on the flux of carbon precursors from AA and, therefore, is related to the rates at which gluconeogenic AA can be degraded by their specific enzymes. The homeostasis of blood glucose requires this process to be sensitive to the dietary and hormonal status as has been shown for many enzymes of AA catabolism (Pestana 1969). It was reported that PPARα plays a role in balancing glucose homeostasis by regulating the expression of glucokinase (Gck) as well as the pyruvate kinase liver and red blood cell (Pklr) (Oosterveer et al. 2009; Pan et al. 2000; Peeters and Baes 2010). Furthermore, the increase in the catabolism of gluconeogenic AA in Ppara-null mice was accompanied by an increase in the expression of Pck1 after consumption of the 50/33 % diet, which indicates that de novo glucose synthesis was increased. However, there was no effect on either blood glucose or the accumulation of hepatic glycogen. It is known that Ppara-null mice show a significant increase in the rate of whole-body and adipose tissue glucose utilization in the fasting state, associated with a large increase in solute carrier family 2 member 4 (GLUT4) expression in adipocytes that are not regulated by the fed-to-fast transition (Knauf et al. 2006). In addition, previous results have shown that FAO disorders lead to fasting hypoglycemia by uncompensated increased demand of peripheral glucose (Houten et al. 2013). Further kinetic studies are needed to assess the flux of AA carbon skeletons in the gluconeogenesis and glycogenesis pathways.
The functional analysis of metabolic pathways in livers of WT mice showed that the most important activated pathways where those associated with AA and fatty acid metabolism in fasting and refeeding. In contrast, in Ppara-null mice, there was a dramatic change in the pathways activated by diet involving apoptotic processes and cell proliferation and growth, in agreement with the histological findings. Differentially expressed genes associated with a functional network related to AA and lipid metabolism were identified, which revealed that PPARα regulates proteins involved in AA metabolism, and this may occur indirectly, mediated through HNF4α. Previous studies have reported the established ability of PPARα to down-regulate the expression of HNF4α (Hertz et al. 1995, 1996; Marrapodi and Chiang 2000; Shin et al. 2006) and demonstrated an association between HNF4α and the expression of some AACE (Nitsch et al. 1993; Odom et al. 2004; Shin et al. 2006).
In fact, our study indicates that PPARα regulates the expression of AACE genes through modulation of HNF4α trans-activation activity, as shown in the Sds promoter, rather than via direct down-regulation of AACE promoters. This is supported by previous evidence showing weak PPARα binding to the putative responsive elements in the promoter of AACE by EMSA analysis (Aleman et al. 2013b). In addition, our data showed that PPARα activated by its synthetic ligand Wy-14,643 was able to significantly reduce the HNF4α activity, leading in the attenuation of SDS expression. Taking together, these findings indicate that PPARα represents an important metabolic switch that modulates the use of energy substrates by up-regulating the expression of genes involved in FAO and reducing those related to AA oxidation, with the main aim of preserving body nitrogen.
Electronic supplementary material
Acknowledgments
We thank Rafael Hernández and Mónica Guevara from the Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (INCMNSZ) for animal care and Guillermo Ordaz from INCMNSZ for assistance in the handling of mice. This work is part of the Doctoral Thesis of A.V.C., who is student in the Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM). This work was supported by Consejo Nacional de Ciencia y Tecnología, Grant 54939 (to A.R.T.) and the Instituto Nacional de Medicina Genómica, Grant 16/2011/E (to A.V.C.).
Conflict of interest
Alejandra V. Contreras, Claudia Rangel-Escareño, Nimbe Torres, Gabriela Alemán-Escondrillas, Victor Ortiz, Lilia G. Noriega, Ivan Torre-Villalvazo, Omar Granados, Laura A. Velázquez-Villegas, Sandra Tobon-Cornejo, Diana González-Hirschfeld, Félix Recillas-Targa, Elizabeth Tejero-Barrera, Frank J. Gonzalez and Armando R. Tovar declare that they have no conflict of interest.
References
- Alberti KG, Johnston DG, Piniewska-Hulas M, Whittaker J. Disturbances of metabolic homeostasis is liver disease. Acta Med Port Suppl. 1981;(Suppl 2):35–47. [PubMed] [Google Scholar]
- Albrecht PP, et al. A species difference in the peroxisome proliferator-activated receptor alpha-dependent response to the developmental effects of perfluorooctanoic acid. Toxicol Sci. 2013;131:568–582. doi: 10.1093/toxsci/kfs318. [DOI] [PubMed] [Google Scholar]
- Aleman G, Torres N, Bourges H, Tovar AR. Regulation of histidase gene expression by glucagon, hydrocortisone and protein-free/high carbohydrate diet in the rat. Life Sci. 1998;63:1663–1672. doi: 10.1016/S0024-3205(98)00435-4. [DOI] [PubMed] [Google Scholar]
- Aleman G, et al. Hepatic amino acid-degrading enzyme expression is downregulated by natural and synthetic ligands of PPAR alpha in rats. J Nutr. 2013 doi: 10.3945/jn.113.176354. [DOI] [PubMed] [Google Scholar]
- Aleman G, et al. Hepatic amino acid-degrading enzyme expression is downregulated by natural and synthetic ligands of PPARalpha in rats. J Nutr. 2013;143:1211–1218. doi: 10.3945/jn.113.176354. [DOI] [PubMed] [Google Scholar]
- Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, Gonzalez FJ. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor alpha (PPARalpha) J Biol Chem. 1998;273:5678–5684. doi: 10.1074/jbc.273.10.5678. [DOI] [PubMed] [Google Scholar]
- Berry MN, Friend DS. High-yield preparation of isolated rat liver parenchymal cells: a biochemical and fine structural study. J Cell Biol. 1969;43:506–520. doi: 10.1083/jcb.43.3.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Carroll NV, Longley RW, Roe JH. The determination of glycogen in liver and muscle by use of anthrone reagent. J Biol Chem. 1956;220:583–593. [PubMed] [Google Scholar]
- Chondrogianni N, Gonos ES. Proteasome function determines cellular homeostasis and the rate of aging. In: Tavernarakis N, editor. Protein metabolism and homeostasis in aging. New York: Springer; 2010. pp. 38–46. [DOI] [PubMed] [Google Scholar]
- Contreras AV, Torres N, Tovar AR. PPAR-alpha as a key nutritional and environmental sensor for metabolic adaptation. Adv Nutr. 2013;4:439–452. doi: 10.3945/an.113.003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corton JC, et al. Mode of action framework analysis for receptor-mediated toxicity: the peroxisome proliferator-activated receptor alpha (PPARalpha) as a case study. Crit Rev Toxicol. 2014;44:1–49. doi: 10.3109/10408444.2013.835784. [DOI] [PubMed] [Google Scholar]
- Costet P, Legendre C, More J, Edgar A, Galtier P, Pineau T. Peroxisome proliferator-activated receptor alpha-isoform deficiency leads to progressive dyslipidemia with sexually dimorphic obesity and steatosis. J Biol Chem. 1998;273:29577–29585. doi: 10.1074/jbc.273.45.29577. [DOI] [PubMed] [Google Scholar]
- da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Delimaris I. Adverse effects associated with protein intake above the recommended dietary allowance for adults. ISRN Nutr. 2013;2013:126929. doi: 10.5402/2013/126929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromentin C, et al. Dietary proteins contribute little to glucose production, even under optimal gluconeogenic conditions in healthy humans. Diabetes. 2013;62:1435–1442. doi: 10.2337/db12-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas MJ, Pitot HC. Glucocorticoids stimulate CREB binding to a cyclic-AMP response element in the rat serine dehydratase gene. Arch Biochem Biophys. 1999;362:317–324. doi: 10.1006/abbi.1998.1044. [DOI] [PubMed] [Google Scholar]
- Harper AE. Genetic factors in nutrition. California: Academic Press Inc; 1984. Biological factors influencing the utilization of amino acids; pp. 243–267. [Google Scholar]
- Hertz R, Bishara-Shieban J, Bar-Tana J. Mode of action of peroxisome proliferators as hypolipidemic drugs. Suppression of apolipoprotein C-III. J Biol Chem. 1995;270:13470–13475. doi: 10.1074/jbc.270.22.13470. [DOI] [PubMed] [Google Scholar]
- Hertz R, Seckbach M, Zakin MM, Bar-Tana J. Transcriptional suppression of the transferrin gene by hypolipidemic peroxisome proliferators. J Biol Chem. 1996;271:218–224. doi: 10.1074/jbc.271.1.218. [DOI] [PubMed] [Google Scholar]
- Houten SM, et al. Impaired amino acid metabolism contributes to fasting-induced hypoglycemia in fatty acid oxidation defects. Hum Mol Genet. 2013 doi: 10.1093/hmg/ddt382. [DOI] [PubMed] [Google Scholar]
- Kaloyianni M, Freedland RA. Contribution of several amino acids and lactate to gluconeogenesis in hepatocytes isolated from rats fed various diets. J Nutr. 1990;120:116–122. doi: 10.1093/jn/120.1.116. [DOI] [PubMed] [Google Scholar]
- Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J Clin Invest. 1999;103:1489–1498. doi: 10.1172/JCI6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten S, Mandard S, Escher P, Gonzalez FJ, Tafuri S, Desvergne B, Wahli W. The peroxisome proliferator-activated receptor alpha regulates amino acid metabolism. FASEB J. 2001;15:1971–1978. doi: 10.1096/fj.01-0147com. [DOI] [PubMed] [Google Scholar]
- Knauf C, et al. Peroxisome proliferator-activated receptor-alpha-null mice have increased white adipose tissue glucose utilization, GLUT4, and fat mass: role in liver and brain. Endocrinology. 2006;147:4067–4078. doi: 10.1210/en.2005-1536. [DOI] [PubMed] [Google Scholar]
- Lane HW, Keith RE, Strahan S, White MT. The effect of diet, exercise and 7,12-dimethylbenz(a)anthracene on food intake, body composition and carcass energy levels in virgin female BALB/c mice. J Nutr. 1991;121:1876–1882. doi: 10.1093/jn/121.11.1876. [DOI] [PubMed] [Google Scholar]
- Lee SS, et al. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol. 1995;15:3012–3022. doi: 10.1128/mcb.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrapodi M, Chiang JY. Peroxisome proliferator-activated receptor alpha (PPARalpha) and agonist inhibit cholesterol 7alpha-hydroxylase gene (CYP7A1) transcription. J Lipid Res. 2000;41:514–520. [PubMed] [Google Scholar]
- Menezes AL, et al. A low-protein, high-carbohydrate diet increases de novo fatty acid synthesis from glycerol and glycerokinase content in the liver of growing rats. Nutr Res. 2013;33:494–502. doi: 10.1016/j.nutres.2013.04.010. [DOI] [PubMed] [Google Scholar]
- Merrill MJ, Pitot HC. Regulation of ornithine aminotransferase by cyclic AMP and glucose in primary cultures of adult rat hepatocytes. Arch Biochem Biophys. 1985;237:373–385. doi: 10.1016/0003-9861(85)90289-9. [DOI] [PubMed] [Google Scholar]
- Motojima K, Passilly P, Peters JM, Gonzalez FJ, Latruffe N. Expression of putative fatty acid transporter genes are regulated by peroxisome proliferator-activated receptor alpha and gamma activators in a tissue- and inducer-specific manner. J Biol Chem. 1998;273:16710–16714. doi: 10.1074/jbc.273.27.16710. [DOI] [PubMed] [Google Scholar]
- Nitsch D, Boshart M, Schutz G. Activation of the tyrosine aminotransferase gene is dependent on synergy between liver-specific and hormone-responsive elements. Proc Natl Acad Sci USA. 1993;90:5479–5483. doi: 10.1073/pnas.90.12.5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odom DT, et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303:1378–1381. doi: 10.1126/science.1089769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oosterveer MH, et al. Fenofibrate simultaneously induces hepatic fatty acid oxidation, synthesis, and elongation in mice. J Biol Chem. 2009;284:34036–34044. doi: 10.1074/jbc.M109.051052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan DA, Mater MK, Thelen AP, Peters JM, Gonzalez FJ, Jump DB. Evidence against the peroxisome proliferator-activated receptor alpha (PPARalpha) as the mediator for polyunsaturated fatty acid suppression of hepatic L-pyruvate kinase gene transcription. J Lipid Res. 2000;41:742–751. [PubMed] [Google Scholar]
- Patsouris D, Reddy JK, Muller M, Kersten S. Peroxisome proliferator-activated receptor alpha mediates the effects of high-fat diet on hepatic gene expression. Endocrinology. 2006;147:1508–1516. doi: 10.1210/en.2005-1132. [DOI] [PubMed] [Google Scholar]
- Peeters A, Baes M. Role of PPARalpha in hepatic carbohydrate metabolism. PPAR Res. 2010 doi: 10.1155/2010/572405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestana A. Dietary and hormonal control of enzymes of amino acid catabolism in liver. Eur J Biochem/FEBS. 1969;11:400–404. doi: 10.1111/j.1432-1033.1969.tb00787.x. [DOI] [PubMed] [Google Scholar]
- Peters JM, Aoyama T, Cattley RC, Nobumitsu U, Hashimoto T, Gonzalez FJ. Role of peroxisome proliferator-activated receptor alpha in altered cell cycle regulation in mouse liver. Carcinogenesis. 1998;19:1989–1994. doi: 10.1093/carcin/19.11.1989. [DOI] [PubMed] [Google Scholar]
- Phinney SD, Fisler JS, Tang AB, Warden CH. Liver fatty acid composition correlates with body fat and sex in a multigenic mouse model of obesity. Am J Clin Nutr. 1994;60:61–67. doi: 10.1093/ajcn/60.1.61. [DOI] [PubMed] [Google Scholar]
- Reeves PG. Components of the AIN-93 diets as improvements in the AIN-76A diet. J Nutr. 1997;127:838S–841S. doi: 10.1093/jn/127.5.838S. [DOI] [PubMed] [Google Scholar]
- Reiner A, Yekutieli D, Benjamini Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics. 2003;19:368–375. doi: 10.1093/bioinformatics/btf877. [DOI] [PubMed] [Google Scholar]
- Shin M, Kim I, Inoue Y, Kimura S, Gonzalez FJ. Regulation of mouse hepatic alpha-amino-beta-carboxymuconate-epsilon-semialdehyde decarboxylase, a key enzyme in the tryptophan-nicotinamide adenine dinucleotide pathway, by hepatocyte nuclear factor 4alpha and peroxisome proliferator-activated receptor alpha. Mol Pharmacol. 2006;70:1281–1290. doi: 10.1124/mol.106.026294. [DOI] [PubMed] [Google Scholar]
- Smyth GK (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3:Article3 doi:10.2202/1544-6115.1027 [DOI] [PubMed]
- Smyth G. Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and computational biology solutions using R and bioconductor. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- Soemitro S, Block KP, Crowell PL, Harper AE. Activities of branched-chain amino acid–degrading enzymes in liver from rats fed different dietary levels of protein. J Nutr. 1989;119:1203–1212. doi: 10.1093/jn/119.8.1203. [DOI] [PubMed] [Google Scholar]
- Stepien M, Gaudichon C, Fromentin G, Even P, Tome D, Azzout-Marniche D. Increasing protein at the expense of carbohydrate in the diet down-regulates glucose utilization as glucose sparing effect in rats. PLos One. 2011;6:e14664. doi: 10.1371/journal.pone.0014664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Kanamoto R, Miller DA, Ogawa H, Pitot HC. Regulation of the expression of the serine dehydratase gene in the kidney and liver of the rat. Biochem Biophys Res Commun. 1990;170:892–899. doi: 10.1016/0006-291X(90)92175-Y. [DOI] [PubMed] [Google Scholar]
- Torres N, Lopez G, De Santiago S, Hutson SM, Tovar AR. Dietary protein level regulates expression of the mitochondrial branched-chain aminotransferase in rats. J Nutr. 1998;128:1368–1375. doi: 10.1093/jn/128.8.1368. [DOI] [PubMed] [Google Scholar]
- Torres N, Beristain L, Bourges H, Tovar AR. Histidine-imbalanced diets stimulate hepatic histidase gene expression in rats. J Nutr. 1999;129:1979–1983. doi: 10.1093/jn/129.11.1979. [DOI] [PubMed] [Google Scholar]
- Tovar AR, Ascencio C, Torres N. Soy protein, casein, and zein regulate histidase gene expression by modulating serum glucagon. Am J Physiol Endocrinol Metab. 2002;283:E1016–E1022. doi: 10.1152/ajpendo.00398.2001. [DOI] [PubMed] [Google Scholar]
- Young VR, Marchini JS. Mechanisms and nutritional significance of metabolic responses to altered intakes of protein and amino acids, with reference to nutritional adaptation in humans. Am J Clin Nutr. 1990;51:270–289. doi: 10.1093/ajcn/51.2.270. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.