

Abstract
Immune checkpoint regulators are critical modulators of the immune system, allowing the initiation of a productive immune response and preventing the onset of autoimmunity. Co-inhibitory and co-stimulatory immune checkpoint receptors are required for full T-cell activation and effector functions such as the production of cytokines. In autoimmune rheumatic diseases, impaired tolerance leads to the development of diseases such as rheumatoid arthritis, systemic lupus erythematosus, and Sjogren’s syndrome. Targeting the pathways of the inhibitory immune checkpoint molecules CD152 (cytotoxic T lymphocyte antigen-4) and CD279 (programmed death-1) in cancer shows robust anti-tumor responses and tumor regression. This observation suggests that, in autoimmune diseases, the converse strategy of engaging these molecules may alleviate inflammation owing to the success of abatacept (CD152-Ig) in rheumatoid arthritis patients. We review the preclinical and clinical developments in targeting immune checkpoint regulators in rheumatic disease.
Introduction
Rheumatic diseases include inflammatory disorders that cause pain, inflammation, or damage in joints and other organs, resulting in significant morbidity, mortality, and societal costs. Examples that are felt to be the result of autoimmunity include rheumatoid arthritis (RA), juvenile idiopathic arthritis (JIA), systemic lupus erythematosus (SLE), psoriasis, systemic sclerosis (SSc), and Sjogren’s syndrome (SS). The magnitude of an inflammatory response is the net result of molecular pathways that enhance or temper immunity. Both genetic and environmental factors control these pathways and can influence the development and severity of these diseases. Beyond engagement of the innate immune system, the perpetuation and amplification of these pathologic processes requires signaling through the B-cell or T-cell receptor, followed by subsequent ligand interactions delivering co-stimulatory and/or co-inhibitory signals. These secondary signals are critical in determining cellular effector functions and modulating immunity to maintain homeostasis [1]. Co-stimulatory and co-inhibitory molecules belong to the B7/B7 ligand family and the tumor necrosis factor (TNF)/TNF receptor family. Their expression and functions are summarized in Tables 1 and 2.
Table 1.
B7/B7 ligand family members and functions
| Molecule | Expression | Ligand/receptor | Function |
|---|---|---|---|
| CD28 | Resting T cells | CD80 or CD86 on APCs | Lowers TCR threshold |
| Delivers a co-stimulatory signal | |||
| Promotes T-cell proliferation, survival, cytokine production, T cell-dependent B cell functions [2] | |||
| CTLA-4/CD152 | Resting/activating T cells | Outcompetes CD28 for CD80/CD86 binding | Increased TCR threshold |
| Delivers a co-inhibitory signal [2] | |||
| Upregulates indoleamine 2,3-dioxygenase | |||
| CTLA-4–/– mice develop autoimmunity [2] | |||
| PD-1/CD279 | T cells, B cells, DCs, monocytes, NK T cells, exhausted cells and Tregs | PD-L1 on APCs, B cells, and T cells. PD-L2/CD273 on APCs. PD-L1/CD274 can also bind to CD80 | Delivers an inhibitory signal |
| Suppresses Bcl-xl [2] | |||
| ICOS/CD278 | Activated T cells, T follicular helper cells | ICOSL/CD275 on APCs and B cells [2] | Induces proliferation |
| Propagates germinal center reactions | |||
| Upregulates IL-10 production [2] | |||
| BTLA/CD272 | B cells, DCs, Th1 cells, macrophages | Herpes virus entry mediator [2] | BTLA delivers an inhibitory signal via ITIM and ITSM [2] |
| B7-H3/CD276 | B cells, NK cells, T cells, activated monocytes [2] | Unknown | Co-inhibitory and co-stimulatory functions. Suppress antitumor responses [2] |
| B7-H4/B7S1/B7x/Vtcn1 | APCs, cancer cells, and mRNA expression on nonhematopoietic tissue [2] | Unknown | Reduces cell proliferation and T cell interleukin-2 production [2] |
| B7-H6 | Tumor cells [3] | NKp30 | Binds NKp30 on NK cells resulting in interferon gamma and cytotoxic function [2] |
| VISTA/Dies1/Gi24/PD-1H | Highly expressed on murine myeloid cells. Low expression on T cells [2] | Unknown | Inhibits T-cell proliferation, reduces CD44 and CD69 expression [2] |
| Increases cell motility [2] |
APC, antigen presenting cell; BTLA, B and T lymphocyte attenuator; CTLA-4, cytotoxic T lymphocyte antigen-4; DC, dendritic cell; ICOS, inducible co-stimulator; ICOSL, inducible co-stimulator ligand; ITIM, immunoreceptor tyrosine-based inhibition motif; ITSM, immunoreceptor tyrosine-based switch motif; NK, natural killer; PD-1, programmed death-1; PD-L, programmed death ligand; TCR, T-cell receptor; Th, T helper; Treg, regulatory T cell.
Table 2.
Tumor necrosis factor/tumor necrosis factor receptor family members and functions
| Molecule | Expression | Ligand/receptor | Function |
|---|---|---|---|
| DR3 | Lymphocytes | Tumor necrosis factor-like cytokine 1A (TL1A) expressed on APCs [4] | Delivers a co-inhibitory signal, induces cell survival, prevents apoptosis [4,5] |
| 4-1BB/CD137 | Activated T cells, Tregs, DCs and B cells | 4-1BB ligand on DCs and B cells [6] | 4-1BB induces CD8+ T-cell, NK T-cell and B-cell survival [6] |
| Signaling back via 4-1BB ligand induces monocyte activation [6] | |||
| OX40/CD134 | Activated T cells | OX40 ligand/CD252 on B cells, endothelial cells, DCs and macrophages | OX40 increases CD4+ T-cell survival/effector function [6] |
| OX40 impacts immunoregulation by reducing interleukin-10 production by Tr1 and CD4+ Tregs [6] | |||
| CD27 | Naïve T-cells, memory B cells, NK T cells, NK cells [6] | CD70 on activated lymphocytes and DCs | CD27–CD70 signaling on B cells propagates germinal center formation and plasma cell activities |
| Signaling on T cells results in proliferation and cytokine production [6] | |||
| CD40 | B cells | CD154/CD40 ligand on T cells, T follicular helper cells, endothelial and epithelial cells, B cells or APCs [7] | CD154+ T cells permit germinal center formation |
| Signaling via CD40 on B cells induces B-cell differentiation, isotype switching and proliferation [7] | |||
| Signaling via CD40 on APCs, increases CD80 and CD86 expression [7] |
APC, antigen presenting cell; DC, dendritic cell; NK, natural killer; Treg, regulatory T cell.
The ability to interfere with the inhibitory function of checkpoint receptors CD152 (cytotoxic T lymphocyte antigen-4) and CD279 (programmed death-1) in oncology has proved successful. In 2011 the US Food and Drug Administration (FDA) approved ipilimumab, an αCD152 monoclonal antibody (Ab), for use in the clinic. In patients with metastatic melanoma, ipilimumab was found to effectively prolong survival and reduce metastases [8,9]. Manipulating the CD279 pathway has been shown to have remarkable efficacy in cancer patients. In patients with melanoma, nonsmall cell lung cancer, and renal cell carcinoma, treatment with BMS-936558, a CD279 blocking Ab, promoted anti-tumor responses [8]. Similarly, CT-011 (Cure Tech Ltd, Yavne, Israel), a humanized αCD279 IgG1 monoclonal Ab, safely induced remission in a subset of patients with hematologic malignancies [8]. In patients with solid cancers, tumor regression was noted following therapy with MDX-1106 (Medarex, Princeton, NJ, USA), an αCD279 IgG4 Ab, further demonstrating that the CD279 pathway plays a crucial role in cancer progression [2]. In addition to targeting CD279, there are ongoing phase 1 clinical trials investigating the role of CD279 ligands: CD274/programmed death ligand (PD-L)-1 in patients with solid tumors [ClinicalTrials.gov:NCT00729664] and CD273/PD-L2 in stage IV melanoma patients [ClinicalTrials.gov:NCT00658892]. The successes in manipulating CD152 and CD279/CD279 ligands in cancer provide proof of concept that targeting these molecules can have profound effects on the human immune response [8].
In contrast to the cancer studies, delivering an inhibitory signal or blocking a stimulatory signal to achieve endogenous immunosuppression is critical in autoimmune diseases. This was first shown in 2005 when the FDA approved the humanized fusion protein CD152-IgG1 (abatacept) as a treatment for RA [10]. The aim of this review is to discuss the function of co-stimulatory and co-inhibitory molecules in the pathogenesis of SLE, RA, JIA, SS, psoriasis, and SSc, as well as their potential use as therapeutic targets.
Systemic lupus erythematosus
SLE is a chronic inflammatory disease targeting multiple organs including the skin, joints, kidneys, lungs, and central nervous system. During disease, autoantibodies to a spectrum of self-antigens, including nuclear antigens, develop and form immune complexes in various tissues. In the kidneys this complex formation results in glomerulonephritis (GN) [11-13]. Current therapies in SLE focus on both B-cell and T-cell targets. The αCD20 Ab rituximab (Genentech, South San Francisco, CA, USA) has been effective anecdotally, but failed to achieve significant benefit above background therapy in separate clinical trials of nonrenal and renal SLE [14,15]. However, it is now thought that these trials may have failed because the high dosage of glucocorticoids used could have masked the effect of rituximab [16,17]. In addition, drug efficacy is hindered by the lack of a definitive target antigen, impaired T-cell homeostasis by reduced CD4+ regulatory T-cell function, an altered T-cell repertoire that in turn promotes autoantibody synthesis [18], inflammation driven by T helper (Th)1 and Th2 responses [19], and soluble mediators such as interferon (IFN) alpha [20].
Positive co-stimulation through CD28 and negative regulation through CD152 are a major focus in designing immunotherapeutic agents to treat lupus patients. There is a wealth of evidence in murine models identifying interactions between CD28 and CD152 and their receptors, CD80 and CD86, as attractive clinical targets. In the spontaneous lupus-prone murine model NZBWF-1, where females develop fatal GN, CD152-Ig therapy effectively reduces autoantibody production [21]. In combination with cyclophosphamide, CD152-Ig effectively reverses nephritis and prolongs survival in NZBWF-1 mice [22]. Furthermore, prenephritic mice treated with CD152-Ig and αCD154 Ab have delayed disease onset and reduced anti-double-stranded DNA Abs [23]. Administered as a triple therapy combining CD152-Ig, cyclophosamide, and αCD154 Ab, prolonged remission and the presence of renal type II activated macrophages that serve as a biomarker of remission have been noted in this model [24]. In contrast to the success of CD152-Ig in murine studies, the humanized CD152-IgG1 fusion protein abatacept has been disappointing in the clinic due to an increase in serious adverse events found in a subset of patients in a phase IIb study [25]. A clinical trial investigating the efficacy of the combination of abatacept and cyclophosamide in human lupus nephritis is ongoing [ClinicalTrials.gov:NCT00774852].
There are several other observations that the CD28–CD152 axis is involved in the pathogenesis of lupus. Elevated levels of soluble CD28 are found in serum from lupus patients, which can inhibit T-cell proliferation in vitro [26]. Whether these levels are active in tempering disease is unknown. There is also an association between polymorphisms in the CD152 gene and SLE susceptibility in some ethnic groups [27]. How this polymorphism impacts disease progression is unclear because CD152 expression itself does not appear to be aberrant in SLE patients. However, studies have shown that CD152 may be functionally impaired in SLE, perhaps as a result of αCD152 autoantibodies [28]. Recently, one study has suggested that abatacept might be linked to regulatory T-cell repopulation [29]. Expression of CD152 ligands also appears to be relevant in SLE. High CD80 expression on CD4+ T cells correlates with disease severity [30], and treatment with αCD80 Ab reduces disease severity in the pristine-induced murine model of disease [31].
The CD278 (inducible co-stimulator)–CD275 (inducible co-stimulator ligand) co-stimulatory pathway may play a role in SLE pathogenesis. In SLE patients, CD278 is expressed on renal lymphocytes and peripheral blood T cells whereas CD275 is highly expressed on B cells but reduced on memory B cells, possibly due to recent interactions with CD278+ T cells [32,33]. In vitro, CD278 and CD3 stimulation leads to increased autoantibody synthesis from SLE peripheral blood mononuclear cells, indicating that CD278 contributes to disease and perturbed B-cell Ab responses [34]. This is further supported in vivo by reduced autoantibody production by CD278–/– lupus-prone MRL/lpr mice [35]. In NZBWF-1 mice, prophylactic and therapeutic treatment with αCD275 Ab significantly reduced disease pathology [36], indicative that both the receptor and ligand are involved in perpetuating inflammation. The role of CD278 as a therapeutic target in human SLE is currently being evaluated in a phase Ib trial with AMG557, an αCD275 Ab [ClinicalTrials.gov:NCT00774943].
CD279 is an inhibitory receptor expressed on activated T cells that upon binding to CD274 (PD-L1) or CD273 (PD-L2) delivers a negative signal into the T cells [37]. In SLE patients, polymorphisms in the CD279 gene are associated with disease susceptibility [38]. In vivo, the role of CD279 is evident by the development of GN in CD279–/– mice bred onto the lupus-prone strain lpr/lpr [39]. In NZBWF-1 mice, CD274 blockade increases CD4+CD279+ T cells and clinical pathology, demonstrating that blockade of the CD279:CD274 axis exacerbates disease [40]. In contrast, one study showed that αCD279 Ab treatment is associated with increases in CD8+ and CD4+ regulatory T cells and protection from disease in NZBWF-1 mice [41,42]. Because of these contradictory results, how the CD279 pathway signals in SLE remains unknown. One explanation is that the αCD274 Ab acted as a blocking Ab, which in turn would explain the increase in effector CD279+CD4+ T cells that produce IFNγ and perpetuate disease [40]. It is also possible that αCD279 Ab may delete CD279+ effector T cells, resulting in disease remission.
CD154 and its receptor, CD40, regulate both humoral and cellular immunity. CD154+ activated T cells can trigger B-cell activation, germinal center formation, long-lived Ab responses, and dendritic cell activation to facilitate the development of CD4+ and CD8+ T-cell responses [7]. Blocking this central co-stimulatory signal is an attractive target in lupus. An increase in CD154 expression on SLE lymphocytes correlates with disease severity in pediatric patients [43]. In active SLE, CD154 is highly expressed on lymphocytes and diminished in remission patients [44]. In a separate study, CD154 expression on T cells was found to induce CD80 on B cells, thus increasing their activation status and further propagating disease [45]. High levels of soluble CD154 have been detected in the serum of patients with advanced disease [46]. In vivo, αCD154 Ab therapy in NZBWF-1 mice prevents the onset of GN [47] and, when administered prior to the establishment of GN, diminishes renal immune complex deposition and prolongs survival [48]. Therapeutic intervention with αCD154 Ab in this disease model also reduces disease severity and resolves proteinuria [48].
In spite of the success of αCD154 Ab in murine models, the results of treatment in clinical trials are inconclusive [49]. In patients with active SLE, treatment with IDEC-131, a humanized αCD154 Ab, did not significantly improve disease scores or suppress anti-double-stranded DNA Ab or complement consumption [49]. However, this finding is confounded by the high placebo response in this trial due to the heterogeneity of SLE [49]. In a phase II trial in patients with proliferative lupus nephritis, BG9588 (Biogen, Inc., Cambridge, MA, USA), another αCD154 Ab, ameliorated disease activity, including proteinuria, and diminished CD38+ Ab-secreting cells [50]. Unfortunately, hematuria and thromboembolic events were reported in two subjects leading to concerns over the safety of targeting CD154 and to early termination of the trial [51]. Later studies have since reported that αCD154 Ab contributes to atherosclerosis and prothrombotic events [52]. This has prompted the reengineering of αCD154 Ab for safe use in SLE preclinical trials. Recently, researchers have shown that an αCD154 Ab containing a mutation in the IgG1 domain impairs Fc effector function and reduces thromboembolism in NZBWF-1 mice [53]. This observation suggests that further development of the αCD154 Abs with these modifications could increase their safety.
CD134 (OX40), a member of the TNF receptor superfamily, is a co-stimulatory molecule expressed on activated T cells and has been examined in SLE. CD134 expression on peripheral blood T cells from patients with lupus nephritis correlates with disease severity [30]. In the glomerular wall of lupus nephritis patients, CD134 expression on T cells and CD252 (OX40 ligand) expression on renal cells have been reported [54]. To understand the function of these molecules, in vitro assays have been performed to examine the function of both CD134 and CD252. For example, treatment of splenocytes from lupus-prone BXSB mice with αCD252 Ab, in combination with CD152-Ig, suppresses autoantibody production and proinflammatory cytokines [55]. Similarly, in vitro treatment of peripheral blood mononuclear cells from SLE patients with an αCD134 Ab reduces interleukin (IL)-4 and IL-10 and enhances IFNγ production whereas CD134-Ig reduces both Th1 and Th2 cytokines [56]. The method of targeting CD134 can exert different outcomes and warrants further investigation. For example, αCD134 Ab controls inflammation in lymph nodes while CD134-Ig prevented the onset of GN [56]. Collectively, these studies show that the CD134–CD252 pathway is involved in regulating inflammation by reducing the production of cytokines such as IL-4 and IL-10, known to perpetuate inflammation in SLE. To date, no clinical trials targeting the CD134 pathway have been conducted.
The CD70–CD27 and CD137 (4-1BB)–CD137 ligand (4-1BB ligand) co-stimulatory pathways belong to the TNF/TNF receptor family and signal on activated T cells. In SLE patients, impaired DNA methylation of CD70 on T cells is associated with disease progression [57] and expression of CD27 on memory SLE B cells and plasma cells correlates with disease severity [58,59]. At present, the CD70–CD27 pathway has yet to be extensively examined in murine lupus models. Several in vivo studies have investigated the role of CD137–CD137 ligand in SLE. CD137–/– mice bred on a MRL/lpr background have increased autoantibody production, pathogenic T cells, and reduced survival [60]. Additionally, treatment of MRL/lpr mice with αCD137 Ab reduced CD4+ T cells, GN, and germinal center formation, as well as prolonging survival [61]. Similarly, αCD137 Ab therapy reduces disease severity in the NZBWF-1 model [62]. No clinical trials have been reported with either pathway in SLE.
Rheumatoid arthritis
RA is a chronic systemic inflammatory disease characterized by destructive synovitis that, left undiagnosed, results in significant pain, deformity, and disability. RA predominately manifests in females and affects about 0.24% of the population [63]. Tissue inflammation and damage is mediated through several cell types, including T cells, B cells, monocytes, macrophages, and osteoclasts. Treatment of RA has been revolutionized by the use of biologics, including TNF inhibitors, rituximab, abatacept, and others beyond standard therapy, generally consisting of methotrexate.
One of the main pathways examined in RA involves CD28 and CD152 interactions with their binding partners, CD80 and CD86. Certain CD152 polymorphisms are associated with an increased risk of developing RA [64]. Soluble CD152 expression also correlates with disease severity, and membrane expression of CD152 on regulatory T cells is reduced [65,66]. Abatacept is effective in treating disease alone or in combination with methotrexate. Patients with low baseline levels of CD8+CD28– T cells are more likely to achieve full remission when treated with abatacept [67]. Additionally, the frequency of CD4+CD28– T cells decreases with treatment but it is uncertain whether this is a direct effect [68].
Other inhibitory B7 family members are also relevant in RA. CD279 polymorphisms are associated with increased susceptibility to disease, and soluble and membrane expression of CD279 is decreased in RA patients [69,70]. In mouse RA models, deficiency of CD279 or CD274 exacerbated disease [71,72]. Furthermore, treatment of mice with a CD274-Ig fusion protein marginally attenuates collagen-induced arthritis (CIA) [73]. High levels of soluble B7-H4, a B7 family inhibitory ligand, are associated with disease severity in RA patients as well as in murine models [74,75]. Both deficiency of B7-H4 and transgenically increased soluble expression of B7-H4 in CIA leads to accelerated disease, and treatment with a B7-H4-Ig fusion protein attenuates CIA [75]. AMP-110, a B7-H4-Ig fusion protein, is currently in a phase I study for use in RA [ClinicalTrials.gov:NTC01878123]. Co-inhibitory ligand CD272 (BTLA) and its receptor HVEM are found in affected synovium of RA patients [76,77]. Treatment of mice with HVEM-Ig increased disease severity in CIA [78]. No clinical trials involving BTLA and HVEM have been documented in RA.
The CD40 and CD154 pathway is implicated in RA pathogenesis. Some CD40 gene polymorphisms are associated with increased RA susceptibility. In addition, CD154 expression is increased in blood and synovial fluid T cells from RA patients [79,80]. IgM and IgG Abs against rheumatoid factor also correlate with increased levels of soluble CD154 [81]. Females with disease have greater expression of CD154 on CD4 T cells in comparison with healthy female controls, and the CD154 promoter is hypomethylated in females, but not males, with RA [82]. In CIA, αCD40 Ab treatment exacerbates disease, and αCD154 Ab and siRNA silencing of CD40 attenuates disease [83-86].
Other TNF superfamily checkpoint regulators are associated with RA. Soluble CD137 and CD137 ligand are also increased in RA patients in comparison with healthy controls, and αCD137 Ab treatment decreases CIA severity [87-89]. In addition, T cells expressing CD134 accumulate in effected synovial joints of RA patients. In CIA, both αCD134 Ab treatment and CD134-Ig treatment can attenuate disease [90-93]. This observation suggests that blocking engagement of CD134 with its ligand is responsible for this effect. CD70 is also overexpressed on CD4+CD28– T cells from RA patients, and treatment of CIA with αCD70 Ab reduced autoantibody titers and disease severity [94,95]. Currently there are no clinical trials of these molecules in RA.
Juvenile idiopathic arthritis
Immune checkpoint regulators are also implicated in the pathogenesis of JIA. Patients have an increased proportion of activated CD4+ and CD8+ T cells expressing CD25 and CD69, and lacking CD28 expression. There is a concomitant decrease in the naïve T-cell pool with shortened telomere length, suggestive of premature aging of this cell population [96]. In the CD8 T-cell compartment, CD28 negative cells have enhanced CD31 expression, allowing T cell receptor-independent activation of these cells [97]. In comparison with healthy controls, JIA patients have reduced numbers of dendritic cells in the blood and increased numbers in the synovial fluid. The dendritic cells in the synovial fluid express high levels of CD80, CD86, and CD40 in comparison with those in the blood, suggesting they are actively promoting inflammation in the joint [98]. Abatacept is FDA approved for the treatment of JIA alone and in combination with methotrexate on the basis of a double-blind withdrawal study [99]. Additional potential targets in this disease are B7-H4, identified as a susceptibility gene, and CD154, whose soluble levels are elevated in JIA [100,101].
Sjogren’s syndrome
SS is an autoimmune systemic disorder that can occur by itself or in combination with other connective tissue diseases. Clinically, SS is characterized by dry eyes and dry mouth. This results from impaired exocrine function of the lacrimal and salivary glands, respectively, due to damage from a predominantly CD4+ T-cell infiltration [102]. The serologic hallmarks of SS include autoantibodies directed against ribonucleoprotein components SS-A (Ro) and/or SS-B (La) [103].
CD28 and CD152 interactions with CD80 and CD86 represent the best studied immune checkpoint regulators in SS. In SS patients, high levels of soluble CD28 are found in the serum and loss of expression on T cells is detected [26,104] and, in vitro, soluble CD28 inhibits T-cell proliferation in response to αCD3 [26]. This is further complimented in vivo in NFS/sld mice, a model of SS where CD4+CD28– T cells in predisease mice express mRNA for IL-4, IL-10, and transforming growth factor beta, and prevent autoimmune lesion formation when adoptively transferred [105]. In SS, CD152 polymorphisms are linked to disease susceptibility and autoantibody production [106,107]. At the transcriptional level CD152 mRNA is found in salivary gland tissue, and αCD152 Abs are detected in the serum of SS patients suggesting the presence of this protein [28,108].
In addition to normal expression on antigen presenting cells, CD80 and CD86 are expressed on salivary gland epithelial cells and duct cells, and can be further upregulated in patients with sialoadentis with IFNγ [109,110]. On duct cells, CD80 and CD86 expression is associated with Th1 cytokines (IL-2 and IFNγ) and Th2 cytokines (IL-4 and IL-5), respectively [111]. This suggests that duct cells and other nonhematopoietic cells in the salivary glands express checkpoint regulator proteins that influence the development of inflammation. Therapeutically, NFS/sld mice treated with αCD86 Ab show reduced autoantibody production, T-cell activation, and a lack of autoimmune lesions, suggesting that the therapy eliminates CD28 co-stimulation [112]. In C57BL/6.NOD-Aec1Aec2 mice, another model for SS, treatment with an adeno-associated virus-2 vector encoding a CD152-Ig fusion protein reduced cell infiltration and Th1 and Th17 cytokine production [113], probably by binding to CD80 and CD86 and preventing co-stimulation through CD28. At present, a phase II study is currently recruiting inflammatory arthritis patients with SS to investigate the efficacy of abatacept [ClinicalTrials.gov:NCT02027298].
A few additional B7 family members have been examined in SS. For example, increased CD279 mRNA levels have been found in SS salivary gland tissue [108]. T cells in inflamed salivary glands express elevated levels of CD279, and CD274 expression on epithelial cells can be induced by IFNγ [114]. Since expression is intact, one speculation is that signaling through this co-inhibitory molecule pair is defective, because this potent negative checkpoint regulator pathway would be expected to control inflammatory T-cell responses in such a setting. One study has found in SS patients that salivary gland epithelial cells expressing CD275 can differentiate CD4+ T cells into T-follicular helper cells [115]. In one murine model, increased cell infiltration into the salivary glands has been reported in CD272–/– mice [116]. These B7 molecules therefore play a role in determining the outcome of T-cell responses in SS and warrant further preclinical studies.
TNF superfamily members are also implicated in SS. Soluble CD154 is increased in the serum of SS patients and CD40:CD154 engagement can induce apoptosis of salivary gland cells [117,118]. However, treatment of nonobese diabetic mice with a vector containing CD40-Ig showed no significant reduction in inflammation in the salivary glands [119]. One study has linked CD252 polymorphisms with B-cell activation in patients with primary SS [120]. Similarly, the CD27–CD70 pathway is associated with B-cell abnormalities in SS. This association is demonstrated by the presence of CD27+ memory B cells in inflamed salivary glands [121,122] and overexpression of CD70 on CD4+ T cells due to hypomethylation of the CD70 promoter in SS patients [123].
Psoriasis
Psoriasis is a chronic inflammatory skin disease characterized by hyperproliferation of kertinocytes, affecting approximately 2% of the population [124]. Some patients also have associated psoriatic arthritis, a destructive spondyloarthropathy. Treatments for psoriasis vary considerably depending on disease severity and the presence or absence of arthritis. These treatments include topical or systemic agents such as corticosteroids, retinoids, vitamin D analogs, ultraviolet light, methotrexate, cyclosporin, TNF inhibitors, and αIL-12/23 (p40) [125]. Little work has been done examining the role of co-stimulatory and co-inhibitory molecules during disease. However, studies with abatacept have shown that treatment can result in clinical improvement in patients with severe psoriasis and psoriatic arthritis [126-130]. In addition, elevated levels of CD154 are found on T cells from both psoriasis and psoriatic arthritis patients, suggesting that αCD154 Ab treatment may be effective in this disease [131,132].
Systemic sclerosis
SSc is a complex autoimmune disease characterized by inflammation, vascular abnormalities, and fibrosis in the skin and internal organs and is more common in females than males. Early skin lesions demonstrate infiltration of activated CD4+ T cells, CD8+ T cells, monocytes, and macrophages [133-135]. In addition, autoantibodies are present that can directly promote fibrosis, and the pattern of antinuclear antibodies present can predict disease progression [136-138]. TNF superfamily co-stimulatory molecules have been associated with disease. Demethylation of CD154 regulatory elements in females is associated with CD154 overexpression on CD4+ T cells [139]. Patients also exhibit elevated plasma levels of soluble CD154. In bleomycin-induced skin sclerosis, a murine model of the disease, prophylactic αCD154 Ab treatment attenuated disease [140]. In addition, CD70 is overexpressed in T cells from patients with SSc and this correlates with demethylation of its promoter region [141]. Gene polymorphisms in CD134 are associated with increased susceptibility to SSc [142]. These observations suggest that therapies targeting CD154, CD70, and CD134 may be worth further investigation for the treatment of disease.
Conclusions
In summary, checkpoint regulators represent viable immunotherapeutic targets for the treatment of both autoimmunity and cancer. A wealth of both mouse and human data indicate that co-stimulatory and co-inhibitory molecules are critical in a number of autoimmune rheumatic diseases (Figure 1). In addition, interventions in multiple murine models utilizing several of these pathways, either by blocking co-stimulatory receptors or by engaging inhibitory receptors, have profound therapeutic effects. However, abatacept is the only agent to achieve FDA approval in the autoimmune human diseases RA and JIA. To date, these findings have not translated into safe and effective therapy for other diseases. In light of the veritable explosion of effective treatments for RA and the approval of more agents for the treatment of psoriatic arthritis, a particular disappointment is the limited progress on this front in SLE. Expectations are still high that this will be achieved within the next few years. The current leading contenders for a successful agent in SLE from a checkpoint regulator perspective are αCD275, abatacept, or a modified αCD154 Ab. Perhaps SS and certainly SSc remain even more daunting disease targets. The intense ongoing investigation in this already fruitful area will undoubtedly produce more candidates for clinical trials in SLE and other rheumatic diseases in the near future.
Figure 1.
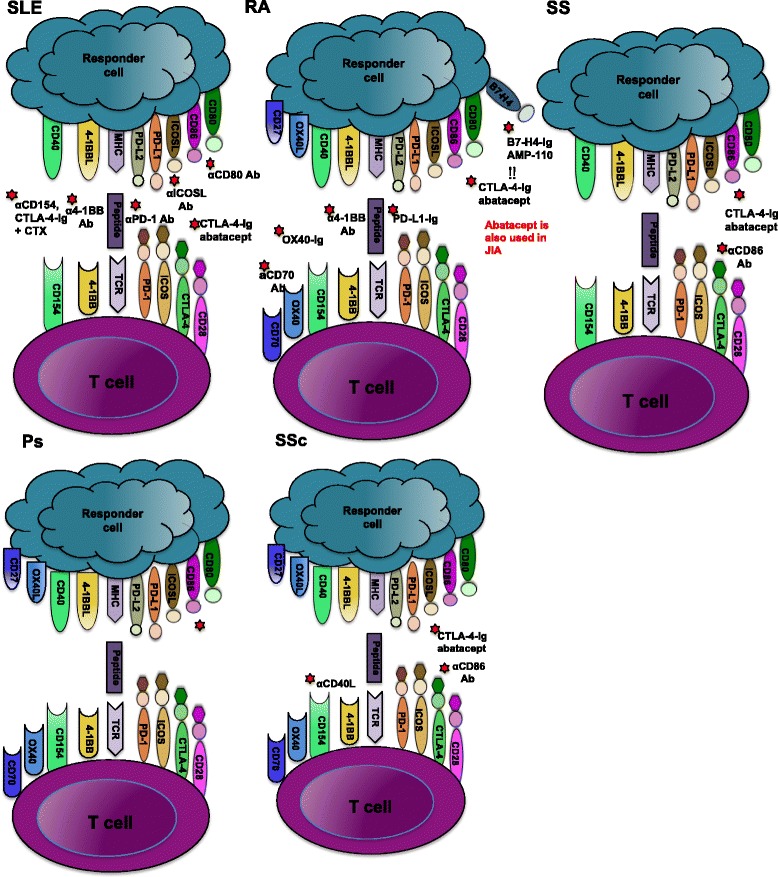
Current in vivo immune checkpoint receptor therapies in rheumatic diseases. T-cell activation requires two signals. The first is via the T-cell receptor (TCR), where peptide is presented by the major histocompatibility complex (MHC) on responder cells. The second involves a network of co-inhibitory and co-stimulatory molecules pathways such as CD80/CD86–CD28/cytotoxic T lymphocyte antigen-4 (CTLA-4), inducible co-stimulator (ICOS)–ICOS ligand (ICOSL), programmed death-1 (PD-1), programme death ligand-1/2 (PD-L1/PD-L2), 4-1BB–4-1BB ligand (4-1BBL), CD40–CD154 ligand, OX40–OX40 ligand and CD27–CD70. This diagram summarizes current therapies for manipulating these pathways to suppress disease in systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), Sjogren’s syndrome (SS), psoriasis (Ps), and systemic sclerosis (SSc). Ab, antibody; CTX, cyclophosphamide; JIA, juvenile idiopathic arthritis.
Acknowledgements
This research was supported by the National Institutes of Health (R01AI098007), the Wellcome Trust (principle research fellowship to RJN), and the National Institute for Health Research Biomedical Research Centre based at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London. The views expressed are those of the authors and not necessarily those of the National Institutes of Health, the National Health Service, the National Institute for Health Research, or the Department of Health.
Abbreviations
- Ab
Antibody
- BTLA
B and T lymphocyte attenuator
- CIA
Collagen-induced arthritis
- FDA
US Food and Drug Administration
- GN
Glomerulonephritis
- HVEM
Herpes virus entry mediator
- IFN
Interferon
- Ig
Immunoglobulin, IL, interleukin
- JIA
Juvenile idiopathic arthritis
- PD-L
Programmed death ligand
- RA
Rheumatoid arthritis
- SLE
Systemic lupus erythematosus
- SS
Sjogren’s syndrome
- SSc
Systemic sclerosis
- Th
T helper
- TNF
Tumor necrosis factor
Footnotes
Competing interests
RJN is CSO, has a commercial research grant, other commercial research support, and ownership interest (including patents), and is a consultant/advisory board member of ImmuNext. The remaining authors declare that they have no competing interests.
Contributor Information
Sabrina Ceeraz, Email: sabrina.c.delong@dartmouth.edu.
Elizabeth C Nowak, Email: elizabeth.c.nowak@dartmouth.edu.
Christopher M Burns, Email: christopher.m.burns@hitchcock.org.
Randolph J Noelle, Email: rjn@dartmouth.edu.
References
- 1.Jenkins MK. The ups and downs of T cell costimulation. Immunity. 1994;1:443–446. doi: 10.1016/1074-7613(94)90086-8. [DOI] [PubMed] [Google Scholar]
- 2.Ceeraz S, Nowak EC, Noelle RJ. B7 family checkpoint regulators in immune regulation and disease. Trends Immunol. 2013;34:556–563. doi: 10.1016/j.it.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brandt CS, Baratin M, Yi EC, Kennedy J, Gao Z, Fox B, Haldeman B, Ostrander CD, Kaifu T, Chabannon C, Moretta A, West R, Xu W, Vivier E, Levin SD. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J Exp Med. 2009;206:1495–1503. doi: 10.1084/jem.20090681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meylan F, Richard AC, Siegel RM. TL1A and DR3, a TNF family ligand-receptor pair that promotes lymphocyte costimulation, mucosal hyperplasia, and autoimmune inflammation. Immunol Rev. 2011;244:188–196. doi: 10.1111/j.1600-065X.2011.01068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Migone TS, Zhang J, Luo X, Zhuang L, Chen C, Hu B, Hong JS, Perry JW, Chen SF, Zhou JX, Cho YH, Ullrich S, Kanakaraj P, Carrell J, Boyd E, Olsen HS, Hu G, Pukac L, Liu D, Ni J, Kim S, Gentz R, Feng P, Moore PA, Ruben SM, Wei P. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity. 2002;16:479–492. doi: 10.1016/S1074-7613(02)00283-2. [DOI] [PubMed] [Google Scholar]
- 6.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 7.Elgueta R, Benson MJ, de Vries VC, Wasiuk A, Guo Y, Noelle RJ. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229:152–172. doi: 10.1111/j.1600-065X.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pardoll D, Drake C. Immunotherapy earns its spot in the ranks of cancer therapy. J Exp Med. 2012;209:201–209. doi: 10.1084/jem.20112275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbé C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alvarez-Quiroga C, Abud-Mendoza C, Doniz-Padilla L, Juarez-Reyes A, Monsivais-Urenda A, Baranda L, Gonzalez-Amaro R. CTLA-4-Ig therapy diminishes the frequency but enhances the function of Treg cells in patients with rheumatoid arthritis. J Clin Immunol. 2011;31:588–595. doi: 10.1007/s10875-011-9527-5. [DOI] [PubMed] [Google Scholar]
- 11.Wen Z, Xu L, Chen X, Xu W, Yin Z, Gao X, Xiong S. Autoantibody induction by DNA-containing immune complexes requires HMGB1 with the TLR2/microRNA-155 pathway. J Immunol. 2013;190:5411–5422. doi: 10.4049/jimmunol.1203301. [DOI] [PubMed] [Google Scholar]
- 12.Swigris JJ, Fischer A, Gillis J, Meehan RT, Brown KK. Pulmonary and thrombotic manifestations of systemic lupus erythematosus. Chest. 2008;133:271–280. doi: 10.1378/chest.07-0079. [DOI] [PubMed] [Google Scholar]
- 13.Deane KD, El-Gabalawy H. Pathogenesis and prevention of rheumatic disease: focus on preclinical RA and SLE. Nat Rev Rheumatol. 2014;10:212–228. doi: 10.1038/nrrheum.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramos-Casals M, Soto MJ, Cuadrado MJ, Khamashta MA. Rituximab in systemic lupus erythematosus: a systematic review of off-label use in 188 cases. Lupus. 2009;18:767–776. doi: 10.1177/0961203309106174. [DOI] [PubMed] [Google Scholar]
- 15.Merrill JT, Neuwelt CM, Wallace DJ, Shanahan JC, Latinis KM, Oates JC, Utset TO, Gordon C, Isenberg DA, Hsieh HJ, Zhang D, Brunetta PG. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010;62:222–233. doi: 10.1002/art.27233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eisenstein M. Approval on a knife edge. Nat Biotechnol. 2012;30:26–29. doi: 10.1038/nbt.2084. [DOI] [PubMed] [Google Scholar]
- 17.Townsend MJ, Monroe JG, Chan AC. B-cell targeted therapies in human autoimmune diseases: an updated perspective. Immunol Rev. 2010;237:264–283. doi: 10.1111/j.1600-065X.2010.00945.x. [DOI] [PubMed] [Google Scholar]
- 18.Desai-Mehta A, Mao C, Rajagopalan S, Robinson T, Datta SK. Structure and specificity of T cell receptors expressed by potentially pathogenic anti-DNA autoantibody-inducing T cells in human lupus. J Clin Invest. 1995;95:531–541. doi: 10.1172/JCI117695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valencia X, Yarboro C, Illei G, Lipsky PE: Deficient CD4+CD25highT regulatory cell function in patients with active systemic lupus erythematosus.J Immunol 2007, 178:2579–2588. [DOI] [PubMed]
- 20.Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. 2007;8:492–502. doi: 10.1038/sj.gene.6364408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finck BK, Linsley PS, Wofsy D. Treatment of murine lupus with CTLA4Ig. Science. 1994;265:1225–1227. doi: 10.1126/science.7520604. [DOI] [PubMed] [Google Scholar]
- 22.Daikh DI, Wofsy D. Cutting edge: reversal of murine lupus nephritis with CTLA4Ig and cyclophosphamide. J Immunol. 2001;166:2913–2916. doi: 10.4049/jimmunol.166.5.2913. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Huang W, Mihara M, Sinha J, Davidson A. Mechanism of action of combined short-term CTLA4Ig and anti-CD40 ligand in murine systemic lupus erythematosus. J Immunol. 2002;168:2046–2053. doi: 10.4049/jimmunol.168.4.2046. [DOI] [PubMed] [Google Scholar]
- 24.Schiffer L, Bethunaickan R, Ramanujam M, Huang W, Schiffer M, Tao H, Madaio MP, Bottinger EP, Davidson A. Activated renal macrophages are markers of disease onset and disease remission in lupus nephritis. J Immunol. 2008;180:1938–1947. doi: 10.4049/jimmunol.180.3.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merrill JT, Burgos-Vargas R, Westhovens R, Chalmers A, D’Cruz D, Wallace DJ, Bae SC, Sigal L, Becker JC, Kelly S, Raghupathi K, Li T, Peng Y, Kinaszczuk M, Nash P. The efficacy and safety of abatacept in patients with non-life-threatening manifestations of systemic lupus erythematosus: results of a twelve-month, multicenter, exploratory, phase IIb, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2010;62:3077–3087. doi: 10.1002/art.27601. [DOI] [PubMed] [Google Scholar]
- 26.Hebbar M, Jeannin P, Magistrelli G, Hatron PY, Hachulla E, Devulder B, Bonnefoy JY, Delneste Y. Detection of circulating soluble CD28 in patients with systemic lupus erythematosus, primary Sjogren’s syndrome and systemic sclerosis. Clin Exp Immunol. 2004;136:388–392. doi: 10.1111/j.1365-2249.2004.02427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee YH, Harley JB, Nath SK. CTLA-4 polymorphisms and systemic lupus erythematosus (SLE): a meta-analysis. Hum Genet. 2005;116:361–367. doi: 10.1007/s00439-004-1244-1. [DOI] [PubMed] [Google Scholar]
- 28.Matsui T, Kurokawa M, Kobata T, Oki S, Azuma M, Tohma S, Inoue T, Yamamoto K, Nishioka K, Kato T. Autoantibodies to T cell costimulatory molecules in systemic autoimmune diseases. J Immunol. 1999;162:4328–4335. [PubMed] [Google Scholar]
- 29.Shin JI, Park SJ, Saleem MA. The beneficial effect of abatacept on lupus nephritis: the role of podocyte beta1-integrin stabilization and Tregs repopopulation? Arthritis Rheumatol. 2014;66:2913–2914. doi: 10.1002/art.38740. [DOI] [PubMed] [Google Scholar]
- 30.Patschan S, Dolff S, Kribben A, Durig J, Patschan D, Wilde B, Specker C, Philipp T, Witzke O: CD134 expression on CD4+T cells is associated with nephritis and disease activity in patients with systemic lupus erythematosus.Clin Exp Immunol 2006, 145:235–242. [DOI] [PMC free article] [PubMed]
- 31.Shi Q, Gao ZY, Xie F, Wang LF, Gu YP, Yang TJ, Huang L, Qian QH, Qiu YH. A novel monoclonal antibody against human CD80 and its immune protection in a mouse lupus-like disease. Int J Immunopathol Pharmacol. 2011;24:583–593. doi: 10.1177/039463201102400304. [DOI] [PubMed] [Google Scholar]
- 32.Hutloff A, Buchner K, Reiter K, Baelde HJ, Odendahl M, Jacobi A, Dorner T, Kroczek RA. Involvement of inducible costimulator in the exaggerated memory B cell and plasma cell generation in systemic lupus erythematosus. Arthritis Rheum. 2004;50:3211–3220. doi: 10.1002/art.20519. [DOI] [PubMed] [Google Scholar]
- 33.Her M, Kim D, Oh M, Jeong H, Choi I. Increased expression of soluble inducible costimulator ligand (ICOSL) in patients with systemic lupus erythematosus. Lupus. 2009;18:501–507. doi: 10.1177/0961203308099176. [DOI] [PubMed] [Google Scholar]
- 34.Yang JH, Zhang J, Cai Q, Zhao DB, Wang J, Guo PE, Liu L, Han XH, Shen Q. Expression and function of inducible costimulator on peripheral blood T cells in patients with systemic lupus erythematosus. Rheumatology (Oxford) 2005;44:1245–1254. doi: 10.1093/rheumatology/keh724. [DOI] [PubMed] [Google Scholar]
- 35.Tada Y, Koarada S, Tomiyoshi Y, Morito F, Mitamura M, Haruta Y, Ohta A, Nagasawa K. Role of inducible costimulator in the development of lupus in MRL/lpr mice. Clin Immunol. 2006;120:179–188. doi: 10.1016/j.clim.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 36.Iwai H, Abe M, Hirose S, Tsushima F, Tezuka K, Akiba H, Yagita H, Okumura K, Kohsaka H, Miyasaka N, Azuma M. Involvement of inducible costimulator–B7 homologous protein costimulatory pathway in murine lupus nephritis. J Immunol. 2003;171:2848–2854. doi: 10.4049/jimmunol.171.6.2848. [DOI] [PubMed] [Google Scholar]
- 37.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prokunina L, Castillejo-Lopez C, Oberg F, Gunnarsson I, Berg L, Magnusson V, Brookes AJ, Tentler D, Kristjansdottir H, Grondal G, Bolstad AI, Svenungsson E, Lundberg I, Sturfelt G, Jönssen A, Truedsson L, Lima G, Alcocer-Varela J, Jonsson R, Gyllensten UB, Harley JB, Alarcón-Segovia D, Steinsson K, Alarcón-Riquelme ME. A regulatory polymorphism in PDCD1 is associated with susceptibility to systemic lupus erythematosus in humans. Nat Genet. 2002;32:666–669. doi: 10.1038/ng1020. [DOI] [PubMed] [Google Scholar]
- 39.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–151. doi: 10.1016/S1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 40.Kasagi S, Kawano S, Okazaki T, Honjo T, Morinobu A, Hatachi S, Shimatani K, Tanaka Y, Minato N, Kumagai S: Anti-programmed cell death 1 antibody reduces CD4+PD-1+T cells and relieves the lupus-like nephritis of NZB/W F1 mice.J Immunol 2010, 184:2337–2347. [DOI] [PubMed]
- 41.Wong M, La Cava A, Hahn BH: Blockade of programmed death-1 in young (New Zealand Black × New Zealand White) F1 mice promotes the suppressive capacity of CD4+regulatory T cells protecting from lupus-like disease.J Immunol 2013, 190:5402–5410. [DOI] [PMC free article] [PubMed]
- 42.Wong M, La Cava A, Singh RP, Hahn BH: Blockade of programmed death-1 in young (New Zealand black × New Zealand white) F1 mice promotes the activity of suppressive CD8+T cells that protect from lupus-like disease.J Immunol 2010, 185:6563–6571. [DOI] [PubMed]
- 43.Mehta J, Genin A, Brunner M, Scalzi LV, Mishra N, Beukelman T, Cron RQ. Prolonged expression of CD154 on CD4 T cells from pediatric lupus patients correlates with increased CD154 transcription, increased nuclear factor of activated T cell activity, and glomerulonephritis. Arthritis Rheum. 2010;62:2499–2509. doi: 10.1002/art.27554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Desai-Mehta A, Lu L, Ramsey-Goldman R, Datta SK. Hyperexpression of CD40 ligand by B and T cells in human lupus and its role in pathogenic autoantibody production. J Clin Invest. 1996;97:2063–2073. doi: 10.1172/JCI118643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koshy M, Berger D, Crow MK. Increased expression of CD40 ligand on systemic lupus erythematosus lymphocytes. J Clin Invest. 1996;98:826–837. doi: 10.1172/JCI118855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kato K, Santana-Sahagun E, Rassenti LZ, Weisman MH, Tamura N, Kobayashi S, Hashimoto H, Kipps TJ. The soluble CD40 ligand sCD154 in systemic lupus erythematosus. J Clin Invest. 1999;104:947–955. doi: 10.1172/JCI7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Early GS, Zhao W, Burns CM. Anti-CD40 ligand antibody treatment prevents the development of lupus-like nephritis in a subset of New Zealand black × New Zealand white mice. Response correlates with the absence of an anti-antibody response. J Immunol. 1996;157:3159–3164. [PubMed] [Google Scholar]
- 48.Quezada SA, Eckert M, Adeyi OA, Schned AR, Noelle RJ, Burns CM. Distinct mechanisms of action of anti-CD154 in early versus late treatment of murine lupus nephritis. Arthritis Rheum. 2003;48:2541–2554. doi: 10.1002/art.11230. [DOI] [PubMed] [Google Scholar]
- 49.Kalunian KC, Davis JC, Jr, Merrill JT, Totoritis MC, Wofsy D, IDEC-131 Lupus Study Group Treatment of systemic lupus erythematosus by inhibition of T cell costimulation with anti-CD154: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46:3251–3258. doi: 10.1002/art.10681. [DOI] [PubMed] [Google Scholar]
- 50.Grammer AC, Slota R, Fischer R, Gur H, Girschick H, Yarboro C, Illei GG, Lipsky PE. Abnormal germinal center reactions in systemic lupus erythematosus demonstrated by blockade of CD154–CD40 interactions. J Clin Invest. 2003;112:1506–1520. doi: 10.1172/JCI200319301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boumpas DT, Furie R, Manzi S, Illei GG, Wallace DJ, Balow JE, Vaishnaw A, BG9588 Lupus Nephritis Trial Group A short course of BG9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum. 2003;48:719–727. doi: 10.1002/art.10856. [DOI] [PubMed] [Google Scholar]
- 52.Hausding M, Jurk K, Daub S, Kroller-Schon S, Stein J, Schwenk M, Oelze M, Mikhed Y, Kerahrodi JG, Kossmann S, Jansen T, Schulz E, Wenzel P, Reske-Kunz AB, Becker C, Munzel T, Grabbe S, Daiber A. CD40L contributes to angiotensin II-induced pro-thrombotic state, vascular inflammation, oxidative stress and endothelial dysfunction. Basic Res Cardiol. 2013;108:386. doi: 10.1007/s00395-013-0386-5. [DOI] [PubMed] [Google Scholar]
- 53.Xie JH, Yamniuk AP, Borowski V, Kuhn R, Susulic V, Rex-Rabe S, Yang X, Zhou X, Zhang Y, Gillooly K, Brosius R, Ravishankar R, Waggie K, Mink K, Price L, Rehfuss R, Tamura J, An Y, Cheng L, Abramczyk B, Ignatovich O, Drew P, Grant S, Bryson JW, Suchard S, Salter-Cid L, Nadler S, Suri A. Engineering of a novel anti-CD40L domain antibody for treatment of autoimmune diseases. J Immunol. 2014;192:4083–4092. doi: 10.4049/jimmunol.1303239. [DOI] [PubMed] [Google Scholar]
- 54.Aten J, Roos A, Claessen N, Schilder-Tol EJ, Ten Berge IJ, Weening JJ. Strong and selective glomerular localization of CD134 ligand and TNF receptor-1 in proliferative lupus nephritis. J Am Soc Nephrol. 2000;11:1426–1438. doi: 10.1681/ASN.V1181426. [DOI] [PubMed] [Google Scholar]
- 55.Zhou YB, Ye RG, Li YJ, Xie CM, Wu YH. Effect of anti-CD134L mAb and CTLA4Ig on ConA-induced proliferation, Th cytokine secretion, and anti-dsDNA antibody production in spleen cells from lupus-prone BXSB mice. Autoimmunity. 2008;41:395–404. doi: 10.1080/08916930802002240. [DOI] [PubMed] [Google Scholar]
- 56.Zhou YB, Ye RG, Li YJ, Xie CM. Targeting the CD134–CD134L interaction using anti-CD134 and/or rhCD134 fusion protein as a possible strategy to prevent lupus nephritis. Rheumatol Int. 2009;29:417–425. doi: 10.1007/s00296-008-0697-2. [DOI] [PubMed] [Google Scholar]
- 57.Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 1990;33:1665–1673. doi: 10.1002/art.1780331109. [DOI] [PubMed] [Google Scholar]
- 58.Wei C, Anolik J, Cappione A, Zheng B, Pugh-Bernard A, Brooks J, Lee EH, Milner EC, Sanz I. A new population of cells lacking expression of CD27 represents a notable component of the B cell memory compartment in systemic lupus erythematosus. J Immunol. 2007;178:6624–6633. doi: 10.4049/jimmunol.178.10.6624. [DOI] [PubMed] [Google Scholar]
- 59.Jacobi AM, Odendahl M, Reiter K, Bruns A, Burmester GR, Radbruch A, Valet G, Lipsky PE, Dorner T. Correlation between circulating CD27high plasma cells and disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2003;48:1332–1342. doi: 10.1002/art.10949. [DOI] [PubMed] [Google Scholar]
- 60.Vinay DS, Kim JD, Asai T, Choi BK, Kwon BS. Absence of 4 1BB gene function exacerbates lacrimal gland inflammation in autoimmune-prone MRL-Faslpr mice. Invest Ophthalmol Vis Sci. 2007;48:4608–4615. doi: 10.1167/iovs.07-0153. [DOI] [PubMed] [Google Scholar]
- 61.Sun Y, Chen HM, Subudhi SK, Chen J, Koka R, Chen L, Fu YX. Costimulatory molecule-targeted antibody therapy of a spontaneous autoimmune disease. Nat Med. 2002;8:1405–1413. doi: 10.1038/nm1202-796. [DOI] [PubMed] [Google Scholar]
- 62.Foell J, Strahotin S, O’Neil SP, McCausland MM, Suwyn C, Haber M, Chander PN, Bapat AS, Yan XJ, Chiorazzi N, Hoffmann MK, Mittler RS. CD137 costimulatory T cell receptor engagement reverses acute disease in lupus-prone NZB x NZW F1 mice. J Clin Invest. 2003;111:1505–1518. doi: 10.1172/JCI200317662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cross M, Smith E, Hoy D, Carmona L, Wolfe F, Vos T, Williams B, Gabriel S, Lassere M, Johns N, Buchbinder R, Woolf A, March L. The global burden of rheumatoid arthritis: estimates from the Global Burden of Disease 2010 study. Ann Rheum Dis. 2014;73:1316–1322. doi: 10.1136/annrheumdis-2013-204627. [DOI] [PubMed] [Google Scholar]
- 64.Plenge RM, Padyukov L, Remmers EF, Purcell S, Lee AT, Karlson EW, Wolfe F, Kastner DL, Alfredsson L, Altshuler D, Gregersen PK, Klareskog L, Rioux JD. Replication of putative candidate-gene associations with rheumatoid arthritis in >4,000 samples from North America and Sweden: association of susceptibility with PTPN22, CTLA4, and PADI4. Am J Hum Genet. 2005;77:1044–1060. doi: 10.1086/498651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Flores-Borja F, Jury EC, Mauri C, Ehrenstein MR. Defects in CTLA-4 are associated with abnormal regulatory T cell function in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2008;105:19396–19401. doi: 10.1073/pnas.0806855105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cao J, Zou L, Luo P, Chen P, Zhang L. Increased production of circulating soluble co-stimulatory molecules CTLA-4, CD28 and CD80 in patients with rheumatoid arthritis. Int Immunopharmacol. 2012;14:585–592. doi: 10.1016/j.intimp.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 67.Scarsi M, Ziglioli T, Airo P. Baseline numbers of circulating CD28-negative T cells may predict clinical response to abatacept in patients with rheumatoid arthritis. J Rheumatol. 2011;38:2105–2111. doi: 10.3899/jrheum.110386. [DOI] [PubMed] [Google Scholar]
- 68.Gomez-Garcia L, Ramirez-Assad C, Vargas A, Masso F, Sanchez-Munoz F, Marquez-Velasco R, Amezcua-Guerra LM, Bojalil R: Reduced numbers of circulating CD28-negative CD4+cells in patients with rheumatoid arthritis chronically treated with abatacept.Int J Rheum Dis 2013, 16:469–471. [DOI] [PubMed]
- 69.Kong EK, Prokunina-Olsson L, Wong WH, Lau CS, Chan TM, Alarcon-Riquelme M, Lau YL. A new haplotype of PDCD1 is associated with rheumatoid arthritis in Hong Kong Chinese. Arthritis Rheum. 2005;52:1058–1062. doi: 10.1002/art.20966. [DOI] [PubMed] [Google Scholar]
- 70.Li S, Liao W, Chen M, Shan S, Song Y, Zhang S, Song H, Yuan Z: Expression of programmed death-1 (PD-1) on CD4+and CD8+T cells in rheumatoid arthritis.Inflammation 2014, 37:116–121. [DOI] [PubMed]
- 71.Raptopoulou AP, Bertsias G, Makrygiannakis D, Verginis P, Kritikos I, Tzardi M, Klareskog L, Catrina AI, Sidiropoulos P, Boumpas DT. The programmed death 1/programmed death ligand 1 inhibitory pathway is up-regulated in rheumatoid synovium and regulates peripheral T cell responses in human and murine arthritis. Arthritis Rheum. 2010;62:1870–1880. doi: 10.1002/art.27500. [DOI] [PubMed] [Google Scholar]
- 72.Hamel KM, Cao Y, Wang Y, Rodeghero R, Kobezda T, Chen L, Finnegan A. B7-H1 expression on non-B and non-T cells promotes distinct effects on T- and B-cell responses in autoimmune arthritis. Eur J Immunol. 2010;40:3117–3127. doi: 10.1002/eji.201040690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang G, Hu P, Yang J, Shen G, Wu X. The effects of PDL-Ig on collagen-induced arthritis. Rheumatol Int. 2011;31:513–519. doi: 10.1007/s00296-009-1249-0. [DOI] [PubMed] [Google Scholar]
- 74.Kamimura Y, Kobori H, Piao J, Hashiguchi M, Matsumoto K, Hirose S, Azuma M. Possible involvement of soluble B7-H4 in T cell-mediated inflammatory immune responses. Biochem Biophys Res Commun. 2009;389:349–353. doi: 10.1016/j.bbrc.2009.08.144. [DOI] [PubMed] [Google Scholar]
- 75.Azuma T, Zhu G, Xu H, Rietz AC, Drake CG, Matteson EL, Chen L. Potential role of decoy B7-H4 in the pathogenesis of rheumatoid arthritis: a mouse model informed by clinical data. PLoS Med. 2009;6:e1000166. doi: 10.1371/journal.pmed.1000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shang Y, Guo G, Cui Q, Li J, Ruan Z, Chen Y. The expression and anatomical distribution of BTLA and its ligand HVEM in rheumatoid synovium. Inflammation. 2012;35:1102–1112. doi: 10.1007/s10753-011-9417-2. [DOI] [PubMed] [Google Scholar]
- 77.Guo G, Shang Y, Zhu G, Bao X, Xu S, Chen Y. The expression and distribution of immunomodulatory proteins B7-H1, B7-DC, B7-H3, and B7-H4 in rheumatoid synovium. Clin Rheumatol. 2012;31:271–281. doi: 10.1007/s10067-011-1815-1. [DOI] [PubMed] [Google Scholar]
- 78.Pierer M, Schulz A, Rossol M, Kendzia E, Kyburz D, Haentzschel H, Baerwald C, Wagner U. Herpesvirus entry mediator-Ig treatment during immunization aggravates rheumatoid arthritis in the collagen-induced arthritis model. J Immunol. 2009;182:3139–3145. doi: 10.4049/jimmunol.0713715. [DOI] [PubMed] [Google Scholar]
- 79.Garcia-Bermudez M, Gonzalez-Juanatey C, Lopez-Mejias R, Teruel M, Corrales A, Miranda-Filloy JA, Castaneda S, Balsa A, Fernandez-Gutierrez B, Gonzalez-Alvaro I, omez-Vaquero C, Blanco R, Llorca J, Martin J, Gonzalez-Gay MA. Study of association of CD40-CD154 gene polymorphisms with disease susceptibility and cardiovascular risk in Spanish rheumatoid arthritis patients. PLoS One. 2012;7:e49214. doi: 10.1371/journal.pone.0049214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.MacDonald KP, Nishioka Y, Lipsky PE, Thomas R. Functional CD40 ligand is expressed by T cells in rheumatoid arthritis. J Clin Invest. 1997;100:2404–2414. doi: 10.1172/JCI119781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tamura N, Kobayashi S, Kato K, Bando H, Haruta K, Oyanagi M, Kuriyama M, Kipps TJ, Hashimoto H. Soluble CD154 in rheumatoid arthritis: elevated plasma levels in cases with vasculitis. J Rheumatol. 2001;28:2583–2590. [PubMed] [Google Scholar]
- 82.Liao J, Liang G, Xie S, Zhao H, Zuo X, Li F, Chen J, Zhao M, Chan TM, Lu Q. CD40L demethylation in CD4 (+) T cells from women with rheumatoid arthritis. Clin Immunol. 2012;145:13–18. doi: 10.1016/j.clim.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 83.Kyburz D, Carson DA, Corr M. The role of CD40 ligand and tumor necrosis factor alpha signaling in the transgenic K/BxN mouse model of rheumatoid arthritis. Arthritis Rheum. 2000;43:2571–2577. doi: 10.1002/1529-0131(200011)43:11<2571::AID-ANR26>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 84.Tellander AC, Michaelsson E, Brunmark C, Andersson M. Potent adjuvant effect by anti-CD40 in collagen-induced arthritis. Enhanced disease is accompanied by increased production of collagen type-II reactive IgG2a and IFN-gamma. J Autoimmun. 2000;14:295–302. doi: 10.1006/jaut.2000.0374. [DOI] [PubMed] [Google Scholar]
- 85.Andreakos E, Rauchhaus U, Stavropoulos A, Endert G, Wendisch V, Benahmed AS, Giaglis S, Karras J, Lee S, Gaus H, Bennett CF, Williams RO, Sideras P, Panzner S. Amphoteric liposomes enable systemic antigen-presenting cell-directed delivery of CD40 antisense and are therapeutically effective in experimental arthritis. Arthritis Rheum. 2009;60:994–1005. doi: 10.1002/art.24434. [DOI] [PubMed] [Google Scholar]
- 86.Zheng X, Suzuki M, Zhang X, Ichim TE, Zhu F, Ling H, Shunnar A, Wang MH, Garcia B, Inman RD, Min WP. RNAi-mediated CD40-CD154 interruption promotes tolerance in autoimmune arthritis. Arthritis Res Ther. 2010;12:R13. doi: 10.1186/ar2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Michel J, Langstein J, Hofstadter F, Schwarz H. A soluble form of CD137 (ILA/4-1BB), a member of the TNF receptor family, is released by activated lymphocytes and is detectable in sera of patients with rheumatoid arthritis. Eur J Immunol. 1998;28:290–295. doi: 10.1002/(SICI)1521-4141(199801)28:01<290::AID-IMMU290>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 88.Foell JL, Diez-Mendiondo BI, Diez OH, Holzer U, Ruck P, Bapat AS, Hoffmann MK, Mittler RS, Dannecker GE. Engagement of the CD137 (4-1BB) costimulatory molecule inhibits and reverses the autoimmune process in collagen-induced arthritis and establishes lasting disease resistance. Immunology. 2004;113:89–98. doi: 10.1111/j.1365-2567.2004.01952.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Seo SK, Choi JH, Kim YH, Kang WJ, Park HY, Suh JH, Choi BK, Vinay DS, Kwon BS. 4-1BB-mediated immunotherapy of rheumatoid arthritis. Nat Med. 2004;10:1088–1094. doi: 10.1038/nm1107. [DOI] [PubMed] [Google Scholar]
- 90.Brugnoni D, Bettinardi A, Malacarne F, Airo P, Cattaneo R: CD134/OX40 expression by synovial fluid CD4+T lymphocytes in chronic synovitis.Br J Rheumatol 1998, 37:584–585. [DOI] [PubMed]
- 91.Passacantando A, Parzanese I, Rascente M, Petrucci C, Minisola G, Tonietti G. Synovial fluid OX40T lymphocytes of patients with rheumatoid arthritis display a Th2/Th0 polarization. Int J Immunopathol Pharmacol. 2006;19:499–505. doi: 10.1177/039463200601900305. [DOI] [PubMed] [Google Scholar]
- 92.Boot EP, Koning GA, Storm G, Wagenaar-Hilbers JP, van Eden W, Everse LA, Wauben MH: CD134 as target for specific drug delivery to auto-aggressive CD4+T cells in adjuvant arthritis.Arthritis Res Ther 2005, 7:R604–R615. [DOI] [PMC free article] [PubMed]
- 93.Findlay EG, Danks L, Madden J, Cavanagh MM, McNammee K, McCann F, Snelgrove RJ, Shaw S, Feldmann M, Taylor PC, Horwood NJ, Hussell T. OX40L blockade is therapeutic in arthritis, despite promoting osteoclastogenesis. Proc Natl Acad Sci U S A. 2014;111:2289–2294. doi: 10.1073/pnas.1321071111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee WW, Yang ZZ, Li G, Weyand CM, Goronzy JJ. Unchecked CD70 expression on T cells lowers threshold for T cell activation in rheumatoid arthritis. J Immunol. 2007;179:2609–2615. doi: 10.4049/jimmunol.179.4.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Oflazoglu E, Boursalian TE, Zeng W, Edwards AC, Duniho S, McEarchern JA, Law CL, Gerber HP, Grewal IS. Blocking of CD27–CD70 pathway by anti-CD70 antibody ameliorates joint disease in murine collagen-induced arthritis. J Immunol. 2009;183:3770–3777. doi: 10.4049/jimmunol.0901637. [DOI] [PubMed] [Google Scholar]
- 96.Almanzar G, Zlamy M, Koppelstaetter C, Brunner A, Jeller V, Duftner C, Dejaco C, Brunner J, Prelog M: Increased replication of CD4+naive T cells and changes in T cell homeostasis in a case of acute exacerbation of juvenile idiopathic arthritis: a case comparison study.J Med Case Rep 2013, 7:135. [DOI] [PMC free article] [PubMed]
- 97.Dvergsten JA, Mueller RG, Griffin P, Abedin S, Pishko A, Michel JJ, Rosenkranz ME, Reed AM, Kietz DA, Vallejo AN: Premature cell senescence and T cell receptor-independent activation of CD8+T cells in juvenile idiopathic arthritis.Arthritis Rheum 2013, 65:2201–2210. [DOI] [PMC free article] [PubMed]
- 98.Smolewska E, Stanczyk J, Brozik H, Biernacka-Zielinska M, Cebula B, Robak T, Smolewski P. Distribution and clinical significance of blood dendritic cells in children with juvenile idiopathic arthritis. Ann Rheum Dis. 2008;67:762–768. doi: 10.1136/ard.2007.077669. [DOI] [PubMed] [Google Scholar]
- 99.Goldzweig O, Hashkes PJ. Abatacept in the treatment of polyarticular JIA: development, clinical utility, and place in therapy. Drug Des Devel Ther. 2011;5:61–70. doi: 10.2147/DDDT.S16489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Albers HM, Reinards TH, Brinkman DM, Kamphuis SS, van Rossum MA, Hoppenreijs EP, Girschick HJ, Wouters C, Saurenmann RK, Bakker E, Verduijn W, Slagboom P, Huizinga TW, Toes RE, Houwing-Duistermaat JJ, ten Cate R, Schilham MW. Genetic variation in VTCN1 (B7-H4) is associated with course of disease in juvenile idiopathic arthritis. Ann Rheum Dis. 2014;73:1198–1201. doi: 10.1136/annrheumdis-2013-204466. [DOI] [PubMed] [Google Scholar]
- 101.Prahalad S, Martins TB, Tebo AE, Whiting A, Clifford B, Zeft AS, McNally B, Bohnsack JF, Hill HR. Elevated serum levels of soluble CD154 in children with juvenile idiopathic arthritis. Pediatr Rheumatol Online J. 2008;6:8. doi: 10.1186/1546-0096-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bolstad AI, Jonsson R. Genetic aspects of Sjogren’s syndrome. Arthritis Res. 2002;4:353–359. doi: 10.1186/ar599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gordon TP, Bolstad AI, Rischmueller M, Jonsson R, Waterman SA. Autoantibodies in primary Sjogren’s syndrome: new insights into mechanisms of autoantibody diversification and disease pathogenesis. Autoimmunity. 2001;34:123–132. doi: 10.3109/08916930109001960. [DOI] [PubMed] [Google Scholar]
- 104.Smolenska Z, Pawlowska J, Zdrojewski Z, Daca A, Bryl E: Increased percentage of CD8+CD28–T cells correlates with clinical activity in primary Sjogren’s syndrome.Cell Immunol 2012, 278:143–151. [DOI] [PubMed]
- 105.Saegusa K, Ishimaru N, Yanagi K, Haneji N, Nishino M, Azuma M, Saito I, Hayashi Y: Autoantigen-specific CD4+CD28lowT cell subset prevents autoimmune exocrinopathy in murine Sjogren’s syndrome.J Immunol 2000, 165:2251–2257. [DOI] [PubMed]
- 106.Downie-Doyle S, Bayat N, Rischmueller M, Lester S. Influence of CTLA4 haplotypes on susceptibility and some extraglandular manifestations in primary Sjogren’s syndrome. Arthritis Rheum. 2006;54:2434–2440. doi: 10.1002/art.22004. [DOI] [PubMed] [Google Scholar]
- 107.Gottenberg JE, Loiseau P, Azarian M, Chen C, Cagnard N, Hachulla E, Puechal X, Sibilia J, Charron D, Mariette X, Miceli-Richard C. CTLA-4 + 49A/G and CT60 gene polymorphisms in primary Sjogren syndrome. Arthritis Res Ther. 2007;9:R24. doi: 10.1186/ar2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bolstad AI, Eiken HG, Rosenlund B, Alarcon-Riquelme ME, Jonsson R. Increased salivary gland tissue expression of Fas, Fas ligand, cytotoxic T lymphocyte-associated antigen 4, and programmed cell death 1 in primary Sjogren’s syndrome. Arthritis Rheum. 2003;48:174–185. doi: 10.1002/art.10734. [DOI] [PubMed] [Google Scholar]
- 109.Manoussakis MN, Dimitriou ID, Kapsogeorgou EK, Xanthou G, Paikos S, Polihronis M, Moutsopoulos HM. Expression of B7 costimulatory molecules by salivary gland epithelial cells in patients with Sjogren’s syndrome. Arthritis Rheum. 1999;42:229–239. doi: 10.1002/1529-0131(199902)42:2<229::AID-ANR4>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 110.Matsumura R, Umemiya K, Goto T, Nakazawa T, Kagami M, Tomioka H, Tanabe E, Sugiyama T, Sueishi M. Glandular and extraglandular expression of costimulatory molecules in patients with Sjogren’s syndrome. Ann Rheum Dis. 2001;60:473–482. doi: 10.1136/ard.60.5.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tsunawaki S, Nakamura S, Ohyama Y, Sasaki M, Ikebe-Hiroki A, Hiraki A, Kadena T, Kawamura E, Kumamaru W, Shinohara M, Shirasuna K. Possible function of salivary gland epithelial cells as nonprofessional antigen-presenting cells in the development of Sjogren’s syndrome. J Rheumatol. 2002;29:1884–1896. [PubMed] [Google Scholar]
- 112.Saegusa K, Ishimaru N, Yanagi K, Haneji N, Nishino M, Azuma M, Saito I, Hayashi Y. Treatment with anti-CD86 costimulatory molecule prevents the autoimmune lesions in murine Sjogren’s syndrome (SS) through up-regulated Th2 response. Clin Exp Immunol. 2000;119:354–360. doi: 10.1046/j.1365-2249.2000.01121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yin H, Nguyen CQ, Samuni Y, Uede T, Peck AB, Chiorini JA. Local delivery of AAV2-CTLA4IgG decreases sialadenitis and improves gland function in the C57BL/6NOD-Aec1Aec2 mouse model of Sjogren’s syndrome. Arthritis Res Ther. 2012;14:R40. doi: 10.1186/ar3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kobayashi M, Kawano S, Hatachi S, Kurimoto C, Okazaki T, Iwai Y, Honjo T, Tanaka Y, Minato N, Komori T, Maeda S, Kumagai S. Enhanced expression of programmed death-1 (PD-1)/PD-L1 in salivary glands of patients with Sjogren’s syndrome. J Rheumatol. 2005;32:2156–2163. [PubMed] [Google Scholar]
- 115.Gong YZ, Nititham J, Taylor K, Miceli-Richard C, Sordet C, Wachsmann D, Bahram S, Georgel P, Criswell LA, Sibilia J, Mariette X, Alsaleh G, Gottenberg JE. Differentiation of follicular helper T cells by salivary gland epithelial cells in primary Sjogren’s syndrome. J Autoimmun. 2014;51:57–66. doi: 10.1016/j.jaut.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 116.Oya Y, Watanabe N, Owada T, Oki M, Hirose K, Suto A, Kagami S, Nakajima H, Kishimoto T, Iwamoto I, Murphy TL, Murphy KM, Saito Y. Development of autoimmune hepatitis-like disease and production of autoantibodies to nuclear antigens in mice lacking B and T lymphocyte attenuator. Arthritis Rheum. 2008;58:2498–2510. doi: 10.1002/art.23674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Goules A, Tzioufas AG, Manousakis MN, Kirou KA, Crow MK, Routsias JG. Elevated levels of soluble CD40 ligand (sCD40L) in serum of patients with systemic autoimmune diseases. J Autoimmun. 2006;26:165–171. doi: 10.1016/j.jaut.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 118.Ping L, Ogawa N, Zhang Y, Sugai S, Masaki Y, Weiguo X. p38 mitogen-activated protein kinase and nuclear factor-kappaB facilitate CD40-mediated salivary epithelial cell death. J Rheumatol. 2012;39:1256–1264. doi: 10.3899/jrheum.110097. [DOI] [PubMed] [Google Scholar]
- 119.Roescher N, Vosters JL, Lai Z, Uede T, Tak PP, Chiorini JA. Local administration of soluble CD40:Fc to the salivary glands of non-obese diabetic mice does not ameliorate autoimmune inflammation. PLoS One. 2012;7:e51375. doi: 10.1371/journal.pone.0051375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nordmark G, Kristjansdottir G, Theander E, Appel S, Eriksson P, Vasaitis L, Kvarnstrom M, Delaleu N, Lundmark P, Lundmark A, Sjowall C, Brun JG, Jonsson MV, Harboe E, Goransson LG, Johnsen SJ, Soderkvist P, Eloranta ML, Alm G, Baecklund E, Wahren-Herlenius M, Omdal R, Ronnblom L, Jonsson R, Syvanen AC. Association of EBF1, FAM167A (C8orf13)-BLK and TNFSF4 gene variants with primary Sjogren’s syndrome. Genes Immun. 2011;12:100–109. doi: 10.1038/gene.2010.44. [DOI] [PubMed] [Google Scholar]
- 121.Hansen A, Odendahl M, Reiter K, Jacobi AM, Feist E, Scholze J, Burmester GR, Lipsky PE, Dorner T. Diminished peripheral blood memory B cells and accumulation of memory B cells in the salivary glands of patients with Sjogren’s syndrome. Arthritis Rheum. 2002;46:2160–2171. doi: 10.1002/art.10445. [DOI] [PubMed] [Google Scholar]
- 122.Hansen A, Gosemann M, Pruss A, Reiter K, Ruzickova S, Lipsky PE, Dorner T. Abnormalities in peripheral B cell memory of patients with primary Sjogren’s syndrome. Arthritis Rheum. 2004;50:1897–1908. doi: 10.1002/art.20276. [DOI] [PubMed] [Google Scholar]
- 123.Yin H, Zhao M, Wu X, Gao F, Luo Y, Ma L, Liu S, Zhang G, Chen J, Li F, Zuo X, Lu Q: Hypomethylation and overexpression of CD70 (TNFSF7) in CD4+T cells of patients with primary Sjogren’s syndrome.J Dermatol Sci 2010, 59:198–203. [DOI] [PubMed]
- 124.Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361:496–509. doi: 10.1056/NEJMra0804595. [DOI] [PubMed] [Google Scholar]
- 125.Rahman M, Alam K, Ahmad MZ, Gupta G, Afzal M, Akhter S, Kazmi I, Jyoti, Ahmad FJ, Anwar F. Classical to current approach for treatment of psoriasis: a review. Endocr Metab Immune Disord Drug Targets. 2012;12:287–302. doi: 10.2174/187153012802002901. [DOI] [PubMed] [Google Scholar]
- 126.Abrams JR, Lebwohl MG, Guzzo CA, Jegasothy BV, Goldfarb MT, Goffe BS, Menter A, Lowe NJ, Krueger G, Brown MJ, Weiner RS, Birkhofer MJ, Warner GL, Berry KK, Linsley PS, Krueger JG, Ochs HD, Kelley SL, Kang S. CTLA4Ig-mediated blockade of T-cell costimulation in patients with psoriasis vulgaris. J Clin Invest. 1999;103:1243–1252. doi: 10.1172/JCI5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Abrams JR, Kelley SL, Hayes E, Kikuchi T, Brown MJ, Kang S, Lebwohl MG, Guzzo CA, Jegasothy BV, Linsley PS, Krueger JG. Blockade of T lymphocyte costimulation with cytotoxic T lymphocyte-associated antigen 4-immunoglobulin (CTLA4Ig) reverses the cellular pathology of psoriatic plaques, including the activation of keratinocytes, dendritic cells, and endothelial cells. J Exp Med. 2000;192:681–694. doi: 10.1084/jem.192.5.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mease P, Genovese MC, Gladstein G, Kivitz AJ, Ritchlin C, Tak PP, Wollenhaupt J, Bahary O, Becker JC, Kelly S, Sigal L, Teng J, Gladman D. Abatacept in the treatment of patients with psoriatic arthritis: results of a six-month, multicenter, randomized, double-blind, placebo-controlled, phase II trial. Arthritis Rheum. 2011;63:939–948. doi: 10.1002/art.30176. [DOI] [PubMed] [Google Scholar]
- 129.Altmeyer MD, Kerisit KG, Boh EE. Therapeutic hotline. Abatacept: our experience of use in two patients with refractory psoriasis and psoriatic arthritis. Dermatol Ther. 2011;24:287–290. doi: 10.1111/j.1529-8019.2011.01405.x. [DOI] [PubMed] [Google Scholar]
- 130.Ursini F, Naty S, Russo E, Grembiale RD. Abatacept in psoriatic arthritis: case report and short review. J Pharmacol Pharmacother. 2013;4:S29–S32. doi: 10.4103/0976-500X.120943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ohta Y, Hamada Y. In situ expression of CD40 and CD40 ligand in psoriasis. Dermatology. 2004;209:21–28. doi: 10.1159/000078582. [DOI] [PubMed] [Google Scholar]
- 132.Daoussis D, Antonopoulos I, Andonopoulos AP, Liossis SN. Increased expression of CD154 (CD40L) on stimulated T-cells from patients with psoriatic arthritis. Rheumatology (Oxford) 2007;46:227–231. doi: 10.1093/rheumatology/kel229. [DOI] [PubMed] [Google Scholar]
- 133.Fleischmajer R, Perlish JS, Reeves JR. Cellular infiltrates in scleroderma skin. Arthritis Rheum. 1977;20:975–984. doi: 10.1002/art.1780200410. [DOI] [PubMed] [Google Scholar]
- 134.Prescott RJ, Freemont AJ, Jones CJ, Hoyland J, Fielding P. Sequential dermal microvascular and perivascular changes in the development of scleroderma. J Pathol. 1992;166:255–263. doi: 10.1002/path.1711660307. [DOI] [PubMed] [Google Scholar]
- 135.Roumm AD, Whiteside TL, Medsger TA, Jr, Rodnan GP. Lymphocytes in the skin of patients with progressive systemic sclerosis. Quantification, subtyping, and clinical correlations. Arthritis Rheum. 1984;27:645–653. doi: 10.1002/art.1780270607. [DOI] [PubMed] [Google Scholar]
- 136.Hamaguchi Y. Autoantibody profiles in systemic sclerosis: predictive value for clinical evaluation and prognosis. J Dermatol. 2010;37:42–53. doi: 10.1111/j.1346-8138.2009.00762.x. [DOI] [PubMed] [Google Scholar]
- 137.Fineschi S, Cozzi F, Burger D, Dayer JM, Meroni PL, Chizzolini C. Anti-fibroblast antibodies detected by cell-based ELISA in systemic sclerosis enhance the collagenolytic activity and matrix metalloproteinase-1 production in dermal fibroblasts. Rheumatology (Oxford) 2007;46:1779–1785. doi: 10.1093/rheumatology/kem241. [DOI] [PubMed] [Google Scholar]
- 138.Fineschi S, Goffin L, Rezzonico R, Cozzi F, Dayer JM, Meroni PL, Chizzolini C. Antifibroblast antibodies in systemic sclerosis induce fibroblasts to produce profibrotic chemokines, with partial exploitation of toll-like receptor 4. Arthritis Rheum. 2008;58:3913–3923. doi: 10.1002/art.24049. [DOI] [PubMed] [Google Scholar]
- 139.Lian X, Xiao R, Hu X, Kanekura T, Jiang H, Li Y, Wang Y, Yang Y, Zhao M, Lu Q: DNA demethylation of CD40l in CD4+T cells from women with systemic sclerosis: a possible explanation for female susceptibility.Arthritis Rheum 2012, 64:2338–2345. [DOI] [PubMed]
- 140.Kawai M, Masuda A, Kuwana M. A CD40–CD154 interaction in tissue fibrosis. Arthritis Rheum. 2008;58:3562–3573. doi: 10.1002/art.23994. [DOI] [PubMed] [Google Scholar]
- 141.Jiang H, Xiao R, Lian X, Kanekura T, Luo Y, Yin Y, Zhang G, Yang Y, Wang Y, Zhao M, Lu Q: Demethylation of TNFSF7 contributes to CD70 overexpression in CD4+T cells from patients with systemic sclerosis.Clin Immunol 2012, 143:39–44. [DOI] [PubMed]
- 142.Gourh P, Arnett FC, Tan FK, Assassi S, Divecha D, Paz G, McNearney T, Draeger H, Reveille JD, Mayes MD, Agarwal SK. Association of TNFSF4 (OX40L) polymorphisms with susceptibility to systemic sclerosis. Ann Rheum Dis. 2010;69:550–555. doi: 10.1136/ard.2009.116434. [DOI] [PMC free article] [PubMed] [Google Scholar]