

Abstract
Background
Corynebacterium pseudotuberculosis biovar ovis is a facultative intracellular pathogen, and the etiological agent of caseous lymphadenitis in small ruminants. During the infection process, the bacterium is subjected to several stress conditions, including nitrosative stress, which is caused by nitric oxide (NO). In silico analysis of the genome of C. pseudotuberculosis ovis 1002 predicted several genes that could influence the resistance of this pathogen to nitrosative stress. Here, we applied high-throughput proteomics using high definition mass spectrometry to characterize the functional genome of C. pseudotuberculosis ovis 1002 in the presence of NO-donor Diethylenetriamine/nitric oxide adduct (DETA/NO), with the aim of identifying proteins involved in nitrosative stress resistance.
Results
We characterized 835 proteins, representing approximately 41% of the predicted proteome of C. pseudotuberculosis ovis 1002, following exposure to nitrosative stress. In total, 102 proteins were exclusive to the proteome of DETA/NO-induced cells, and a further 58 proteins were differentially regulated between the DETA/NO and control conditions. An interactomic analysis of the differential proteome of C. pseudotuberculosis in response to nitrosative stress was also performed. Our proteomic data set suggested the activation of both a general stress response and a specific nitrosative stress response, as well as changes in proteins involved in cellular metabolism, detoxification, transcriptional regulation, and DNA synthesis and repair.
Conclusions
Our proteomic analysis validated previously-determined in silico data for C. pseudotuberculosis ovis 1002. In addition, proteomic screening performed in the presence of NO enabled the identification of a set of factors that can influence the resistance and survival of C. pseudotuberculosis during exposure to nitrosative stress.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-1065) contains supplementary material, which is available to authorized users.
Keywords: Corynebacterium pseudotuberculosis, Caseous lymphadenitis, Proteomics, Label-free proteomics, Nitrosative stress, Nitric oxide
Background
Corynebacterium pseudotuberculosis is a Gram-positive, facultative, intracellular pathogen belonging to the Corynebacterium, Mycobacterium, Nocardia, or CMN, group. This group belongs to the phylum Actinobacteria. The defining characteristics of the CMN group are a specific cell wall organization, consisting of peptidoglycan, arabinogalactan, and mycolic acids, and a high chromosomal G + C content [1]. C. pseudotuberculosis ovis is the etiological agent of the chronic infectious disease caseous lymphadenitis, which affects small ruminants worldwide. As a result, C. pseudotuberculosis ovis is responsible for significant economic losses in the goat and sheep industries, mainly stemming from decreased meat, wool, and milk production, reproductive disorders, and carcass contamination [1, 2]. Bacterial factors that contribute to the virulence of C. pseudotuberculosis include phospholipase D [3], toxic cell wall lipids [4], and the iron transporter fagABC complex [5].
In silico analysis of the genome of C. pseudotuberculosis ovis 1002 [6], as well as the pan-genome analysis of 15 other strains of C. pseudotuberculosis[7], identified genes involved in the response of this pathogen to different types of stress. Recently, the functional genome of C. pseudotuberculosis ovis 1002 was evaluated at the transcriptional level following exposure to different types of abiotic stress, including heat, osmotic, and acid stresses [8]. This allowed the characterization of several genes involved in distinct biological processes that favor the survival of the pathogen under the given stress condition.
However, during the infection process, C. pseudotuberculosis encounters nitrosative stress, caused by nitric oxide (NO), in the macrophage intracellular environment. A reactive nitrogen species (RNS) found in mammalian systems, NO is produced from L-arginine by NO synthases (NOS), and is present in three isoforms: endothelial NOS, neuronal NOS, involved in blood pressure control and neural signaling, and inducible NOS, associated with host defenses [9, 10]. The NO produced during bacterial infection has antimicrobial properties, killing pathogens by causing damage to DNA, RNA, and proteins [11]. However, several pathogens contain pathways that allow bacterial survival under nitrosative stress conditions, including NO-sensitive transcriptional regulators [12], DNA and protein repair systems [13], and antioxidant systems [14].
Currently, little is known about the factors involved in the resistance of C. pseudotuberculosis to nitrosative stress. Pacheco et al. [15] showed that the alternative sigma (σ) factor, σE, plays a role in the survival of C. pseudotuberculosis in the presence of RNS. A σE null strain showed increased susceptibility to nitric oxide compared with the wild-type, and, in an in vivo assay, was unable to persist in mice. However, in iNOS-deficient mice, the mutant strain maintained its virulence [15]. In the same study, the extracellular proteome of C. pseudotuberculosis was analyzed in response to nitrosative stress, allowing the characterization of proteins that contribute to the adaptive processes of the pathogen in this environment [15].
To complement the results obtained in previous studies, and to identify factors involved in the survival of C. pseudotuberculosis under nitrosative stress conditions, we applied high-throughput proteomics using an liquid chromatograph high definition mass spectrometry (LC-HDMSE) (data-independent acquisition, in ion mobility mode) approach to evaluate the global expression of the functional genome of C. pseudotuberculosis ovis 1002 at the protein level under nitrosative stress conditions.
Methods
Bacterial strain and growth conditions
C. pseudotuberculosis biovar ovis strain 1002, isolated from a goat, was maintained in brain heart infusion broth (BHI; HiMedia Laboratories Pvt. Ltd., Mumbai, India) at 37°C. For stress-resistance assays, strain 1002 was cultivated in a chemically-defined medium (CDM), containing Na2HPO4.7H2O (12.93 g/l), KH2PO4 (2.55 g/l), NH4Cl (1 g/l), MgSO4.7H2O (0.20 g/l), CaCl2 (0.02 g/l), 0.05% (v/v) Tween 80, 4% (v/v) MEM vitamin solution (Invitrogen, Gaithersburg, MD, USA), 1% (v/v) MEM amino acid solution (Invitrogen), 1% (v/v) MEM non-essential amino acid solution (Invitrogen), and 1.2% (w/v) glucose, at 37°C [16].
Nitric oxide assay and preparation of whole bacterial lysates
Diethylenetriamine/nitric oxide adduct (DETA/NO) resistance of C. pseudotuberculosis was characterized as previously described [15]. When strain 1002 reached exponential growth phase (OD600 = 0.6) in the chemically-defined medium, the culture was divided into two aliquots (control condition, strain 1002_Ct; NO exposure, strain 1002_DETA/NO), and DETA/NO was added to the appropriate aliquot to a final concentration of 0.5 mM. The growth of strain 1002 in the presence of DETA/NO was then evaluated for 10 h. For proteomic analysis, protein was extracted after 1 h of exposure to DETA/NO. Both the control and DETA/NO cultures were centrifuged at 4,000 × g for 10 min at 4°C. The cell pellets were washed in phosphate buffered saline and then resuspended in 1 ml of lysis buffer (7 M urea, 2 M thiourea, 4% (w/v) CHAPS, and 1 M dithiothreitol (DTT)). The cells were then sonicated using five 1-min cycles on ice. The resulting lysates were centrifuged at 14,000 × g for 30 min at 4°C. The extracted proteins were then submitted to centrifugation at 13,000 × g for 10 min using a spin column with a threshold of 10 kDa (Millipore, Billerica, USA). Proteins were denatured with (0.1% (w/v) RapiGEST SF surfactant at 60°C for 15 min (Waters, Milford, CA, USA), reduced using 10 mM DTT for 30 min at 60°C, and alkylated with 10 mM iodoacetamide in a dark chamber at 25°C for 30 min. Next, the proteins were enzymatically digested with 1:50 (w/w) trypsin at 37°C for 16 hours (sequencing grade modified trypsin; Promega, Madison, WI, USA). The digestion process was stopped by adding 10 μl of 5% (v/v) Trifluoroacetic acid (TFA) (Fluka, Buchs, Germany). Glycogen phosphorylase was added to the digests to a final concentration of 20 fmol/μl as an internal standard for normalization prior to each replicate injection. Analysis was carried out using a two-dimensional reversed phase (2D RP-RP) nanoUPLC-MS (Nano Ultra Performance Liquid Chromatography) approach, using multiplexed HDMSE label-free quantitation as described previously [17].
LC-HDMSE analysis and data processing
Qualitative and quantitative by 2D nanoUPLC tandem nanoESI-HDMSE (Nano Electrospray High Definition Mass Spectrometry) experiments were conducted using a 1-h reversed phase (RP) acetonitrile (0.1% v/v formic acid) gradient (7–40% (v/v)) at 500 nl/min on a nanoACQUITY UPLC 2D RP × RP Technology system [18]. A nanoACQUITY UPLC High Strength Silica (HSS) T3 1.8 μm 75 μm × 15 cm column (pH 3) was used in conjunction with a RP XBridge BEH130 C18 5 μm 300 μm × 50 mm nanoflow column (pH 10). Typical on-column sample loads were 250 ng of the total protein digests for each of the five fractions (250 ng/fraction/load). For all measurements, the mass spectrometer was operated in resolution mode, with a typical effective m/z conjoined ion-mobility resolving power of at least 1.5 M FWHM, an ion mobility cell filled with nitrogen gas, and a cross-section resolving power at least 40 Ω/ΔΩ. All analyses were performed using nano-electrospray ionization in the positive ion mode nanoESI (+), and a NanoLockSpray (Waters) ionization source. The lock mass channel was sampled every 30 s. The mass spectrometer was calibrated with a MS/MS spectrum of [Glu1]-fibrinopeptide B (Glu-Fib) human solution (100 fmol/μl) delivered though the reference sprayer of the NanoLockSpray source. The double-charged ion ([M + 2H]2+ = 785.8426) was used for initial single-point calibration, and MS/MS fragment ions of Glu-Fib were used to obtain the final instrument calibration. Multiplexed data-independent scanning with added specificity and selectivity of a non-linear “T-wave” ion mobility (HDMSE) experiments were performed using a Synapt G2-S HDMS mass spectrometer (Waters). The mass spectrometer was set to switch automatically between standard MS (3 eV) and elevated collision energies HDMSE (19–45 eV) applied to the transfer “T-wave” collision-induced dissociation cell with argon gas. The trap collision cell was adjusted for 1 eV using a millisecond scan time adjusted based on the linear velocity of the chromatography peak delivered though nanoACQUITY UPLC, to obtain a minimum of 20 scan points for each single peak at both low-energy and high-energy transmission, followed by an orthogonal acceleration time-of-flight from 50–2000 m/z. The radio frequency (RF) offset (MS profile) was adjusted so that the nanoUPLC-HDMSE data were effectively acquired from an m/z range of 400–2000, which ensured that any masses observed in the high energy spectra of less than 400 m/z arose from dissociations in the collision cell.
Data processing
Protein identification and quantitative data packaging were generated using dedicated algorithms [19, 20], and by searching against a C. pseudotuberculosis database with default parameters for ion accounting [21]. The databases were reversed “on-the fly” during the database query searches, and appended to the original database to assess the false positive rate of identification. For proper processing of spectra and database searching conditions, ProteinLynxGlobalServer v.2.5.2 (PLGS) with IdentityE and ExpressionE informatics v.2.5.2 (Waters) were used. UniProtKB (release 2013_01) with manually-reviewed annotations was also used, and the search conditions were based on taxonomy (C. pseudotuberculosis), maximum missed cleavages by trypsin allowed up to one, and variable carbamidomethyl, acetyl N-terminal, phosphoryl, and oxidation (M) modifications [21, 22]. The IdentityE algorithm with Hi3 methodology was used for protein quantitation. The search threshold for accepting each individual spectrum was set to the default value, with a false-positive value of 4%. Biological variability was addressed by analyzing each culture three times. Normalization was performed using the ExpressionE tool with a housekeeping protein that showed no significant difference in abundance across all injections. The proteins obtained were organized by the PLGS ExpressionE tool algorithm into a statistically significant list corresponding to increased and decreased regulation ratios among the different groups. The quantitation values were averaged over all of the samples, and the quoted standard deviations at p ≤ 0.05 in the ExpressionE software refer to the differences between biological replicates. Only proteins with a differential expression log2 ratio between the two conditions greater than or equal to 1.2 were considered [23].
Bioinformatics analysis
The identified proteins were analyzed using the prediction tools SurfG+ v1.0 [24], to predict sub-cellular localization, and Blast2GO, to predict gene ontology functional annotations [25]. The PIPS software predicted proteins present in pathogenicity islands [26]. The protein-protein interaction network was constructed using interolog mapping methodology and metrics according to Rezende et al. [27]. A preview of the interaction network was generated using Cytoscape version 2.8.3 [28], with a spring-embedded layout. CMRegNet was used to predict gene regulatory networks [29].
Results
Effects of nitric oxide on the growth of C. pseudotuberculosis
In this study, we examined the exponential growth of C. pseudotuberculosis strain 1002 under nitrosative stress. The growth and cell viability of strain 1002 was monitored for 10 h with and without DETA/NO supplementation (Figure 1). The control culture reached stationary phase by 5 h post-inoculation, while the culture containing DETA/NO did not reach stationary phase until approximately 10 h post-inoculation. However, these results showed that although DETA/NO (0.5 mM) affected the growth rate, C. pseudotuberculosis likely contains factors that promote survival in the presence of RNS.
Figure 1.
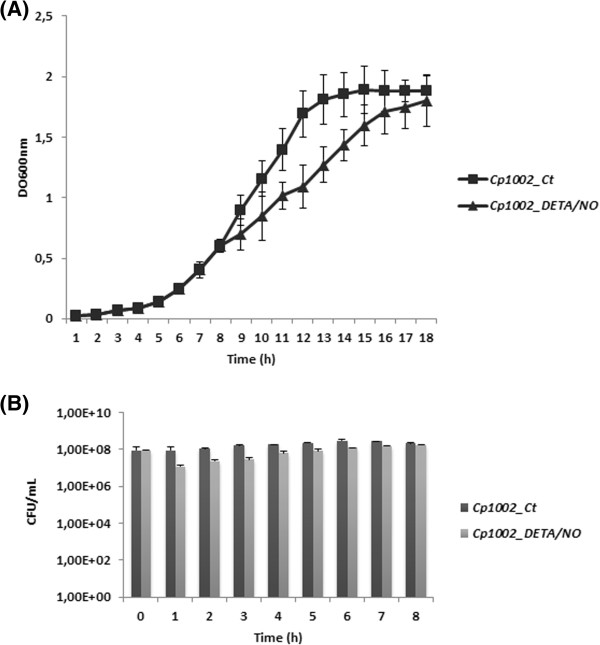
Growth and survival profile of C. pseudotuberculosis during NO exposure. (A) Growth of C. pseudotuberculosis after 10 h exposure to 0.5 mM DETA/NO. (B) Survival of C. pseudotuberculosis evaluated by colony forming units. The results shown in A and B represent an average of three independent experiments.
Label-free proteomic analysis of C. pseudotuberculosisgrown under nitrosative stress conditions
Total proteome digests from three biological replicates of each individual condition were subjected to LC/MSE. In total, we identified more than 31,000 peptides, with a normal distribution of 10 ppm error of the total identified peptides. Peptides as source fragments, peptides with a charge state of at least [M + 2H]2+, and the absence of decoys were factors considered to increase data quality. A combined total of 2,063 proteins were present in at least two of the three biological replicates for the two conditions tested, with an average of 15 peptides per protein, and a false discovery rate (FDR) of 0% when decoy detection was set at agreement of two out of three replicates. The proteins referred to as exclusive to one condition or another was only identified in one condition within the detection limits of the experiment (LOD). The dynamic range of the quantified proteins is about 3 logs, and proteins unique to one condition or another were only observed above the LOD of the experiment, which was determined by the sample normalization prior to injection. Therefore, in our study, all samples were normalized using “scouting runs” taking into account the stoichiometry between the intensity and molarity proportion prior to the replicate runs per condition. The dynamic range was similar for each sample, and the total amount of sample used in fmol was nearly the same. We generate a graph of protein amounts of the identified proteins from all samples against protein ranks (Figure 2A).
Figure 2.
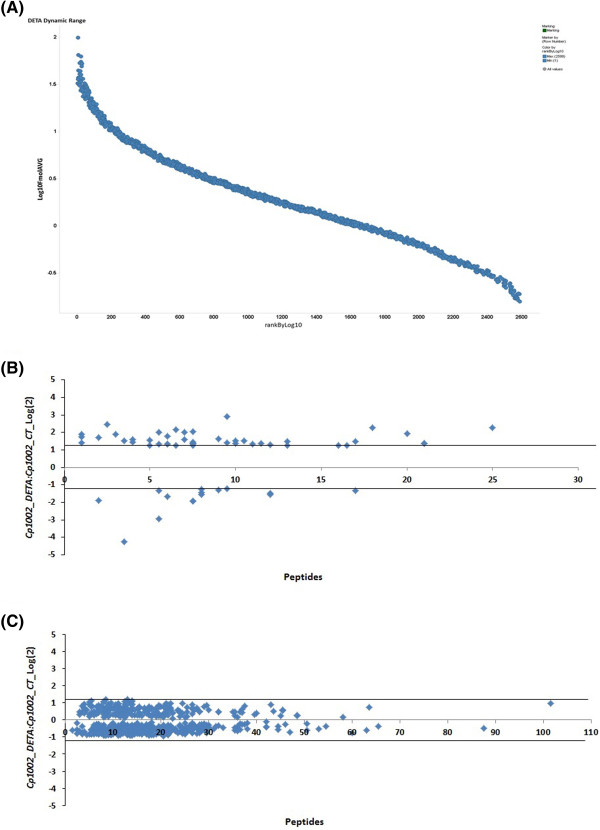
2D nanoUPLC HDMSE analysis showing: (A) Dynamic range of the method based on protein abundance estimates, data points derived from LC-HDMS E analysis. (B and C) Proteins that were significantly differentially-regulated during NO exposure. The distribution of identified proteins with p < 0.05, and differentially-regulated proteins with an I:C log2 ratio < 1.2 in relation to the number of peptides identified for each protein. (B) Proteins with p < 0.05 and an I:C log2 ratio < 1.2. (C) Proteins with p < 0.05 and an I:C log2 ratio > 1.2.
After, analysis by PLGS v2.5.2 software, the 2,063 proteins originally identified in two out of three replicates were narrowed down to 699 proteins with p ≤ 0.05. Among these proteins, 44 were up-regulated in the presence of DETA/NO, while 14 proteins were down-regulated (Table 1, Figure 2B and C). The remaining 641 proteins with p ≤ 0.05 and log2 < 1.2 that were common to the two treatments are summarized in Additional file 1. In addition to the 699 identified proteins that were present under both control and stress conditions, 34 proteins were exclusively expressed under the control conditions, and 102 proteins were exclusively expressed in response to DETA/NO stress (Additional files 2 and 3). Thus, our final list of proteins is composed of 835 proteins from C. pseudotuberculosis.
Table 1.
Proteins identified as differentially-expressed following exposure to nitrosative stress
| Uniprot access | Proteins | Score | Peptides | log 2 DETA:CT (a) | p -value (a) | Subcellular localization (c) | Gene name | Genome (b) |
|---|---|---|---|---|---|---|---|---|
| Transport | ||||||||
| F9Y2Z3_CORP1 | Cell wall channel | 5321.88 | 4 | 1.42 | 1 | CYT | porH | Shared |
| Cell division | ||||||||
| D9Q7G2_CORP1 | Hypothetical protein | 2417.8 | 21 | 1.34 | 1 | CYT | Cp1002_0716 | Core |
| DNA synthesis and repair | ||||||||
| D9Q5V6_CORP1 | Nucleoid-associated protein | 2327.08 | 5 | 1.52 | 1 | CYT | ybaB | Core |
| D9Q923_CORP1 | Methylated-DNA-protein-cysteine methyltransferase | 6332.83 | 8 | 1.22 | 1 | CYT | ada | Core |
| D9Q4P0_CORP1 | 7,8-dihydro-8-oxoguanine-triphosphatase | 1640.23 | 8 | -1.97 | 0 | CYT | mutT | Core |
| Transcription | ||||||||
| D9Q8W2_CORP1 | LexA repressor | 800.31 | 6 | -1.37 | 0.04 | CYT | lexA | Shared |
| D9Q5L4_CORP1 | ECF family sigma factor k | 364.82 | 8 | -1.58 | 0 | CYT | sigK | Core |
| Translation | ||||||||
| D9Q753_CORP1 | Fkbp-type peptidyl-prolyl cis-trans isomerase | 7113.34 | 3 | 2.43 | 1 | CYT | fkbP | Core |
| D9Q830_CORP1 | 50S ribosomal protein L35 | 2271.66 | 1 | 1.36 | 1 | CYT | rpmI | Core |
| D9Q7W1_CORP1 | Aspartyl glutamyl-tRNA amidotransferase subunit C | 3100.8 | 7 | 1.24 | 0.99 | CYT | gatC | Core |
| D9Q582_CORP1 | 50S ribosomal protein L9 | 41082.46 | 10 | -1.25 | 0 | CYT | rplI | |
| D9Q6H6_CORP1 | 30S ribosomal protein S8 | 45333.23 | 9 | -1.34 | 0 | CYT | rpsH | Core |
| Cell communication | ||||||||
| D9Q559_CORP1 | Hypothetical protein | 1402.27 | 6 | 1.99 | 1 | PSE | Cp1002_2005 | Core |
| D9Q5U9_CORP1 | Thermosensitive gluconokinase | 2068.35 | 7 | 1.96 | 0.99 | CYT | gntK | Core |
| D9Q668_CORP1 | Sensory transduction protein RegX3 | 2540.92 | 13 | 1.45 | 1 | CYT | regX3 | Core |
| Detoxification | ||||||||
| D9Q7U6_CORP1 | Thioredoxin | 1835.7 | 11 | 1.50 | 1 | CYT | trxA | Core |
| D9Q4E5_CORP1 | Glutathione peroxidase | 1426.27 | 10 | 1.47 | 1 | CYT | Cp1002_1731 | Core |
| D9Q5T5_CORP1 | Glyoxalase bleomycin resistance protein dihydroxybiphenyl dioxygenase | 2417.77 | 11 | 1.28 | 1 | CYT | Cp1002_0124 | Shared |
| D9Q5N2_CORP1 | NADH dehydrogenase | 7030.94 | 12 | 1.25 | 1 | CYT | noxC | Shared |
| D9Q680_CORP1 | Glutaredoxin-like domain protein | 292.69 | 2 | -1.91 | 0 | CYT | Cp1002_0272 | Core |
| Glycolysis pathways | ||||||||
| D9Q5B6_CORP1 | N-Acetylglucosamine kinase | 228.69 | 6 | 1.74 | 0.98 | CYT | nanK | Core |
| D9Q4U9_CORP1 | Alcohol dehydrogenase | 236.02 | 17 | 1.22 | 1 | CYT | adhA | Shared |
| Iron-sulfur clusters | ||||||||
| D9Q7L6_CORP1 | Ferredoxin | 36927.57 | 7 | 2.10 | 1 | CYT | fdxA | Core |
| Antibiotic resistance | ||||||||
| D9Q827_CORP1 | Metallo-beta-lactamase superfamily protein | 657.33 | 6 | -2.95 | 0 | CYT | Cp1002_0937 | Core |
| Amino acid metabolism | ||||||||
| D9Q622_CORP1 | Phosphoserine phosphatase | 949.15 | 9 | 1.58 | 0.99 | PSE | serB | Core |
| D9Q4N1_CORP1 | Carboxylate-amine ligase | 205.54 | 16 | 1.24 | 1 | CYT | Cp1002_1819 | Core |
| D9Q6H4_CORP1 | L-serine dehydratase I | 284.11 | 17 | -1.37 | 0 | MEM | sdaA | Core |
| Lipid metabolism | ||||||||
| D9Q520_CORP1 | Glycerophosphoryl diester phosphodiesterase | 2417.8 | 21 | 1.34 | 1 | PSE | glpQ | Core |
| Oxidative phosphorylation | ||||||||
| D9Q8I5_CORP1 | Cytochrome aa3 controlling protein | 676.2 | 6 | 1.28 | 1 | MEM | Cp1002_1095 | Core |
| Specific metabolic pathways | ||||||||
| D9Q5M9_CORP1 | Inositol-3-phosphate synthase | 7473.38 | 18 | 2.25 | 1 | CYT | ino1 | Core |
| D9Q721_CORP1 | Hypothetical protein | 4602.9 | 17 | 1.44 | 1 | SEC | Cp1002_0573 | Core |
| D9Q689_CORP1 | 3-Hydroxyisobutyrate dehydrogenase | 2137.24 | 12 | 1.34 | 1 | CYT | mmsB | Core |
| D9Q4X1_CORP1 | Urease accessory protein UreG | 1532.39 | 12 | -1.6 | 0 | CYT | ureG | Core |
| Nucleotide metabolism | ||||||||
| D9Q4S2_CORP1 | Orotate phosphoribosyltransferase | 2618.52 | 8 | -1.26 | 0 | CYT | pyrE | Core |
| Unknown function | ||||||||
| D9Q6Y9_CORP1 | Hypothetical protein | 491.89 | 10 | 2.87 | 1 | CYT | Cp1002_0540 | Core |
| D9Q6C7_CORP1 | Hypothetical protein | 689.6 | 25 | 2.25 | 1 | PSE | Cp1002_0320 | Core |
| D9Q3P3_CORP1 | Hypothetical protein | 5703.38 | 3 | 1.87 | 1 | CYT | Cp1002_1474 | Core |
| D9Q5V4_CORP1 | Hypothetical protein | 994.52 | 1 | 1.7 | 1 | CYT | Cp1002_0143 | Core |
| D9Q610_CORP1 | Hypothetical protein | 27217.36 | 2 | 1.67 | 1 | CYT | Cp1002_0202 | Core |
| D9Q8D8_CORP1 | Hypothetical protein | 2324.12 | 7 | 1.57 | 0.98 | CYT | Cp1002_1048 | Shared |
| D9Q6W1_CORP1 | Hypothetical protein | 9303.91 | 4 | 1.54 | 1 | CYT | Cp1002_0512 | Core |
| D9Q6V5_CORP1 | Hypothetical protein | 1346.2 | 4 | 1.5 | 0.99 | CYT | Cp1002_0506 | Core |
| D9Q5R7_CORP1 | Hypothetical protein | 2090.7 | 8 | 1.42 | 1 | CYT | Cp1002_0105 | Core |
| D9Q917_CORP1 | Hypothetical protein | 555.89 | 10 | 1.37 | 1 | PSE | Cp1002_1281 | Core |
| D9Q3P5_CORP1 | Hypothetical protein | 1121.7 | 6 | 1.29 | 1 | SEC | Cp1002_1476 | Core |
| D9Q7U5_CORP1 | Hypothetical protein | 517.06 | 8 | 1.28 | 1 | CYT | Cp1002_0852 | Core |
| D9Q7L1_CORP1 | Hypothetical protein | 15693.97 | 6 | 1.28 | 1 | SEC | Cp1002_0766 | Core |
| D9Q3P6_CORP1 | Hypothetical protein | 1729.59 | 5 | 1.22 | 1 | CYT | Cp1002_1477 | Core |
| D9Q6Z7_CORP1 | Hypothetical protein | 1835.7 | 13 | 1.22 | 1 | CYT | Cp1002_0548 | Core |
| D9Q8V8_CORP1 | Hypothetical protein | 293.23 | 8 | -1.48 | 0 | Cp1002_1221 | Core | |
| D9Q6C8_CORP1 | Hypothetical protein | 413.31 | 12 | -1.52 | 0 | PSE | Cp1002_0321 | Core |
| D9Q5H0_CORP1 | Hypothetical protein | 12376.2 | 6 | -1.71 | 0 | CYT | Cp1002_0007 | Core |
| D9Q4D5_CORP1 | Hypothetical protein | 10161.64 | 4 | -4.29 | 0 | CYT | Cp1002_1721 | Shared |
| Others | ||||||||
| D9Q5N5_CORP1 | Iron-regulated MEM protein | 992.54 | 8 | 2.01 | 0 | PSE | piuB | Core |
| D9Q922_CORP1 | CobW/HypB/UreG, nucleotide-binding | 1771.22 | 20 | 1.88 | 1 | CYT | Cp1002_1286 | Core |
| D9Q8C4_CORP1 | Prokaryotic ubiquitin-like protein Pup | 2194.86 | 1 | 1.84 | 1 | CYT | pup | Core |
| D9Q7B8_CORP1 | Ribosomal-protein-alanine n-acetyltransferase | 2791.1 | 10 | 1.34 | 1 | CYT | rimJ | Shared |
| D9Q7K9_CORP1 | Arsenate reductase | 5147.54 | 8 | 1.32 | 1 | CYT | arsC | Core |
(a) Ratio values to: strain 1002_DETA/NO:strain 1002_Ct, Log(2) Ratio > 1.5, p > 0.95 = up-regulation, p < 0.05 = down-regulation.
(b) Core-genome analysis of 15 strains of C. pseudotuberculosis: shared = present in two or more strains; core = present in 15 strains of C. pseudotuberculosis.
(c) CYT =cytoplasmic, MEM = membrane, PSE = potentially surface-exposed, SEC = secreted.
In silicoanalysis of LC-HDMSEdata
The 835 proteins were then analyzed using the SurfG+ tool to predict sub-cellular localization. According with SurfG+, our data set included approximately 41% of the predicted proteome of strain 1002 (Figure 3A). In addition, we characterized proteins belonging to the following cell fractions: cytoplasmic (CYT) (668 proteins), membrane (MEM) (59 proteins), potentially surface-exposed (PSE) (69 proteins), and secreted (SEC) (39 proteins) (Figure 3B).
Figure 3.
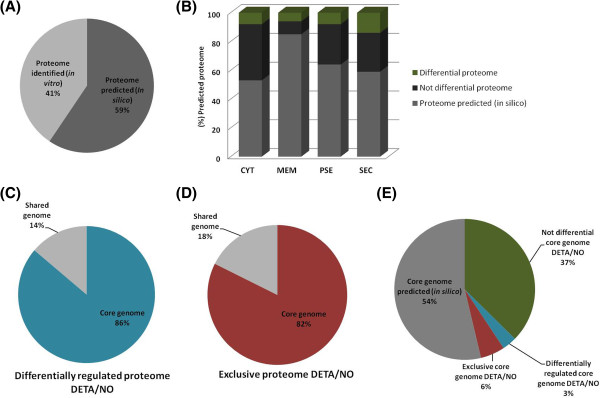
Correlation of in silico predicted data with proteome results. (A) Percentage of coverage of the C. pseudotuberculosis 1002 in silico proteome. (B) Prediction of the subcellular localization of the proteins identified by LC/MS. (C) Analysis of the differentially-regulated proteins of cells exposed to DETA/NO in relation to the core genome of C. pseudotuberculosis (shared genome: present in only two strains; core genome: present in 15 strains of C. pseudotuberculosis). (D) Analysis of the exclusive proteome of cells exposed to DEA/NO in relation to the core-genome of C. pseudotuberculosis (shared genome: present in only two strains; core genome: present in 15 strains of C. pseudotuberculosis). (E) Percent coverage of the core-genome of C. pseudotuberculosis in relation to the characterized proteome in vitro.
To evaluate whether the proteins identified in our proteomic analysis could represent a protein set expressed by C. pseudotuberculosis during exposure to nitrosative stress, we correlated our proteomic data with the predicted core-genomes of 15 C. pseudotuberculosis strains [7]. Of the open reading frames (ORFs) coding for the differentially-regulated proteins and exclusive proteome of DETA/NO-exposed cells, 86% (50/58 proteins) and 82% (84/102 proteins) were identified, respectively, in the core-genome of C. pseudotuberculosis (Figure 3C and D). In addition, of the 835 total proteins identified from the proteome of strain 1002 following exposure to nitrosative stress, 83% (696 proteins) of the ORFs coding for these proteins were present in the core-genome of C. pseudotuberculosis, this result correspond approximately 46% of the predicted core-genome of C. pseudotuberculosis (Figure 3E).
Functional classification of the proteome of C. pseudotuberculosisexpressed under exposure to nitrosative stress
The strain 1002 proteome was functionally classified using the Blast2Go tool [24]. A large proportion of the differentially-regulated proteins and those exclusive to one condition were identified as hypothetical proteins. According to the biological function prediction, 18 biological processes were classified as differentially regulated (Figure 4A). In addition, the analysis of the exclusive proteome of each condition revealed 12 common processes between the control and stress conditions (Figure 4B). However, seven biological processes were identified only in stress-exposed cells. These processes were antibiotic metabolism (six proteins), nucleotide metabolism (five proteins), oxidative phosphorylation (three proteins), translation (three proteins), glycolysis pathways (one protein), iron-sulfur clusters (one protein), and starch and sucrose metabolism (one protein). Among all processes identified, DNA synthesis and repair proteins (14 proteins) were most common. An overview of the C. pseudotuberculosis response to nitrosative stress according with the proteins identified is shown in Figure 5.
Figure 4.
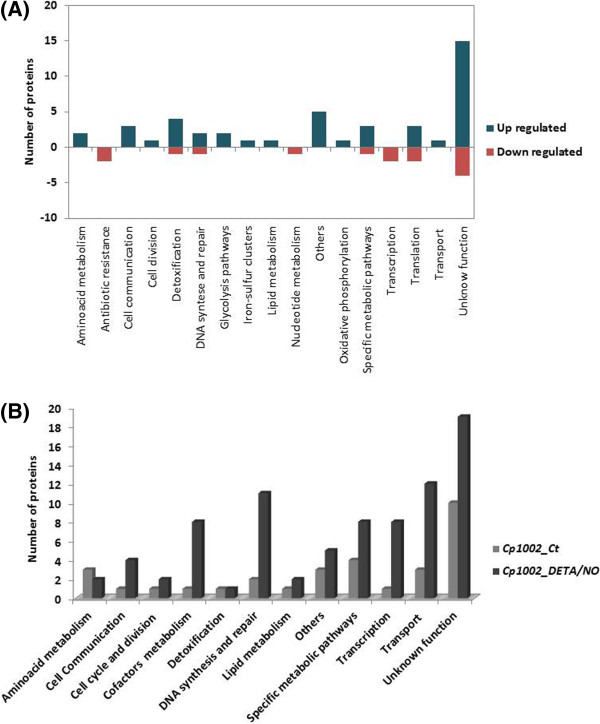
Comparison of biological processes between control and DETA/NO conditions. A representation of the biological processes in relation to a set list of proteins identified as (A) differentially-regulated in DETA/NO-stressed cells and (B) comparison of exclusive biological process between the two test conditions.
Figure 5.
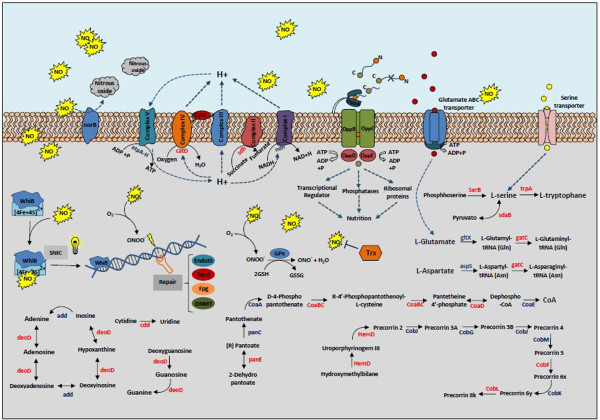
Overview of C. pseudotuberculosis response to nitrosative stress. All proteins detected by proteomic analysis are marked in red (differentially-regulated proteins or exclusive to the proteome of DETA/NO-stressed cells).
The proteins that were grouped into of transcriptional process were evaluated by CMRegNet and among regulators identified; we identified the GntR- family regulatory protein (D9Q5B7_CORP1), genes regulated by GntR-type regulators are usually involved in carbohydrate metabolism. The CMRegNet analysis showed that of the four genes under the control of this regulator, the N-acetylglucosamine kinase (D9Q5B6_CORP1) protein was highly expressed by C. pseudotuberculosis in response to DETA/NO. We identified other regulator the LexA repressor (D9Q8W2_CORP1) that was down regulated in the DETA/NO condition. According with CMRegNet, two proteins regulated by this repressor were detected in the DETA/NO proteome specific, pyridoxal biosynthesis lyase (PdxS; D9Q5T9_CORP1) and DNA translocase (D9Q8Z6_CORP1). Others proteins under the control of this repressor was detected, however not presented significant differential regulation like RecA protein
Protein-protein interaction network
To investigate the interactions among the proteins identified as exclusive and differentially regulated in cells exposed to DETA/NO, we generated a protein interaction network using Cytoscape. The interactome analysis revealed 67 protein-protein interactions (Figure 6). DnaB/DNA helicase (D9Q578_CORP1), identified in the exclusive proteome for strain 1002_DETA/NO, and PyrE/orotate phosphoribosyltransferase (D9Q4S2_CORP1), which was down-regulated in strain 1002_DETA/NO, showed the greatest number of interactions with other proteins (eight interactions each). Moreover, both of these proteins interact with proteins that are involved in metabolic processes, DNA processes, antibiotic metabolism, cell cycling, and translation.
Figure 6.
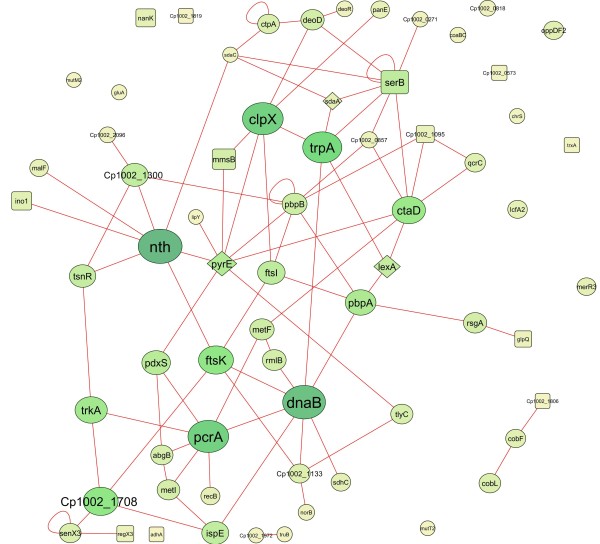
Protein-protein interactions. Protein-protein interactions of the proteins identified in DETA/NO-exposed cells. Exclusive proteome, circle; up-regulated, square; and down-regulated, rhombus. The sizes of the nodes represent the degree of interaction for each gene/protein; the major nodes demonstrate greater interactions. The colors of nodes and lines are in an increasing gradient scale from yellow to green to blue. The networks were visualized using Cytoscape.
Discussion
C. pseudotuberculosis is exposed to different forms of oxidative and nitrosative stress during the infection process. A previous study showed that C. pseudotuberculosis resists nitrosative stress generated by the NO-donor DETA/NO, and that a low concentration of DETA/NO (100 μM) induces a change in the extracellular proteome this pathogen [15]. To better understand the physiology of C. pseudotuberculosis in response to nitrosative stress, we analyzed the proteome of whole bacterial lysates of C. pseudotuberculosis in response to exposure to DETA/NO (0.5 mM).
The strain 1002 proteome under nitrosative stress reveals proteins involved in bacterial defense against DNA damage
Proteomic analysis identified proteins involved in DNA repair systems in both the exclusive proteome of DETA/NO-exposed cells and in the differentially-regulated proteome. We detected the proteins formamidopyrimidine-DNA glycosylase (Fpg) (D9Q598_CORP1), RecB (D9Q8C9_CORP1), and methylated-DNA-protein-cysteine methyltransferase (Ada) (D9Q923_CORP1), the genes for which were previously identified in a transcriptome analysis of strain 1002 in response to different abiotic stresses [8]. Activation of these proteins in response to nitrosative stress confirms that they belong a group of general stress-response proteins in C. pseudotuberculosis.
The expression of Fpg was up-regulated in response to acid stress [8]. We also identified endonuclease III (Endo III) (D9Q615_CORP1), which, in addition to Fpg, is involved in the base excision repair (BER) system of various bacteria. This system cleaves N-glycosidic bonds from damaged bases, allowing their excision and replacement. In Salmonella enterica serovar Typhimurium, the BER system repairs DNA damaged by exposure to NO. In addition, an S. Typhimurium strain defective in Fpg demonstrated reduced virulence in a murine model [30]. Our interactome analysis showed that Endo III had one of the highest numbers of interactions with other proteins, including interactions with proteins involved in DNA replication such zinc metalloprotease (D9Q378_CORP1) and DNA translocase (D9Q8Z6_CORP1), suggesting that this protein could play an important role in the defense pathway against RNS.
The Ada and RecB protein were up-regulated in response to osmotic stress [8]. Ada is involved in the repair of DNA-methylation damage, this protein have plays important in the pathway DNA damage [31]. RecB is a component of the RecBC system, which is part of the SOS response the more regulatory network encoded by prokaryotic involved in DNA repair [32]. The RecBC system acts in the recombination or degradative repair of arrested DNA replication forks. Studies in S. Typhimurium showed that recBC mutant strains are more attenuated than recA mutants in a murine model of infection [33]. In addition, unlike recA mutants, recBC mutants were susceptible to RNS [34], indicating that RecBC is highly important in the bacterial response to nitrosative stress. The LexA repressor (D9Q8W2_CORP1), which forms part of the general SOS system along with RecA [35], was down-regulated in C. pseudotuberculosis cells exposed to DETA/NO. We also detected the RecA protein (D9Q8Y3_CORP1); however, despite having a p-value <0.05, the fold-change of -0.50 showed that this protein was not activated under the experimental conditions. Studies performed in Mycobacterium tuberculosis showed that recA was not induced until cells had been exposed to DETA/NO (0.5 mM) for 4 h, but that hydrogen peroxide induced the immediate expression of recA[36], suggesting that RecA is involved in the later stages of the nitrosative stress response. Nevertheless, CMRegNet analysis identified other proteins that are regulated by LexA in the DETA/NO-specific proteome, including pyridoxal biosynthesis lyase (PdxS; D9Q5T9_CORP1) and DNA translocase (D9Q8Z6_CORP1).
NO-sensitive transcriptional regulators are activated in the presence of NO
To activate these DNA repair systems, it is essential that bacteria can detect ROS and RNS, and concomitantly activate the transcriptional regulators needed for the expression of genes involved in protection against these compounds. In the DETA/NO-specific proteome, we detected the transcription factor WhiB (D9Q6Y2_CORP1). The WhiB transcriptional family is composed of iron-sulfur (Fe-S) cluster proteins. These proteins are O2- and NO-sensitive, and allow the sensing of both external environmental signals and the redox state for intracellular bacteria [37, 38]. In M. tuberculosis, the reaction of the iron-sulfur cluster of WhiB3 with NO generates a dinitrosyl iron complex (DNIC), which activates a sensing mechanism in response to the NO, consequently activating a system of defense against nitrosative stress [12]. In addition, other in vivo and in vitro studies have also demonstrated that WhiB regulators play a role in the adaptation and survival of M. tuberculosis during exposure to redox environments [12, 39–41].
We identified other regulators that are activated in response to environmental stimuli, such as a MerR-family transcriptional regulator (D9Q889_CORP1) and a LysR-type transcriptional regulator (LTTR) (D9Q7H8_CORP1). This regulator was also highly expressed in the transcriptional response of C. pseudotuberculosis 1002 to acid stress [8]. MerR-type regulators have been described in the detoxification of toxic metal in several pathogenic and non-pathogenic bacteria [42]. Other studies have shown that this class of regulator plays a role in bacterial resistance to oxidative and nitrosative stress [43, 44]. LTTRs are associated with the regulation of several biological processes, as well as in the adaptive response of bacteria to different types of stress [45]. In Vibrio cholerae, LTTRs are associated with efflux pump regulation, which contribute to antimicrobial resistance, and are involved in colonization of the human host [46]. In pathogens like E. coli[47], Enterococcus faecalis[48], S. enterica[49], and Pseudomonas aeruginosa[50], LTTRs are involved in resistance to oxidative stress.
The detoxification pathways of C. pseudotuberculosisfollowing NO exposure
Our proteomic analysis identified proteins specifically expressed by cells exposed to DETA/NO that are involved in the detoxification process. Two of these proteins were thioredoxin (trxA) (D9Q7U6_CORP1) and glutathione peroxidase (D9Q4E5_CORP1). The thioredoxin and glutathione systems play major roles in thiol and disulfide balance, respectively [14]. In pathogens such as Helicobacter pylori, Streptococcus pyogenes, and M. tuberculosis, this system is of great importance in combating the presence of ROS/RNS [36, 51, 52]. A glyoxalase/dioxygenase (D9Q5T5_CORP1) was identified in the differential proteome of cells exposed to DETA/NO. This protein was previously detected in the proteome of C. pseudotuberculosis strain 1002 in response to 0.1 mM DETA/NO [15]. The presence of this protein suggests that glyoxalase/dioxygenase plays a role in the resistance of this pathogen to nitrosative stress.
Nevertheless, unlike P. aeruginosa, which contains a complete denitrification pathway [53], the predicted genome of C. pseudotuberculosis ovis 1002 revealed a truncated denitrification pathway. However, we detected the nitric-oxide reductase cytochrome b (NorB) (D9Q5T6_CORP1) in the exclusive proteome of DETA/NO-stressed cells. norB, which codes for this nitric-oxide reductase, is organized into the norCBQDEF operon in Paracoccus denitrificans[54], and into the norCBD operon in P. aeruginosa[55]. The C. pseudotuberculosis genome was predicted to only contain norB. Moreover, norB is located in the Cp1002PiCp12 pathogenicity island, suggesting horizontal acquisition of the gene by this pathogen. Nitric-oxide reductase is an important protein in the denitrification process of some bacteria [56]. In P. aeruginosa, NorB plays a role in both the growth of the pathogen in the presence of NO, and in its survival in macrophages [55]. The flavohemoglobin Hmp is involved in the NO detoxification pathway in S. Typhimurium, and levels of Hmp are increased approximately two-fold in macrophages [57]. Interestingly, in N. meningitidis, NorB levels are increased ten-fold in macrophages [58], demonstrating the great power of this protein in the detoxification process.
Metabolic profile of C. pseudotuberculosisin response to nitrosative stress
In addition to the presence of proteins involved in bacterial defense and detoxification pathways, strain 1002 needs to undergo metabolic adaptation to favor bacterial survival. We observed a metabolic readjustment in this pathogen in the proteomic analysis. Of the proteins involved in central carbohydrate metabolism, we detected only phosphoglycerate mutase (D9Q533_CORP1) and N-acetylglucosamine kinase (D9Q5B6_CORP1) in the proteome of DETA/NO-exposed cells. Other essential proteins involved in glycolysis (the Embdem-Meyerhof pathway), the pentose phosphate pathway, and the citric acid cycle were not detected. Similar results were found in a metabolomic study of V. cholerae in response to nitrosative stress [59].
However, we hypothesized that C. pseudotuberculosis uses oxidative phosphorylation to obtain energy. This is supported by the presence of cytochrome C oxidase polypeptide I (D9Q486_CORP1), succinate dehydrogenase cytochrome b556 subunit (D9Q650_CORP1), and ubiquinol-cytochrome C reductase cytochrome C subunit (D9Q3J7_CORP1) in the exclusive proteome of DETA/NO-stressed cells, and by the up-regulation of the cytochrome oxidase assembly protein (D9Q8I5_CORP1) under the same conditions. However, this oxidative phosphorylation may be associated with the bacterial culture conditions used in this work, in which C. pseudotuberculosis was cultivated in the presence of DETA/NO under aerobic conditions. Studies have shown that growing M. tuberculosis in a low concentration of NO with low levels of O2 can induce anaerobic respiration as a result of the inhibition of the respiratory proteins cytochrome c oxidase and NADH reductase by irreversible ligation of NO. The ligation of NO to the respiratory proteins is an effect that may be both short-term reversible and long-term irreversible [60]. Thus, we suggest that activation of the oxidative phosphorylation system may be a more effective pathway for this pathogen to obtain energy [61].
Another metabolic adjustment was observed in relation to amino acid biosynthesis. Transporters and enzymes involved in the synthesis of methionine, tryptophan, and serine were identified. However, the presence of these proteins can be associated with the bioavailability of these amino acids during exposure to NO. In addition, we detected two oligopeptide transport ATP-binding proteins (OppD) (D9Q6G5_CORP1/ D9Q3X0_CORP1) that compose the oligopeptide permease system (Opp). This complex is associated with the internalization of peptides from the extracellular environment to be used as a source of carbon and nitrogen in bacterial nutrition [62]. We also identified proteins that are cofactors of metabolism, such as CoaBC (D9Q8L2_CORP1), phosphopantetheine adenylyltransferase (D9Q809_CORP1), and 2-dehydropantoate 2-reductase (D9Q7J9_CORP1). The presence of these proteins demonstrates activity in pantothenic acid metabolism and the biosynthesis of coenzyme A (CoA). Studies performed in species such as Corynebacterium diphtheriae[63], Streptococcus haemolyticus[64], and M. tuberculosis[65] showed that pantothenic acid and CoA could have an important role in the growth and viability of these pathogens.
Conclusions
In this work, we applied high-throughput proteomics to characterize the proteome of C. pseudotuberculosis ovis 1002 following exposure to NO. Our proteomic analysis generated two profiles, which together validated findings from previous in silico analyses of C. pseudotuberculosis ovis 1002. The proteomic profile generated after the addition of the NO-donor, DETA/NO (0.5 mM), revealed a set of proteins that are involved in distinct biological process. We detected proteins related to both the general stress response and to a more specific nitrosative stress response, which together form a network of factors that promote the survival of this pathogen under stress conditions. However, more detailed studies are needed to assess the true role of these proteins in response to nitrosative stress in C. pseudotuberculosis. In conclusion, this functional analysis of the genome of C. pseudotuberculosis shows the versatility of this pathogen in the presence of NO. Moreover, the results presented in this study provide insights into the processes of resistance of C. pseudotuberculosis during exposure to nitrosative stress.
Electronic supplementary material
Additional file 1: Table S1: Complete list of proteins identified as significantly altered (p < 0.05). (XLSX 169 KB)
Additional file 2: Table S2: Unique proteins identified in strain 1002_DETA/NO. (XLSX 21 KB)
Additional file 3: Table S3: Unique proteins identified in strain 1002 control condition. (XLSX 14 KB)
Acknowledgements
This work was supported by the Genomics and Proteomics Network of the State of Pará of the Federal University of Pará, the Amazon Research Foundation (FAPESPA), the National Council for Scientific and Technological Development (CNPq), the Brazilian Federal Agency for the Support and Evaluation of Graduate Education (CAPES), the Minas Gerais Research Foundation (FAPEMIG), and Waters Corporation, Brazil. Yves Le Loir is the recipient of a PVE grant (71/2013) from Programa Ciências sem Fronteiras.
Abbreviations
- G
Guanine
- C
Citosine
- NO
Nitric oxide
- RNS
Reactive nitrogen species
- NOS
Nitric oxide synthases
- LC-HDMSE
Liquid chromatograph high definition mass spectrometry
- LC/MS
Liquid chromatograph mass spectrometry
- CDM
Chemically-defined médium
- DETA/NO
Diethylenetriamine/nitric oxide adduct
- DTT
Dithiothreitol
- 2D-RP
Two-dimensional reversed phase
- nanoUPLC
Nano Ultra performance liquid chromatography
- nanoESI-HDMS
Nano electrospray high definition mass spectrometry
- HSS
High strength silica
- PLGS
Protein lynx global server
- FDR
False discovery rate.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
WMS, RDC, and IFSB performed microbiological analyses and sample preparation for proteomic analysis. GHMFS and WMS conducted the proteomic analysis. SCS and ELF performed bioinformatics analysis of the data. YLL and AM contributed substantially to data interpretation and revisions. VA and AS participated in all steps of the project as coordinators, and critically reviewed the manuscript. All authors read and approved the final manuscript.
Contributor Information
Wanderson M Silva, Email: silvamarques@yahoo.com.br.
Rodrigo D Carvalho, Email: rodrigodoc2@gmail.com.
Siomar C Soares, Email: siomars@gmail.com.
Isabela FS Bastos, Email: isabelafbastos@yahoo.com.br.
Edson L Folador, Email: edson.folador@gmail.com.
Gustavo HMF Souza, Email: Gustavo_Souza@waters.com.
Yves Le Loir, Email: yves.leloir@rennes.inra.fr.
Anderson Miyoshi, Email: miyoshi@icb.ufmg.br.
Artur Silva, Email: asilva@ufpa.br.
Vasco Azevedo, Email: vasco@icb.ufmg.br.
References
- 1.Dorella FA, Pacheco LG, Oliveira SC, Miyoshi A, Azevedo V. Corynebacterium pseudotuberculosis: microbiology, biochemical properties, pathogenesis and molecular studies of virulence. Vet Res. 2006;37:201–218. doi: 10.1051/vetres:2005056. [DOI] [PubMed] [Google Scholar]
- 2.Baird GJ, Fontaine MC. Corynebacterium pseudotuberculosis and its role in ovine caseous lymphadenitis. J Comp Pathol. 2007;137:179–210. doi: 10.1016/j.jcpa.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Hodgson AL, Bird P, Nisbet IT. Cloning, nucleotide sequence, and expression in Escherichia coli of the phospholipase D gene from Corynebacterium pseudotuberculosis. J Bacteriol. 1990;172:1256–1261. doi: 10.1128/jb.172.3.1256-1261.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hard GC. Comparative toxic effect of the surface lipid of Corynebacterium ovis on peritoneal macrophages. Infect Immun. 1975;12:1439–1449. doi: 10.1128/iai.12.6.1439-1449.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Billington SJ, Esmay PA, Songer JG, Jost BH. Identification and role in virulence of putative iron acquisition genes from Corynebacterium pseudotuberculosis. J Bacteriol. 2002;180:3233–3236. doi: 10.1111/j.1574-6968.2002.tb11058.x. [DOI] [PubMed] [Google Scholar]
- 6.Ruiz JC, D'Afonseca V, Silva A, Ali A, Pinto AC, Santos AR, Rocha AA, Lopes DO, Dorella FA, Pacheco LG, Costa MP, Turk MZ, Seyffert N, Moraes PM, Soares SC, Almeida SS, Castro TL, Abreu VA, Trost E, Baumbach J, Tauch A, Schneider MP, McCulloch J, Cerdeira LT, Ramos RT, Zerlotini A, Dominitini A, Resende DM, Coser EM, Oliveira LM, et al. Evidence for reductive genome evolution and lateral acquisition of virulence functions in two Corynebacterium pseudotuberculosis strains. PLoS One. 2011;18:e18551. doi: 10.1371/journal.pone.0018551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soares SC, Silva A, Trost E, Blom J, Ramos R, Carneiro A, Ali A, Santos AR, Pinto AC, Diniz C, Barbosa EG, Dorella FA, Aburjaile F, Rocha FS, Nascimento KK, Guimarães LC, Almeida S, Hassan SS, Bakhtiar SM, Pereira UP, Abreu VA, Schneider MP, Miyoshi A, Tauch A, Azevedo V. The pan-genome of the animal pathogen Corynebacterium pseudotuberculosis reveals differences in genome plasticity between the biovar ovis and equi strains. PLoS One. 2013;8:e53818. doi: 10.1371/journal.pone.0053818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pinto AC, de Sá PH, Ramos RT, Barbosa S, Barbosa HP, Ribeiro AC, Silva WM, Rocha FS, Santana MP, de Paula Castro TL, Miyoshi A, Schneider MP, Silva A, Azevedo V. Differential transcriptional profile of Corynebacterium pseudotuberculosis in response to abiotic stresses. BMC Genomics. 2014;9:14. doi: 10.1186/1471-2164-15-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marletta MA. Nitric oxide synthase: aspects concerning structure and catalysis. Cell. 1994;23:927–930. doi: 10.1016/0092-8674(94)90268-2. [DOI] [PubMed] [Google Scholar]
- 10.Griffith OW, Stueh DJ. Nitric oxide synthases: properties and catalytic mechanism. Annu Rev Physiol. 1995;57:707–736. doi: 10.1146/annurev.ph.57.030195.003423. [DOI] [PubMed] [Google Scholar]
- 11.Nathan C, Shiloh MU. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci USA. 2000;1:8841–8848. doi: 10.1073/pnas.97.16.8841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh A, Guidry L, Narasimhulu KV, Mai D, Trombley J, Redding KE, Giles GI, Lancaster JR, Jr, Steyn AJ. Mycobacterium tuberculosis WhiB3 responds to O2 and nitric oxide via its [4Fe-4S] cluster and is essential for nutrient starvation survival. Proc Natl Acad Sci USA. 2007;10:11562–11567. doi: 10.1073/pnas.0700490104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ehrt S, Schnappinger D. Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell Microbiol. 2009;11:1170–1178. doi: 10.1111/j.1462-5822.2009.01335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu J, Holmgren A. The thioredoxin antioxidant system. Free Radic Biol Med. 2013;8:75–87. doi: 10.1016/j.freeradbiomed.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 15.Pacheco LG, Castro TL, Carvalho RD, Moraes PM, Dorella FA, Carvalho NB, Slade SE, Scrivens JH, Feelisch M, Meyer R, Miyoshi A, Oliveira SC, Dowson CG, Azevedo V. A role for sigma factor σ(E) in Corynebacterium pseudotuberculosis resistance to nitric oxide/peroxide stress. Front Microbiol. 2012;3:126. doi: 10.3389/fmicb.2012.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moura-Costa LF, Paule BJA, Freire SM, Nascimento I, Schaer R, Regis LF, Vale VLC, Matos DP, Bahia RC, Carminati R, Meyer R. Chemically defined synthetic medium for Corynebacterium pseudotuberculosis culture. Rev Bras Saúde Prod An. 2002;3:1–9. [Google Scholar]
- 17.Silva JC, Gorenstein MV, Li GZ, Vissers JP, Geromanos SJ. Absolute quantification of proteins by LC/MSE: a virtue of parallel MS acquisition. Mol Cell Proteomics. 2006;5:144–156. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- 18.Gilar M, Olivova P, Daly AE, Gebler JC. Two-dimensional separation of peptides using RP-RP-HPLC system with different pH in first and second separation dimensions. J Sep Sci. 2005;28:1694–1703. doi: 10.1002/jssc.200500116. [DOI] [PubMed] [Google Scholar]
- 19.Silva JC, Denny R, Dorschel CA, Gorenstein M, Kass IJ, Li GZ, McKenna T, Nold MJ, Richardson K, Young P, Geromanos S. Quantitative proteomic analysis by accurate mass retention time pairs. Anal Chem. 2005;77:2187–2000. doi: 10.1021/ac048455k. [DOI] [PubMed] [Google Scholar]
- 20.Geromanos SJ, Vissers JP, Silva JC, Dorschel CA, Li GZ, Gorenstein MV, Bateman RH, Langridge JI. The detection, correlation, and comparison of peptide precursor and product ions from data independent LC-MS with data dependant LC-MS/MS. Proteomics. 2009;9:1683–1695. doi: 10.1002/pmic.200800562. [DOI] [PubMed] [Google Scholar]
- 21.Li GZ, Vissers JP, Silva JC, Golick D, Gorenstein MV, Geromanos SJ. Database searching and accounting of multiplexed precursor and product ion spectra from the data independent analysis of simple and complex peptide mixtures. Proteomics. 2009;9:1696–1719. doi: 10.1002/pmic.200800564. [DOI] [PubMed] [Google Scholar]
- 22.Curty N, Kubitschek-Barreira PH, Neves GW, Gomes D, Pizzatti L, Abdelhay E, Souza GH, Lopes-Bezerra LM. Discovering the infectome of human endothelial cells challenged with Aspergillus fumigatus applying a mass spectrometry label-free approach. J Proteomics. 2014;31:126–140. doi: 10.1016/j.jprot.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 23.Levin Y, Hadetzky E, Bahn S. Quantification of proteins using data-independent analysis (MSE) in simple and complex samples: a systematic evaluation. Proteomics. 2011;11:3273–3287. doi: 10.1002/pmic.201000661. [DOI] [PubMed] [Google Scholar]
- 24.Barinov A, Loux V, Hammani A, Nicolas P, Langella P, Ehrlich D, Maguin E, van de Guchte M. Prediction of surface exposed proteins in Streptococcus pyogenes, with a potential application to other Gram-positive bacteria. Proteomics. 2009;9:61–73. doi: 10.1002/pmic.200800195. [DOI] [PubMed] [Google Scholar]
- 25.Conesa A, Gotz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;15:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 26.Soares SC, Abreu VA, Ramos RT, Cerdeira L, Silva A, Baumbach J, Trost E, Tauch A, Hirata R, Jr, Mattos-Guaraldi AL, Miyoshi A, Azevedo V. PIPs: pathogenicity island prediction software. PLoS One. 2012;7:e30848. doi: 10.1371/journal.pone.0030848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rezende AM, Folador EL, Resende Dde M, Ruiz JC. Computational prediction of protein-protein interactions in Leishmania predicted proteomes. PLoS One. 2012;7:e51304. doi: 10.1371/journal.pone.0051304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pauling J, Röttger R, Tauch A, Azevedo V, Baumbach J. CoryneRegNet 6.0 -Updated database content, new analysis methods and novel features focusing on community demands. Nucleic Acids Res. 2012;40:D610–614. doi: 10.1093/nar/gkr883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richardson AR, Soliven KC, Castor ME, Barnes PD, Libby SJ, Fang FC. The base excision repair system of Salmonella enterica serovar typhimurium counteracts DNA damage by host nitric oxide. PLoS Pathog. 2009;5:e1000451. doi: 10.1371/journal.ppat.1000451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sedgwick B. Repairing DNA-methylation damage. Nat Rev Mol Cell Biol. 2004;5:148–157. doi: 10.1038/nrm1312. [DOI] [PubMed] [Google Scholar]
- 32.Baharoglu Z, Mazel D. SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol Rev. 2014;38:1126–1145. doi: 10.1111/1574-6976.12077. [DOI] [PubMed] [Google Scholar]
- 33.Cano DA, Pucciarelli MG, García-del Portillo F, Casadesús J. Role of the recBCD recombination pathway in Salmonella virulence. J Bacteriol. 2002;184:592–595. doi: 10.1128/JB.184.2.592-595.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koskiniemi S, Andersson DI. Translesion DNA polymerases are required for spontaneous deletion formation in Salmonella typhimurium. Proc Natl Acad Sci U S A. 2009;23:10248–10253. doi: 10.1073/pnas.0904389106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Butala M, Zgur-Bertok D, Busby SJ. The bacterial LexA transcriptional repressor. Cell Mol Life Sci. 2009;66:82–93. doi: 10.1007/s00018-008-8378-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Voskuil MI, Bartek IL, Visconti K, Schoolnik GK. The response of Mycobacterium tuberculosis to reactive oxygen and nitrogen species. Front Microbiol. 2011;13:105. doi: 10.3389/fmicb.2011.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Green J, Paget MS. Bacterial redox sensors. Nat Rev Microbiol. 2004;2:954–966. doi: 10.1038/nrmicro1022. [DOI] [PubMed] [Google Scholar]
- 38.Green J, Rolfe MD, Smith LJ. Transcriptional regulation of bacterial virulence gene expression by molecular oxygen and nitric oxide. Virulence. 2014;4:5(4). doi: 10.4161/viru.27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh A, Crossman DK, Mai D, Guidry L, Voskuil MI, Renfrow MB, Steyn AJ. Mycobacterium tuberculosis WhiB3 maintains redox homeostasis by regulating virulence lipid anabolism to modulate macrophage response. PLoS Pathog. 2009;5:e1000545. doi: 10.1371/journal.ppat.1000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chawla M, Parikh P, Saxena A, Munshi M, Mehta M, Mai D, Srivastava AK, Narasimhulu KV, Redding KE, Vashi N, Kumar D, Steyn AJ, Singh A. Mycobacterium tuberculosis WhiB4 regulates oxidative stress response to modulate survival and dissemination in vivo. Mol Microbiol. 2012;85:1148–1165. doi: 10.1111/j.1365-2958.2012.08165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larsson C, Luna B, Ammerman NC, Maiga M, Agarwal N, Bishai WR. Gene expression of Mycobacterium tuberculosis putative transcription factors WhiB1-7 in redox environments. PLoS One. 2012;7:e37516. doi: 10.1371/journal.pone.0037516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hobman JL. MerR family transcription activators: similar designs, different specificities. Mol Microbiol. 2007;63:1275–1278. doi: 10.1111/j.1365-2958.2007.05608.x. [DOI] [PubMed] [Google Scholar]
- 43.McEwan AG, Djoko KY, Chen NH, Couñago RL, Kidd SP, Potter AJ, Jennings MP. Novel bacterial MerR-like regulators their role in the response to carbonyl and nitrosative stress. Adv Microb Physiol. 2011;58:1–22. doi: 10.1016/B978-0-12-381043-4.00001-5. [DOI] [PubMed] [Google Scholar]
- 44.Brown NL, Stoyanov JV, Kidd SP, Hobman JL. The MerR family of transcriptional regulators. FEMS Microbiol Rev. 2003;27:145–163. doi: 10.1016/S0168-6445(03)00051-2. [DOI] [PubMed] [Google Scholar]
- 45.Maddocks SE, Oyston PC. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology. 2008;154:3609–3623. doi: 10.1099/mic.0.2008/022772-0. [DOI] [PubMed] [Google Scholar]
- 46.Chen S, Wang H, Katzianer DS, Zhong Z, Zhu J. LysR family activator-regulated major facilitator superfamily transporters are involved in Vibrio cholerae antimicrobial compound resistance and intestinal colonisation. Int J Antimicrob Agents. 2013;41:188–192. doi: 10.1016/j.ijantimicag.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 47.Gonzalez-Flecha B, Demple B. Role for the oxyS gene in regulation of intracellular hydrogen peroxide in Escherichia coli. J Bacteriol. 1999;181:3833–3836. doi: 10.1128/jb.181.12.3833-3836.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Verneuil N, Rincé A, Sanguinetti M, Posteraro B, Fadda G, Auffray Y, Hartke A, Giard JC. Contribution of a PerR-like regulator to the oxidative-stress response and virulence of Enterococcus faecalis. Microbiology. 2005;151:3997–4004. doi: 10.1099/mic.0.28325-0. [DOI] [PubMed] [Google Scholar]
- 49.Lahiri A, Das P, Chakravortty D. The LysR-type transcriptional regulator Hrg counteracts phagocyte oxidative burst and imparts survival advantage to Salmonella enterica serovar Typhimurium. Microbiology. 2008;154:2837–2846. doi: 10.1099/mic.0.2008/017574-0. [DOI] [PubMed] [Google Scholar]
- 50.Reen FJ, Haynes JM, Mooij MJ, O'Gara F. A non-classical LysR-type transcriptional regulator PA2206 is required for an effective oxidative stress response in Pseudomonas aeruginosa. PLoS One. 2013;8:e54479. doi: 10.1371/journal.pone.0054479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Comtois SL, Gidley MD, Kelly DJ. Role of the thioredoxin system and the thiol-peroxidases Tpx and Bcp in mediating resistance to oxidative and nitrosative stress in Helicobacter pylori. Microbiology. 2003;149:121–129. doi: 10.1099/mic.0.25896-0. [DOI] [PubMed] [Google Scholar]
- 52.Brenot A, King KY, Janowiak B, Griffith O, Caparon MG. Contribution of glutathione peroxidase to the virulence of Streptococcus pyogenes. Infect Immun. 2004;72:408–413. doi: 10.1128/IAI.72.1.408-413.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kalkowski I, Conrad R. Metabolism of nitric oxide in denitrifying Pseudomonas aeruginosa and nitrate-respiring Bacillus cereus. FEMS Microbiol Lett. 1991;15:107–111. doi: 10.1111/j.1574-6968.1991.tb04848.x. [DOI] [PubMed] [Google Scholar]
- 54.de Boer AP, van der Oost J, Reijnders WN, Westerhoff HV, Stouthamer AH, van Spanning RJ. Mutational analysis of the nor gene cluster which encodes nitric-oxide reductase from Paracoccus denitrificans. Eur J Biochem. 1996;15:592–600. doi: 10.1111/j.1432-1033.1996.0592r.x. [DOI] [PubMed] [Google Scholar]
- 55.Kakishima K, Shiratsuchi A, Taoka A, Nakanishi Y, Fukumori Y. Participation of nitric oxide reductase in survival of Pseudomonas aeruginosa in LPS-activated macrophages. Biochem Biophys Res Commun. 2007;6:587–591. doi: 10.1016/j.bbrc.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 56.Hendriks J, Oubrie A, Castresana J, Urbani A, Gemeinhardt S, Saraste M. Nitric oxide reductases in bacteria. Biochim Biophys Acta. 2000;15:266–273. doi: 10.1016/S0005-2728(00)00161-4. [DOI] [PubMed] [Google Scholar]
- 57.Stevanin TM, Poole RK, Demoncheaux EA, Read RC. Flavohemoglobin Hmp protects Salmonella enterica serovar Typhimurium from nitric oxide-related killing by human macrophages. Infect Immun. 2002;70:4399–4405. doi: 10.1128/IAI.70.8.4399-4405.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stevanin TM, Moir JW, Read RC. Nitric oxide detoxification systems enhance survival of Neisseria meningitidis in human macrophages and in nasopharyngeal mucosa. Infect Immun. 2005;73:3322–3329. doi: 10.1128/IAI.73.6.3322-3329.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stern AM, Liu B, Bakken LR, Shapleigh JP, Zhu J. A novel protein protects bacterial iron-dependent metabolism from nitric oxide. J Bacteriol. 2013;195:4702–4708. doi: 10.1128/JB.00836-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brown GC. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome C oxidase. Biochim Biophys Acta. 2001;1:46–57. doi: 10.1016/S0005-2728(00)00238-3. [DOI] [PubMed] [Google Scholar]
- 61.Kadenbach B. Intrinsic and extrinsic uncoupling of oxidative phosphorylation. Biochim Biophys Acta. 2003;5:77–94. doi: 10.1016/S0005-2728(03)00027-6. [DOI] [PubMed] [Google Scholar]
- 62.Payne JW, Smith MW. Peptide transport by micro-organisms. Adv Microb Physiol. 1994;36:1–80. doi: 10.1016/S0065-2911(08)60176-9. [DOI] [PubMed] [Google Scholar]
- 63.Mueller JH, Klotz AW. Pantothenic acid as a growth factor for the diphtheria bacillus. J Am Chem Soc. 1938;60:3086–3087. doi: 10.1021/ja01279a510. [DOI] [Google Scholar]
- 64.Mcllwain H. Pantothenic acid and the growth of Streptococcus haemolyticus. Br J Exp Pathol. 1939;20:330–333. [Google Scholar]
- 65.Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1: Complete list of proteins identified as significantly altered (p < 0.05). (XLSX 169 KB)
Additional file 2: Table S2: Unique proteins identified in strain 1002_DETA/NO. (XLSX 21 KB)
Additional file 3: Table S3: Unique proteins identified in strain 1002 control condition. (XLSX 14 KB)