

Abstract
Autosomal dominant familial hypercholesterolemia (FH) is a monogenic life-threatening disease. We tested the efficacy of low-density lipoprotein receptor (LDLR) gene therapy using helper-dependent adenoviral vector (HDAd) in a nonhuman primate model of FH, comparing intravenous injection versus intrahepatic arterial injection in the presence of balloon catheter-based hepatic venous occlusion. Rhesus monkeys heterozygous for mutant LDLR gene (LDLR+/−) developed hypercholesterolemia while on a high cholesterol diet. We treated them with HDAd-LDLR either by intravenous delivery, or by catheter-based intra-hepatic artery injection. Intravenous injection of ≤1.1×1012 viral particles (vp)/kg failed to have any effect on plasma cholesterol. Increasing the dose to 5×1012 vp/kg led to a 59% lowering of the plasma cholesterol that lasted for 30 days before it returned to pretreatment levels by day 40. A further increase in dose to 8.4×1012 vp/kg resulted in severe lethal toxicity. In contrast, direct hepatic artery injection following catheter-based hepatic venous occlusion enabled the use of a reduced HDAd-LDLR dose of 1×1012 vp/kg that lowered plasma cholesterol within a week, and reached a nadir of 59% pretreatment level on days 20 to 48 after injection. Serum alanine aminotransaminase (ALT) remained normal until day 48 when it went up slightly and stayed mildly elevated on day 72 before it returned to normal on day 90. In this monkey, the HDAd-LDLR-induced trough of hypocholesterolemia started trending upwards on day 72 and returned to pretreatment levels on day 120. We measured the LDL apolipoprotein B turnover rate at 10 days before, and again 79 days after, HDAd-LDLR treatment in two monkeys that exhibited a cholesterol lowering response. HDAd-LDLR therapy increased the LDL fractional catabolic rate by 78% and 50%, respectively, in the two monkeys, coincident with an increase in hepatic LDLR mRNA expression. In conclusion, HDAd-mediated LDLR gene delivery to the liver using a balloon catheter occlusion procedure is effective in reversing hypercholesterolemia in a nonhuman primate FH model; however, the unsustainability of the hypocholesterolemic response during 3–4 months of follow up and heterogeneous response to the treatment remains a challenge.
Keywords: helper-dependent adenovirus, familial hypercholesterolemia, LDL receptor, nonhuman primate
Introduction
Autosomal dominant familial hypercholesterolemia (FH) is caused by mutations in the low-density lipoprotein receptor (LDLR).1 Homozygous FH patients present with massively elevated LDL cholesterol and cardiovascular disease. They have severe atherosclerosis and die of ischemic heart disease usually in their third decade of life. The majority of homozygous and a substantial proportion of heterozygous patients are refractory to conventional pharmacological therapy. Therapeutic options for these resistant patients are limited to LDL apheresis, portacaval anastomosis, or liver transplantation.2 Gene therapy has been explored as an alternative treatment. The liver is the main target organ for FH gene therapy because of its capacity to dispose of excess cholesterol by diverting it into bile acids; it is also accessible to gene delivery via the intravenous (i.v.) route or the hepatic artery. A number of studies have shown that hepatic reconstitution of LDLR expression ex vivo can reverse hypercholesterolemia, including promising results in a rabbit model of FH.3 In the only clinical gene therapy trial for FH to date, Grossman et al. isolated hepatocytes from FH patients, transduced them ex vivo with retroviral vector expressing LDLR, and reimplanted them into the liver of the patients.4,5 Only marginal therapeutic benefit was achieved in this study. It was difficult to determine whether the reduction of LDL cholesterol level was the direct result of the gene transfer or other factors were involved. Plasma LDL level is determined by LDL production and removal. For example, the decline of LDL cholesterol after portacaval anastomosis is caused by a decreased secretion of very-low-density lipoprotein (VLDL), a precursor of LDL, not by an enhanced LDL removal.6 In this clinical trial, LDL turnover was not measured, which led to the comment, “a modest 17% fall in plasma cholesterol after 25% hepatectomy and re-infusion of hepatocytes infected with a retrovirus might have been due to either diminished lipoprotein production or to enhanced activity of the patient’s own receptor”.7 The focus has shifted to in vivo gene therapy thereafter. Helper-dependent adenoviral vector (HDAd) is devoid of all viral protein genes and has shown considerable promise for liver-directed gene transfer with long-term transgene expression, which lasted a life-time in mice.8 In a previous study in LDLR−/− mice, we showed that a single injection of HDAd expressing monkey LDLR reduced plasma cholesterol over 2 years and attenuated atherosclerotic lesion progression.9 We also demonstrated that LDLR gene therapy induces the regression of established atherosclerosis in LDLR−/− mice.10 Despite promising results of gene therapy in small animal models, its efficacy in large animal models has not been tested; there are important differences in physiology and in immune responses between rodents and humans. This issue is particularly relevant in gene therapy for lipid disorders.11
A nonhuman primate model of FH has been described in rhesus monkeys,12,13 which carried a heterozygous nonsense mutation involving codon Trp28314 of the LDLR. Extensive cross-breeding of the affected monkeys failed to yield any homozygotes, indicating that the mutation may be linked to a lethal mutation. With the availability only of the heterozygous (LDLR+/−) rhesus monkey, we will be modeling heterozygous FH in humans, a relatively common genetic disorder that affects about 1 in 500 people in most ethnic groups.15 Heterozygous LDLR-deficient monkeys displayed elevated plasma cholesterol (5.17–6.47 mmol/l or 200–250 mg/dl) compared with unaffected monkeys (2.59–3.36 mmol/l or 100–130 mg/dl); the plasma cholesterol level further increased to 12.93–20.69 mmol/l (500–800 mg/dl) when the animals were fed a high-cholesterol diet.16 In this study, we tested the efficacy of HDAd-based monkey LDLR gene therapy in high cholesterol diet-fed LDLR+/− rhesus monkeys. We compared the effect of intravenous (i.v.) injection of HDAd-LDLR to that of a balloon catheter-based procedure developed by Brunetti-Pierri et al.17 We found that a single i.v. injection of HDAd-LDLR into LDLR+/− monkeys produced a >50% lowering of plasma cholesterol that lasted about a month. We next tested a modified percutaneous catheter-based gene delivery strategy also developed by Brunetti-Pierri et al.18 In this refinement, the HDAd-LDLR was injected directly into the hepatic artery in the presence of increased intrahepatic pressure induced by transient blockage of hepatic venous drainage by a balloon catheter positioned in the inferior vena cava (IVC). The optimized gene delivery strategy was highly efficacious in reducing the vector dose while substantially prolonging the therapeutic hypocholesterolemic response to the treatment regimen.
Results
Intravenous Injection of HDAd-LDLR
We treated four LDLR+/− monkeys as study subjects with a single i.v. injection of escalating doses of HDAd-LDLR.9 We first treated monkey #8796 with 20 ml of saline and found no significant changes in plasma cholesterol levels after treatment (Figure 1). As expected, we also failed to detect any change in plasma cholesterol when we treated another LDLR+/− monkey #9908 with an empty vector HDAd-0 [0.8×1012 viral particles (vp)/kg]. We next injected i.v. HDAd-LDLR into a third LDLR+/− monkey #7139 at a dose of 1.1×1012 vp/kg, an HDAd dose that is 10-fold higher than the dose of HDAd-α-fetoprotein that stimulated significant elevation in α-fetoprotein secretion in serum in baboons17 and again failed to observe any change in plasma cholesterol level in monkey #7139. We then treated a fourth monkey #13090 at an even higher i.v. dose of 5×1012 vp/kg of HDAd-LDLR. The treatment was well-tolerated by the monkey and led to a 60% reduction of plasma cholesterol from a baseline of 14.95 mmol/l (578 mg/dl) to 5.90 mmol/l (229 mg/dl) on day 7. The plasma cholesterol lowering persisted until day 21, when it went up to 10.70 mmol/l (413 mg/dl)] on day 28, and towards pretreatment levels on day 42. These results indicate that a dose higher than 1.1×1012 vp/kg was needed to reverse hypercholesterolemia in LDLR+/− monkeys, and a dose of 5×1012 vp/kg significantly restored normal plasma cholesterol in a heterozygous FH monkey, an effect that lasted for about a month. We next treated a fifth monkey #11226 with an even higher dose of 8.4×1012 vp/kg, which was modestly below a dose that had previously proven to be lethal19 and observed severe acute toxicity and lethality within a day of treatment. The clinical picture and necropsy revealed hemorrhagic shock syndrome likely resulting from the high dose of HDAd vector used.
Figure 1. Efficacy of intravenous injection of HDAd expressing monkey LDLR in heterozygous LDLR-deficient rhesus monkeys.
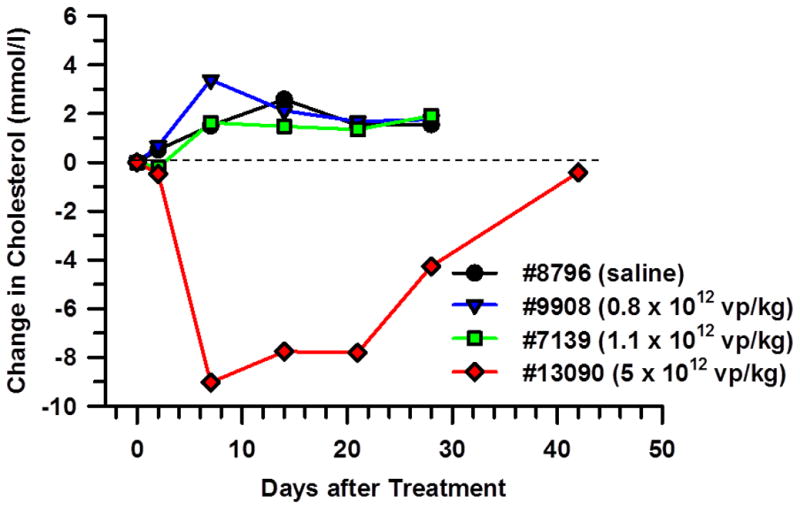
Four heterozygous LDLR-deficient monkeys were treated with a single intravenous injection of saline (#8796), empty vector at a dose of 0.8×1012 vp/kg (#9908), or HDAd-LDLR at a dose of 1.1×1012 vp/kg (#7139) or 5×1012 vp/kg (#13090). Baseline cholesterol levels were 18.0 mmol/l (696 mg/dl) in monkey #8796, 9.5 mmol/l (368 mg/dl) in monkey #9908, 8.0 mmol/l (308 mg/dl) in monkey #7139 and 15.0 mmol/l (578 mg/dl) in monkey #13090, respectively. The broken line shows pretreatment cholesterol levels.
Balloon Occlusion-Based HDAd Delivery into Hepatic Artery
To improve upon i.v. vector injection as a delivery method, Brunetti-Perri et al developed a protocol17,18 to deliver the vector via an intrahepatic arterial catheter. Simultaneously, under fluoroscopic guidance, they inserted a balloon catheter into the inferior vena cava (IVC) via the femoral vein and positioned it over the hepatic venous outflow (Figure 2a). Intrahepatic arterial HDAd injection when the balloon was inflated led a10-fold increase efficiency in transgene expression (Figure 2b and c). The IVC occlusion was also monitored by the venous pressure (Figure. 2d). We performed the same procedure in rhesus monkeys and injected the HDAd vector (2 ml) within a minute via a hepatic artery catheter immediately after the balloon was inflated.
Figure 2. Balloon catheter-based hepatic artery injection.

(a). Schematic diagram of hepatic artery injection. The liver circulation is isolated by inserting a balloon catheter via the femoral vein and placing it in the inferior vena cava (IVC). A second IV catheter is inserted into the hepatic artery through the contralateral femoral artery. The placement of the catheter is visualized using fluoroscopy. Once occlusion of the hepatic circulation has been established via the balloon catheter in the IVC, the vector is injected via the arterial catheter. The occlusion is confirmed by monitoring hepatic venous pressure through the third catheter inserted into the femoral vein. BD: bile duct; HA: hepatic artery; HV: hepatic vein; PV: portal vein. (b) Fluoroscopy image to confirm the position of a balloon-catheter. (c) Fluoroscopy after the balloon inflated. Contrast reagent was injected to confirm that the catheter was placed at the IVC. (d) Venous pressure. Occlusion was monitored by venous pressure.
The monkeys used for this procedure are summarized in Table 1. We first performed the procedure in a chow-fed (Purina LabDiet5LEO, St. Louis, MO) normal LDLR+/+ (#19254) and a heterozygous LDLR+/− (#19499) monkey. The injection was done immediately after the balloon was deflated but while hepatic venous pressure remained high. As reported previously17,18, systemic blood pressure fell significantly when the balloon was inflated. We found that serum IL-6 level increased 30 minutes after injection and peaked at 2 hours (Figure 3a) but decreased to non-detectable levels by 72 hours. The procedure also led to transient and inconsistent changes in plasma liver enzymes (Figure 3b and c). Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels peaked at about 24 hours; the increase was mild and resolved by day 5. Plasma total cholesterol levels in the LDLR+/− (#19499) monkey decreased from a baseline of 5.70 mmol/l (219 mg/dl) to 3.90 mmol/l (150 mg/dl) within 24 hours. It gradually went back up over the next few days returning to baseline by day 5. The plasma cholesterol level did not change in the non-FH (LDLR+/+) (#19254) monkey (Figure 3d).
Table 1.
Summary of rhesus monkeys used for balloon-catheter occlusion
| ID | Sex | Age (Years) | Bodyweight (kg) | Genotype | Dose (VP/kg) |
|---|---|---|---|---|---|
| #19254 | F | 3 | 3.9 | N | Saline |
| #19499* | M | 3 | 3.4 | H | Saline |
| #21588 | F | 2 | 2.6 | N | 2.0×1012 |
| #19360 | F | 3 | 4.0 | N | 2.0×1012 |
| #19251 | F | 3 | 4.2 | H | 2.0×1012 |
| #19498 | M | 3 | 5.0 | H | 2.0×1012 |
| #18340 | M | 5 | 5.7 | H | Saline |
| #19269 | F | 4 | 5.0 | H | 1×1012 |
| #19255 | M | 4 | 5.7 | H | Saline |
| #19536 | F | 4 | 4.4 | H | 0.3×1012 |
| #20031* | F | 3 | 3.4 | N |
N: normal; H: heterozygous LDLR-deficient.
Monkeys were used as donors for LDL turnover study.
Figure 3. Acute toxicity measurements associated with balloon catheter based hepatic artery injection.

One normal LDLR+/+ (#19254) and one heterozygous LDLR+/− (#19499) monkeys on normal chow were treated by an injection of saline after and a complete blood test and IL-6 measurement were performed. (a) Plasma IL-6 levels. (b) Serum alanine aminotransferase (ALT) levels. (c) Serum aspartate aminotransferase (AST) levels. (d) Plasma cholesterol levels.
We next fed monkeys with a Rhesus Western diet (Texas Biomedical Research Institute, San Antonio, TX) for 7 weeks prior to treatment and were kept on the diet afterwards. We injected HDAd-LDLR (2×1012 vp/kg) into 4 monkeys immediately after the balloon was deflated. The plasma cholesterol did not change in two wild-type LDLR+/+ monkeys (#19360, and #21588) suggesting that the gene delivery does not have an effect on the cholesterol dynamics in monkeys that express normal amounts of LDLR. Of the two heterozygous LDLR+/− monkeys, one (#19251) showed no change in plasma cholesterol (Figure 4A, green line), whereas another LDLR+/− monkey (#19498) exhibited a 57% drop in plasma cholesterol level from 8.15 mmol/l (315 mg/dl) to 3.25 mmol/l (126 mg/dl) at day 7 (Figure 4a, red line). So there was a heterogeneous response in heterozygous FH monkeys treated at this dose of HDAd-LDLR. The cholesterol lowering effect of HDAd-LDLR in the LDLR+/− (#19498) monkey that responded to the treatment was sustained for about 100 days. The plasma lowering effect reached its nadir 7 days, and stayed at or near the nadir for another three weeks. Afterwards, it gradually rose to 5.09 mmol/l (197 mg/dl) at day 78, and then to above the pretreatment level (9.30 mmol/l, or 361 mg/dl) by day 105 (Figure 4a, red line). The two wild-type LDLR+/+ monkeys maintained normal serum ALT throughout the observation period of 120 days. The LDLR+/− monkey (#19251) that did not showed a hypocholesterolemic response also maintained normal ALT levels for 67 days, end of the observation period for this monkey. In contrast, the serum ALT of the LDLR+/− monkey (#19498) that showed a hypocholesterolemic response maintained a normal ALT level during the first 3 weeks of treatment when the plasma cholesterol showed an excellent response (Figure 4a, red line). The ALT began to edge above normal to 70 U/l on day 36, and continued to go up to peak at 144 U/l on day 72, before it started trending down, eventually returning to normal on day 120 (Figure 4b, red line). It is noteworthy that this monkey that had responded to the treatment developed liver enzyme elevation late, and the delayed increase in serum ALT coincided with the onset of loss of the cholesterol lowering effect of the treatment. While the significance of the timing is unclear, we note that a similar pattern is evident in an experiment involving another LDLR+/− monkey (#19269, see below).
Figure 4. Plasma cholesterol level of rhesus monkeys after balloon-catheter based hepatic artery delivery.
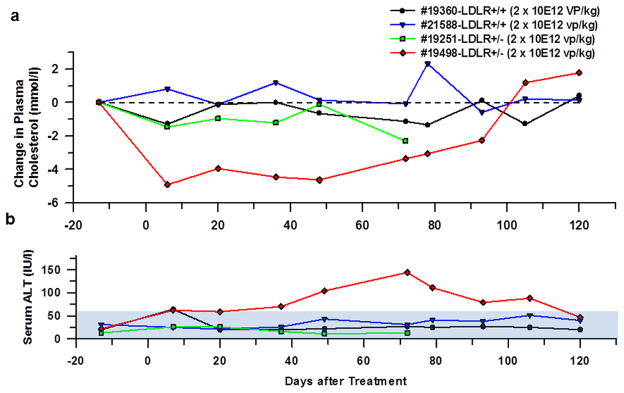
HDAd-LDLR was injected into monkeys in a volume of 10 ml/kg at a dose of 2×1012 vp/kg. The vector was injected after the balloon was deflated. #19360 and #21588 were normal LDLR+/+ monkeys and #19251 and #19498 were heterozygous LDLR+/− monkeys. (a) Changes in plasma cholesterol levels. Pretreatment plasma cholesterol levels were 7.55 mmol/l (292 mg/dl) in #19360, 7.25 mmol/l (281 mg/dl) in #21588, 10.30 mmol/l (399 mg/dl) in #19251 and 8.15 mmol/l (315 mg/dl) in #19498, respectively. (b) Plasma ALT levels. Normal ALT range (5-61 IU/l) is shown by filled area. #19251 had a low hematocrit level at day 78 and blood analyses were not performed. Because this animal did not show any effects of gene therapy, further follow up was deemed unnecessary and #19251 was removed from the study.
Optimized Balloon Occlusion Protocol Increases Efficacy of HDAd-LDLR Therapy
The HDAd-LDLR-mediated hypocholesterolemic response was encouraging in monkey #19498. However, the dose used (2.2×1012 vp/kg) was only ~4 fold lower than a lethal dose (8.4×1012 vp/kg, lethal for monkey #11226). In an attempt to increase the efficiency of HDAd-LDLR treatment so as to obtain comparable results with a lower dose of HDAd, we decided to modify the protocol by keeping the balloon inflated (and thus intrahepatic pressure maintained at a high level) throughout the vector injection. We applied the modified protocol on LDLR+/− monkey (#19269, fed a Rhesus Western diet)) at half the dose used in the last group of monkeys, i.e., at 1 ×1012 vp/kg, immediately after the balloon was inflated. The injection line was flushed with 20 mL of saline and the IVC balloon was kept inflated for an additional 5 min before it was deflated (Figure 2d). Despite the use of a lower dose, the plasma cholesterol of this monkey (#19269) decreased from 10.40 mmol/l (402 mg/dl) to 5.75 mmol/l (222 mg/dl) at day 7; at day 20, it decreased further to 4.30 mmol/l (165 mg/dl), constituting a 59% reduction from the pretreatment level. The plasma cholesterol level stayed at the same level (4.30 mmol/l) until day 48 (Figure 5a), when ALT level went up modestly to 81 U/l and stayed mildly elevated on day 72 (89 U/l) (Figure 5b). At day 72, plasma cholesterol level started trending upwards to 6.00 mmol/l (232 mg/dl) and returned to the pretreatment level by day 120.
Figure 5. Plasma cholesterol and serum ALT levels in heterozygous LDLR-deficient monkey treated by an optimal procedure.
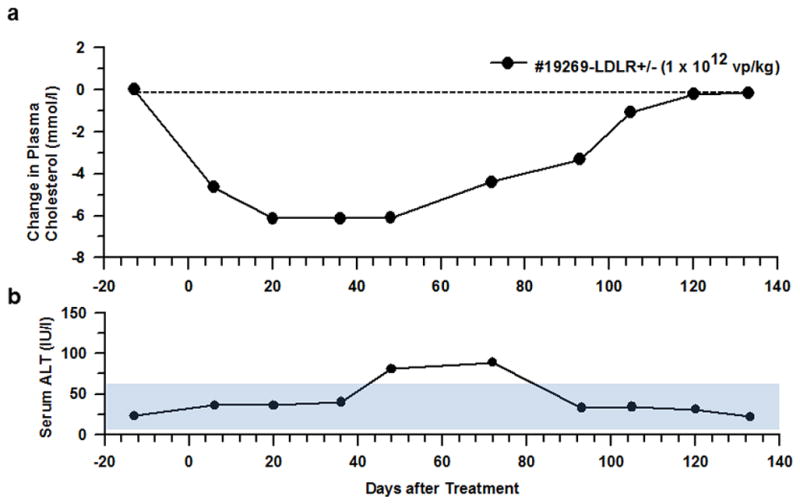
Monkey #19269 was treated with a single injection of HDAd-LDLR at a dose of 1×1012 vp/kg while the balloon was inflated. (a) Plasma cholesterol level. Plasma cholesterol level was 10.40 mmol/l (402 mg/dl) before gene transfer. (b) Serum ALT levels. Normal range (5-61 IU/l) is shown by filled area.
Although intrahepatic delivery of HDAd-LDLR at a dose of 1×1012 vp/kg was effective in reversing hypercholesterolemia, the beneficial effect of the treatment did not last beyond ~100 days. Contrary to the transient nature (lasting up to 3–4 months only) of the cholesterol lowering effect of HDAd-LDLR in LDLR+/− rhesus monkeys, wild-type baboons that had been treated with a low dose (3×1010 vp/kg) HDAd-α-fetoprotein vector were reported to exhibit a much more prolonged expression of α-fetoprotein.17 One possible explanation is that there is heightened host immune responses to HDAd vector because the dose we used in monkeys was 30 times higher than that in baboons. Interestingly, observations similar to the current study have been reported in hemophilia B patients treated with adeno-associated virus (AAV) expressing factor IX in which transgene factor IX expression dropped precipitously at days 50–60 after treatment, an effect attributed to preexisting immunity against AAV vector.20 To examine the possibility of preexisting memory T-cells against Ad by HDAd administration, we tested the effect of a dose of 0.3×1012 vp/kg, which is 10-fold higher than the effective dose reported in baboons17 but 3-fold lower than the dose that induced the increase in ALT in our study. Two LDLR+/− monkeys were treated with either 0.3×1012 vp/kg of vector (#19536) or saline (#19255). HDAd administration at this dose did not lower plasma cholesterol or increase liver enzymes (data not shown). We collected peripheral blood mononuclear cells (PBMCs) at days −21, +34 and +70 and measured cytotoxic T-cell (CTL) activity as reflected by interferon-gamma secretion by lymphocytes upon stimulation with Ad peptides.21 There was no significant increase in CTL activity in either monkey (Figure S1), suggesting that hepatic arterial injection of HDAd at a dose of 0.3×1012 vp/kg did not stimulate preexisting memory T-cells. We also measured neutralizing antibodies before and after vector injection. Plasma collected from monkey #19522 treated with saline did not have any significant neutralizing antibodies at any time of sampling, while plasma from monkey #19536 treated with HDAd-LDLR inhibited the infection of 116 cells with HDAd-EGFP at 1:80 dilution at day +34 and +70 but not at day −21.
Efficacy of LDLR gene therapy
In addition to evaluating the plasma cholesterol as the downstream response to LDLR gene therapy, we also monitored the functional activity of the LDLR gene transfer 10 days before, and 79 days after, HDAd-mediated LDLR gene transfer. Hepatic LDLR gene delivery in LDLR+/− monkeys, #19498 (Figure 4, red line) and #19269 (Figure 5), increased LDLR mRNA levels by 10- and 27-fold, respectively (Table 2, Figure 6). It markedly lowered LDL cholesterol (by 47% and 67%, respectively, Table 2), and raised the LDL apoB Fractional Catabolic Rate (FCR) in both monkeys by 78% and 50%, respectively, indicating that the hypocholesterolemic response to the gene therapy was the result of markedly increased LDLR activity in vivo (Table 2 and Figure S2).
Table 2.
Effects of gene therapy on plasma cholesterol and LDL metabolism.
| #19498 | Pre-treatment | Post-treatment |
|---|---|---|
| Plasma cholesterol (mmol/l) | 7.60 | 4.75 |
| LDL cholesterol (mmol/l) | 5.90 | 3.10 |
| HDL cholesterol (mmol/l) | 1.75 | 1.75 |
| Relative LDLR mRNA expression | 1.37 | 14.32 |
| LDL apoB FCR (pool/day) | 0.230 | 0.410 |
| LDL apoB production rate (mg/kg/day) | 11.28 | 11.52 |
| #19269 | ||
| Plasma cholesterol (mmol/l) | 9.60 | 4.50 |
| LDL cholesterol (mmol/l) | 6.80 | 2.25 |
| HDL cholesterol (mmol/l) | 2.75 | 2.30 |
| Relative LDLR mRNA expression | 0.47 | 12.64 |
| LDL apoB FCR (pool/day) | 0.276 | 0.415 |
| LDL apoB production rate (mg/kg/day) | 10.32 | 10.80 |
The pre-treatment studies were done 10 days before gene therapy and the post-treatment studies were performed 79 days after gene transfer. Both were heterozygous LDLR+/− monkeys. #19498 was injected by 2×1012 vp/kg HDAd-LDLR after balloon was deflated, whereas #19269 was treated by 1×1012 vp/kg while balloon was inflated.
Figure 6. Relative LDLR mRNA expression in rhesus monkeys treated with HDAd-LDLR.
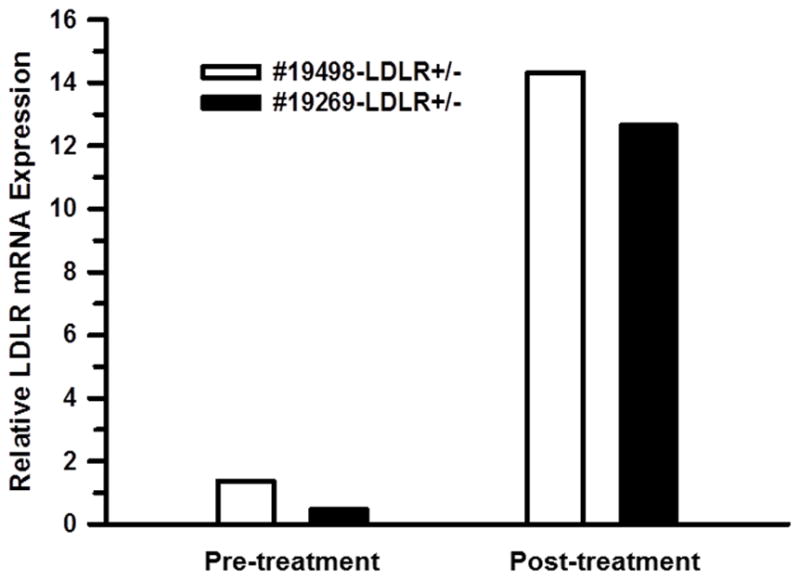
Monkey LDLR mRNA levels in needle biopsies of liver were quantified by TaqMan RT-PCR and normalized to β-actin mRNA. The biopsy was performed 10 days before and 79 days after HDAd-LDLR administration.
Discussion
FH is the most common and severe form of monogenic lipid disorder. Because of its severity and limited availability of conventional therapeutic options, homozygous FH has been an important candidate disease for gene therapy. We and others reported that in a mouse model of homozygous FH, hepatic delivery of LDLR gene by i.v. HDAd or AAV inhibits atherosclerotic lesion progression and may effect lesion regression.9,10,22,23 Since a nonhuman primate model of LDLR deficiency exists only in the heterozygous form, we used the LDLR+/− monkeys a model to test the feasibility of LDLR gene therapy in heterozygous FH, a much more common but serious disorder. Initially, we delivered the HDAd via i.v. injection, a minimally invasive and preferred route of administration. However, i.v. injection requires a high dose, approximately half the lethal dose in baboons,19 to be effective (Figure 1). At 5×1012 vp/kg, i.v. HDAD-LDLR led to a 60% reduction of plasma cholesterol on day 7. However, the hypocholesterolemic effect was short-lived and by day 42 plasma cholesterol level had returned to pre-injection levels. A higher dose (8.4×1012 vp/kg) caused severe acute toxicity, as reported in a baboon that received a similar lethal dose.19
In order to increase hepatic transduction efficiency and reduce dose-dependent toxicity, Brunetti-Pierri et al. developed a catheter-based balloon occlusion method in baboons.17,18,24 We applied this approach to our rhesus monkey model. We first tested the procedure by injecting saline into two monkeys (one normal LDLR+/+ and one LDLR+/− monkey) to determine their response to the procedure. The animals tolerated the procedure well. An acute increase in IL-6 was found in a LDLR+/+ monkey as previously documented in baboons17 but not in a LDLR+/− monkey, suggesting mild but variable responses of animals to the procedure itself. We then treated two LDLR+/+ and two LDLR+/− monkeys that had been fed a Rhesus Western (high cholesterol) diet for 2.5 months. Only one monkey showed a therapeutic response. In this monkey, when we injected the HDAd immediately after the IVC balloon was deflated, the plasma cholesterol went down by 57% at day 6. Therefore, balloon catheter occlusion method appears to be ~2.5-fold more effective than peripheral i.v. injection (2×1012 vp/kg vs. 5×1012 vp/kg) in reducing plasma cholesterol (Figure 4). Unexpectedly, the LDLR+/− monkey (#19498) that showed a cholesterol-lowering response developed a delayed rise in plasma ALT level in at day 45 after the gene therapy, which lasted until day ~100. This transient rise in ALT was followed by the attenuation of the HDAd-LDLR-induced plasma cholesterol normalization and return of hypercholesterolemia. Contrary to our observations in rhesus monkeys, the previous study in baboons using a similar balloon procedure led to prolonged transgene expression that was detectable for at least 963 days at a dose of 3×1010 vp/kg17 that is 70-fold lower than the dose used here in rhesus monkeys. We considered the unforeseen side effects of the high dose HDAd and heterogeneous response possibly resulting from the intrahepatic venous pressure not being maintained during vector injection, and reduced the dose to1×1012 vp/kg but kept the balloon inflated to maintain elevated intrahepatic venous pressure during vector injection. This modification in protocol led to a slightly more intense cholesterol-lowering effect using half the HDAd-LDLR dose (Figure 5). Again, however, we observed a mild transient serum ALT elevation from day 50 to day 70, followed by a gradual return of the previously normalized plasma cholesterol to pretreatment (elevated) levels by day 120.
It is not clear what causes the increase of liver enzymes and subsequent loss of LDLR gene transfer effects. We induced overexpression of the monkey LDLR gene in LDLR+/− monkeys so humoral immunity to the expressed LDLR would not be an issue, although it is possible that there are individual variations in antigen (or transgene product) processing and presentation. Interestingly, Brunetti-Pierri et al. reported that rhesus monkeys treated with HDAd expressing human factor IX (hFIX) at a dose of 1×1012 vp/kg expressed hFIX for up to 1,029 days despite development of neutralizing antibodies.18 There is a fundamental difference in the nature of therapeutic proteins and levels required to reverse phenotype between FH and hemophilia B individuals. LDLR is a membrane protein and the LDLR activity must be over 50% of normal activity to achieve therapeutic effects, whereas FIX is a secretory protein and as little as 1% of normal levels can substantially correct the propensity for bleeding in hemophilia B.25 Although humoral immune responses to therapeutic proteins are most likely not relevant to our findings, we cannot completely exclude such a possibility.
Host immune responses to the HDAd vector itself could be another factor in the delayed failure of treatment. A similar silencing of transgene expression following an asymptomatic increase of transaminases has been reported in clinical trials of Factor IX gene therapy using AAV.20 It was attributed to pre-existing T cells to AAV capsids, which were reactivated upon AAV-mediated gene transfer, eliminating the transduced cells. This response appears to be dose-dependent.25 We studied the responses of PBMCs against immunogenic Ad hexon peptides21 before and after HDAd administration. We did not find any significant T cell responses. The dose used in this experiment did not influence plasma cholesterol levels, which suggests that this dose is below the minimum effective dose for LDLR gene therapy. At such a low dose, HDAd vector does not re-stimulate preexisting memory T cells despite elevated neutralizing antibodies. In support of this explanation, the frequent presence of memory T cells against human adenovirus has been reported in humans,26 while nonhuman primates have very low frequency of adenovirus-specific T cells in PBMCs.27 However, high frequency of pre-existing adenovirus-specific T cells in livers has been reported after active immunization.28 Therefore, the responses of T cells isolated from PBMCs may not be sufficient to detect preexisting cellular immunity against Ad in rhesus monkeys. However, we exhausted available animals after the last experiment and there are no more LDLR+/− monkeys available to further evaluate the possibility of cellular immunity by treating LDLR+/− monkeys at a dose of 1×1012 vp/kg or higher, which could replicate the increase of liver enzymes preceding diminished effects of LDLR gene therapy. Therefore, the cause of the transient nature of the efficacy of LDLR gene therapy in this study remains speculative. Nonetheless, if the cause of our findings is related to the preexisting cellular immunity against HDAd vector, a possible solution is suggested by a recent clinical trial of AAV-mediated transfer of Factor IX for Hemophilia B, where short term glucocorticoid administration normalized liver enzymes and maintained Factor IX level.25
Ad vectors are recognized by the host innate immune system during viral entry and replication in host cells.29 We did not measure cytokine levels in the two monkeys (#19498 and #19269) when they showed asymptomatic increase of ALT. Although the innate immune response reactions have been reported in early phase but not in late phase toxicity associated with Ad 30,31, we cannot completely exclude such a possibility.
Despite the disappointment from the unexpected transient elevation of ALT followed by the loss of efficacy of LDLR gene therapy, we showed that HDAd-mediated LDLR gene therapy works in a nonhuman primate model of FH. We performed functional assay for LDLR activity 10 days before and 79 days after gene transfer. The two monkeys that showed a good cholesterol lowering response displayed markedly higher hepatic LDLR mRNA expression concomitant with an accelerated LDL fractional catabolic rate, which supports a substantial functional enhancement. It is important to note that we took advantage of a natural nonhuman primate model of heterozygous FH after our initial experiments in FH mouse models.9 Not only are there differences in immune responses between rodents and humans, there are also major differences in lipoprotein physiology between these species.11 Critically important are hemodynamic forces, which cause vascular site-specific effects on atherosclerosis.32 Thus, it is difficult to extrapolate the effect of gene therapy on atherosclerosis development in rodent models to that in nonhuman primates and humans.22,23,33
Proprotein convertase subtilisin-like/kexin type 9 (PCSK9), a secreted protease that mediates degradation of LDLR,34 has attracted much attention as a therapeutic target for treating hypercholesterolemia. Both monoclonal antibodies and siRNA have been reported to reduce LDL cholesterol.35–37 Recently, the phase 2 trial targeting PCSK9 using a monoclonal antibody was reported to have achieved substantial further LDL-C reduction in patients with heterozygous FH who were treated with high-dose statins38 and raised the question whether inhibition of PSCSK9 in homozygous FH patients respond to the treatment.39 Homozygous FH patients showed some responses to drug treatment in part via upregulation of the LDLR.40 However, PCSK9 facilitates LDLR degradation by binding to LDLR and preventing its recycling.34 Therefore, the inhibition of PSCK9 may not achieve the targeting LDL-C levels in homozygous FH patients with residual or no LDLR activity. More studies are needed to determine whether targeting PSCK9 will be a treatment of choice for FH. ApoB100, the major protein component of LDL, is another potential therapeutic target. The use of lipid encapsulated siRNA targeting apoB100 was found to silence apoB mRNA in rodents and nonhuman primates.41,42. Alternatively, AAV expressing apoB mRNA-specific shRNA produced long-term apoB silencing and LDL cholesterol lowering in mice43,44. It is unclear which of these therapeutic approaches will turn out to be the most safe and efficacious therapies to lower plasma lipids in FH patients.
In summary, we have found that a single intrahepatic arterial injection of HDAd expressing LDLR accompanied by balloon catheter-based hepatic venous occlusion method corrects hypercholesterolemia in nonhuman primate model of heterozygous FH. Nevertheless, the invasive nature of the procedure, the narrow margin between the effective and the toxic dose, and the delayed immune response which could be associated with a delayed treatment failure remain a significant challenge.
Materials and methods
Recombinant helper-dependent adenoviral vector
Seed stock of HDAd expressing rhesus monkey LDLR was prepared as described9 and large scale vector production was carried out using a suspension system.45 Helper virus contamination and potential rearrangement were determined by quantitative PCR (qPCR) using SYBR Green and Southern blot analysis. The infectious titer of HDAd was defined by relative infectivity to an Adenovirus Type 5 Reference Material (VR-1516, ATCC) in competition to infect HEK293 cells45 except quantification of vectors by qPCR.46 Helper virus contamination measured by real time PCR was 0.05–0.01% and the ratio of viral particles and infectious particles was approximately 15:1. Endotoxin levels tested by Limulus Amebocyte Lysate was <0.05 EU/ml.
Nonhuman primates
Normal LDLR+/+ and heterozygous LDLR+/− rhesus monkeys were housed at the Southwest Foundation for Biomedical Research. Animals of both sex between 3 and 6 kg body weight (Table 1) were fed rhesus western type diet (40% calories from saturated fat and 0.3 mg/Kcal cholesterol) for 7 weeks prior to initiating the sampling schedule and maintained on the diet through the experiment. Animals used for balloon-occlusion based injection into hepatic artery are summarized in Table 1. All animal protocols were performed according to the guidelines of Institutional Animal Care and Usage Committee at the Texas Biomedical Research Institute.
Direct vector delivery into hepatic artery
HDAd was directly delivered to hepatic artery after hepatic venous flow occlusion (Figure 2a) as described by Brunetti-Pieri et al.17 In brief, a 4 French sheath was placed in the right femoral vein, an 11 French sheath in the left femoral vein and a 4 French sheath in the left femoral artery (FA) by standard percutaneous technique. A 20-gauge arterial catheter was placed in the femoral artery for continuous monitoring blood pressure. The custom made 8×3 cm2 balloon occlusion catheter (NuMED, Hopkinton, NY) was introduced into the right femoral vein sheath and positioned in the inferior vena cava (IVC) with the tip just within the IVC-right arterial junction. The placement of the balloon was visualized using fluoroscopy after inflating the balloon catheter (Figure. 2b, c). HDAd or saline in a volume of 2 ml were injected at a rate of 0.5 ml/15 seconds while balloon was inflated through a catheter placed in the hepatic artery. The catheter was flushed with 20 ml of saline; the balloon remained inflated for additional 5 min and then deflated. The occlusion was monitored by venous pressure (Figure 2d).
Assay for cytotoxic T lymphocytes (CTL)
CTL activity was measured by INF-γ secretion by lymphocytes upon stimulation with adenoviral peptides.47,48 Blood (5 mL) was collected with preservative-free heparin at −21, 34 and 70 days posttreatment and lymphocytes and plasma were isolated using Lymphocyte separation medium (Lymphoprep, Axis-Shield). Lymphocytes were frozen at −80°C in freezing medium until use. Cells were thawed and incubated overnight. Pools of 188 overlapping 20 mer peptides derived from immunodominant virion protein, hexon (JPT peptide Technologies)21 were added to the culture next day, and the secreted IFN-γ was captured by the immobilized antibody using a kit from R&D Systems Inc. (cat#EL961). After the formation of colored spots, the membrane was sent to ZellNet Consulting, Inc. for the analysis.
Assay for neutralizing antibodies
The neutralizing antibody titer was determined by an in vitro transduction-inhibition assay. In brief, cells (116 cell line) were plated in a 96-well plate at the density of 1×105 cells/well 2 days prior to infection. Plasma was heat-inactivated at 55°C for 30 min and serially diluted into a 96-well plate (0.1 ml/well). HDAd vector expressing EGFP under elongation factor-1 promoter (HDAd-EGFP) was diluted to 2×108 vp/mL and 0.1 ml of the diluted HDAd vector/well was added to the 96-well plate containing diluted plasma. The plate was incubated at 37°C for 1 hour and then 0.1 ml of plasma/HDAd-EGFP mixture was transferred to wells of a 96-well plate containing the 116 cells. After 30 minutes, 0.1 ml of growth medium was added and incubated in CO2 incubator for 20 hours and the fluorescence was measured by FLUO Star Omega microplate reader (BMG Labtech Inc., Durham, NC).
Kinetic analysis
LDL (d=1.019−1.063) was isolated from donor monkeys, iodinated and intravenously injected into vector treated monkeys.49 LDL apoB turnover data collected for plasma and urinary radioactivity at designated times were analyzed using a two-compartment model, which is characterized by a plasma compartment and an extravascular exchange compartment.49
Other procedures
Serum IL-6 concentrations were determined by Specialty Laboratories (Santa Monica, CA). LDLR mRNA levels in needle biopsies of liver were quantified by TaqMan RT-PCR and normalized to β-actin using human probes (Life Technologies, NY).
Supplementary Material
Acknowledgments
We thank Drs. M. L. Brenner and S. L. Samson for valuable discussion and Dr. M. A. Law, Dr. G. E. Stapleton, E.A. Nour, S. Cormier, R. Razook, G. Cody and S. Wang for technical assistance. This work was supported by HL059314, HL51586, and P30-DK079638.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supplementary information is available at Gene Therapy’s website.
References
- 1.Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest. 2003;111:1795–1803. doi: 10.1172/JCI18925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hovingh GK, Davidson MH, Kastelein JJ, O’Connor AM. Diagnosis and treatment of familial hypercholesterolaemia. Eur Heart J. 2013;34:962–971. doi: 10.1093/eurheartj/eht015. [DOI] [PubMed] [Google Scholar]
- 3.Chowdhury JR, Grossman M, Gupta S, Chowdhury NR, Baker JR, Jr, Wilson JM. Long-term improvement of hypercholesterolemia after ex vivo gene therapy in LDLR-deficient rabbits. Science. 1991;254:1802–1805. doi: 10.1126/science.1722351. [DOI] [PubMed] [Google Scholar]
- 4.Grossman M, Raper SE, Kozarsky K, Stein EA, Engelhardt JF, Muller D, et al. Successful ex vivo gene therapy directed to liver in a patient with familial hypercholesterolaemia. Nat Genet. 1994;6:335–341. doi: 10.1038/ng0494-335. [DOI] [PubMed] [Google Scholar]
- 5.Grossman M, Rader DJ, Muller DW, Kolansky DM, Kozarsky K, Clark BJ, 3rd , et al. A pilot study of ex vivo gene therapy for homozygous familial hypercholesterolaemia. Nat Med. 1995;1:1148–1154. doi: 10.1038/nm1195-1148. [DOI] [PubMed] [Google Scholar]
- 6.Bilheimer DW, Goldstein JL, Grundy SM, Brown MS. Reduction in cholesterol and low density lipoprotein synthesis after portacaval shunt surgery in a patient with homozygous familial hypercholesterolemia. J Clin Invest. 1975;56:1420–1430. doi: 10.1172/JCI108223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown MS, Goldstein JL, Havel RJ, Steinberg D. Gene therapy for cholesterol. Nat Genet. 1994;7:349–350. doi: 10.1038/ng0794-349. [DOI] [PubMed] [Google Scholar]
- 8.Kim IH, Jozkowicz A, Piedra PA, Oka K, Chan L. Lifetime correction of genetic deficiency in mice with a single injection of helper-dependent adenoviral vector. Proc Natl Acad Sci U S A. 2001;98:13282–13287. doi: 10.1073/pnas.241506298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nomura S, Merched A, Nour E, Dieker C, Oka K, Chan L. Low-density lipoprotein receptor gene therapy using helper-dependent adenovirus produces long-term protection against atherosclerosis in a mouse model of familial hypercholesterolemia. Gene Ther. 2004;11:1540–1548. doi: 10.1038/sj.gt.3302310. [DOI] [PubMed] [Google Scholar]
- 10.Li R, Chao H, Ko KWS, Cormier S, Dieker C, Nour EA, et al. Gene Therapy Targeting LDL Cholesterol but not HDL Cholesterol Induces Regression of Advanced Atherosclerosis in a Mouse Model of Familial Hypercholesterolemia. Journal of Genetic Syndromes & Gene Therapy. 2011;2:1000106. doi: 10.4172/2157-7412.1000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yin W, Carballo-Jane E, McLaren DG, Mendoza VH, Gagen K, Geoghagen NS, et al. Plasma lipid profiling across species for the identification of optimal animal models of human dyslipidemia. J Lipid Res. 2011;53:51–65. doi: 10.1194/jlr.M019927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morris MD, Fitch CD. Spontaneous hyperlipidemia in rhesus monkey. Biochem Med. 1968;2:209–215. [Google Scholar]
- 13.Lee JA, Morris MD. Characterization of the serum low-density lipoproteins of normal and two rhesus monkeys with spontaneous hyperbeta-lipoproteinemia. Biochem Med. 1974;10:245–257. doi: 10.1016/0006-2944(74)90028-3. [DOI] [PubMed] [Google Scholar]
- 14.Hummel M, Li ZG, Pfaffinger D, Neven L, Scanu AM. Familial hypercholesterolemia in a rhesus monkey pedigree: molecular basis of low density lipoprotein receptor deficiency. Proc Natl Acad Sci U S A. 1990;87:3122–3126. doi: 10.1073/pnas.87.8.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29:431–438. doi: 10.1161/ATVBAHA.108.179564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scanu AM, Khalil A, Neven L, Tidore M, Dawson G, Pfaffinger D, et al. Genetically determined hypercholesterolemia in a rhesus monkey family due to a deficiency of the LDL receptor. J Lipid Res. 1988;29:1671–1681. [PubMed] [Google Scholar]
- 17.Brunetti-Pierri N, Stapleton GE, Law M, Breinholt J, Palmer DJ, Zuo Y, et al. Efficient, long-term hepatic gene transfer using clinically relevant HDAd doses by balloon occlusion catheter delivery in nonhuman primates. Mol Ther. 2009;17:327–333. doi: 10.1038/mt.2008.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brunetti-Pierri N, Liou A, Patel P, Palmer D, Grove N, Finegold M, et al. Balloon Catheter Delivery of Helper-dependent Adenoviral Vector Results in Sustained, Therapeutic hFIX Expression in Rhesus Macaques. Mol Ther. 2012;20:1863–1870. doi: 10.1038/mt.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brunetti-Pierri N, Palmer DJ, Beaudet AL, Carey KD, Finegold M, Ng P. Acute toxicity after high-dose systemic injection of helper-dependent adenoviral vectors into nonhuman primates. Hum Gene Ther. 2004;15:35–46. doi: 10.1089/10430340460732445. [DOI] [PubMed] [Google Scholar]
- 20.Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 21.Leen AM, Christin A, Khalil M, Weiss H, Gee AP, Brenner MK, et al. Identification of hexon-specific CD4 and CD8 T-cell epitopes for vaccine and immunotherapy. J Virol. 2008;82:546–554. doi: 10.1128/JVI.01689-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kassim SH, Li H, Vandenberghe LH, Hinderer C, Bell P, Marchadier D, et al. Gene therapy in a humanized mouse model of familial hypercholesterolemia leads to marked regression of atherosclerosis. PLoS One. 2010;5:e13424. doi: 10.1371/journal.pone.0013424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen SJ, Sanmiguel J, Lock M, McMenamin D, Draper C, Limberis MP, et al. Biodistribution of AAV8 vectors expressing human low-density lipoprotein receptor in a mouse model of homozygous familial hypercholesterolemia. Hum Gene Ther Clin Dev. 2013;24:154–160. doi: 10.1089/humc.2013.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brunetti-Pierri N, Stapleton GE, Palmer DJ, Zuo Y, Mane VP, Finegold MJ, et al. Pseudo-hydrodynamic delivery of helper-dependent adenoviral vectors into non-human primates for liver-directed gene therapy. Mol Ther. 2007;15:732–740. doi: 10.1038/sj.mt.6300102. [DOI] [PubMed] [Google Scholar]
- 25.Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perreau M, Kremer EJ. Frequency, proliferation, and activation of human memory T cells induced by a nonhuman adenovirus. J Virol. 2005;79:14595–14605. doi: 10.1128/JVI.79.23.14595-14605.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calcedo R, Vandenberghe LH, Roy S, Somanathan S, Wang L, Wilson JM. Host immune responses to chronic adenovirus infections in human and nonhuman primates. J Virol. 2009;83:2623–2631. doi: 10.1128/JVI.02160-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCoy K, Tatsis N, Korioth-Schmitz B, Lasaro MO, Hensley SE, Lin SW, et al. Effect of preexisting immunity to adenovirus human serotype 5 antigens on the immune responses of nonhuman primates to vaccine regimens based on human- or chimpanzee-derived adenovirus vectors. J Virol. 2007;81:6594–6604. doi: 10.1128/JVI.02497-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shayakhmetov DM, Di Paolo NC, Mossman KL. Recognition of virus infection and innate host responses to viral gene therapy vectors. Mol Ther. 2010;18:1422–1429. doi: 10.1038/mt.2010.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cerullo V, Seiler MP, Mane V, Brunetti-Pierri N, Clarke C, Bertin TK, et al. Toll-like receptor 9 triggers an innate immune response to helper-dependent adenoviral vectors. Mol Ther. 2007;15:378–385. doi: 10.1038/sj.mt.6300031. [DOI] [PubMed] [Google Scholar]
- 31.Appledorn DM, Patial S, McBride A, Godbehere S, Van Rooijen N, Parameswaran N, et al. Adenovirus vector-induced innate inflammatory mediators, MAPK signaling, as well as adaptive immune responses are dependent upon both TLR2 and TLR9 in vivo. J Immunol. 2008;181:2134–2144. doi: 10.4049/jimmunol.181.3.2134. [DOI] [PubMed] [Google Scholar]
- 32.VanderLaan PA, Reardon CA, Getz GS. Site specificity of atherosclerosis: site-selective responses to atherosclerotic modulators. Arterioscler Thromb Vasc Biol. 2004;24:12–22. doi: 10.1161/01.ATV.0000105054.43931.f0. [DOI] [PubMed] [Google Scholar]
- 33.Kassim SH, Li H, Bell P, Somanathan S, Lagor W, Jacobs F, et al. Adeno-associated virus serotype 8 gene therapy leads to significant lowering of plasma cholesterol levels in humanized mouse models of homozygous and heterozygous familial hypercholesterolemia. Hum Gene Ther. 2013;24:19–26. doi: 10.1089/hum.2012.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horton JD, Cohen JC, Hobbs HH. PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res. 2009;50 (Suppl):S172–177. doi: 10.1194/jlr.R800091-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koren MJ, Lundqvist P, Bolognese M, Neutel JM, Monsalvo ML, Yang J, et al. Anti-PCSK9 Monotherapy for Hypercholesterolemia: The MENDEL-2 Randomized, Controlled Phase III Clinical Trial of Evolocumab. J Am Coll Cardiol. 2014;63:2531–2540. doi: 10.1016/j.jacc.2014.03.018. [DOI] [PubMed] [Google Scholar]
- 36.Stroes E, Colquhoun D, Sullivan D, Civeira F, Rosenson RS, Watts GF, et al. Anti-PCSK9 Antibody Effectively Lowers Cholesterol in Patients With Statin Intolerance: The GAUSS-2 Randomized, Placebo-Controlled Phase 3 Clinical Trial of Evolocumab. J Am Coll Cardiol. 2014;63:2541–2548. doi: 10.1016/j.jacc.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 37.Robinson JG, Nedergaard BS, Rogers WJ, Fialkow J, Neutel JM, Ramstad D, et al. Effect of evolocumab or ezetimibe added to moderate- or high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: the LAPLACE-2 randomized clinical trial. JAMA. 2014;311:1870–1882. doi: 10.1001/jama.2014.4030. [DOI] [PubMed] [Google Scholar]
- 38.Stein EA, Gipe D, Bergeron J, Gaudet D, Weiss R, Dufour R, et al. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet. 2012;380:29–36. doi: 10.1016/S0140-6736(12)60771-5. [DOI] [PubMed] [Google Scholar]
- 39.Stein EA, Swergold GD. Potential of proprotein convertase subtilisin/kexin type 9 based therapeutics. Curr Atheroscler Rep. 2013;15:310–323. doi: 10.1007/s11883-013-0310-3. [DOI] [PubMed] [Google Scholar]
- 40.Marais AD, Raal FJ, Stein EA, Rader DJ, Blasetto J, Palmer M, et al. A dose-titration and comparative study of rosuvastatin and atorvastatin in patients with homozygous familial hypercholesterolaemia. Atherosclerosis. 2008;197:400–406. doi: 10.1016/j.atherosclerosis.2007.06.028. [DOI] [PubMed] [Google Scholar]
- 41.Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–178. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 42.Zimmermann TS, Lee AC, Akinc A, Bramlage B, Bumcrot D, Fedoruk MN, et al. RNAi-mediated gene silencing in non-human primates. Nature. 2006;441:111–114. doi: 10.1038/nature04688. [DOI] [PubMed] [Google Scholar]
- 43.Koornneef A, Maczuga P, van Logtenstein R, Borel F, Blits B, Ritsema T, et al. Apolipoprotein B knockdown by AAV-delivered shRNA lowers plasma cholesterol in mice. Mol Ther. 2011;19:731–740. doi: 10.1038/mt.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maczuga P, Verheij J, van der Loos C, van Logtenstein R, Hooijer G, Martier R, et al. Therapeutic expression of hairpins targeting apolipoprotein B100 induces phenotypic and transcriptome changes in murine liver. Gene Ther. 2014;21:60–70. doi: 10.1038/gt.2013.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palmer D, Ng P. Improved system for helper-dependent adenoviral vector production. Mol Ther. 2003;8:846–852. doi: 10.1016/j.ymthe.2003.08.014. [DOI] [PubMed] [Google Scholar]
- 46.Li R, Paul A, Ko KW, Sheldon M, Rich BE, Terashima T, et al. Interleukin-7 induces recruitment of monocytes/macrophages to endothelium. Eur Heart J. 2012;33:3114–3123. doi: 10.1093/eurheartj/ehr245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leen AM, Sili U, Vanin EF, Jewell AM, Xie W, Vignali D, et al. Conserved CTL epitopes on the adenovirus hexon protein expand subgroup cross-reactive and subgroup-specific CD8+ T cells. Blood. 2004;104:2432–2440. doi: 10.1182/blood-2004-02-0646. [DOI] [PubMed] [Google Scholar]
- 48.Leen AM, Myers GD, Sili U, Huls MH, Weiss H, Leung KS, et al. Monoculture-derived T lymphocytes specific for multiple viruses expand and produce clinically relevant effects in immunocompromised individuals. Nat Med. 2006;12:1160–1166. doi: 10.1038/nm1475. [DOI] [PubMed] [Google Scholar]
- 49.Kushwaha RS, Barrett PH, Reardon CA, Lewis DS, Carey KD, Getz GS, et al. Relationships of plasma and hepatic variables with rates of plasma low-density lipoprotein apolipoprotein B metabolism in baboons fed low- and high-fat diets. Metabolism. 1995;44:1058–1066. doi: 10.1016/0026-0495(95)90105-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.