

Summary
Parkinson’s disease (PD) is a common neurodegenerative disorder. Functional interactions between some PD genes, like PINK1 and parkin, have been identified, but whether other ones interact remains elusive. Here we report an unexpected genetic interaction between two PD genes, VPS35 and EIF4G1. We provide evidence that EIF4G1 upregulation causes defects associated with protein misfolding. Expression of a sortilin protein rescues these defects, downstream of VPS35, suggesting a potential role for sortilins in PD. We also show interactions between VPS35, EIF4G1 and α–synuclein, a protein with a key role in PD. We extend our findings from yeast to an animal model and show these interactions are conserved in neurons and in transgenic mice. Our studies reveal unexpected genetic and functional interactions between two seemingly unrelated PD genes and functionally connect them to α–synuclein pathobiology in yeast, worms, and mouse. Finally, we provide a resource of candidate PD genes for future interrogation.
Introduction
Parkinson’s disease (PD) is a debilitating neurodegenerative disorder that affects more than 2% of adults over 60 (Jankovic, 2008; Massano and Bhatia, 2012). It is mostly a sporadic disease, but the rare genetic forms have provided key insight into disease mechanisms that are also relevant to sporadic PD. With the emergence of next generation sequencing strategies, together with traditional genetic linkage approaches, and genome-wide association studies (GWAS), risk genes associated with PD are being rapidly discovered (Do et al., 2011; Hamza et al., 2010; International Parkinson Disease Genomics et al., 2011; International Parkinson's Disease Genomics and Wellcome Trust Case Control, 2011; Li et al., 2013; Lill et al., 2012; Nalls et al., 2014a; Satake et al., 2009; Simon-Sanchez et al., 2009; Trinh and Farrer, 2013; Wan et al., 2014) and are beginning to illuminate cellular pathways and functions as critical to disease pathogenesis (Nalls et al., 2014b; Trinh and Farrer, 2013). As the list of PD disease genes grows, it will be important to define if these genes interact with one another or if they function independently. Indeed, compelling evidence points to genetic interactions between some PD genes (e.g., parkin and PINK1 (Clark et al., 2006; Park et al., 2006), ATP13A2 and α–synuclein (Gitler et al., 2009), LRRK2 and parkin (Smith et al., 2005) and glucocerebrosidase and α–synuclein (Mazzulli et al., 2011)). Being able to group some PD genes into related functional networks will facilitate the development of more effective pathway-based targeted therapeutic interventions.
Many of the fundamental cellular pathologies associated with PD, including mitochondrial dysfunction (Wan et al., 2014) and vesicle-trafficking defects (Miklavc et al., 2014), involve impairments in highly conserved cellular pathways. Therefore, simple model organisms have emerged as powerful experimental systems for studying the basic biology underpinning even complicated human neurodegenerative diseases like PD. One simple, yet surprisingly effective model system has been the budding yeast Saccharomyces cerevisiae. Yeast models engineered to express exogenous human PD risk genes (e.g., α–synuclein or LRRK2) recapitulate key cellular phenotypes (Outeiro and Lindquist, 2003; Xiong et al., 2010) and have been used to perform unbiased genetic and chemical screens for phenotypic modifiers.
Two PD genes, conserved from yeast to human, are EIF4G1 and VPS35. Mutations in EIF4G1 and VPS35 were recently identified in familial PD (Chartier-Harlin et al., 2011; Vilarino-Guell et al., 2011). EIF4G1 is a large scaffold protein that functions in translation initiation factor complex assembly (Villa et al., 2013). VPS35 is a component of the retromer complex, which functions to sort cellular cargo within the endocytic system. Cargos retained on endosomes are delivered to the vacuole (lysosomes in mammalian cells) for degradation. Retromer functions to orchestrate the retrieval of some of these proteins from endosomes back to the trans-golgi network (TGN) (Harrison et al., 2014). The exact mechanisms by which these genes contribute to PD are not fully understood. Moreover, EIF4G1 is still somewhat controversial as a bona fide PD gene. Since the initial identification of EIF4G1 mutations in PD, there have been several negative results (Blanckenberg et al., 2014; Lesage et al., 2012; Nishioka et al., 2014; Siitonen et al., 2013; Sudhaman et al., 2013) and some suggestions that the original published mutation is found in controls and therefore it might be a rare but benign polymorphism (Tucci et al., 2012). Therefore, additional functional insight into potential interactions with other PD genes and disease-relevant pathways could be informative.
The yeast VPS35 homolog is also called VPS35 and EIF4G1 has two yeast homologs, TIF4631 and TIF4632. Since TIF4631 is more closely related to human EIF4G1 and deletion of TIF4631, but not TIF4632, causes global alterations in translation initiation rates (Clarkson et al., 2010) we focused on TIF4631. We performed two separate genetic screens with vps35Δ and tif4631Δ yeast strains. We discovered a powerful genetic and functional interaction between VPS35 and EIF4G1, which we extend from yeast to an animal model. We define the mechanism by which these genes interact and connect VPS35 to another PD protein, α–synuclein (α–syn). We show that VPS35 is able to protect against α–syn-induced neurodegeneration in a mouse model. Together, these results reveal robust genetic and functional interactions between two known PD genes and illuminate novel connections to α–syn pathobiology.
Results
We used synthetic genetic array (SGA) (Tong and Boone, 2006) to introduce either the VPS35 or the TIF4631 deletion, by mating, to a collection of ~4900 yeast strains harboring single deletions in non-essential genes (Supplemental Fig. S1A). After performing the vps35Δ and tif4631Δ synthetic lethal screens independently (each screen replicated three times), we scored each of the double deletions for growth. We identified 144 genes synthetic sick or lethal with tif4631Δ and 59 synthetic sick or lethal with vps35Δ. (Supplemental Fig. S1 B,C, Supplemental Table 1). We used GOTermFinder to identify significant gene ontology (GO) terms enriched as hits in each screen (Boyle et al., 2004). The major functional categories for the vps35Δ screen were protein targeting (P=1.99E-05) and vacuolar transport (P=2.56E-05). Since Vps35 has established roles in protein transport, these functional categories were expected and serve as proof of principle that the screen was effective. For the tif4631Δ screen, the major categories of genes enriched include transcription and translation (P=6.40E-05). These too were expected, based on the known functions of this translation initiation factor. However, we found a surprising overlap in functional categories between both screens (Supplemental Fig. S1B). Enriched categories of deletions unexpected for the tif4631Δ screen included genes involved in endosomal transport (P=1.6E-04), and protein targeting (P=7.44E-05) (Supplemental Fig. S1B).
In addition to similar categories of hits, we also identified fourteen synthetic lethal interactions that were common to both vps35Δ and tif4631Δ screens (Fig. S1C; p < 0.0031). Among these common interactors, the most prevalent categories enriched were genes involved in protein localization (p=3E-04) and protein transport (p=6E-04). One hit common to both screens, GCS1, is a yeast ortholog of ARFGAP1, a gene encoding a GTPase activating protein known to physically and genetically interact with the PD gene LRRK2 (Xiong et al., 2010). Remarkably, one of the strongest hits from the tif4631Δ screen, INP53, is the yeast homolog of SYNJ1 in humans. Mutations in SYNJ1 were recently identified as a cause of early-onset parkinsonism (Krebs et al., 2013; Quadri et al., 2013). We suggest that future PD sequencing projects might benefit from using the additional hits from these screens as candidates to predict new PD risk genes (Supplemental Table 1).
Even though both screens enriched overlapping categories of hits (Supplemental Fig. S1B), we did not identify a synthetic lethal interaction between tif4631Δ and vps35Δ. This prompted us to consider other types of potential genetic interactions between the two genes – we performed a series of experiments upregulating one gene while deleting the other and vice versa. This led us to discover a surprising and remarkably potent interaction. Upregulation of TIF4631 in wild type yeast cells had no effect on growth, however it was highly toxic in vps35Δ cells (Fig. 1A). Upregulation of human EIF4G1 had the same effect (Fig. 1B), indicating that this function is conserved from the yeast to human protein. Likewise, co-expression of either yeast or human VPS35 could rescue the phenotype (Fig. 1B), providing further evidence for the robust and highly conserved nature of the functional interaction between VPS35 and EIF4G1. To test the specificity of the TIF4631-VPS35 genetic interaction even further, we performed an unbiased genomewide screen in yeast where we overexpressed 5,500 yeast genes in the vps35Δ yeast cells, looking for genes that caused toxicity in vps35Δ but not WT cells (i.e., similar to the effects of TIF4631 upregulation). Out of 5,500 genes tested, we only found 25 that caused specific growth defects when upregulated in vps35Δ cells (Supplemental Table S2). If vps35Δ were simply a sick strain and/or sensitive to many different perturbations we would expect to uncover many genes. In contrast, we find a very small number of genes as hits in this screen, including TIF4631, the EIF4G1 homolog. Importantly, we did not identify any other yeast translation initiation factors in this screen, suggesting the effects are specific to TIF4631. Because there are many reasons why some genes might be missed in a large screen and to further test specificity, we directly tested four other yeast translation initiation factors, including TIF4632, and only TIF4631 upregulation was toxic in vps35Δ cells (Supplemental Fig. S2A). Finally, we tested specific mutations in Tif4631 that interfere with its association with eIF4E (L459,460A) or eIF4A (W579A) and therefore impair its function in translation initiation (Park et al., 2011; Schutz et al., 2008). These were less toxic in vps35Δ cells than WT Tif4631 (Supplemental Fig. S2B). Thus, the effect on vps35Δ cells seems to be specific to the EIF4G1 and its function in translation initiation.
Figure 1.

Genetic interaction between α-syn, VPS35 and EIF4G1 in yeast. A) Yeast spotting assays showing that TIF4631 upregulation (under control of a galactose-inducible promoter) is toxic in cells harboring a VPS35 deletion (vps35Δ). Fivefold serial dilution of yeast cells spotted onto glucose (TIF4631 “off”) or galactose (TIF4631 “on”). B) Yeast spotting assays demonstrating both yeast (TIF4631) and human (EIF4G1) translation initiation factor upregulation is toxic in vps35Δ cells. Toxicity is suppressed by co-expression of either human or yeast VPS35. C) A schematic showing the yeast retromer complex comprised of a cargo recognition trimer (Vps35, Vps26, and Vps29) and a trimer recruitment dimer (Vps5 and Vps17). D) Genetic interactions between translation initiation factor and other components of the retromer complex. Overexpression of TIF4631 is toxic in cells lacking several components of the retromer complex (Vps26 and Vps17). E) The PD-linked VPS35 mutation, D620N, impairs the ability of VPS35 to rescue toxicity from EIF4G1 upregulation in vps35Δ cells. See also Figures S1, S2.
Vps35 is a component of the retromer complex (Fig. 1C), which consists of a well-conserved cargo recognition trimer, comprised of Vps26, Vps29, and Vps35 as well as a trimer recruitment dimer. The trimer recruitment dimer, comprised of Vps5 and Vps17 in yeast has multiple orthologs in mammalian cells known as sorting nexins (Seaman et al., 1998). Deficiencies in other retromer subunits (vps26Δ and vps17Δ) also conferred sensitivity to TIF4631 upregulation (Fig. 1D). Because PD-linked mutations and a genetic deletion of VPS35 may behave differently, we also tested the effects of expression of mutant VPS35 in this assay. Whereas expression of WT human VPS35 rescued toxicity from EIF4G1 upregulation in vps35Δ cells, expression of the PD-linked D620 VPS35 mutation failed to rescue toxicity (Fig. 1E). This result is consistent with the disease-causing mutation in VPS35 impairing its function. Thus, two seemingly unrelated PD genes, VPS35 and EIF4G1, are able to interact in a powerful way and, in a broader sense, retromer deficiency sensitizes cells to effects of altering translation initiation factor function.
To extend our studies from yeast and to validate the VPS35–EIF4G1 genetic interaction in the nervous system, we used the nematode C. elegans. The C. elegans cholinergic motor neuron DA9 is an established system to study synapse formation in vivo (Ou et al., 2010). The DA9 axon forms approximately 25 en passant presynapses within a discrete and stereotyped location along its axon, which can be visualized by GFP-tagged synaptic vesicle proteins such as RAB-3 (Fig. 2 A,B). vps-35 single loss-of-function mutants have very subtle effects on the density and size of RAB-3 puncta (Fig. 2 D, G,H), providing a sensitized genetic background within which an enhancement in synapse formation defects can be readily visualized. In this sensitized background, overexpression of human EIF4G1 in the DA9 resulted in significantly reduced number of RAB-3 puncta and dramatic increases in the puncta size (Fig. 2 E, G,H). Overexpression of human EIF4G1 in the DA9 neurons of wild type nematodes did not lead to obvious synaptic phenotypes (Fig. 2 C, G,H). These results demonstrate a synthetic effect between the vps-35 mutation and overexpression of EIF4G1 in neurons and indicate that the VPS35-EIF4G1 genetic interaction that we discovered in yeast is conserved and functional in the nervous system.
Figure 2.

EIF4G1, vps-35 and α–syn genetic interactions in C.elegans neurons. A) A schematic showing the morphology and synaptic pattern of the C.elegans DA9 neuron. A, anterior; D, dorsal. (B–E) Confocal images of the pattern of synaptic vesicle associated protein GFP::RAB-3 in wildtype (B), wyEx6554 (Pitr-1::EIF4G1) (C), vps-35 mutant (D), and vps-35, wyEx6554 (E). (F–G) Quantifications of the RAB-3 puncta density (F) and the average puncta size (G). n=10–15 worms in each genotype. *: P<0.05; ***: P<0.001. One-way ANOVA. Mutant α-syn (A53T) enhances the effects of vps-35 in synapse formation in DA9 neurons. (H–K) Confocal images of SV-associated protein GFP::RAB-3 pattern in wildtype (H), wyEx5613 (Pitr-1::α-syn(A53T)mCherry) (I), vps-35 (J), and vps-35, wyEx6513 (k). (L–M) Quantifications of the RAB-3 puncta density (L) and the average puncta size (M). n=10–15 worms in each genotype. **: P<0.01; ***: P<0.001. One-way ANOVA. Scale bar, 10µm.
We next investigated the mechanistic basis for the potent genetic interaction between VPS35 and EIF4G1. In other words, why would vps35Δ and retromer deficiency sensitize cells to translation initiation factor upregulation? Since EIF4G1 is a scaffold protein for the assembly of a multi-subunit translation initiation complex (Villa et al., 2013), its upregulation might cause stoichiometric imbalances leading to defects in translation leading to an accumulation of misfolded and unfolded proteins. Impairments in retromer function would potentially impede the cell’s ability to effectively handle these misfolded proteins. To test this hypothesis, we used Hsp104-GFP as a misfolded protein sensor. Hsp104 is a protein disaggregase (Shorter, 2008), diffusely localized throughout the cytoplasm and nucleus under normal conditions but accumulates at sites of protein misfolding and aggregation. The appearance of Hsp104-GFP foci indicates that cells are under proteotoxic stress (Oromendia et al., 2012). In WT cells, vps35Δ cells or cells with TIF4631 upregulated, Hsp104-GFP was diffusely localized throughout the cell with only occasional cells harboring one or two Hsp104-GFP foci (Fig. 3A), consistent with the cell’s normal ability to cope with the consequences of translation initiation factor upregulation. In contrast, in vps35Δ cells, TIF4631 upregulation resulted in a dramatic increase in the percentage of cells with Hsp104-GFP foci and in the number of foci per cell (Fig. 3A). Restoring VPS35 function to vps35Δ cells was sufficient to reduce the number of Hsp104-GFP foci caused by TIF4631 upregulation (Fig. 3A). Thus, retromer deficiency, in situations when EIF4G1 is upregulated, leads to the accumulation of protein aggregates, which could be due to problems in protein folding and/or impairments in the ability to effectively handle or clear protein aggregates.
Figure 3.

Translation initiation factor upregulation results in accumulation of misfolded proteins and activation of UPR and is normally antagonized by Vps35 and the retromer complex via Vps10/sortilin. A) Localization of the misfolded protein sensor Hsp104-GFP in indicated yeast strains. Hsp104-GFP is distributed in a mostly diffuse pattern in WT or vps35Δ cells and in WT cells over-expressing TIF4631. However, abundant Hsp104-GFP foci form as a result of overexpression of TIF4631 vps35Δ cells, indicating the presence of unfolded or misfolded proteins. Restoring Vps35 function to these cells is sufficient to reduce Hsp104-GFP foci. B) Yeast spotting assays showing that deletion of mediators of the UPR, IRE1 and HAC1, impair the cellular response to upregulation of TIF4631. TIF4631 upregulation is toxic ire1Δ and hac1Δ yeast cells. C) Upregulation of TIF4631 in vps35Δ cells leads to a 3-fold increase in the activation of the UPR, measured using a LacZ reporter construct under control of the UPR element (UPRE). (D,E) VPS10 functions downstream of VPS35 to mediate protection against upregulation TIF4631. D) Upregulation of galactose-inducible yeast or human translation initiation factors (TIF4631 or EIF4G1) is toxic in vps10Δ yeast cells. E) Co-expression of VPS10 is able to fully rescue the toxicity of TIF4631 upregulation in vps35Δ cell. See also Figures S3
Because vps35Δ cells accumulated misfolded proteins upon TIF4631 upregulation, and since the endoplasmic reticulum (ER) is required for the proper folding of most secreted and membrane proteins in eukaryotic cells, we next asked whether defects in folding of these proteins causes ER stress. To combat protein misfolding, cells activate the unfolded protein response (UPR), a cellular program to boost protein folding, promote the degradation of misfolded proteins, and attenuate protein translation (Walter and Ron, 2011). We used two independent approaches to test if TIF4631 upregulation in vps35Δ cells activated the UPR. First, we used a LacZ reporter fused to unfolded protein response elements (UPRE) (Patil and Walter, 2001). Expression of TIF4631 in vps35Δ caused a threefold increase in ER stress relative to control cells (Fig. 3C). Next, we upregulated TIF4631 in hac1Δ or ire1Δ cells. Hac1 and Ire1 are critical mediators of the UPR. Upregulation of TIF4631 or EIF4G1 inhibited growth in hac1Δ or ire1Δ cells (Fig. 3B), indicating that a functional UPR is required to deal with misfolded proteins that accumulate in vps35Δ cells expressing TIF4631 or EIF4G1. Taken together, these data demonstrate that Vps35 and retromer are able to mitigate the deleterious effects of protein misfolding caused by TIF4631/EIF4G1.
Another way that misfolded proteins are normally processed is by the sortilin family of proteins, which includes the yeast sortilin homolog Vps10 (Dennes et al., 2002). Vps10 helps sort misfolded proteins from the trans-golgi network (TGN) to prevacuolar endosomes for degradation (Reitz, 2012). Vps35 and the retromer complex help recycle Vps10 back from endosomes to the TGN where it can continue its surveillance for misfolded proteins and, if necessary, initiate further rounds of protein sorting (Seaman et al., 1997). Indeed, Vps35 was originally discovered in yeast based on its role in endosome-to-Golgi retrieval of Vps10 (Seaman et al., 1997). We next tested if the protective effects of Vps35 in dealing with misfolded proteins were mediated by Vps10. In other words, we hypothesized that Vps10 could facilitate the sorting of misfolded proteins towards the vacuole for degradation and Vps35 would help to retrieve Vps10 to the Golgi for further rounds of protein quality control. Consistent with this hypothesis, upregulation of yeast TIF4631 or human EIF4G1 was toxic in yeast cells lacking VPS10 (Fig. 3D). Remarkably, overexpression of VPS10 was able to fully suppress the toxicity associated with TIF4631 upregulation in vps35Δ cells (Fig. 3E). These results strongly suggest that Vps10/sortilin acts downstream or in parallel to Vps35 to mediate protection against TIF4631. Emerging roles for sortilin proteins in other neurodegenerative diseases, such as FTLD and Alzheimer’s disease (Lane et al., 2012; Reitz et al., 2013) and our results linking sortilin to Vps35 and PD now warrant further examination of a role for sortilin proteins in PD. Our data are consistent with the hypothesis that Tif4631 upregulation causes alterations in translation in the absence of Vps35, which could lead to protein misfolding and impairments in sortilin to cope with these misfolded proteins. See Supplemental Results and Supplemental Fig. S3, where we test this hypothesis by measuring protein synthesis directly with polysome-profiling and ribosome-profiling experiments.
Given the strong interactions between two PD genes VPS35 and EIF4G1 in both yeast cells and C. elegans neurons we next investigated whether these genes might also interact with α–synuclein (α–syn). Point mutations or multiplications in the α–syn gene are a cause of some rare forms of PD and α–syn is the major component of Lewy bodies, the hallmark pathological lesion in PD (Lee and Trojanowski, 2006). To test for interactions between α–syn and EIF4G1 or VPS35, we used the yeast α–syn model. Numerous studies have shown that expression of α–syn is toxic in yeast cells in a dose-dependent manner (Outeiro and Lindquist, 2003). Deletion of yeast VPS35 enhanced α–syn toxicity whereas the deletion of TIF4631 did not (Fig. 4A). The lack of toxicity enhancement in tif4631Δ could be due to the ability of the related gene TIF4632 to compensate for loss of TIF4631 (Clarkson et al., 2010). Compared to its normal localization on the plasma membrane with occasional localization to cytoplasmic vesicular inclusions in WT cells, α–syn formed more numerous cytoplasmic inclusions in vps35Δ cells and also frequently accumulated in the vacuole (Fig. 4B). Even though deletion of TIF4631 did not enhance α–syn toxicity, upregulation of TIF4631 or human EIF4G1 suppressed toxicity ((Yeger-Lotem et al., 2009); Fig. 4C). Interestingly, the PD-associated EIF4G1variant R1205H was impaired in its ability to suppress α–syn toxicity (Fig. 4C). EIF4G1 is still somewhat controversial as a bona fide PD gene (Blanckenberg et al., 2014; Lesage et al., 2012; Nishioka et al., 2014; Siitonen et al., 2013; Sudhaman et al., 2013; Tucci et al., 2012). Our results functionally connecting EIF4G1 to two other PD genes (VPS35 and α–syn) and the reduced ability of a PD-associated EIF4G1 variant to suppress α–syn toxicity in yeast, provides support for a role of EIF4G1 in PD. Nevertheless, further sequencing of EIF4G1 in additional PD patients and control subjects will be required to resolve the extent to which EIF4G1 mutations cause PD.
Figure 4.

Genetic interaction between α-syn, VPS35 and EIF4G1 in yeast. A) α-Syn toxicity is enhanced in yeast cells lacking VPS35. B) α-Syn-YFP subcellular localization is also altered in vps35Δ cells compared to its localization in WT cells. C) Upregulation of galactose-inducible yeast or human translation initiation factors (TIF4631 or EIF4G1) is sufficient to suppress α-syn toxicity. A PD-linked EIF4G1 variant (R1205H) is impaired in its ability to suppress α-syn toxicity in this yeast assay.
We next sought to validate the genetic interaction between α–syn and VPS35 in neurons and we again used C. elegans (Fig. 2 H–M). Indeed, similar to the findings in yeast, overexpression of mutant α-syn (A53T) in DA9 in the vps-35 mutants dramatically decreased the RAB-3 puncta density and increased the puncta size (Fig. 2 K–M), whereas α-syn (A53T) alone did not affect the DA9 synapse pattern (Fig. 2 I,L,M). The effects of wild type α-syn in the vps-35 mutant background were less severe than the A53T mutant (Fig. 2 L,M). These results confirm that the genetic interaction between VPS35 and α–syn that we identified in yeast functions in a similar way in the nervous system.
We next determined if VPS35 could functionally impact neurodegeneration mediated by α–syn and if PD-linked VPS35 mutations compromise this function. We analyzed neurodegeneration in an established transgenic (tg) mouse synucleinopathy model ((Masliah et al., 2000; Rockenstein et al., 2002) and see Supplemental Results for further characterization of VPS35 and EIF4G1 protein levels and localization in mouse and human brain). Compared to non-tg controls, in α-syn tg mice, we found reduced NeuN positive cells in the CA3 region of the hippocampus (Fig. 5 A,B). We bilaterally injected brains of α-syn tg mice with lentiviral constructs that either upregulated human VPS35 (WT or a PD-linked P316S mutant (Vilarino-Guell et al., 2011)) or inhibited endogenous VPS35 by shRNA-mediated knockdown. Strikingly, upregulation of WT VPS35 was able to almost completely rescue the α–syn-induced neurodegeneration (Fig. 5 A,B). In contrast, upregulation of the PD-linked mutant (VPS35 P316S) or knock-down of endogenous VPS35 resulted in more marked neuronal loss in the hippocampus of the α-syn tg mice (Fig. 5 A,B). We obtained similar results with another PD-linked VPS35 mutant (D620N; data not shown). Upregulation of WT VPS35 also markedly reduced neuro-inflammation in α–syn tg mice. Upregulation of WT VPS35 reduced GFAP immunoreactivity to levels comparable to non-tg sections (Fig. 5 C,D). However, VPS35 P316S upregulation caused a significant increase in GFAP immunoreactivity (Fig. 5 C,D). These data indicate that VPS35 is able to functionally antagonize α–syn-mediated neurodegeneration and extend our findings from yeast and worms to mouse.
Figure 5.
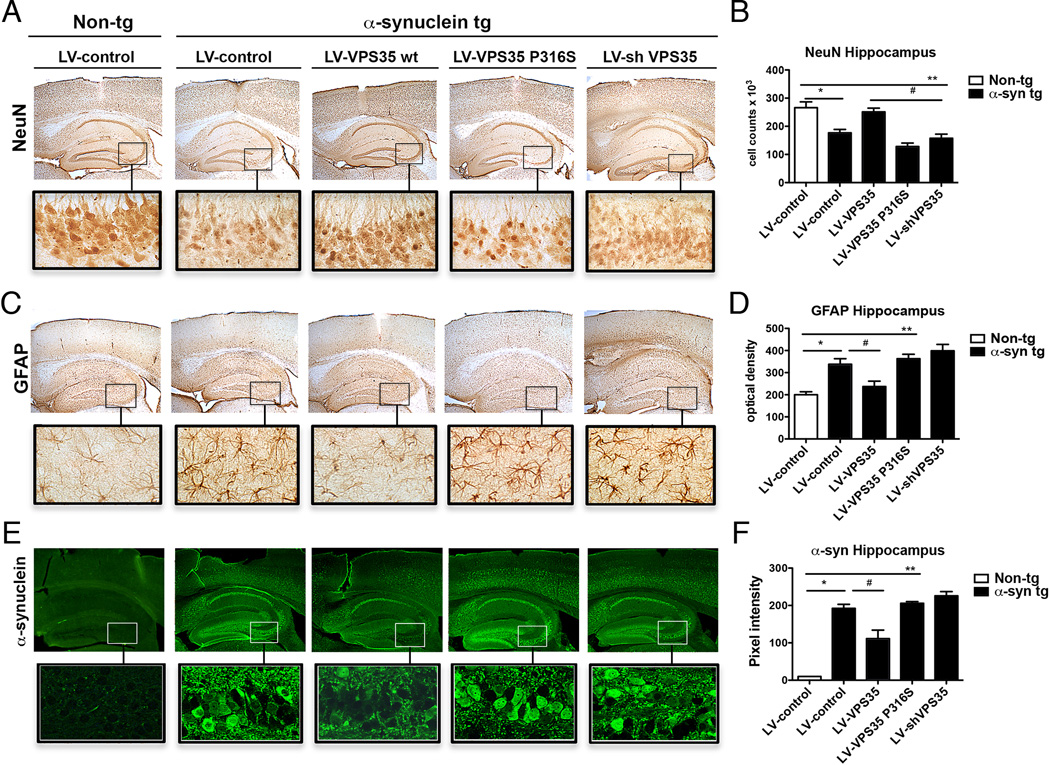
VPS35 increased levels are neuroprotective against α-synuclein mediated toxicity acting on α-syn accumulation in neurons and protecting against neuronal loss and astrogliosis. Proteins immunoreactivity in hippocampus of non-tg vehicle mice (Non-tg, LV-control) and α-syn tg mice injected with vehicle (LV-control), lentivirus VPS35 wt (LV-VPS35 wt), VPS35 mutant (LV-P316S) and silenced VPS35 (shVPS35) has been analyzed. A,B) NeuN immunostaining to evaluate the neuronal loss in hippocampus of non-tg vehicle mice and α-syn tg mice overexpressing VPS35 wt, mutant, VPS silencing and relative signal quantification. C,D) GFAP immunostaining to evaluate astrogliosis in hippocampus of non-tg vehicle mice and α-syn tg mice overexpressing VPS35 wt, mutant, VPS silencing and relative signal quantification. E,F) α-syn immunostaining in hippocampus of non-tg vehicle mice and α-syn tg mice overexpressing VPS35 wt, mutant, VPS35 silencing and relative signal quantification. See also Figures S4–S7.
We investigated the mechanism by which VPS35 protected against neurodegeneration in α–syn tg mice. Because α–syn protein levels are closely linked to PD (multiplication of the SNCA locus causes early onset PD (Singleton et al., 2003) and common variants near the SNCA locus are associated with sporadic PD (Lill et al., 2012)) we tested the hypothesis that VPS35 upregulation protects against α–syn accumulation. Upregulation of WT VPS35 reduced the intra-neuronal accumulation of α-syn in tg mice to levels comparable to non-tg mice (Fig. 5 E,F), whereas upregulation of VPS35 P316S or VPS35 silencing using shRNA further increased the accumulation of α–syn (Fig. 5 E,F). These findings suggest that VPS35 functions, at least in part, to suppress α–syn accumulation while knockdown or expression of PD-linked mutant VPS35 displays defective α–syn clearance resulting in widespread accumulation. Together, these results suggest that increased levels of VPS35 are neuroprotective against α–syn-mediated toxicity. VPS35 controls α–syn accumulation in neurons and protects brain tissues against neuronal loss and astrogliosis.
We also tested VPS35 in another synucleinopathy model. Emerging evidence indicates that pathological forms of α-syn can spread from neuron-to-neuron in a prion-like manner (Luk et al., 2012) and there is evidence that exogenous α-syn may utilize endosomal transport as part of the cell-to-cell spreading process (Volpicelli-Daley et al., 2011). Given the role of VPS35 and retromer in endosomal trafficking (Burd and Cullen, 2014), we studied the effect of VPS35 modulation on the uptake of exogenous α–syn fibrils into rodent cortical neurons. We infected cortical neurons after 7DIV for 5 days with lentiviruses encoding WT VPS35 or two VPS35 mutations genetically linked in humans to PD (P316S and D620N). We also lowered VPS35 levels with siRNA. Subsequently, neurons were exposed to 1 µ M of exogenous human recombinant α–syn seeds (see Supplemental Fig. S6 for detailed characterization of the recombinant α-syn seeds) for 24 hours. We found that intracellular levels of exogenous α–syn seeds were significantly decreased in cortical neurons transduced with WT VPS35 expressing lentivirus compared to the control infected neurons (Supplemental Fig. S7). Expressing the two PD-linked VPS35 mutants or lowering VPS35 levels with siRNA had the opposite effect (Supplemental Fig. S7). These data suggest that endosomal dysfunction caused by defects in VPS35 and the retromer complex impair the ability of neurons to cope with the accumulation of α–syn seeds, which could hasten the prion-like spread of PD pathology. It also suggests the possibility that approaches to boost retromer function might help to mitigate α-syn accumulation and propagation. Indeed a small molecule stabilizer of the retromer complex decreases pathogenic processing of amyloid precursor protein in Alzheimer’s disease (Mecozzi et al., 2014).
Discussion
Using two genome-wide screens in yeast combined with mechanistic studies in yeast and functional validation in an animal model, we provide evidence that two distinct PD risk genes, VPS35 and EIF4G1, interact genetically and functionally. We connect these two genes to a cellular pathway involved in the surveillance and processing of misfolded proteins and show how impairments in this pathway result in the accumulation of protein misfolding and proteotoxic stress and how this can exacerbate the effects of α–syn accumulation. We provide evidence that VPS35 can protect against neurodegeneration and α–syn accumulation in transgenic mice and in a primary neuron α–syn prion-like seeding model.
The toxicity caused by translation initiation factor upregulation in the absence of a functional retromer complex can be a result of altered translational landscape in the cell. Because eIF4G1 is a scaffold protein on which translation initiation complexes assemble, alterations in complex stoichiometry could result in increased or decreased translation or even alterations in the profile of mRNAs translated. Such alterations in translation would result in numerous defects in protein targeting, sorting, and folding, which are revealed when protein quality control machinery is compromised (e.g., retromer or sortilin deficiency, defects in UPR machinery, or even decline of proteostasis machinery with aging (Clarkson et al., 2010)). Future studies will be aimed at quantitatively defining the contribution of alterations in translation per se to neurodegenerative disease phenotypes.
We also demonstrate that a sortilin protein acts downstream of Vps35 to mediate the protection against translation initiation factor upregulation. SORT1, the human ortholog of yeast VPS10, is associated with other neurodegenerative diseases, including Alzheimer’s disease and frontotemporal dementia (Hu et al., 2010; Lewis and Golde, 2010), and our studies now suggest a potential role for this gene in PD pathogenesis. Beyond sortilin, we propose that the human homologs of the other genes from the vps35Δ and tif4631Δ screens may help to suggest novel PD susceptibility genes. Indeed, one of the strongest hits from the tif4631Δ screen, INP53, is the yeast homolog of human SYNJ1, which was recently linked to parkinsonism (Krebs et al., 2013; Quadri et al., 2013). Thus, in a way, the screen has already worked – producing another PD susceptibility gene and it now warrants closer examination of the other hits from the yeast screens (Supplemental Table 1) for connections to PD in humans. Large-scale exome sequencing projects are underway (Novarino et al., 2014) and promise to expand and clarify the genetic landscape of neurodegenerative diseases, including PD (Bonifati, 2014). Novel functional approaches to assess biological functions of candidate disease genes and risk factors will hopefully aid in the interpretation and analysis of the human sequencing data.
The functional links between Vps35 and α–synuclein reported here in yeast, worms, and mouse underscore a role of perturbations in vesicle trafficking pathways as a component of PD pathogenesis (Cooper et al., 2006; Gitler, 2008; MacLeod et al., 2013). The unexpected link between translation initiation factor eIF4G1 and Vps35, including the role of protein misfolding and activation of the unfolded protein response, illuminates translation regulation and protein quality control pathways as well. Protein misfolding and impairments in protein degradation lie at the heart of neurodegenerative diseases, including PD (Forman et al., 2004). These results are also strikingly consistent with a recent report of alterations in translational control mediating neurodegeneration in PD caused by LRRK2 mutations (Martin et al., 2014).
Our findings that the retromer complex can normally combat the accumulation of unfolded and misfolded proteins, masking the toxic effects of alterations in levels of translation initiation factor expression, helps to explain how mutations in Vps35 could lead to PD and how other genetic or environmental susceptibilities that might lead to misregulated translation could also result in neurodegeneration (Lee and Trojanowski, 2006). Because expression of Vps35 or sortilin proteins can suppress the toxicity associated with eIF4G1 upregulation, screening for compounds that boost retromer or sortilin levels or function could be an effective therapeutic strategy. The yeast models presented here should provide a tractable system for such screens.
Experimental Procedures
Yeast experiments
We generated the vps35Δ and tif4631Δ query strains by replacing the VPS35 and TIF4631 coding region with the natMX resistance cassette in strain Y7092; MAT can1Δ::STE2pr-Sp_his5 lyp1Δ his3Δ1 leu2Δ0 ura3Δ0 met15Δ0. Strains were manipulated and media prepared using standard techniques. We obtained the yeast VPS35 and TIF4631 entry clone constructs from the Yeast FLEXGene library (Harvard Institute of Proteomics). For the UPRE-LacZ β-galactosidase activity assays, yeast strains were grown to mid-log phase, induced with 2% Galactose (or repressed with 2% Glucose) for 4 hours, and then lysed with Y-PER Plus Extraction Reagent (Thermo, #78999) with 1X Halt Protease Inhibitor Cocktail. Afterwards, protocol was followed as per the Yeast β-Galactosidase Activity Kit (Thermo, #75768).
vps35Δ and tif4631Δ synthetic lethality screens
We used synthetic genetic array (SGA) analysis to identify non-essential yeast deletions that are synthetic lethal with vps35Δ or tif4631Δ. We performed this screen essentially as described in (Tong and Boone, 2006), with some modifications, using a Singer RoToR HDA (Singer Instruments, Somerset, UK). We mated the MAT vps35Δ or tif4631Δ query strain to the yeast haploid deletion collection of non-essential genes (MATa, each gene deleted with KanMX cassette (confers resistance to G418). Following diploid selection and sporulation, we selected double-mutant haploids. Colony sizes were measured using the ht-colony-measurer software (Collins et al., 2006). The raw values were normalized dividing them by the median colony size of the plate. We performed the entire screen three independent times. The data from three rounds were averaged and the difference between double and single mutant colony size was calculated and used to call a synthetic sick/lethal interaction. We used GOTermFinder to identify significant gene ontology (GO) terms enriched as hits in each screen. A description of how P-values are calculated is presented in (Boyle et al., 2004).
Worm experiments
Worms were raised on NGM plates at 20°C, using OP50 E.coli as a food source. N2 Bristol was used as the wild-type reference strain. The following mutant strains were obtained through the Caenorhabditics Genetics Center: KN555 vps-35(hu68) II. Expression clones were made in the pSM vector, a derivative of pPD49.26 (A kind gift from A. Fire) with extra cloning sites. The wyIs85 integrated worm strain was described as previous (Klassen and Shen, 2007). The transgenic strains were generated as the following: wyEx6554 (Pitr-1::EIF4G1): The human EIF4G1 cDNA was amplified from Gateway® entry clone vector containing full length cDNA of EIF4G1 and subcloned into the Pmig-13 Δ pSM between the AscI and EcoRV sites. The Pmig-13 promoter was then replaced by Pitr-1 promoter between SphI and AscI sites. The plasmid was then injected into vps-35;wyIs85 at 4.0 ng µL−1, with 40 ng µL−1 Podr-1::GFP. Two extrachromosomal arrays were generated, and all showed similar effects.wyEx5609 (Pitr- 1::α-synuclein::mcherry), wyEx5609 (Pitr-1::α-synucleinA53T::mcherry): The human α-synuclein wildtype and A53T cDNA was subcloned from pDONR-α–syn and pDONR-α–synA53T, respectively, into worm expression vector Pitr-1::mcherry Δ pSM. The plasmids were injected into wyIs85 at 50 ng µL−1, with 40 ng µL−1 Podr-1::GFP. Images of fluorescently tagged fusion proteins were captured in live C. elegans using a plan-Apchromat 40×1.3 objective on a Zeiss LSM710 confocal microscope, using identical image and laser setting for each genotype. Young adult hermaphrodites were immobilized using 3mM levamisole (sigma) dissolved in M9 buffer. Straightened DA9 dorsal cords were extracted from the confocal images using the “straighten to line ” in the “ROI” plug-in program of ImageJ, then aligned along the anteroposterior axis using the DA9 commisure turning-point as a reference mark. The puncta number and size were calculated using the “analyze particles” program of Image J.
Transgenic mouse lines and injections of lentiviral vectors
For this study, mice over-expressing α-synuclein from the Thy1 promoter (Line 61) were utilized (Rockenstein et al., 2002). This model was selected because mice from this line develop intraneuronal α-synuclein aggregates distributed through the brain including the thalamus and substantia nigra similar to what has been described in LBD. A cohort of n=10 non-tg and 40 tg (LV-control, LV-VPS35 wt, LV-VPS35 P316S, LV-shVPS35, n=10 mice per group) were injected with 3 µL of the lentiviral preparations (2.5 × 107 TU) into the hippocampus (using a 5 µL Hamilton syringe). Briefly, mice were placed under anesthesia on a Koft stereotaxic apparatus and coordinates (hippocampus: AP 2.0 mm, lateral 1.5 mm, depth 1.3 mm) were determined as per the Franklin and Paxinos atlas. The lentiviral vectors were delivered using a Hamilton syringe connected to a hydraulic system to inject the solution at a rate of 1 µL every 2 min. To allow diffusion of the solution into the brain tissue, the needle was left for an additional 5 min after the completion of the injection. Mice survived for 3 months after the lentiviral injection. Brains and peripheral tissues were removed and divided sagitally. The right hemibrain was post-fixed in phosphate-buffered 4% PFA (pH 7.4) at 4°C for 48 hours for neuropathological analysis, while the left hemibrain was snap-frozen and stored at −70°C for subsequent protein analysis.
Immunohistochemical and image analysis
Analysis was performed using free-floating, 40 µm thick, vibratome, blind-coded sections, as previously described (Games et al., 2013). Briefly, sections were incubated overnight at 4°C with antibodies against total α-syn (1:500, affinity purified rabbit polyclonal, Millipore) (Masliah et al., 2000), mouse monoclonal α-syn (clone SYN 211 Life Technologies, 1:300) and VPS35 (1:500; Millipore), followed by biotin-tagged antirabbit or anti-mouse IgG1 (1:100, Vector Laboratories, Inc., Burlingame, CA) secondary antibodies, Avidin D-HRP (1:200, ABC Elite, Vector), and visualized with diaminobenzidine (DAB). Sections were scanned with a digital Olympus bright field digital microscope (BX41). Analysis of neurodegenerative pathology was performed in vibratome sections immunolabeled with antibodies against the pan-neuronal marker NeuN (1:500, Millipore) and the astroglial marker glial fibrillary acidic protein (GFAP, 1:1000, Millipore) (Games et al., 2013; Masliah et al., 2000). Sections reacted with biotinylated secondary antibodies, Avidin D-HRP, and visualized with DAB. GFAP was analyzed with a digital Olympus bright field digital microscope (BX41). For each case a total of three sections (4 digital images per section at 400 × magnification) were obtained from the CA1 and CA3 region of the hippocampus and analyzed as previously described with the image J program (NIH) to obtain optical density, levels were corrected to background. Sections labeled with Iba-1, were analyzed utilizing the Image-Pro Plus program (Media Cybernetics, Silver Spring, MD) (10 digital images per section at 400 × magnification) and analyzed in order to estimate the average number of immuno-labeled cells per unit area (mm2) and the average intensity of the immunostaining (corrected optical density). Additional analysis of α-syn was performed in vibratome sections immunolabeled with the SYN211 monoclonal antibodies and visualized with FITC and laser scanning confocal microscopy as previously described (Games et al., 2013).
The numbers of NeuN-immunoreactive neurons were estimated utilizing unbiased stereological methods (Overk et al., 2009). Hemi-sections containing the neocortex, hippocampus and striatum were outlined using an Olympus BX51 microscope running StereoInvestigator 8.21.1 software (Micro-BrightField, Cochester, VT). Grid sizes for the hippocampal CA3 and CA1 pyramidal layers were: 300 × 300 µm and the counting frames were and 50 × 50 µm, respectively. The average coefficient of error for each region was 0.9. Sections were analyzed using a 100 × 1.4 PlanApo oil-immersion objective. A 5 µm high dissector, allowed for 2 µm top and bottom guard-zones.
Preparation and treatment of mouse primary cortical neurons
Primary cortical neuronal cultures were prepared from the neocortices of 1-day-old mice (P1) as previously described (Hirling et al., 2000). Cortical hemispheres were isolated in Hank’s balanced salt solution (HBSS, Invitrogen) and incubated in 0.1% papain (Sigma-Aldrich). After 10 minutes, the papain was removed, and the reaction was blocked using 30% Fetal Calf Serum (FCS, Invitrogen). Individual cells were obtained by trituration (8×) and then the cells were resuspended in MEM medium 1× (Invitrogen) supplemented with 0.5mM L-Glutamine 200mM (Invitrogen), 5% heat-inactivated fetal calf serum (FCS, Invitrogen), and 2% Penicilin/Streptomycin (Pen/Strep, Invitrogen) for 4 hours. Cells were seeded at density of 3.5×105 cells on 35mm dish and in 12 well plates a density of 1×105 with miscrope cover glass (Fisherbrand) both previously treated with 0.01% poly-L-Lysine (Sigma) for 1 hour. After 4 hours the media was removed and changed with NeuroBasal (NB) medium (Invitogen) supplemented with 1× B27 (Invitrogen), 0.5mM Glutamine and 2% Pen/Strep. After 10 days in vitro (DIV), the differentiated cortical neurons were treated following the experimental procedures.
Lentivirus infection and gene Silencing
The VPS35 wt and mutants cDNA was PCR amplified and cloned into the lentivector to generate LV-VPS35wt, P316S and D620N as previously described (Spencer et al., 2009) to generate LV-shVPS35. The control shRNA lentivector (LV-shLuc) contains an shRNA directed against firefly luciferase. The lentivirus vector expressing the human wild-type α-syn has been previously described (Bar-On et al., 2008). Lentiviruses were prepared by transient transfection in 293T cells. VPS35 gene was silenced by transfection with siRNA VPS35 (Silencer Select, Ambion, Life Technologies). The siRNA VPS35 was co-incubated with Lipofectamine RNAiMAX reagent (Invitrogen) and then added at the cells. Lipofectamine and siRNA were added at the cells cultured in medium without serum and antibiotic for 5 hours and then shifted back with complete medium. For siRNA negative control (Silencer Select, Ambion, Life Technologies).
Supplementary Material
Highlights.
Discovery of unexpected connections between PD genes EIF4G1, VPS35 and α–synuclein
Sortilin functions downstream of VPS35 to protect against misfolded proteins
VPS35 loss-of-function enhances α–synuclein toxicity in yeast, worms and mouse.
Hits from VPS35 and EIF4G1 genetic screens predict new candidate genes for Parkinson’s disease, including SYNJ1
Acknowledgments
We thank Chris Burd for helpful suggestions and discussions about retromer and protein trafficking and advice on experiments. We thank Martin Duennwald (Western University) for the yeast UPRE-LacZ plasmid. This work was supported by NIH grants 1R01NS065317 and 1R01NS07366 (A.D.G.) and 1R01AG18440 (E.M.). N.D. is supported by NIH grant K12-GM088033: Institutional Research and Academic Career Development Postdoctoral Award (IRACDA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bar-On P, Crews L, Koob AO, Mizuno H, Adame A, Spencer B, Masliah E. Statins reduce neuronal alpha-synuclein aggregation in in vitro models of Parkinson's disease. J. Neurochem. 2008;105:1656–1667. doi: 10.1111/j.1471-4159.2008.05254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanckenberg J, Ntsapi C, Carr JA, Bardien S. EIF4G1 R1205H and VPS35 D620N mutations are rare in Parkinson's disease from South Africa. Neurobiol. Aging. 2014;35:445 e441–445 e443. doi: 10.1016/j.neurobiolaging.2013.08.023. [DOI] [PubMed] [Google Scholar]
- Bonifati V. Genetics of Parkinson's disease--state of the art, 2013. Parkinsonism & related disorders. 2014;20(Suppl 1):S23–S28. doi: 10.1016/S1353-8020(13)70009-9. [DOI] [PubMed] [Google Scholar]
- Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, Sherlock G. GO::TermFinder--open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004;20:3710–3715. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd C, Cullen PJ. Retromer: a master conductor of endosome sorting. Cold Spring Harb. Perspect. Biol. 2014;6:a016774. doi: 10.1101/cshperspect.a016774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Dachsel JC, Vilarino-Guell C, Lincoln SJ, Lepretre F, Hulihan MM, Kachergus J, Milnerwood AJ, Tapia L, Song MS, et al. Translation initiator EIF4G1 mutations in familial Parkinson disease. Am. J. Hum. Genet. 2011;89:398–406. doi: 10.1016/j.ajhg.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Clarkson BK, Gilbert WV, Doudna JA. Functional overlap between eIF4G isoforms in Saccharomyces cerevisiae. PLoS One. 2010;5:e9114. doi: 10.1371/journal.pone.0009114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SR, Schuldiner M, Krogan NJ, Weissman JS. A strategy for extracting and analyzing large-scale quantitative epistatic interaction data. Genome Biol. 2006;7:R63. doi: 10.1186/gb-2006-7-7-r63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennes A, Madsen P, Nielsen MS, Petersen CM, Pohlmann R. The yeast Vps10p cytoplasmic tail mediates lysosomal sorting in mammalian cells and interacts with human GGAs. J. Biol. Chem. 2002;277:12288–12293. doi: 10.1074/jbc.M112295200. [DOI] [PubMed] [Google Scholar]
- Do CB, Tung JY, Dorfman E, Kiefer AK, Drabant EM, Francke U, Mountain JL, Goldman SM, Tanner CM, Langston JW, et al. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson's disease. PLoS Genet. 2011;7:e1002141. doi: 10.1371/journal.pgen.1002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman MS, Trojanowski JQ, Lee VM. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat. Med. 2004;10:1055–1063. doi: 10.1038/nm1113. [DOI] [PubMed] [Google Scholar]
- Games D, Seubert P, Rockenstein E, Patrick C, Trejo M, Ubhi K, Ettle B, Ghassemiam M, Barbour R, Schenk D, et al. Axonopathy in an alpha-synuclein transgenic model of Lewy body disease is associated with extensive accumulation of C-terminal-truncated alpha-synuclein. Am. J. Pathol. 2013;182:940–953. doi: 10.1016/j.ajpath.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler AD. Beer and Bread to Brains and Beyond: Can Yeast Cells Teach Us about Neurodegenerative Disease? Neurosignals. 2008;16:52–62. doi: 10.1159/000109759. [DOI] [PubMed] [Google Scholar]
- Gitler AD, Chesi A, Geddie ML, Strathearn KE, Hamamichi S, Hill KJ, Caldwell KA, Caldwell GA, Cooper AA, Rochet JC, et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 2009;41:308–315. doi: 10.1038/ng.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, Kay DM, Doheny KF, Paschall J, Pugh E, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson's disease. Nat. Genet. 2010;42:781–785. doi: 10.1038/ng.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison MS, Hung CS, Liu TT, Christiano R, Walther TC, Burd CG. A mechanism for retromer endosomal coat complex assembly with cargo. Proc. Natl. Acad. Sci. USA. 2014;111:267–272. doi: 10.1073/pnas.1316482111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirling H, Steiner P, Chaperon C, Marsault R, Regazzi R, Catsicas S. Syntaxin 13 is a developmentally regulated SNARE involved in neurite outgrowth and endosomal trafficking. Eur. J. Neurosci. 2000;12:1913–1923. doi: 10.1046/j.1460-9568.2000.00076.x. [DOI] [PubMed] [Google Scholar]
- Hu F, Padukkavidana T, Vaegter CB, Brady OA, Zheng Y, Mackenzie IR, Feldman HH, Nykjaer A, Strittmatter SM. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010;68:654–667. doi: 10.1016/j.neuron.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Parkinson Disease Genomics, C. Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM, Saad M, Simon-Sanchez J, Schulte C, Lesage S, et al. Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet. 2011;377:641–649. doi: 10.1016/S0140-6736(10)62345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Parkinson's Disease Genomics, C. and Wellcome Trust Case Control, C. A two-stage meta-analysis identifies several new loci for Parkinson's disease. PLoS Genet. 2011;7:e1002142. doi: 10.1371/journal.pgen.1002142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovic J. Parkinson's disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry. 2008;79:368–376. doi: 10.1136/jnnp.2007.131045. [DOI] [PubMed] [Google Scholar]
- Krebs CE, Karkheiran S, Powell JC, Cao M, Makarov V, Darvish H, Di Paolo G, Walker RH, Shahidi GA, Buxbaum JD, et al. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Human Mutat. 2013;34:1200–1207. doi: 10.1002/humu.22372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane RF, St George-Hyslop P, Hempstead BL, Small SA, Strittmatter SM, Gandy S. Vps10 family proteins and the retromer complex in aging-related neurodegeneration and diabetes. J. Neurosci. 2012;32:14080–14086. doi: 10.1523/JNEUROSCI.3359-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Trojanowski JQ. Mechanisms of Parkinson's disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron. 2006;52:33–38. doi: 10.1016/j.neuron.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Lesage S, Condroyer C, Klebe S, Lohmann E, Durif F, Damier P, Tison F, Anheim M, Honore A, Viallet F, et al. EIF4G1 in familial Parkinson's disease: pathogenic mutations or rare benign variants? Neurobiol. Aging. 2012;33:2233 e2231–2233 e2235. doi: 10.1016/j.neurobiolaging.2012.05.006. [DOI] [PubMed] [Google Scholar]
- Lewis J, Golde TE. Sorting out frontotemporal dementia? Neuron. 2010;68:601–603. doi: 10.1016/j.neuron.2010.11.014. [DOI] [PubMed] [Google Scholar]
- Li H, Teo YY, Tan EK. Patterns of linkage disequilibrium of LRRK2 across different races: implications for genetic association studies. PLoS One. 2013;8:e75041. doi: 10.1371/journal.pone.0075041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill CM, Roehr JT, McQueen MB, Kavvoura FK, Bagade S, Schjeide BM, Schjeide LM, Meissner E, Zauft U, Allen NC, et al. Comprehensive research synopsis and systematic meta-analyses in Parkinson's disease genetics: The PDGene database. PLoS Genet. 2012;8:e1002548. doi: 10.1371/journal.pgen.1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, Marder KS, Honig LS, Clark LN, Small SA, et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson's disease risk. Neuron. 2013;77:425–439. doi: 10.1016/j.neuron.2012.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin I, Kim JW, Lee BD, Kang HC, Xu JC, Jia H, Stankowski J, Kim MS, Zhong J, Kumar M, et al. Ribosomal Protein s15 Phosphorylation Mediates LRRK2 Neurodegeneration in Parkinson's Disease. Cell. 2014;157:472–485. doi: 10.1016/j.cell.2014.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- Massano J, Bhatia KP. Clinical approach to Parkinson's disease: features, diagnosis, and principles of management. Cold Spring Harb. Perspect. Med. 2012;2:a008870. doi: 10.1101/cshperspect.a008870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecozzi VJ, Berman DE, Simoes S, Vetanovetz C, Awal MR, Patel VM, Schneider RT, Petsko GA, Ringe D, Small SA. Pharmacological chaperones stabilize retromer to limit APP processing. Nat. Chem. Biol. 2014;10:443–449. doi: 10.1038/nchembio.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklavc P, Ehinger K, Thompson KE, Hobi N, Shimshek DR, Frick M. Surfactant Secretion in LRRK2 Knock-Out Rats: Changes in Lamellar Body Morphology and Rate of Exocytosis. PLoS One. 2014;9:e84926. doi: 10.1371/journal.pone.0084926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M, et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat. Genet. 2014a;46:989–993. doi: 10.1038/ng.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Saad M, Noyce AJ, Keller MF, Schrag A, Bestwick JP, Traynor BJ, Gibbs JR, Hernandez DG, Cookson MR, et al. Genetic comorbidities in Parkinson's disease. Hum. Mol. Genet. 2014b;23:831–841. doi: 10.1093/hmg/ddt465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka K, Funayama M, Vilarino-Guell C, Ogaki K, Li Y, Sasaki R, Kokubo Y, Kuzuhara S, Kachergus JM, Cobb SA, et al. EIF4G1 gene mutations are not a common cause of Parkinson's disease in the Japanese population. Parkinsonism Relat. Disord. 2014;20:659–661. doi: 10.1016/j.parkreldis.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novarino G, Fenstermaker AG, Zaki MS, Hofree M, Silhavy JL, Heiberg AD, Abdellateef M, Rosti B, Scott E, Mansour L, et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science. 2014;343:506–511. doi: 10.1126/science.1247363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oromendia AB, Dodgson SE, Amon A. Aneuploidy causes proteotoxic stress in yeast. Genes Dev. 2012;26:2696–2708. doi: 10.1101/gad.207407.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou CY, Poon VY, Maeder CI, Watanabe S, Lehrman EK, Fu AK, Park M, Fu WY, Jorgensen EM, Ip NY, et al. Two cyclin-dependent kinase pathways are essential for polarized trafficking of presynaptic components. Cell. 2010;141:846–858. doi: 10.1016/j.cell.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro TF, Lindquist S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science. 2003;302:1772–1775. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overk CR, Kelley CM, Mufson EJ. Brainstem Alzheimer's-like pathology in the triple transgenic mouse model of Alzheimer's disease. Neurobiol. Dis. 2009;35:415–425. doi: 10.1016/j.nbd.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EH, Walker SE, Lee JM, Rothenburg S, Lorsch JR, Hinnebusch AG. Multiple elements in the eIF4G1 N-terminus promote assembly of eIF4G1*PABP mRNPs in vivo. EMBO J. 2011;30:302–316. doi: 10.1038/emboj.2010.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Patil C, Walter P. Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr. Opin. Cell Biol. 2001;13:349–355. doi: 10.1016/s0955-0674(00)00219-2. [DOI] [PubMed] [Google Scholar]
- Quadri M, Fang M, Picillo M, Olgiati S, Breedveld GJ, Graafland J, Wu B, Xu F, Erro R, Amboni M, et al. Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Human Mutat. 2013;34:1208–1215. doi: 10.1002/humu.22373. [DOI] [PubMed] [Google Scholar]
- Reitz C. The role of intracellular trafficking and the VPS10d receptors in Alzheimer's disease. Future Neurol. 2012;7:423–431. doi: 10.2217/fnl.12.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitz C, Tosto G, Vardarajan B, Rogaeva E, Ghani M, Rogers RS, Conrad C, Haines JL, Pericak-Vance MA, Fallin MD, et al. Independent and epistatic effects of variants in VPS10-d receptors on Alzheimer disease risk and processing of the amyloid precursor protein (APP) Transl. Psychiatry. 2013;3:e256. doi: 10.1038/tp.2013.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Hashimoto M, Song D, Shults CW, Lang I, Masliah E. Differential neuropathological alterations in transgenic mice expressing alpha-synuclein from the platelet-derived growth factor and Thy-1 promoters. J. Neurosci. Res. 2002;68:568–578. doi: 10.1002/jnr.10231. [DOI] [PubMed] [Google Scholar]
- Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat. Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- Schutz P, Bumann M, Oberholzer AE, Bieniossek C, Trachsel H, Altmann M, Baumann U. Crystal structure of the yeast eIF4A-eIF4G complex: an RNA-helicase controlled by protein-protein interactions. Proc. Natl. Acad. Sci. USA. 2008;105:9564–9569. doi: 10.1073/pnas.0800418105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MN, Marcusson EG, Cereghino JL, Emr SD. Endosome to Golgi retrieval of the vacuolar protein sorting receptor, Vps10p, requires the function of the VPS29, VPS30, and VPS35 gene products. J. Cell Biol. 1997;137:79–92. doi: 10.1083/jcb.137.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MN, McCaffery JM, Emr SD. A membrane coat complex essential for endosome-to-Golgi retrograde transport in yeast. J. Cell Biol. 1998;142:665–681. doi: 10.1083/jcb.142.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J. Hsp104: a weapon to combat diverse neurodegenerative disorders. Neurosignals. 2008;16:63–74. doi: 10.1159/000109760. [DOI] [PubMed] [Google Scholar]
- Siitonen A, Majounie E, Federoff M, Ding J, Majamaa K, Singleton AB. Mutations in EIF4G1 are not a common cause of Parkinson's disease. Eur. J. Neurol. 2013;20:e59. doi: 10.1111/ene.12051. [DOI] [PubMed] [Google Scholar]
- Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat. Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Smith WW, Pei Z, Jiang H, Moore DJ, Liang Y, West AB, Dawson VL, Dawson TM, Ross CA. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc. Natl. Acad. Sci. USA. 2005;102:18676–18681. doi: 10.1073/pnas.0508052102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson's and Lewy body diseases. J. Neurosci. 2009;29:13578–13588. doi: 10.1523/JNEUROSCI.4390-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhaman S, Behari M, Govindappa ST, Muthane UB, Juyal RC, Thelma BK. VPS35 and EIF4G1 mutations are rare in Parkinson's disease among Indians. Neurobiol. Aging. 2013;34:2442 e2441–2442 e2443. doi: 10.1016/j.neurobiolaging.2013.04.025. [DOI] [PubMed] [Google Scholar]
- Tong AH, Boone C. Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods Mol. Biol. 2006;313:171–192. doi: 10.1385/1-59259-958-3:171. [DOI] [PubMed] [Google Scholar]
- Trinh J, Farrer M. Advances in the genetics of Parkinson disease. Nat. Rev. Neurol. 2013;9:445–454. doi: 10.1038/nrneurol.2013.132. [DOI] [PubMed] [Google Scholar]
- Tucci A, Charlesworth G, Sheerin UM, Plagnol V, Wood NW, Hardy J. Study of the genetic variability in a Parkinson's Disease gene: EIF4G1. Neurosci. Lett. 2012;518:19–22. doi: 10.1016/j.neulet.2012.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, Soto-Ortolaza AI, Cobb SA, Wilhoite GJ, Bacon JA, et al. VPS35 mutations in Parkinson diseaseA. J. Hum. Genet. 2011;89:162–167. doi: 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa N, Do A, Hershey JW, Fraser CS. Human eukaryotic initiation factor 4G (eIF4G) protein binds to eIF3c, -d, and -e to promote mRNA recruitment to the ribosome. J. Biol. Chem. 2013;288:32932–32940. doi: 10.1074/jbc.M113.517011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wan JY, Edwards KL, Hutter CM, Mata IF, Samii A, Roberts JW, Agarwal P, Checkoway H, Farin FM, Yearout D, et al. Association mapping of the PARK10 region for Parkinson's disease susceptibility genes. Parkinsonism Relat. Disord. 2014;20:93–98. doi: 10.1016/j.parkreldis.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Coombes CE, Kilaru A, Li X, Gitler AD, Bowers WJ, Dawson VL, Dawson TM, Moore DJ. GTPase activity plays a key role in the pathobiology of LRRK2. PLoS Genet. 2010;6:e1000902. doi: 10.1371/journal.pgen.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeger-Lotem E, Riva L, Su LJ, Gitler AD, Cashikar AG, King OD, Auluck PK, Geddie ML, Valastyan JS, Karger DR, et al. Bridging high-throughput genetic and transcriptional data reveals cellular responses to alpha-synuclein toxicity. Nat. Genet. 2009;41:316–323. doi: 10.1038/ng.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.