

Abstract
Background
Heart failure (HF), despite continuing progress, remains a leading cause of mortality and morbidity. P2X4 receptors (P2X4R) have emerged as potentially important molecules in regulating cardiac function and as potential targets for HF therapy. Transgenic P2X4R overexpression can protect against HF, but this does not explain the role of native cardiac P2X4R. Our goal is to define the physiological role of endogenous cardiac myocyte P2X4R under basal conditions and during HF induced by myocardial infarction or pressure overload.
Methods and Results
Mice established with conditional cardiac-specific P2X4R knockout were subjected to left anterior descending coronary artery ligation–induced postinfarct or transverse aorta constriction–induced pressure overload HF. Knockout cardiac myocytes did not show P2X4R by immunoblotting or by any response to the P2X4R-specific allosteric enhancer ivermectin. Knockout hearts showed normal basal cardiac function but depressed contractile performance in postinfarct and pressure overload models of HF by in vivo echocardiography and ex vivo isolated working heart parameters. P2X4R coimmunoprecipitated and colocalized with nitric oxide synthase 3 (eNOS) in wild-type cardiac myocytes. Mice with cardiac-specific P2X4R overexpression had increased S-nitrosylation, cyclic GMP, NO formation, and were protected from postinfarct and pressure overload HF. Inhibitor of eNOS, L-N5-(1-iminoethyl)ornithine hydrochloride, blocked the salutary effect of cardiac P2X4R overexpression in postinfarct and pressure overload HF as did eNOS knockout.
Conclusions
This study establishes a new protective role for endogenous cardiac myocyte P2X4R in HF and is the first to demonstrate a physical interaction between the myocyte receptor and eNOS, a mediator of HF protection.
Keywords: heart failure, myocytes, cardiac, purines
Adenosine (P1) and P2Y12 receptors are known targets for cardiovascular disease. Increasingly, these receptors are understood to communicate via purinergic signaling,1,2 with evidence that extracellular ATP acting via its P2 purinergic receptors mediates a growing number of biological functions.3–5 In adult ventricular myocytes from healthy wild-type (WT) mice, agonist 2-methylthioadenosine-5′-triphosphate (2-mesATP) causes an increase in a nonselective cation current partially insensitive to antagonism by suramin and pyridoxalphosphate-6-azophenyl-2′,4′-disulphonic acid, characteristic of P2X4 receptors (P2X4R).6 This current’s reversal potential is similar to the cloned P2X4R.6 Interestingly, mice with cardiac-specific P2X4R overexpression exhibit a modest increase in basal cardiac contraction without cardiac hypertrophy or failure as they age.7 These mice are protected from heart failure (HF) with improved cardiac function and survival in postinfarct and calsequestrin overexpression–induced HF.8,9 This effect has also been described in the myocardium of other mammals, including humans.10 Although transgenic mice studies suggest that cardiac P2X4R is important, the high level (>20-fold) of receptor expression makes these mice unsuitable to address the physiological role of P2X4R. No direct evidence points to a role for the receptor in health or disease. Although cardiac transgenic overexpression of P2X4R is capable of protecting against dilated postinfarct and calsequestrin HF, nothing is known about the function of endogenous cardiac myocyte P2X4R under basal condition or during HF. To study this question, we created a tamoxifen-responsive Cre-mediated knockout (KO) of P2X4R in cardiac myocytes. KO mice showed a worse HF phenotype after left left anterior descending coronary artery (LAD) ligation or pressure overload by transverse aorta constriction (TAC) compared with WT mice. To study the underlying protective mechanism, endothelial nitric oxide synthase (eNOS) was coimmunoprecipitated and colocalized with eNOS in cardiac myocytes from both WT and P2X4R-overexpressing transgenic hearts. Pharmacological inhibition of eNOS or eNOS KO blocked the salutary effect of P2X4R overexpression in transgenic mice subjected to postinfarction and pressure overload HF. PX4R-overexpressing transgenic hearts showed enhanced eNOS activation, with higher levels of S-nitrosylation and cyclic GMP as well as increased NO formation by agonist. The present study elucidates a new physiological function of endogenous cardiac myocyte P2X4R and demonstrates for the first time the mechanism by which the receptor interacts physically with eNOS in exerting its salutary effect.
Methods
Mice
The University of Connecticut Health Center Animal Care Committee, in compliance with Animal Welfare Assurance, approved all mice-handling procedures.
P2X4 gene targeting and production of targeted embryonic stem cells were performed as described.11 loxP sites were inserted in introns 1 and 4, and homozygous P2X4floxed/floxed mice were obtained as described (see Methods in the Data Supplement). Homozygous P2X4floxed/floxed mice were then bred with transgenic mice (Myh6-cre/Esr1*; Jax stock no. 005650 in BL6 background), and offspring tamoxifen treatment12 was performed to delete cardiac P2X4R. Jackson Laboratory supplied NOS3 or eNOS KO mice (B6.129P2-Nos3tm1Unc/J) in C57BL/6J background.
Isolation and Measurement of Contraction Shortening of Adult Mouse Cardiac Ventricular Myocytes
Ventricular myocytes were obtained from 3-month-old P2X4R transgenic, KO, or respective WT mice as described previously.6,7 Contraction shortening was elicited by field stimulation at 0.5 Hz and was measured by a video edge-detector device (Crescent Electronics, Sandy, UT) as previously described.13 Five traces were averaged, and amplitude parameters were analyzed with Clampfit 9.0. The amplitude of contraction shortening was expressed as a percentage of resting cell length.
LAD Ligation, TAC, Isolated Working Heart, and Echocardiography
Permanent LAD ligation was performed as previously described.8 Cardiac function by echocardiography was studied at 7, 30, and 90 days after ligation. Ex vivo working heart preparation was then performed 90 days after ligation. For TAC, constriction was created by placing a 27-gauge needle in transverse aorta between innominate and left carotid arteries as described.14,15 A ligature of 7-0 Ethilon nylon suture was tied around the needle and the aorta, after which the needle was removed. The increase in pressure proximal to the constriction was confirmed by an increased Doppler jet velocity across the aortic banding site16 and by directly measuring right and left carotid artery blood pressure difference.16 After euthanasia, the integrity of the banding was confirmed by inspection of the surgical constriction and by visualization of differences in the caliber of the right and left carotid arteries. At 3 weeks, we observed moderate left ventricular (LV) hypertrophy with a reduced cardiac fractional shortening (FS) but no overt HF or arrhythmias. All groups of mice had similar pressure gradients across the aortic banding (data not shown). In studying the effect of TAC on cardiac function in P2X4R KO mice, we focused on early phase (2 and 7 days after banding) because of the early manifestation of adverse effects of TAC.
Cardiac function was determined by echocardiography in vivo8 and by isolated working heart preparation ex vivo.17 Mice were euthanized and, when relevant, infarct size was determined as previously described.8
Immunoprecipitation, Immunoblotting, Immunohistochemistry, and Histology
Immunoprecipitation was performed as described.18 Histological staining, immunoblotting, and immunohistochemistry were performed as described in Methods in the Data Supplement.
Determination of NO Production, Cyclic GMP, and Protein S-Nitrosylation Levels
Cardiac myocyte NO production19 and intact heart cyclic GMP and S-nitrosylation levels were determined as described in Methods in the Data Supplement.
Statistics
Statistical differences were assessed by 2-tailed Mann–Whitney test, Student t test, 1-way ANOVA followed by Newman–Keuls comparison or repeated-measures ANOVA. The Kolmogorov–Smirnov test was used to test for equality of intensity distributions for biotin switch confocal data. We considered P values <0.05 statistically significant. Standard errors were shown.
Materials
Primary immunoblotting antibodies used included mouse monoclonal eNOS (BD Bioscience; 1:1000 dilution) and rabbit polyclonal P2X4 (Alomone Laboratories; 1:1000 dilution). For immunoprecipitation, we used mouse monoclonal eNOS (BD Bioscience; 1–3 μg) and rabbit polyclonal P2X4 (Alomone; 2 μg). L-N5-(1-iminoethyl) ornithine hydrochloride (L-NIO; Sigma-Aldrich) was administered via intraperitonel route at 40 mg/kg per day20 beginning at 3 days before coronary ligation or TAC until cardiac function determinations at 7 days postsurgery.
Results
Cardiac Myocyte–Specific KO of P2X4R and Cardiac Function Under Basal Condition and During Ischemic HF
Conditional Cre-loxP–mediated KO resulted in an absence of P2X4R by immunoblotting (Figure 1, inset). Cardiac myocytes from KO hearts have similar basal contraction shortening amplitude (expressed percent resting cell length; 4.509±0.419%; n=8) as WT cardiac myocytes (4.465±0.450%; n=15) and similar cell length and width (Tables 1 to 3). WT mice were nonfloxed but were Myh6Tg, similarly treated with tamoxifen. Consistent with KO of P2X4R, KO ventricular myocytes could not mount increased contraction in response to the P2X4-specific allosteric enhancer ivermectin (Figure 1A–1C). The KO mice showed apparently normal basal cardiac function by echocardiography in vivo and by isolated working heart parameters ex vivo (Tables 1 to 3). Trichrome, picosirius red, and hematoxylin staining demonstrated apparently normal cardiac histological features at baseline (data not shown).
Figure 1.

Characterization of heart functions in cardiac-specific knockout (KO) of P2X4 receptors (P2X4R). A, Homozygous P2X4floxed/floxed mice were bred with transgenic mice (Myh6-cre/Esr1*) to generate tamoxifen-inducible cardiac myocyte–specific P2X4R KO. Cardiac ventricular myocytes from control (nonfloxed Myh6-cre/Esr1*) mice (15 cells from 10 mice) showed an increase in contraction shortening in response to P2X agonist 2-methylthioadenosine-5′-triphosphate (2-meSATP; P<0.05 vs basal) and a further increase by the P2X4-specific allosteric enhancer ivermectin (*P<0.05 vs 2-meSATP; repeated-measures ANOVA). B, KO myocytes (8 cells from 6 mice) had a response to 2-meSATP alone (P<0.05 vs basal) but did not show further increase by ivermectin. The percent increase over basal by 2-meSATP alone or by 2-meSATP plus ivermectin was less in KO than in control myocytes (P<0.05). KO myocytes did not have detectable P2X4R by immunoblotting as compared with control myocytes (inset). C, Representative myocyte tracings are shown. Multiple cells within the same mouse were assumed to be independent.
Table 1.
Basal Cardiac Function in Cardiac-Specific P2X4 KO Mice—In Vivo Echo
| IVS-D | LVID-D | LVPW-D | IVS-S | LVID-S | LVPW-S | FS, % | HR, beats/min | |
|---|---|---|---|---|---|---|---|---|
| WT (n=9) | 0.412±0.0040 | 3.794±0.0614 | 0.412±0.0040 | 0.528±0.0049 | 2.819±0.0499 | 0.527±0.0049 | 25.70±0.73 | 482±13 |
| KO (n=17) | 0.415±0.0042 | 3.808±0.0631 | 0.415±0.0092 | 0.523±0.0043 | 2.771±0.056 | 0.523±0.0043 | 27.25±0.84 | 474±17 |
Mice with conditional cardiac-specific knockout (KO) of P2X4 receptors (P2x4rflox/MerCreMer) and wild-type (WT; MerCreMer) mice were characterized for basal cardiac function by both in vivo echocardiography and ex vivo isolated working heart preparation 3 wk after tamoxifen treatment as described in the Methods section. There was no statistically significant difference in any of the cardiac function parameters between KO and WT mice. D indicates diastole; FS, fractional shortening; HR, heart rate; IVS, interventricular septum; LVID, left ventricular internal dimension at systole; LVPW, LV posterior wall; and S, systole.
Table 3.
Basal Cardiac Function in Cardiac-Specific P2X4 KO Mice—Isolated Ventricular Cardiomyocytes
| Resting Cell Length, μm | Resting Cell Width, μm | |
|---|---|---|
| WT (57 cells from 9 mice) | 118.9±2.3 | 15.3±0.4 |
| KO (45 cells from 9 mice) | 120.7±3.1 | 15.6±0.4 |
Mice with conditional cardiac-specific knockout (KO) of P2X4 receptors (P2x4rflox/MerCreMer) and wild-type (WT; MerCreMer) mice were characterized for basal cardiac function by both in vivo echocardiography and ex vivo isolated working heart preparation 3 wk after tamoxifen treatment as described in the Methods section. There was no statistically significant difference in cardiomyocyte length or width between KO and WT mice.
To study the pathophysiologic role of endogenous cardiac myocyte P2X4R, we examined the cardiac function in KO mice during postinfarct HF. Compared with WT control mice, KO mice showed a reduced +dP/dt, −dP/dt, LV developed pressure, and FS at 90 days after LAD ligation (Figure 2A–2D). In the working heart preparation, KO hearts showed a lower aortic forward flow (5.71±0.25 mL/min; n=39 mice) compared with control hearts (6.50±0.21 mL/min; n=21; P=0.049). Sham-operated KO mice showed similar function in +dP/dt, −dP/dt, LV developed pressure, and FS as nonoperated KO mice (not shown). Sham-operated WT and KO mice had similar cardiac functions, which were significantly better than those in ligated WT or KO mice (Figure 2). The infarct size was similar in both KO (34.2±1.8; n=16) and control (38.4±2.3; n=11; P=0.15) hearts. Ninety days after infarction, they did not differ in LV thickness or heart weight/body weight ratio (not shown), although KO hearts seem more dilated (LV internal dimension at systole trended greater in KOs at 3.21±0.11 mm; n=20, versus controls at 2.91±0.12; n=11; P=0.11). The impaired cardiac FS in KO mice was apparent as early as 7 days after LAD ligation (Figure IA in the Data Supplement). At 30 days after infarction, FS was also more reduced in KO than in control mice (Figure IB in the Data Supplement). These data are the first to show that the endogenous cardiac myocyte P2X4R plays a protective role in maintaining cardiac function during ischemic HF.
Figure 2.

P2X4 receptor (P2X4R) knockout (KO) mice showed a more severe heart failure phenotype after infarction. Wild-type (WT) and KO mice were subjected to sham operation or left anterior descending coronary artery ligation and cardiac functions determined 90 d later. KO hearts had a lower +dP/dt (A), −dP/dt (B), and left ventricular (LV) developed pressure (C) by ex vivo working heart preparation (n=39) as well as a more reduced fractional shortening (FS) by in vivo echocardiography (D; n=20) than WT hearts (n=19 and 11 for ex and in vivo measurements, respectively). Both WT and KO hearts showed lower ±dP/dt and FS than either sham WT (n=10 for ±dP/dt; n=6 for FS) or sham KO (n=19 for ±dP/dt; n=9 for FS) hearts. *P<0.05 ligated WT vs sham WT, sham KO, or ligated KO. **P<0.05 ligated KO vs sham WT, sham KO. P>0.05 sham WT vs sham KO.
Role of Cardiac Myocyte P2X4R During Pressure Overload–Induced HF
The absence of myocyte P2X4R resulted in a more severe phenotype. Compared with WT controls, KO mice subjected to TAC showed impaired +dP/dt and −dP/dt after 7 days (Figure 3A and 3B). KO mice also showed reduction in echocardiography-derived FS during this early phase of pressure overload (Figure 3C). KO animals also exhibited a concomitantly greater degree of adverse remodeling with an increased end-systolic dimension early (2 and 7 days) after aortic banding (Figure II in the Data Supplement and Figure 3D, respectively). We observed no statistically significant difference of LV thickness in KO hearts compared with controls after banding (not shown) although the overall heart weight/body weight was greater in KO (10.17±0.44; n=10) versus control (8.45±0.29; n=5; P=0.019). Sham-operated KO mice showed similar function in +dP/dt, −dP/dt, LV developed pressure, and FS as nonoperated KO mice (not shown). Sham-operated WT and KO mice had similar cardiac functions, which were significantly better than those in banded KO mice. WT mice with banding did not show a decrease in cardiac function compared with sham-operated mice in the ex vivo working heart model (Figure 3A and 3B), although there was a slightly reduced FS by echocardiography (Figure 3C). At this early time after banding, KO but not WT hearts began to show dilatation with an increase in LV internal dimension at systole (Figure 3D).
Figure 3.

P2X4 receptor (P2X4R) knockout (KO) hearts also had a more impaired function after transverse aorta constriction (TAC). Wild-type (WT) and KO mice were subjected to sham operation or TAC and cardiac functions determined 7 d later. KO mice showed a lower +dP/dt (A) and −dP/dt (B) by working heart (n=10), as well as a more reduced fractional shortening (FS; C) and a larger left ventricular internal dimension at systole (LVIDs; D) by echocardiography (n=9) than control WT mice (n=5 for working hearts and n=13 for echocardiography). KO hearts showed lower ±dP/dt, LV developed pressure (LVdevP), and FS compared with either sham WT (n=10 for ±dP/dt and LVdevP; n=19 for FS) or sham KO (n=9 for ±dP/dt and LVdevP; n=13 for FS) hearts. KO hearts also showed more dilated LVIDs compared with sham WT or sham KO hearts. *P<0.05 banded KO vs banded WT, sham WT, or sham KO; P>0.05 sham WT vs KO; P>0.05 banded WT vs sham WT or sham KO in ±dP/dt and LVIDs; P<0.05 banded WT vs sham WT or sham KO in FS comparison.
To further study the protective role and mechanism of cardiac myocyte P2X4R, we investigated the effect of myocyte P2X4R overexpression in pressure overload HF. Transgenic mice with cardiac-specific P2X4R overexpression showed greater levels of +dP/dt, −dP/dt, LV developed pressure, and FS compared with WT control mice at 3 weeks after TAC (Figure IIIA–IIID in the Data Supplement). After TAC, P2X4R transgenic mice also showed less remodeling with smaller LV dimensions at both systole (Figure IIIE in the Data Supplement) and diastole (not shown) compared with nontransgenic WT mice. The protected phenotype of P2X4 transgenic mice was evident early during pressure overload. Seven days after TAC, cardiac FS (Figure IVA in the Data Supplement) was enhanced, and the LV chamber became less dilated (Figure IVB in the Data Supplement). There was also less fibrosis in the P2X4 transgenic than in WT hearts after TAC (Figure IVC and IVD in the Data Supplement).
Role of eNOS in Protection From Postinfarction HF
Because P2X4R can increase calcium, we hypothesized that P2X4R can interact with and activate the calcium-dependent eNOS. Data summarized in Figure 4 show that P2X4R is present in eNOS immunoprecipitates obtained from cardiac myocytes of both WT (Figure 4A) and P2X4R transgenic (Figure 4B) hearts. P2X4R coimmunoprecipitates with eNOS in both WT and P2X4 transgenic cardiac myocytes. The absence of P2X4R in immunocomplexes captured using an IgG-matched control antibody demonstrates the specificity of this physical interaction.
Figure 4.

Endothelial nitric oxide synthase (eNOS) and P2X4 receptor (P2X4R) coimmunoprecipitated in cardiac ventricular myocytes of wild-type (WT) and P2X4-Tg hearts. A, WT myocyte lysates were incubated overnight with anti-eNOS antibody or with nonspecific IgG as control. The isolated complex was subjected to Western blot (WB) analysis and probed with eNOS (top) and P2X4 (bottom) antibodies. IP indicates immunoprecipitation; and Tg, transgenic. As a control, eNOS coimmunoprecipitated itself. Coimmunoprecipitation of P2X4R with eNOS antibody (lane 3) but not with control IgG (lane 2) is shown. B, Same experiment as in A conducted in P2X4-Tg myocytes. C, Immunostaining of eNOS (green), P2X4R (red), and merged image is shown for a P2X4R-overexpressing Tg cardiac myocyte.
To further support the association between P2X4R and eNOS, we immunostained P2X4 transgenic cardiac myocytes. Both proteins showed a sarcolemmal staining pattern, in addition to diffuse cellular staining (Figure 4C). When plotted as staining intensity versus distance, the 2 proteins colocalized over a specific region in the myocyte (Figure 4C, inset with the line plot). P2X4R and eNOS colocalized in the merged staining. Both proteins also showed sarcolemmal staining in WT myocytes, although P2X4R staining was considerably fainter (Figure V in the Data Supplement), likely reflecting the low level of endogenous expression. Despite this, we observed colocalization with eNOS at the sarcolemma.
To examine the functional consequence of P2X4R–eNOS interaction, we found that myocardial S-nitrosylation levels are greater in P2X4R-overexpressing transgenic hearts than in WT hearts (Figure 5A), consistent with a higher level of stimulation of P2X4R–eNOS–NO in the transgenic heart. This concept was further supported by a higher cyclic GMP level in transgenic (2.56±0.24 fmol/mg; n=13) than in non-transgenic WT (1.86±0.12 fmol/mg; n=8; P=0.027) hearts. Agonist2-meSATP increased the fluorescence of NO-sensitive indicator 4-amino-5-methylamino-2,7-difluorofluorescein diacetate (DAF-FM DA) over vehicle in transgenic cardiomyocytes (Figure 5B). Fluorescence at each time point normalized to baseline was higher in myocytes exposed to 2-meSATP (30 μmol/L; n=43 cells) than in vehicle-treated myocytes (n=33 cells; P=0.008–0.027; Mann–Whitney). After 10 minutes, intensities with agonist normalized to those with vehicle were 109.3±2.6%. The increased eNOS function in P2X4R-overexpressing transgenic mice was not due to an upregulation of eNOS because eNOS protein levels did not change in transgenic (32 087±7508; mean and SEM of intensity normalized to glyceraldehyde-3-phosphate dehydrogenase by ImageJ; n=5) versus WT (41 923±5433; n=5; P=0.42) hearts (Figure VI in the Data Supplement). In studying the function of eNOS as a mediator of P2X4R protection, we sought to inhibit eNOS in transgenic mice during HF. Cardiac transgenic P2X4R overexpression caused a higher cardiac FS after infarction in these mice compared with nontransgenic WT animals.8 We confirmed this finding in data summarized in Figure 6A. Daily injection with L-NIO, used to demonstrate eNOS function in vivo,20 abrogated the improved cardiac FS of P2X4R transgenic mice during ischemic HF (Figure 6A). L-NIO treatment also blocked improved +dP/dt in these transgenic mice after infarction (vehicle: 7552±180.5 mm Hg/sec; n=10 versus L-NIO: 6764±167.0 mm Hg/sec; n=16; P=0.005).
Figure 5.
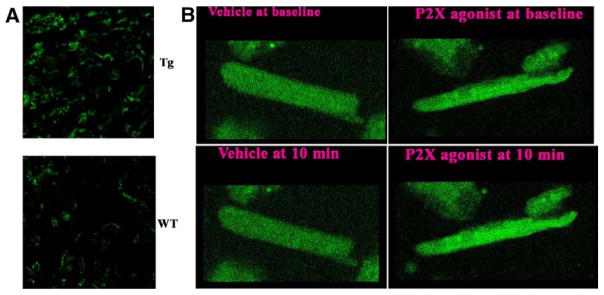
Increased cardiac protein S-nitrosylation levels and nitric oxide formation in P2X4 receptor (P2X4R) transgenic (Tg) animals. In A, a typical example of S-nitrosylated proteins in P2X4R-overexpressing Tg and wild-type (WT) hearts (n=5 for each) is shown as determined using biotin switch method. The counts of total summed intensities produced 1 distribution for each heart and analyzed with Kolmogorov–Smirnov test for equality of distributions. The test is rejected (P<0.001) in favor of the distributions not being the same. B, Typical changes in DAF-FM intensities at baseline vs after exposure to agonist or vehicle. Note the declined intensity in vehicle-treated cells due to photobleaching of fluorescein.
Figure 6.

Pharmacological inhibition by L-N5-(1-iminoethyl)ornithine hydrochloride (L-NIO) and genetic knockout (KO) of endothelial NO synthase (eNOS) abrogated the improved cardiac function by P2X4 receptor (P2X4R) overexpression in heart failure. A, Daily injection of L-NIO 3 d before and 7 d after left anterior descending coronary artery (LAD) ligation in P2X4R-overexpressing transgenic (Tg) mice (n=13) resulted in a reduced fractional shortening (FS) compared with Tg mice not receiving drug (ie, receiving vehicle n=24). Tg mice receiving L-NIO had similar FS as WT mice (n=21 WT) receiving the drug. KO of eNOS in P2X4R Tg mice blocked the improved +dP/dt (B) and FS (C) by P2X4R overexpression in postinfarction heart failure. Thirty days after LAD ligation, P2X4R Tg mice (n=20) showed better +dP/dt compared with P2X4R Tg/eNOS KO (n=22), non-Tg WT (n=9), or eNOS KO mice (n=21). Similarly, P2X4R Tg mice (n=9) had greater FS than any other genotype at 30 d after ligation. Non-Tg WT (n=10), eNOS KO (n=14), P2X4R Tg/eNOS KO mice (n=12) did not differ in FS (ANOVA and post-test comparison). D, Using the same drug injection protocol as in A, P2X4R Tg mice treated with vehicle subjected to transverse aorta constriction (TAC; n=22) had higher FS compared with Tg animals that had received L-NIO (n=11); WT animals (n=11) and Tg mice treated with L-NIO had similar FS. E, 21 days after TAC, P2X4R Tg mice (n=8) showed better FS compared with P2X4R Tg/eNOS KO (n=12) or eNOS KO mice (n= 13). *P<0.05 vs any other group. P>0.05 among groups not marked*.
To further confirm eNOS’s role in protecting myocyte P2X4R, eNOS KO was bred in transgenic mice with cardiac P2X4R overexpression (P2X4 transgenic/eNOS KO). KO of eNOS abrogated the protected phenotype conferred by the P2X4 transgenic genotype in postinfarction HF. Both improved +dP/dt (Figure 6B) and FS (Figure 6C) in P2X4 transgenic mice during HF were lost in P2X4 transgenic/eNOS KO mice. We further studied the role of eNOS in mediating the protective effect of P2X4R overexpression in TAC-subjected P2X4R transgenic mice. L-NIO blocked FS increase in these animals (Figure 6D). KO of eNOS in transgenic animals also abrogated the improved FS in these transgenic mice after TAC (Figure 6E). The data provided functional evidence for P2X4R–eNOS interaction and a key role of eNOS as a mediator of cardiac P2X4 protection in HF.
Discussion
Cell surface purinergic receptors were originally postulated as ATP-responsive P2 or adenosine-responsive P1 receptors in causing nonadrenergic, noncholinergic relaxation of gut smooth muscle.21 Cloning these receptors has allowed classification into metabotropic (P2Y) and ionotropic (P2X) receptor subfamilies.3 Both P1 and P2 purinergic receptors have several important physiological and pathophysiologic roles.4 In adult murine ventricular myocytes, P2X agonist 2-mesATP induced a nonselective cation current with reversal potential similar to that of the cloned P2X4R.6 The P2X4R is selectively potentiated by ivermectin and is an important subunit of the endogenous P2X receptor in cardiac myocytes. Although transgenic mice with cardiac-specific overexpression of human P2X4R are protected from ischemic HF induced by LAD ligation and from a genetic model of dilated cardiomyopathy from the overexpression of calsequestrin, the 20-fold receptor overexpression is artifactual and nonphysiological. The biological role of endogenous myocyte P2X4R was previously unknown. Although the activation of cardiac P2X receptors has been shown to elicit a cyclic AMP–independent increase in cardiac contractile function,13 the mechanism by which these ligand-gated channels protect against HF was also unknown. Using conditional cardiac-specific P2X4R KO and overexpressing mice in various models of HF, we determined a physiological role of this receptor channel and demonstrated eNOS as a physically interacting effector in cardioprotection.
Cardiac Myocyte–Specific KO of P2X4R Showed Normal Basal Cardiac Performance But Depressed Function During HF
Intact cardiac function of P2X4R KO animals was similar to WT control (P2X4floxed/floxed /Myh6 negative) by both in vivo echocardiography-derived functional measures and by ex vivo working heart contractile parameters. The equal loading in a working heart preparation provided a controlled condition to compare contractile performance, complementing the echocardiography measures. KO cardiac ventricular myocytes have similar resting cell length, width, and contraction shortening as controls. KO myocytes did not show P2X4R by immunoblotting or by any response to ivermectin, consistent with its absence in KO hearts. KO hearts were similar to control hearts without any evidence for fibrosis or overt pathology. Thus, cardiac-specific P2X4R KO hearts have normal phenotypes without any hypertrophy, fibrosis, or cardiac myocyte dysfunction. However, when we induced ischemic HF by LAD ligation or pressure overloading by TAC, KO animals showed depressed cardiac function both in vivo and ex vivo. In both types of HF, cardiac function was reduced at the onset of HF and remained depressed as HF progressed. In the pressure overload model, P2X4 KO mice showed a greater heart weight/ body weight ratio, confirming a pathophysiological relevance for the depressed cardiac function. Consistent with cardiac P2X4R protective ability, transgenic overexpression resulted in improved cardiac function in both pressure overload (Figures III and IV in the Data Supplement) and ischemic models of HF.8
P2X4R Interacts With eNOS
P2X4R coimmunoprecipitates with eNOS in isolated cardiac myocytes in both P2X4R-overexpressing transgenic and WT animals. That P2X4R is colocalized with a population, the sarcolemmal eNOS in the P2X4 transgenic cardiac myocyte further supports an interaction of the 2 proteins. The low level of P2X4R immunostaining in WT cardiac myocytes precludes concluding their colocalization in WT myocytes. However, both P2X4R and eNOS showed a similar sarcolemmal appearance in WT cardiac myocytes. Together, the coimmunoprecipitation and colocalization studies support a protein–protein interaction between P2X4R and eNOS. Because P2X4R-overexpressing transgenic hearts were protected from HF, direct measurement of NO production was made in transgenic cardiac myocytes after 2-meSATP stimulation. NO levels, as measured by DAF-FM fluorescence, were increased by the P2X agonist. Furthermore, the levels of S-nitrosylated proteins and cGMP were higher in transgenic than in WT hearts, consistent with a greater stimulation of endogenous NO by the overexpressed P2X4R in transgenic hearts. These findings demonstrate functional evidence for the activation of cardiac P2X4R–eNOS. This activation apparently occurs even as the expression of eNOS in cardiac myocytes is relatively low.22 The data provided a proof of principle for eNOS activation. In murine cardiac myocytes, a modest increase in DAF fluorescence by nitrone had a functional significant impact in reversing contractile dysfunction.19 Our data showed a similar degree of increase in DAF fluorescence after P2X agonist stimulation, and this degree of NO formation may have contributed to its HF protection. In considering another aspect of eNOS activation, phospho-eNOS Ser1177/ total eNOS was not different in P2X4R-overexpressing transgenic versus WT hearts. Phosphorylation of Ser1177 increases its catalytic activity, representing a mechanism of eNOS activation different from that achieved by calmodulin binding.23 We postulated that the mechanism of eNOS activation by P2X4R is from Ca2+ increase and subsequent calmodulin binding and may not involve Ser1177 phosphorylation. In endothelial cells where eNOS is more abundant, P2X4R has also been implicated to stimulate eNOS.24 In the endothelium, the interaction between P2X4R and eNOS remains unstudied; whether inhibiting eNOS could abrogate the vascular P2X4R function is not clear. To understand the significance of P2X4R–eNOS interaction in HF protection, we undertook additional studies.
eNOS Mediates the Cardioprotective Effect of Cardiac P2X4R
If interaction of eNOS with cardiac P2X4R is important in mediating the receptor’s protective effect, one would expect that the inhibition of eNOS could block cardiac P2X4R protective effect, and we indeed found this. Either pharmacological inhibition of eNOS by L-NIO or genetic KO of eNOS abrogated the HF protective effect of cardiac P2X4R overexpression. Cardiac P2X4R overexpression protected from both ischemic and pressure overload forms of HF. The cardioprotective effect of eNOS in ischemia/reperfusion injury and in postinfarct HF has been demonstrated in eNOS-overexpressing transgenic animals.25–27 Upregulating eNOS by H2S has also been shown to protect against pressure overload HF.28 Although eNOS overexpression or upregulation is cardioprotective, eNOS KO has been reported to increase, decrease, or cause no change in ischemia/reperfusion injury.25 The reason for this variable finding may have to do with the presence or absence of a compensatory upregulation of inducible NOS in some of the eNOS KO mice.25 The UNC eNOS−/−, mice from Jackson Laboratory, were shown to have an upregulated iNOS. In the present study, the eNOS KO mice obtained from Jackson Laboratory did not show a more severe HF phenotype after infarction, consistent with their compensated phenotype. Nonetheless, protection provided by the P2X4R–eNOS interaction in the P2X4 transgenic hearts was abrogated by KO of eNOS.
We interpret the data from L-NIO and eNOS KO mice with caution. Although L-NIO is specific for eNOS, it may have other actions that secondarily affect remodeling after pressure overload or infarction as a consequence of its inhibition of eNOS in the endothelium, inducing hypertension and vasoconstriction. The eNOS KO animals exhibit mild hypertension and LV hypertrophy that could in turn confound the salutary effect of cardiac myocyte–specific P2X4R overexpression. A more severe and worse HF could potentially result from pressure overload in eNOS KO animals, which in turn could abrogate the salutary effect of cardiac P2X4R overexpression. However, when the same eNOS KO mice as used here were subjected to abdominal aortic banding, they did not show LV dilatation when compared with the WT control.29 In fact, ejection fraction was greater in eNOS KO than in WT mice after abdominal banding. This apparent protection against abdominal banding may be because of the compensatory upregulaton of iNOS. However, in postinfarct hearts, eNOS KO was not protective because the cardiac function was similarly depressed in both eNOS KO and WT mice (Figure 6C).
That L-NIO or KO of eNOS blocked the salutary effect of cardiac P2X4 transgenic mice supports a cardioprotective role for eNOS. That L-NIO or KO of eNOS could block the P2X4 effect in both postinfarct and post-TAC HF models suggests eNOS as an important mediator of the cardioprotective effect of P2X4R.
The present data are consistent with previously reported protection by eNOS via T-type calcium channel in pressure overload HF.20 Although eNOS is cardioprotective, the downstream effectors mediating eNOS activity are unknown. Possible candidates include cyclic GMP and protein kinase G or cellular protein nitrosylation.5,30 Our study also has implications for the development of P2X4R antagonist as a new therapy to treat pain.31 Because the endogenous cardiac myocyte P2X4R is cardioprotective, its antagonism during pain control may be deleterious to those individuals under cardiac stress, including HF. Although the role of P2X4R in other tissues such as endothelium and monocytes awaits characterization in tissue-specific KO, cardiac-specific activation of eNOS may be more cardioprotective compared with an increased activity of global systemic eNOS.27 Overall, the existence of a physical P2X4R–eNOS interaction is new and its functional significance in HF protection suggests a novel sarcolemmal receptor–enzyme pathway in the heart. A small molecule stimulating this sarcolemmal pathway would represent a new therapy for both ischemic and pressure overloaded HF.
Supplementary Material
Supplemental Table 1 Primer sequences for genotyping
Figure 1. Cardiac KO of P2X4 receptors showed reduced FS as early as 7 days after LAD ligation. (a) Seven days after ligation, KO hearts (n=29) showed a more depressed FS than WT control hearts (n=23, P=0.0064). (b) At 1 month after ligation, KO hearts (n=22) had a lower FS than WT control hearts (n=18, P=0.019). There was no difference in infarct size at any time point between the control and KO hearts.
Figure 2. Cardiac KO of P2X4R showed cardiac dilatation as early as 2 days after TAC. Two days after TAC, KO hearts (n=9) exhibited a more dilated LVID@S than WT hearts (n=4, P=0.019).
Figure 3. Mice with cardiac-specific Tg overexpression of P2X4R had better cardiac function 3 weeks after TAC. Tg mice (n=18) had higher +dP/dt (a) and −dP/dt (b) than WT mice (n=18). Tg hearts (n=16) also had a higher FS (c) and a smaller LVID@S (d) than control WT mice (n=18). *P<0.05 WT subjected to TAC was lower vs. sham WT (n=8), sham Tg (n=8 and 9) or Tg subjected to TAC; **P<0.05 Tg subjected to TAC was different from sham Tg or sham WT.
Figure 4. Cardiac overexpression of P2X4R caused a better preserved FS at 7 days after TAC. Seven days after TAC, (a) FS was greater in P2X4R Tg hearts (n=12) than in WT control hearts (n=11, P=0.046). (b) LVID@S was less in Tg than in WT control hearts (P=0.0077). At 3 weeks after TAC, P2X4 Tg hearts (n=16) showed less fibrosis by (c) picrosirius red (P=0.0008) and (d) trichrome (P=0.0017) staining as compared to WT hearts (n=11).
Figure 5. Immunostaining of P2X4R and eNOS in WT cardiac ventricular myocytes.
Immunostaining was carried out as described in Methods. Both P2X4R (a) and eNOS (b) showed sarcolemmal staining and some diffuse pattern. With only secondary antibodies (goat-α-mouse-FITC from Santa Cruz and goat-α-rabbit- Texas Red from Invitrogen), there was a slight background staining (c). The staining was representative of 6 myocytes from three hearts.
Figure 6. Immunoblotting for eNOS and phospho-eNOS Ser1177 in P2X4R Tg and WT heart homogenates.
Immunoblotting on heart homogenates of P2X4R overexpressing (OE) Tg and WT animals (n=5 for each genotype) was carried out as described in Methods. Upper part of the blot was first probed with antibody against phospho-eNOS Ser1177 and then re-probed with antibody against eNOS after stripping. Lower part of the same blot was cut for probing with antibodies against P2X4R and GAPDH. In P2X4R OE vs. WT comparison, ratios of phospho-eNOS to total eNOS (p1177/eNOS) were similar. Ratios of total eNOS to GAPDH were also not different. Data were plotted as ratios normalized to the average of the WT ratios, as shown in the graph.
Table 2.
Basal Cardiac Function in Cardiac-Specific P2X4 KO Mice—Ex Vivo Working Heart
| +dP/dt, mm Hg/sec | −dP/dt, mm Hg/sec | LVsysP, mm Hg | LVdevP, mm Hg | AF, mL/min | CF, mL/min | CO, mL/min | HR, beats/min | HW/BW mg/g | |
|---|---|---|---|---|---|---|---|---|---|
| WT (n=10) | 7675±193 | 5687±142 | 119.2±2.6 | 121.6±2.7 | 6.4±0.4 | 2.6±0.2 | 9.0±0.3 | 327±12 | 6.0±0.1 |
| KO (n=11) | 7773±254 | 5695±129 | 118.4±3.1 | 119.7±3.3 | 6.6±0.4 | 2.6±0.2 | 9.0±0.4 | 358±10 | 5.8±0.2 |
Mice with conditional cardiac-specific knockout (KO) of P2X4 receptors (P2x4rflox/MerCreMer) and wild-type (WT; MerCreMer) mice were characterized for basal cardiac function by both in vivo echocardiography and ex vivo isolated working heart preparation 3 wk after tamoxifen treatment as described in the Methods section. There was no statistically significant difference in any of the cardiac function parameters between KO and WT mice. AF indicates atrial fibrillation; CF, coronary flow; CO, cardiac output; HR, heart rate; LVdevP, left ventricular developed pressure; and LVsysP, LV systolic pressure.
CLINICAL PERSPECTIVE.
Despite continuing progress, heart failure (HF) remains a leading cause of mortality and morbidity. P2X4 receptors (P2X4R) are ligand-gated ion channels and have emerged as potentially important molecules in regulating cardiac function. We provided evidence for this receptor as a potential new target for HF therapy. Conditional knockout (KO) of this receptor in cardiac myocytes revealed a previously unknown function of the receptor. Mice with cardiac KO of P2X4R showed a more severe HF phenotype in both postinfarct and pressure overload models. The data suggest a new protective role of the native cardiac myocyte channel in HF. As a proof of principle for developing the P2X4R as a new therapy, we showed that mice with cardiac-specific overexpression of the receptor were protected from both pressure overload and postinfarct HF. The mechanism of protection was because of a local production of nitric oxide (NO) arising from a physical interaction between P2X4R and endothelial NO synthase within the cardiomyocyte. The myocyte-intrinsic formation of NO may offer several advantages over NO derived externally from its donor. For example, elevation of this protective molecule in presumably key domain with physical proximity to downstream effector(s) may increase the efficacy of NO. The direct physical interaction of the receptor with the enzyme may also make NO synthase less susceptible to uncoupling during oxidative stress in HF. A strategic localization of the receptor–enzyme pair within the myocyte may ultimately render both more effective in cardioprotection. Increased expression of cardiac P2X4R by gene therapy or stimulation of the receptor by a small-molecule agonist may become a new therapeutic approach to patients with HF.
Acknowledgments
We thank Chunxia Cronin and Carol McGuiness for surgery and echocardiography assistance.
Sources of Funding
This work was supported by National Institutes of Health grant HL48225 and the National Institute of Diabetes and Digestive and Kidney Diseases Intramural Research Program.
Footnotes
Disclosures
None.
References
- 1.Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller C. Nomenclature and classification of adenosine receptors – an update. Pharmacol Rev. 2011;63:1–34. doi: 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oh EY, Abraham T, Saad N, Rapp JH, Vastey FL, Balmir E. A comprehensive comparative review of adenosine diphosphate receptor antagonists. Expert Opin Pharmacother. 2012;13:175–191. doi: 10.1517/14656566.2012.647683. [DOI] [PubMed] [Google Scholar]
- 3.Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden TK, Jacobson KA, Leff P, Williams M. Nomenclature and classification of purinoceptors. Pharmacol Rev. 1994;46:143–156. [PMC free article] [PubMed] [Google Scholar]
- 4.Khakh BS, Burnstock G. The double life of ATP. Sci Am. 2009;301:84–90. 92. doi: 10.1038/scientificamerican1209-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang R, Liang BT. Cardiac P2X4 receptors: targets in ischemia and HF? Circ Res. 2012;111:397–401. doi: 10.1161/CIRCRESAHA.112.265959. [DOI] [PubMed] [Google Scholar]
- 6.Shen JB, Pappano AJ, Liang BT. Extracellular ATP-stimulated current in wild-type and P2X4 receptor transgenic mouse ventricular myocytes: implications for a cardiac physiologic role of P2X4 receptors. FASEB J. 2006;20:277–284. doi: 10.1096/fj.05-4749com. [DOI] [PubMed] [Google Scholar]
- 7.Hu B, Mei QB, Yao XJ, Smith E, Barry WH, Liang BT. A novel contractile phenotype with cardiac transgenic expression of the human P2X4 receptor. FASEB J. 2001;15:2739–2741. doi: 10.1096/fj.01-0445fje. [DOI] [PubMed] [Google Scholar]
- 8.Sonin D, Zhou SY, Cronin C, Sonina T, Wu J, Jacobson KA, Pappano A, Liang BT. Role of P2X purinergic receptors in the rescue of ischemic HF. Am J Physiol Heart Circ Physiol. 2008;295:H1191–H1197. doi: 10.1152/ajpheart.00577.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shen JB, Shutt R, Agosto M, Pappano A, Liang BT. Reversal of cardiac myocyte dysfunction as a unique mechanism of rescue by P2X4 receptors in cardiomyopathy. Am J Physiol Heart Circ Physiol. 2009;296:H1089–H1095. doi: 10.1152/ajpheart.01316.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gergs U, Boknik P, Schmitz W, Simm A, Silber RE, Neumann J. A positive inotropic effect of ATP in the human cardiac atrium. Am J Physiol Heart Circ Physiol. 2008;294:H1716–H1723. doi: 10.1152/ajpheart.00945.2007. [DOI] [PubMed] [Google Scholar]
- 11.Lee EC, Yu D, Martinez de Velasco J, Tessarollo L, Swing DA, Court DL, Jenkins NA, Copeland NG. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics. 2001;73:56–65. doi: 10.1006/geno.2000.6451. [DOI] [PubMed] [Google Scholar]
- 12.Ho VC, Duan L-J, Cronin C, Liang BT, Fong G-H. Elevated VEGF receptor-2 abundance contributes to increased angiogenesis in VEGF receptor-1 deficient mice. Circulation. 2012;126:741–52. doi: 10.1161/CIRCULATIONAHA.112.091603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen JB, Shutt R, Pappano A, Liang BT. Characterization and mechanism of P2X receptor-mediated increase in cardiac myocyte contractility. Am J Physiol Heart Circ Physiol. 2007;293:H3056–H3062. doi: 10.1152/ajpheart.00515.2007. [DOI] [PubMed] [Google Scholar]
- 14.Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, Grueter CE, Qi X, Richardson JA, Hill JA, Katus HA, Bassel-Duby R, Maier LS, Olson EN. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci USA. 2009;106:2342–2347. doi: 10.1073/pnas.0813013106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res. 2006;98:837–845. doi: 10.1161/01.RES.0000215985.18538.c4. [DOI] [PubMed] [Google Scholar]
- 16.Li YH, Reddy AK, Taffet GE, Michael LH, Entman ML, Hartley CJ. Doppler evaluation of peripheral vascular adaptations to transverse aortic banding in mice. Ultrasound Med Biol. 2003;29:1281–1289. doi: 10.1016/s0301-5629(03)00986-4. [DOI] [PubMed] [Google Scholar]
- 17.Zhou SY, Mamdani M, Qanud K, Shen JB, Pappano AJ, Kumar TS, Jacobson KA, Hintze T, Recchia FA, Liang BT. Treatment of heart failure by a methanocarba derivative of adenosine monophosphate: implication for a role of cardiac purinergic P2X receptors. J Pharmacol Exp Ther. 2010;333:920–928. doi: 10.1124/jpet.109.164376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Redden JM, Le AV, Singh A, Federkiewicz K, Smith S, Dodge-Kafka KL. Spatiotemporal regulation of PKC via interactions with AKAP7 isoforms. Biochem J. 2012;446:301–309. doi: 10.1042/BJ20120366. [DOI] [PubMed] [Google Scholar]
- 19.Traynham CJ, Roof SR, Wang H, Prosak RA, Tang L, Viatchenko-Karpinski S, Ho HT, Racoma IO, Catalano DJ, Huang X, Han Y, Kim SU, Gyorke S, Billman GE, Villamena FA, Ziolo MT. Diesterified nitrone rescues nitroso-redox levels and increases myocyte contraction via increased SR Ca(2+) handling. PLoS One. 2012;7:e52005. doi: 10.1371/journal.pone.0052005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakayama H, Bodi I, Correll RN, Chen X, Lorenz J, Houser SR, Robbins J, Schwartz A, Molkentin JD. alpha1G-dependent T-type Ca2+ current antagonizes cardiac hypertrophy through a NOS3-dependent mechanism in mice. J Clin Invest. 2009;119:3787–3796. doi: 10.1172/JCI39724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burnstock G. Purinergic receptors. J Theor Biol. 1976;62:491–503. doi: 10.1016/0022-5193(76)90133-8. [DOI] [PubMed] [Google Scholar]
- 22.Ghafourifar P, Parihar MS, Nazarewicz R, Zenebe WJ, Parihar A. Detection assays for determination of mitochondrial nitric oxide synthase activity; advantages and limitations. Methods Enzymol. 2008;440:317–334. doi: 10.1016/S0076-6879(07)00821-X. [DOI] [PubMed] [Google Scholar]
- 23.Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33:829–837. 837a. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamamoto K, Sokabe T, Matsumoto T, Yoshimura K, Shibata M, Ohura N, Fukuda T, Sato T, Sekine K, Kato S, Isshiki M, Fujita T, Kobayashi M, Kawamura K, Masuda H, Kamiya A, Ando J. Impaired flow-dependent control of vascular tone and remodeling in P2X4-deficient mice. Nat Med. 2006;12:133–137. doi: 10.1038/nm1338. [DOI] [PubMed] [Google Scholar]
- 25.Sharp BR, Jones SP, Rimmer DM, Lefer DJ. Differential response to myocardial reperfusion injury in eNOS-deficient mice. Am J Physiol Heart Circ Physiol. 2002;282:H2422–H2426. doi: 10.1152/ajpheart.00855.2001. [DOI] [PubMed] [Google Scholar]
- 26.Janssens S, Pokreisz P, Schoonjans L, Pellens M, Vermeersch P, Tjwa M, Jans P, Scherrer-Crosbie M, Picard MH, Szelid Z, Gillijns H, Van de Werf F, Collen D, Bloch KD. Cardiomyocyte-specific overexpression of nitric oxide synthase 3 improves left ventricular performance and reduces compensatory hypertrophy after myocardial infarction. Circ Res. 2004;94:1256–1262. doi: 10.1161/01.RES.0000126497.38281.23. [DOI] [PubMed] [Google Scholar]
- 27.Elrod JW, Greer JJ, Bryan NS, Langston W, Szot JF, Gebregzlabher H, Janssens S, Feelisch M, Lefer DJ. Cardiomyocyte-specific overexpression of NO synthase-3 protects against myocardial ischemia-reperfusion injury. Arterioscler Thromb Vasc Biol. 2006;26:1517–1523. doi: 10.1161/01.ATV.0000224324.52466.e6. [DOI] [PubMed] [Google Scholar]
- 28.Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, Murohara T, Predmore BL, Gojon G, Sr, Gojon G, Jr, Wang R, Karusula N, Nicholson CK, Calvert JW, Lefer DJ. H2S protects against pressure overload induced HF via upregulation of endothelial nitric oxide synthase (eNOS) Circulation. 2013;127:1116–1127. doi: 10.1161/CIRCULATIONAHA.112.000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruetten H, Dimmeler S, Gehring D, Ihling C, Zeiher AM. Concentric left ventricular remodeling in endothelial nitric oxide synthase knockout mice by chronic pressure overload. Cardiovasc Res. 2005;66:444–453. doi: 10.1016/j.cardiores.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 30.Haldar SM, Stamler JS. S-nitrosylation: integrator of cardiovascular performance and oxygen delivery. J Clin Invest. 2013;123:101–110. doi: 10.1172/JCI62854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsuda M, Kuboyama K, Inoue T, Nagata K, Tozaki-Saitoh H, Inoue K. Behavioral phenotypes of mice lacking purinergic P2X4 receptors in acute and chronic pain assays. Mol Pain. 2009;5:28. doi: 10.1186/1744-8069-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1 Primer sequences for genotyping
Figure 1. Cardiac KO of P2X4 receptors showed reduced FS as early as 7 days after LAD ligation. (a) Seven days after ligation, KO hearts (n=29) showed a more depressed FS than WT control hearts (n=23, P=0.0064). (b) At 1 month after ligation, KO hearts (n=22) had a lower FS than WT control hearts (n=18, P=0.019). There was no difference in infarct size at any time point between the control and KO hearts.
Figure 2. Cardiac KO of P2X4R showed cardiac dilatation as early as 2 days after TAC. Two days after TAC, KO hearts (n=9) exhibited a more dilated LVID@S than WT hearts (n=4, P=0.019).
Figure 3. Mice with cardiac-specific Tg overexpression of P2X4R had better cardiac function 3 weeks after TAC. Tg mice (n=18) had higher +dP/dt (a) and −dP/dt (b) than WT mice (n=18). Tg hearts (n=16) also had a higher FS (c) and a smaller LVID@S (d) than control WT mice (n=18). *P<0.05 WT subjected to TAC was lower vs. sham WT (n=8), sham Tg (n=8 and 9) or Tg subjected to TAC; **P<0.05 Tg subjected to TAC was different from sham Tg or sham WT.
Figure 4. Cardiac overexpression of P2X4R caused a better preserved FS at 7 days after TAC. Seven days after TAC, (a) FS was greater in P2X4R Tg hearts (n=12) than in WT control hearts (n=11, P=0.046). (b) LVID@S was less in Tg than in WT control hearts (P=0.0077). At 3 weeks after TAC, P2X4 Tg hearts (n=16) showed less fibrosis by (c) picrosirius red (P=0.0008) and (d) trichrome (P=0.0017) staining as compared to WT hearts (n=11).
Figure 5. Immunostaining of P2X4R and eNOS in WT cardiac ventricular myocytes.
Immunostaining was carried out as described in Methods. Both P2X4R (a) and eNOS (b) showed sarcolemmal staining and some diffuse pattern. With only secondary antibodies (goat-α-mouse-FITC from Santa Cruz and goat-α-rabbit- Texas Red from Invitrogen), there was a slight background staining (c). The staining was representative of 6 myocytes from three hearts.
Figure 6. Immunoblotting for eNOS and phospho-eNOS Ser1177 in P2X4R Tg and WT heart homogenates.
Immunoblotting on heart homogenates of P2X4R overexpressing (OE) Tg and WT animals (n=5 for each genotype) was carried out as described in Methods. Upper part of the blot was first probed with antibody against phospho-eNOS Ser1177 and then re-probed with antibody against eNOS after stripping. Lower part of the same blot was cut for probing with antibodies against P2X4R and GAPDH. In P2X4R OE vs. WT comparison, ratios of phospho-eNOS to total eNOS (p1177/eNOS) were similar. Ratios of total eNOS to GAPDH were also not different. Data were plotted as ratios normalized to the average of the WT ratios, as shown in the graph.